

Abstract
Expansions of short nucleotide repeats produce several neurological and neuromuscular disorders including Huntington’s disease, muscular dystrophy and amyotrophic lateral sclerosis. A common pathological feature of these diseases is the accumulation of the repeat containing transcripts into aberrant foci in the nucleus. RNA foci, as well as the disease symptoms, only manifest above a critical number of nucleotide repeats, but the molecular mechanism governing foci formation above this characteristic threshold remains unresolved. Here, we show that repeat expansions create templates for multivalent base-pairing, which causes purified RNA to undergo a sol-gel transition at a similar critical repeat number as observed in the diseases. In cells, RNA foci form by phase separation of the repeat-containing RNA and can be dissolved by agents that disrupt RNA gelation in vitro. Analogous to protein aggregation disorders, our results suggest that the sequence-specific gelation of RNAs could be a contributing factor to neurological disease.
Nucleotide repeat expansion disorders constitute some of the most common inherited diseases1,2. Several of the disease associated repeat expansions comprise of a nucleotide triplet of high G/C content, such as CAG in Huntington’s disease (HD) and spinocerebellar ataxias, and CTG in myotonic dystrophy2,3. Likewise, the expansion of the hexanucleotide GGGGCC in the C9orf72 gene is the most common mutation associated with familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)4,5. Disease-causing repeats can occur in the coding or non-coding regions of RNA transcripts2. Intriguingly, in all of the nucleotide repeat expansion disorders identified to date, disease only manifests beyond a critical number of repeats2,4–6.
Three non-exclusive hypotheses have been put forth to explain how repeat expansions give rise to neurotoxicity. First, expansions could result in a gain or loss of function of the associated gene1. However, mutations or deletion of the gene associated with the repeats do not always elicit the disease phenotype, while expression of the expanded repeats alone is sufficient to replicate the disease pathology in animal models7–9. Second, repeat-containing RNAs can be non-canonically translated10 to produce short peptides that can give rise to neurotoxicity11. Third, the repeat-containing RNA accumulate in the nuclei as aberrant foci4,8,12,13 that sequester various RNA binding proteins14–16, resulting in a potential disruption of cellular homeostasis17,18. The molecular mechanism by which a threshold number of nucleotide repeats results in formation of nuclear foci is not known. Here, we tested the hypothesis that sequence-specific physicochemical properties of repeat-containing RNAs may lead to their coalescence into nuclear foci.
Repeat-containing RNAs form gels in vitro
RNAs associated with many repeat expansion diseases contain a regular pattern of Gs and Cs and can form intramolecular hairpins3. Because of their repeating nature, these sequences also can, in principle, template multivalent intermolecular interactions (Fig. 1a). Such multivalent interactions could potentially lead to the formation of large clusters via liquid-liquid phase separation19,20 or a sol-gel transition21–23. To examine whether repeat-containing RNAs assemble into large clusters, we synthesized fluorescently-labeled RNAs containing 47 triplet repeats of CAG (47xCAG) or CUG (47xCUG). As controls, we used RNAs of equivalent length (~250 bases), but with arbitrary sequences with 30–75% GC content, and RNAs with scrambled sequences but with identical base composition as 47xCAG and 47xCUG. Upon annealing (see Methods), the 47xCAG and 47xCUG RNAs formed micron-sized spherical clusters, while the control RNAs remained soluble (Fig. 1b, Extended Data Fig. 1a, b). Clusters were observable at RNA concentration as low as 25 nM (Extended Data Fig. 1c). These clusters were >100-fold enriched in the RNA as compared to the solution phase and contained nearly half of the RNA in the reaction (Extended Data Fig. 1d). The clusters were dissolved by RNaseA, but not proteinase K or DNase I (Extended Data Fig. 1e, f), confirming that clustering was not mediated by protein or DNA contaminants. RNA clustering required Mg2+ and was inhibited by monovalent cations such as Na+ (Extended Data Fig. 1g, h), suggesting that, besides multivalent base-pairing, electrostatic interactions also play a prominent role in RNA clustering, as has been described for homopolymeric RNAs24.
Figure 1. Triplet repeat containing RNAs undergo gelation in vitro at a critical number of repeats.

(a) Schematic for CAG RNA multimerization. (b) Fluorescence micrographs for indicated RNA and quantitation of inhomogeneity as normalized variance (σ2/μ). (c) Effects of CAG- or CUG- repeat number on RNA gelation. (d) Effects of control ASOs (ASO-2, and non-hybridizing oligo, ASO-1) or 6xCTG on 47xCAG RNA clustering. (e) FRAP trajectories for RNA clusters. Scale bars, 5 μm. Error bars, median and interquartile range. Data are representative of ≥ 3 independent experiments.
Molecules that form multivalent interactions show abrupt phase transitions with increasing valency of interaction19–21. Consistent with this valency dependence, we find that the formation of CAG/CUG RNA clusters occurred only with >30 triplet repeats (Fig. 1c). The intermolecular interactions between the transcripts potentially could be competed by shorter complementary antisense oligonucleotides (ASO). Indeed, a 6xCTG ASO prevented clustering of 47xCAG RNA, while control oligonucleotides did not (Fig. 1d). Collectively, these experiments indicate that intermolecular base-pairing interactions in the CAG/CUG-repeat region can lead to the assembly of RNAs into micron-sized clusters.
We next analyzed the physical properties of the CAG/CUG RNA clusters. Their spherical shapes (aspect ratio 1.05 ± 0.1, mean ± s.d., n = 214) are characteristic of polymers undergoing liquid-liquid phase separation25. Molecules within the liquid phase are mobile and undergo fast internal rearrangement. However, fluorescence recovery after photobleaching (FRAP) experiments revealed little or no fluorescence recovery over ~10 min, indicating that RNA in the clusters is immobile (Fig. 1e, Extended Data Fig. 1i, Supplementary Video 1). Because of their solid-like behavior, we refer to these clusters as “RNA gels”22,23. We hypothesize that these RNAs initially phase separate into spherical liquid-like droplets but then rapidly become cross-linked into gels due to increasing intermolecular base-pairing. Consistent with this idea, we occasionally (~1% of clusters) observe incomplete fusion events where two droplets solidified prior to relaxation to a single spherical geometry (Extended Data Fig. 1j). As further evidence for this model, we found that single-stranded DNA, in the presence of polyvalent cations, formed liquid-like droplets and that incorporation of multivalent base-pairing sites progressively imparted solid-like properties to the DNA droplets (Extended Data Fig. 2, Supplementary Video 2). In summary, multivalent base-pairing interactions lead to the gelation of CAG/CUG repeat-containing RNAs in vitro at a similar critical number of repeats as observed in diseases.
CAG repeat RNAs phase separate in cells
We next sought to establish a live-cell reporter assay in U-2OS cells to visualize repeat-containing RNAs and determine whether they form aberrant nuclear foci, as described in patient cells13. For this purpose, we tagged the RNA with 12xMS2-hairpin loops26 and co-expressed YFP-tagged MS2-coat binding protein (Fig. 2a). Multiple stop codons were incorporated upstream of the repeats to minimize translation of repeat-containing proteins27 (Fig. 2a). Upon induction of 47xCAG or 120xCAG RNA transcription, numerous nuclear foci appeared as early as 1 h post induction (Fig. 2b, Extended Data Fig. 3a–c, Supplementary Video 3). The number of foci per nucleus increased with higher levels of RNA induction (Extended Data Fig. 3d). In contrast, 5xCAG (Fig. 2b, c, Extended Data Fig. 4) or control RNAs with coding or non-coding sequences did not form nuclear puncta (Extended Data Fig. 3e–f) when expressed at similar levels (~10,000 copies per cell; Extended Data Fig. 3g, h). The formation of foci did not induce discernable cell death or impede cell growth over a 7-day period post induction (Extended Data Fig. 3i). Since repeat expansion disorders take years to manifest in patients2, short term toxicity in cells is not necessarily expected.
Figure 2. CAG repeat-containing RNAs coalesce into liquid-like nuclear foci.
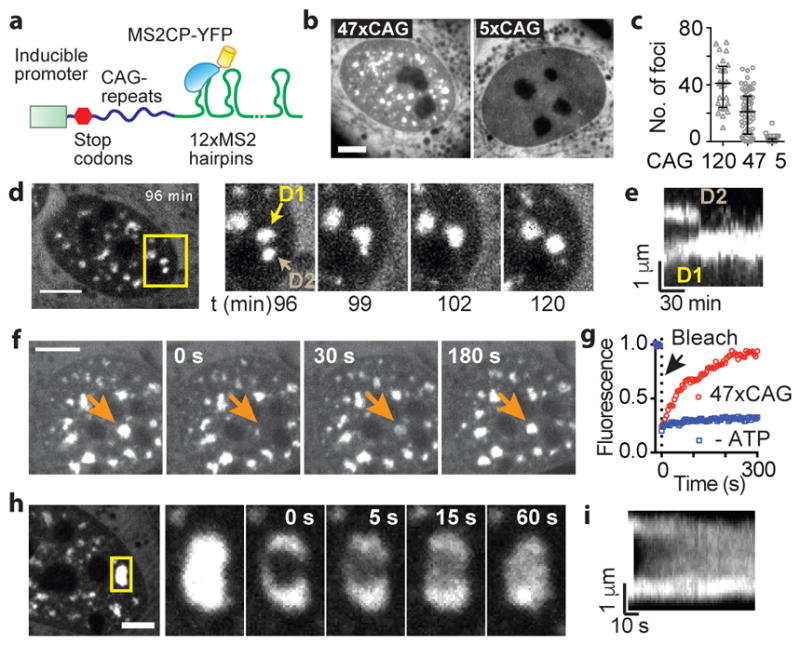
(a) Schematic for RNA visualization. (b) Micrographs of cells expressing 5xCAG or 47xCAG RNA. (c) Quantification of foci as a function of CAG repeat number. Each datum point represents one cell. Error bars, median and interquartile range. (d) Typical fusion event between RNA foci (time post induction indicated). (e) Kymograph corresponding to (d). (f) Images for 47xCAG RNA foci before and after photobleaching (arrow: bleach site). (g). FRAP trajectories for 47xCAG RNA punctum before (47xCAG) and after ATP depletion (−ATP). (h) Similar to (g) after partial photobleaching. (i) Kymograph corresponding to (h). Scale bars, 5 μm. Data are representative of ≥ 3 independent experiments.
The CAG RNA nuclear foci exhibited liquid-like properties. For example, two or more foci could fuse with one another (Fig. 2d, 2e, Supplementary Videos 3, 4), a hallmark of liquid-like behavior28. Upon photobleaching, nuclear foci also exhibited near-complete fluorescence recovery (83 ± 13% recovery, τFRAP = 81 ± 24 s; mean ± s.d., n = 5 foci), indicating that the RNA can move into and out of the foci (Fig. 2f, g, Supplementary Video 5). Upon photobleaching a portion of a 47xCAG RNA punctum, the fluorescence recovered rapidly (τFRAP = 18 ± 5 s, mean ± s.d., n = 5 foci) (Fig. 2h, i, Supplementary Video 6), suggesting that RNA within the foci can undergo internal rearrangement. Thus, unlike their solid-like behavior in vitro (Fig. 1e), CAG RNA foci in cells display liquid-like properties28. We hypothesized that the increased dynamicity might arise from specialized proteins (e.g. helicases) in the nucleoplasm that remodel RNA base-pairing. Consistent with this hypothesis, depletion of cellular ATP significantly reduced fluorescence recovery of the RNA foci after photobleaching (23 ± 7% recovery, mean ± s.d., n = 7, Fig. 2g, Supplementary Video 7).
Similar to the endogenous foci in patient derived fibroblasts29, the induced RNA foci co-localized with the SC-35 marker for nuclear speckles (Fig. 3a, Extended Data Fig. 5), non-membranous bodies that are enriched in pre-mRNA splicing factors30. The foci also recruited endogenous Muscleblind-like-1 (MBNL1) protein (Fig. 3b), sequestration of which has been implicated in CAG/CUG repeat-containing RNA mediated pathogenicity14,31. Using fluorescence in situ hybridization (FISH)32 we found that ~50% of the 47xCAG RNA was retained in the nucleus, compared with <10% of control RNAs (Fig. 3c, d, Extended Data Fig. 3f–h). Thus, CAG repeats cause the RNA to be retained in nucleus within liquid-like bodies that sequester splicing factors.
Figure 3. 47xCAG RNA is retained within the nucleus and co-localizes with nuclear speckles.
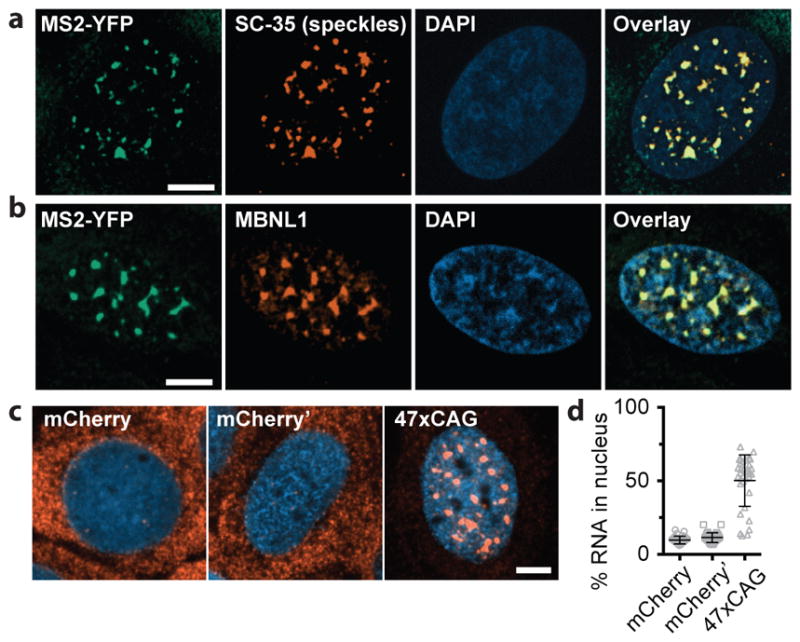
(a) Immunofluorescence images depicting co-localization of 47xCAG RNA foci (MS2-YFP) with nuclear speckles (SC-35). Nuclei are counterstained with DAPI. (b) Similar to (a), staining for MBNL1. (c, d) FISH images (c) and relative RNA abundance per nucleus (d), in cells expressing MS2-tagged 47xCAG, a control coding sequence (mCherry) or its reverse complement (mCherry′). Scale bars, 5 μm. Error bars, mean ± s.d. Data are representative of ≥ 3 independent experiments.
Inhibitors of RNA gelation disrupt foci
Next, we asked if perturbations that prevent RNA gelation in vitro also affect the stability of RNA foci in cells. In vitro, RNA gelation is inhibited by monovalent cations (Extended Data Fig. 1g, h, Fig. 4a). To test the effect of monovalent cations in cells, we used ammonium acetate, which readily permeates into cells and does not perturb intracellular pH33. Strikingly, addition of 100 mM ammonium acetate led to the disappearance of 47xCAG RNA foci within minutes (Fig. 4b, Extended Data Fig. 6a, Supplementary Video 8). Interestingly, nuclear speckles also were disrupted by ammonium acetate, suggesting that the assembly of these ribonucleoprotein bodies depends upon ionic interactions as well (Extended Data Fig. 7). Consistent with previous studies29, we find that the disruption of nuclear speckles with tautomycin also disrupts the 47xCAG RNA foci (Extended Data Fig. 7).
Figure 4. Nuclear foci are reversed by agents that disrupt RNA gelation in vitro.

(a, d) NH4OAc (a) and doxorubicin (Dox) (d) prevent 47xCAG RNA clustering. (b) Images of the same cell before and 300 s after treatment with NH4OAc, and corresponding quantification. (c, e, f) Representative images and quantification of RNA foci upon transfection with indicated ASOs (c), and upon Dox treatment in U-2OS cells expressing exogenous 47xCAG RNA (e) and DM1 fibroblasts (f). Scale bars, 5 μm. Error bars, median and interquartile range. Data are representative of ≥ 3 independent experiments.
Next, we tested agents that might specifically disrupt the base pairing in RNA foci without dissolving nuclear speckles. Transfection of an 8xCTG ASO reduced the number and size of 47xCAG foci as compared against control oligonucleotides (Fig. 4c, Extended Data Fig. 6b). ASOs may disrupt cellular RNA foci either by inhibiting intermolecular base-pairing or by degrading RNA via the RNase H machinery34. To specifically perturb base pairing interactions, we used doxorubicin, a nucleic acid intercalator35. Doxorubicin blocked the formation of CAG RNA gels in vitro and potently dissolved the 47xCAG nuclear foci in cells (Fig. 4d, e, Extended Data Fig. 6c, d) without disrupting nuclear speckles (Extended Data Fig. 7). The concentration of doxorubicin required to disrupt RNA foci in cells (2.5 μM) was lower than that needed for the in vitro experiments (~1 mM), potentially because of the aid of cellular proteins that unwind RNA base pairing and facilitate doxorubicin intercalation. Next, we tested if doxorubicin can alleviate RNA foci derived from an endogenous locus. To this end, we used fibroblasts derived from DM1 patients with a CTG expansion in the DMPK gene, which exhibit RNA foci containing multiple DMPK transcripts (Extended Data Fig. 8 a–c)12. Treatment with doxorubicin (2 μM for 24 h) significantly reduced the number as well as total volume of foci per cell (~65% and ~85% decrease respectively, Fig. 4f, Extended Data Fig. 8d). In summary, agents that inhibit gelation of purified repeat-containing RNAs in vitro also disrupt RNA foci in cells.
ALS/FTD-linked GGGGCC repeats form gels
Besides the canonical Watson-Crick (WC) base pairing, nucleic acids also can form Hoogsteen base-pairs such as in G-quadruplexes (GQ). Interestingly, the GGGGCC repeat found in the C9orf72 locus associated with ALS/FTD forms GQ in vitro and in vivo15,36. A single GQ can bring up to four RNA strands together, but a GGGGCC repeat expansion could potentially give rise to multimolecular RNA complexes36 (Fig. 5a). Indeed, we find that 3xGGGGCC RNA is largely soluble but 5xGGGGCC RNA formed spherical clusters in vitro (Fig. 5b). Longer repeats (10x and 23x) formed an interconnected mesh-like network of aggregated RNA (Fig. 5b). In contrast, 23xCCCCGG RNA, which can form multivalent WC base-pairing but not GQ, was soluble (Fig. 5c). Similar to the CAG/CUG RNA, GGGGCC RNA clusters exhibited solid-like properties, and clustering was inhibited by monovalent cations (Extended Data Fig. 9a, b).
Figure 5. RNA with ALS/FTD-associated GGGGCC repeats form gels in vitro and in cells.

(a) Schematic for GGGGCC RNA multimerization. (b) RNA clusters at indicated number of GGGGCC repeats. (c) 23xCCCCGG is soluble under similar conditions. (d) Images of cells expressing 29xGGGGCC or 29xCCCCGG RNA. (e) Quantification of RNA foci at indicated number of GGGGCC repeats. (f) GGGGCC RNA punctum at indicated time points after photobleaching. (g) Recovery plot corresponding to (f). (h, i) Doxorubicin (h), and NH4OAc (i) disrupt 29xGGGGCC RNA foci. Images in (i) of the same cell before and 300 s after treatment. Scale bars, 5 μm. Error bars, median and interquartile range. Data are representative of ≥ 3 independent experiments. (j) Model for RNA foci formation in repeat expansion disorders.
Cellular expression of 29xGGGGCC, but not 29xCCCCGG, RNA resulted in the formation of nuclear puncta in a dose-dependent manner (Fig. 5d, Extended Data Fig. 9c, d). The number of foci per cell increased with the number of repeats (Fig. 5e, Extended Data Fig. 9e). The threshold number of repeats for disease onset (>23xGGGGCC4) is similar to the repeat length in which most cells exhibit foci (16–29xGGGGCC). This GGGGCC repeat number is higher than that required for RNA gelation in vitro, possibly due to cellular proteins that may unfold GQ37. Interestingly, the 29xGGGGCC RNA foci, as well as those of shorter length, exhibited incomplete FRAP recovery (37 ± 20% recovery, mean ± s.d., n = 10, Fig. 5f, g, Extended Data Fig. 9f,g, Supplementary Video 9), indicating that they are less dynamic than CAG RNA foci. This result suggests a stronger intermolecular interaction between the GGGGCC repeats, which is consistent with intracellular GQ formation (Fig. 5a).
The GGGGCC RNA foci recruited hnRNP H, as previously shown15, as well as MBNL1, and co-localized with nuclear speckles (Extended Data Fig. 10). Like the 47xCAG RNA, the majority of GGGGCC RNA was retained in the nucleus (~60%; Extended Data Fig. 9h). The c9orf72 GGGGCC expansion is located in an intron with ~150 bases upstream and ~7 kb region downstream of the repeats. We found that the incorporation of the endogenous (~150 bases) upstream or a long (~1.7 kb) downstream flanking sequence did not affect the formation of RNA foci (Extended Data Fig. 9i). Intriguingly, incorporation of a longer sequence (~l kb) upstream of the repeats abolished the formation of nuclear puncta (Extended Data Fig. 9i), suggesting that sequences flanking the repeats may influence their assembly into RNA foci. Similar to the CAG RNA foci, the GGGGCC RNA foci were disrupted by ASO transfection, doxorubicin, or ammonium acetate (Fig. 5h, i, Extended Data Fig. 9 j, k, l), indicating that both base-pairing and electrostatic interactions are essential for GGGGCC RNA foci formation.
Conclusion
In summary, we have shown that the propensity of an RNA to form multivalent base-pairing can lead to its gelation without requiring protein components. An emerging body of research has shown that RNP granules, such as stress granules, P-granules and nucleoli, are phase separated liquid-like compartments38. Numerous recent studies have characterized proteins with regard to their ability to phase separate and mediate the assembly of these RNP granules38. Our results demonstrate that sequence-specific base-pairing properties of RNAs can lead to their phase separation and gelation, and raise the possibility that such phenomena could contribute to physiological granule assembly as well.
In the case of repeat expansions diseases, our data suggest that intermolecular base-pairing can result in the aggregation and sequestration of RNA into nuclear foci (Fig. 5j). RNA gelation, which occurs at a boundary condition of increasing valency, might explain why disease appears to be triggered after an expansion of a nucleotide repeat has reached a threshold number (Fig. 5j), a puzzling phenomenon that has lacked a mechanistic explanation thus far2,6. Our results also may explain why placement of distinct repeat expansions in seemingly unrelated genes can result in similar clinical syndromes39,40. RNA gelation and foci formation likely occur upstream of known drivers of cellular toxicity such as sequestration of RNA binding proteins14, nucleolar stress41 and disruption of nucleocytoplasmic transport42,43, although a direct link between foci formation and cellular toxicity remains to be established. Our findings raise the possibility that new strategies for disrupting RNA-RNA base pairing could be effective for treating repeat expansion diseases.
Methods
Cloning
CAG and GGGGCC repeats were cloned via sequential repeat directed elongation as previously described44 in a modified pBluescript vector. Inserts were verified via sequencing from both ends (up to 700 bp read length for CAG/CTG repeats and up to 200 bp read length for GGGGCC repeats) and by verifying the insert size by restriction digestion. All cloning and amplification were performed in E. coli Stbl3 cells (Invitrogen) grown at 30°C. For synthesizing the mammalian expression constructs, repeats were cut directly from the cloning plasmids and ligated at the compatible restriction sites in a modified lentiviral expression vector with tetracycline inducible expression promoter. Plasmids sequences are provided in Supplementary Table 1. We observed that the purified plasmids formed higher order complexes when stored for prolonged periods (> 1 month) at 4°C or −20°C. Stored plasmids when re-transformed in bacteria, often resulted in significant truncations in the repeat region. To avoid such re-transformation associated repeat truncations, we maintained a bacterial stock of each plasmid. The plasmid DNA was freshly purified for each cloning/transfection or RNA transcription experiment and the sequence was verified as described above.
RNA transcription and gelation
Repeat containing RNA were transcribed using T7 or T3 MegaScript kit (Ambion) using manufacturer’s recommendation. Template DNA up to 200 bases long (10xCAG, 20xCAG, 10xCUG, 20xCUG, 3xGGGGCC, 5xGGGGCC and 23xGGGGCC) were purchased from Integrated DNA Technologies, Inc (IDT) as single stranded DNA oligonucleotides. Complementary strand was synthesized using as single complementary primer and a standard polymerase chain reaction kit (Advantage® GC 2 PCR Kit, Clontech Inc.). The double stranded DNA thus generated, were gel purified and used as templates for transcription reactions. Longer templates were obtained by either PCR amplification from plasmids (31xCAG, 47xCAG, 31xCUG, 47xCUG), or by restriction digestion of the repeat containing vectors (66xCAG, 66xCUG) upstream and downstream of the repeat region. Template sequences for transcription reactions are provided in Supplementary Table 2. For synthesis of fluorescently labeled RNA, transcription reactions were doped with Cy3-UTP or Cy5-UTP (Enzo Lifesciences). Free NTPs were removed using lithium chloride precipitation or using RNA purification kit (Zymo Research). Similar results were obtained in both purification schemes. Size of the RNA products was verified using denaturing agarose gel electrophoresis. RNAs were resuspended in water, and either used immediately or aliquoted and flash-frozen in liquid nitrogen and stored at −80°C for up to 1 month.
For phase separation/gelation assays, RNAs were diluted to 0.5 ng/μl – 0.5 μg/μl concentration in 10 mM Tris pH 7.0, 10 mM MgCl2, 25 mM NaCl buffer, unless indicated otherwise. Nuclease free buffer stocks were purchased from Ambion. RNA were denatured at 95°C for 3 min and cooled down at 1–4°C per min to 37°C final temperature in a thermocycler and imaged immediately. Samples were visualized using a custom spinning disk confocal microscope (Nikon Ti-Eclipse equipped with Yokogawa CSU-X spinning disk module) using an x100 1.49 NA oil immersion objective and an air-cooled EM-CCD. Extent of phase separation/gelation was quantified by the index of dispersion (σ2/μ) of fluorescence intensity per pixel (pixel size 83 nm by 83 nm). Briefly, variance in the fluorescence intensity per image was determined, and normalized to the mean fluorescence intensity in the solution phase of the RNA. For dilute solution (< 10% of imaging area occupied with clusters), this parameter reports on the extent of inhomogeneity in the sample. At least 20 independent imaging areas (~1800 μm2 each) were analyzed for each condition to achieve a representative measure across the sample. Each datum point in the bar graphs represents one imaging area. Data shown are representative of 3 or more independent replicates, across 2 or more independent RNA preparations.
For antisense DNA mediated repression of RNA phase separation, 47xCAG RNA (200 ng/μl or 2.4 μM) was incubated with the antisense oligonucleotides at 20 μM final concentration, followed by heat denaturation and annealing as described above. Doxorubicin was purchased from Cell Signaling Technology, Inc (catalog number 5927). For in vitro experiments, doxorubicin was added to pre-formed RNA clusters, and samples were incubated at 37°C for 1 h. Alternatively, doxorubicin and RNA were pre-mixed at indicated concentrations prior to annealing. Similar results were obtained in both cases.
For FRAP experiments, RNA clusters were prepared as described above. RNA clusters were allowed to settle on to the glass surface for ~15 min. A ~1 μm2 region was photobleached using a 405 nm laser modulated by a Rapp UGA-40 photo targeting unit and the fluorescence recovery was monitored over time. The fluorescence recovery was fit to the equation, I = A−I0*exp(−t/τFRAP), and the time constant, τFRAP was determined.
DNA phase separation
DNA oligonucleotides were purchased from IDT. Spermine hydrochloride (Sigma) was resuspended in water and pH was adjusted to 7.5. DNA were heat denatured at 90°C for 2 min to melt secondary structure, incubated on ice for 2 min, and used immediately for phase separation assays. Phase separation was trigged by adding spermine to the DNA solution. All phase separation assays were performed in 10 mM Tris pH 7.0 buffer with the indicated amounts of DNA and salts. DNA clusters were visualized using standard bright field or confocal microscopy as described above. To prevent DNA droplets from fusing on to the glass surface, coverslips were passivated with polyethylene glycol as previously described45. FRAP experiments and analysis were performed as described above.
Cell culture and imaging
U-2OS cells, authenticated by STR profiling, were purchased from the UCSF Cell Culture Facility. We generated a monoclonal U-2OS cell line stably expressing Tet-On 3G transactivator protein (Clontech) and a tandem-dimeric MS2 hairpin binding protein tagged with eYFP (MS2CP-YFP) via sequential lentiviral infection and selection. This stable cell line was transduced with repeat containing plasmids under doxycycline-inducible promoter. Cells were maintained in DMEM with 10% (v/v) tetracycline-free fetal bovine serum (Clontech) and 1X penicillin-streptomycin-glutamine cocktail (Gibco). Cell lines were tested for mycoplasma contamination using a standard PCR kit (LookOut Mycoplasma PCR Detection Kit, Sigma) and verified routinely by live cell DNA staining.
RNA expression was induced by adding 1000 ng/ml doxycycline for 12–48 h, or as indicated. Prior to imaging the culture medium was replaced with DMEM with 25 mM HEPES pH 7.5 or FluoroBrite™ DMEM (Invitrogen) with serum and antibiotics as listed above. For long-term imaging (>2 h) cells were placed in a live cell imaging chamber supplemented with 5% CO2. Cells were imaged using a spinning disk confocal microscope (Nikon Ti-Eclipse equipped with Yokogawa CSU-X spinning disk module) using an x100 1.49 NA oil immersion objective and an air-cooled EM-CCD. For each experimental condition, at least 30 randomly chosen cells were imaged and analyzed. Each datum point in the bar graphs represents one cell. Data shown are representative of 3 or more independent replicates.
ATP depletion was achieved by rinsing cells 2x in DMEM without glucose (Gibco), followed by incubation for 10 min in the ATP depletion medium (DMEM without glucose with 1% (v/v) dialyzed FBS (Gibco), 10 mM sodium azide and 6 mM 2-deoxy-D-glucose). Doxorubicin (stock, 10 mM in DMSO) was diluted to the desired concentration in cell culture medium and added to cells pre-induced with doxycycline for 24 h. Cells were incubated with doxorubicin or an equivalent dilution of DMSO only as control, for 2 h, and imaged as described above. Ammonium acetate (stock, 5 N) was diluted to 200 mM in cell culture medium. This intermediate dilution (2x) was added to cells pre-induced with doxycycline for 24 h to achieve a final concentration of 100 mM of ammonium acetate. Cells were incubated in this medium for 10 min at 37°C. Normal cell culture medium was replaced after treatment, and cell were imaged immediately or 1 h after medium replacement. For treatment with antisense oligonucleotides (ASO), cells pre-induced with doxycycline for 48 h were transfected with 100 nM final concentration of ASO using either Lipofectamine RNAiMAX (Invitrogen) or TransIT-Oligo Transfection Reagent (Mirius Bio) using manufacturer recommended protocols. Similar results were obtained with both transfection reagents. Cells were imaged 12 h post transfection. ASO sequences are provided in Supplementary Table 2.
Analysis of RNA foci
We used a fluorescence intensity and size based threshold to identify RNA foci. Briefly, U-2OS cells expressing the RNA of interest together with MS2CP-YFP were imaged using a spinning disk confocal microscope, and 0.3 μm Z-stacks were acquired. To account for variability in MS2CP-YFP expression levels, we used a cell-intrinsic intensity threshold for foci identification. We manually segmented the nuclei, and determined the mean YFP fluorescence intensity in the nucleus. RNA foci were identified using FIJI 3D Objects Counter plugin, with an intensity threshold as 1.6x the mean fluorescence intensity in the nucleus of the cell, and a size cut-off of >50 adjoining pixels (pixel size, 83 nm x 83 nm). This algorithm faithfully identified the foci as depicted in Extended Data Fig. 4. This method was used to determine the number, volume, surface area and the fluorescence intensity of the foci. Various metrics such as total number of foci per cell, total volume of foci per cell, coefficient of dispersion (variance/mean) and integrated intensity of foci were compared and yielded similar results. Number of foci per cell, and the total volume occupied by the foci per cell were chosen as the parameters of choice to quantify the extent of foci formation. Statistical significance was analyzed using unpaired, two-tailed Mann-Whitney tests. For this analysis, the number of foci per cell in each experiment were assumed to be symmetrically distributed about the median.
Quantification of RNA copy number
To quantify the copy number of RNA in cells, we used two alternative approaches. First, we used NanoString, a proprietary PCR-free RNA quantitation platform, and determined that under our highest induction conditions, the copy number of 47xCAG RNA is about 10-times that of GAPDH or beta-actin RNA, or about 8800 ± 1500 copies per cell (n = 3 independent experiments). Second, we used single molecule FISH to obtain quantitative RNA localization information. We designed fluorescent probes against the MS2 hairpin loop region, such that the 12xMS2 tag can accommodate a maximum of 32 fluorescently labeled probes (probe sequences in Supplementary Table 2). For cells expressing low levels of MS2-tagged control RNAs such as mCherry or 5xCAG RNA, we observed isolated fluorescent spots that exhibit a uniform distribution of intensities, likely arising from single RNA molecules. Similarly, in the cytoplasm of cells expressing 47xCAG or 29xGGGGCC RNA, we observed isolated RNA spots with a similar uniform distribution of fluorescence intensities (Extended Data Fig. 3f, g). We ascribed this intensity value as corresponding to that of a single RNA. Fluorescence spots and corresponding intensities were quantified using ImageJ Spot Counter plugin. The approximate RNA copy number in each cell was then calculated by dividing the total fluorescence intensity of the cell by the fluorescence intensity of a single RNA, after background subtraction. By this method, we have determined that under maximal induction conditions that lead to RNA foci formation, the copy number of 47xCAG is 13,000 ± 7000 copies per cell (mean ± s.d., n = 24 cells), for 29xGGGGCC RNA is 2500 ± 1800 copies per cell (n = 30 cells) and for the control cells expressing mCherry is 20,000 ± 7000 copies per cell (n = 21 cells). The fraction of RNA retained in the nucleus was determined by dividing the fluorescence intensity in the nucleus of the cell by the total fluorescence of the cell.
FRAP experiments and data analysis
To assess the dynamicity of RNA foci, we performed FRAP experiments by bleaching MS2CP-YFP protein. Previous studies have shown that the dimeric MS2CP-YFP is attached with high affinity to the MS2 hairpin sequence and does not dissociate during the observation timescales of a few minutes46 and the fluorescence recovery of MS2CP-YFP can be used to report on the RNA dynamics. To monitor exchange of RNA between foci and the nucleoplasm, an entire punctum, typically a few microns in size, was photobleached and the fluorescence recovery was monitored by time-lapse imaging. To examine internal turn-over, relatively large puncta were manually selected and a region ~1 μm in diameter was photobleached. The fluorescence intensity of the bleached region was normalized and corrected for photobleaching using previously described methods47. To determine fluorescence relaxation time, the recovery curves were fit to the equation I = A−I0*exp(−t/τFRAP) where A and I0 are also fit parameters.
RNA FISH and immunofluorescence
For RNA FISH in U-2OS cells, cells expressing the desired RNA (induced for 24 hours) were fixed with 2% paraformaldehyde for 10 min at room temperature and permeabilized by overnight incubation in 70% ethanol at 4°C. Alternatively, cells were fixed and permeabilized by incubation for 10 min in methanol with 10% (v/v) acetic acid. Similar results were obtained with both fixation protocols. Fixed and permeabilized cells were either used immediately, or stored in the permeabilization medium at −20°C until needed. RNA was detected using Cy3 labeled DNA oligonucleotides designed against the MS2-hairpin sequence. Probe sequences are provided in Supplementary Table 2. Hybridization and wash buffers were purchased from Biosearch Technologies and used per the manufacturer’s protocol. For immunofluorescence detection of proteins, methanol fixed cells were stained using antibodies against muscleblind-like-1 (MBNL1, Abcam, ab45899), hnRNP H (Abcam, ab10374), SC-35 (Abcam, ab11826), Coilin (ab87913), Fibrillarin (Abcam, ab5821) and PML (Abcam, ab179466) and a corresponding Alexa Fluor 647 labeled secondary antibody (Invitrogen, A-21236 or Invitrogen A-21244). Samples were co-stained with an anti-GFP booster antibody (GBA488, Bulldog Bio) to visualize RNA foci. After labeling, samples were mounted in Prolong Gold antifade medium (Thermo Scientific, Inc) and imaged using confocal microscopy as described above.
DM1 fibroblasts
DM1 fibroblasts were obtained from the Coriell Institute (catalog number GM03132 and GM03987). Control fibroblasts (Hs27) were obtained from the UCSF Cell Culture Facility. These cell lines were used without further validation. Cells were maintained in DMEM with 10% (v/v) fetal bovine serum (Clontech) and 1X penicillin-streptomycin-glutamine cocktail (Gibco). For detection of RNA foci, RNA FISH was performed as described above using an 8xCAG oligonucleotide labeled with Atto647N (sequence in Supplementary Table 2) or using a pool of 48 oligonucleotide probes designed against the wild-type DMPK allele obtained as a pooled library from Biosearch Technologies Inc. For disruption of RNA foci, cells were incubated for 24 h with 2 μM doxorubicin or an equivalent dose of DMSO only control. Total volume and the number of RNA foci was quantified using the ImageJ 3D Objects Counter plugin, using an empirically determined fluorescence threshold.
Data availability
The raw data that support the findings of this study are available from the corresponding author upon reasonable request.
Extended Data
Extended Data Figure 1. Disease-associated repeat-containing RNAs form clusters in vitro.

(a) Fluorescence micrographs for RNA with various GC-content compared against RNA with disease-associated repeat expansions. Sequences of the DNA templates used for transcription are provided in Supplementary Table 2. (b) Fluorescence micrographs comparing 47xCUG RNA and a corresponding control RNA (Scr1) with identical base composition but with a scrambled sequence. Similarly, 47xCAG RNA was compared with two different RNAs (Scr2, Scr3) that have same base composition as 47xCAG but the sequences are scrambled. Extent of inhomogeneity is quantified by the index of dispersion (σ2/μ) across >20 independent imaging areas (1800 μm2 each). Each datum point represents an independent imaging area. (c) Representative micrographs of 47xCAG RNA clusters at indicated concentrations. Spherical RNA clusters are observable up to 25 nM RNA concentration. Below this concentration regime, the reaction is reactant limited and the cluster size is below the diffraction limit. RNA clustering at all concentrations was not observed in the presence of 100 mM ammonium acetate. Representative images at indicated RNA concentration in the presence of 100 mM NH4OAc (+ NH4OAc). (d) RNA enrichment in the clusters. (Left) Cy3-labeled 66xCAG RNA was serially diluted in conditions that prevent RNA clustering (10 mM Tris pH 7.0, 10 mM MgCl2, 100 mM NaCl), and the bulk solution fluorescence was calibrated against the RNA concentration. The enrichment of RNA in the clusters was determined by comparing their fluorescence intensity against this calibration. When the input RNA concentration is 100 ng/μl, the concentration in the clusters corresponds to ~16.3 μg/μl, or an enrichment of 163-fold. (Right) RNA clusters were precipitated by centrifugation at 16,000xg for 10 min at room temperature. The concentration of the soluble RNA after centrifugation was determined by measuring absorbance at 260 nm. The concentration of the RNA in the solution phase decreases with the increasing CAG-repeat number. (e, f) 47xCAG RNA clusters were treated with proteinase K (60 U/ml), DNaseI (200 U/ml) or RNaseA (0.7 U/ml) for 10 min at room temperature. Representative micrographs (e) and quantification (f). (g) 47xCAG RNA clustering is inhibited by NaCl. (h) Binary phase diagram for 1.25 μM 47xCAG RNA as a function of MgCl2 and NaCl concentrations. Blue dots represent two-phase regime while the red dots indicate a homogenous single-phase regime. (i) The RNA clusters are in a solid-like state and do not exhibit fluorescence recovery upon photobleaching, as indicated by the representative micrographs for 47xCUG (top) and 47xCAG (bottom) RNA at the indicated time-points. (j) Sample images showing aborted fusion events between 47xCAG RNA clusters suggesting that the clusters were liquid-like initially and later undergo a liquid-to-solid transition. Fusion events are likely aborted as the clusters solidified prior to relaxation to a spherical geometry. Scale bars in (a, b, c, e, g) are 5 μm and in (i, j) are 1 μm. Error bars depict median and interquartile range. Data are representative of ≥ 3 independent experiments across ≥ 2 independent RNA preparations.
Extended Data Figure 2. Base-pairing interactions impart solid-like properties to DNA-spermine complexes.

Electrostatic interactions between polymeric anions (such as nucleic acids) and multivalent cations can lead to phase separation via formation of polyelectrolyte complexes, a phenomenon known as complex coacervation. We find that spermine, a tetravalent cation at pH 7, can induce phase separation of single-stranded DNA oligonucleotides. (a) Mixing 10 mM spermine pH 7 (left tube) with 10 μM T-90 DNA (90-mer polyT DNA oligonucleotide) (right tube) immediately results in a turbid solution (center tube). (b) Examination by brighfield microscopy revealed numerous spherical droplets. Using a fluorescently-labeled T-90, we confirmed that the droplet phase is enriched in DNA. Representative bright field (left), fluorescence (center) and overlay (right) images for the DNA-spermine complexes. (c–f) We investigated the effect of base-pairing interactions on DNA-spermine complexes. The T-90 DNA-spermine complexes are liquid-like, as evidenced by their spherical geometry (c) and a rapid fluorescence recovery after photobleaching (f, 99 ± 1% recovery, τFRAP-T90 = 5 ± 2 s, mean ± s.d., n = 3 droplets). Next, we designed a 90 bases long DNA with five 8-bp long palindromic hybridization sites separated by poly-dT spacers (sequence S1, sequences in Supplementary Table 2). S1 DNA also phase separated and formed spherical liquid-like droplets in the presence of spermine (d). However, the S1-spermine droplets exhibited reduced fluidity, as evidenced by a slower recovery upon photobleaching (f, 90 ± 4% recovery, τFRAP-S1 = 335 ± 41 s, mean ± s.d., n = 5). (e) We performed similar spermine-mediated coacervation experiments with a (dAdT)45 oligonucleotide (AT-45) that may form multivalent A:T base-pairing interactions. AT-45 DNA formed interconnected network-like structures spanning hundreds of microns or gels (e). These AT-45 DNA gels are in a solid-like state as evidenced by lack of fluorescence recovery after photobleaching (f, 14 ± 5% recovery, mean ± s.d., n = 4 clusters). Scale bars represent 5 μm.
Extended Data Figure 3. Disease-associated, repeat-containing RNAs coalesce into nuclear foci in cells.

(a–b) Expression of 47xCAG (a) and 120xCAG (b) RNA leads to the formation of nuclear puncta. Representative images (left) and quantification of the percentage of cells showing RNA foci (right) with and without induction. n: number of cells analyzed. (c) Time-lapse images of 120xCAG RNA accumulation in the nuclei of U-2OS cells. Cells were induced with 1 μg/ml of doxycycline at t = 0. See also, Supplementary Video 3. (d) Number of foci per cell increases with increasing 47xCAG RNA expression levels. The expression levels were controlled by increasing the virus titer. (e) 47xCAG RNA accumulates in the nuclei as puncta, while control RNA with coding (mCherry) or a non-coding sequences (mCherry′, reverse complement of mCherry sequence) do not form nuclear inclusions, as shown in the representative MS2-YFP fluorescence micrographs (left) and quantification of the number of foci per cell (right). (f) U-2OS cells were transduced with the indicated constructs tagged with 12xMS2 hairpins under a tetracycline inducible promoter. RNA was visualized by FISH using Cy3 labeled oligonucleotide probes against MS2-hairpins. Representative micrographs showing the localization of mCherry (top), 47xCAG (middle) and 29xGGGGCC (bottom) RNA with (+ Tet) or without (−Tet) doxycycline induction. The probes do not bind in the absence of induction. Nuclei are counterstained with DAPI (depicted in blue). (g) Intensity distribution for single RNA spots in cells expressing 5xCAG (top) and in the cytoplasm of cells expressing 29xGGGGCC (bottom). (h) RNA copy number was determined by dividing the total Cy3 fluorescence intensity in a cell by that of a single RNA, as determined in (g). 47xCAG RNA copy number corresponds to highest viral titer used in (d). Similar results were obtained using NanoString (8,800 ± 1,500 copies per cell for 47xCAG RNA, mean ± s.d., n = 3 independent experiments). (i) Induction of 47xCAG RNA foci does not cause overt toxicity or a reduction in cell division rates over 7 days. Normalized cell counts in 47xCAG transduced cells with or without doxycycline induction. Cell counts were normalized to control cells (without 47xCAG transduction), grown under corresponding induction conditions. Each datum point in (d, e, h) represents one cell and error bars depict median and interquartile range. Data points in (i) represent technical replicates, and error bars depict mean and s.d. Scale bars represent 5 μm.
Extended Data Figure 4. Identification of RNA foci in live cells.

(a–c). We used a fluorescence intensity and size based threshold to identify RNA foci. Briefly, U-2OS cells expressing the RNA of interest together with MS2CP-YFP were imaged using a spinning disk confocal microscope, and 0.3 μm Z-stacks were acquired (a). To account for variability in MS2CP-YFP expression levels, we used a cell-intrinsic intensity threshold for foci identification. We manually segmented the nuclei (b), and determined the mean fluorescence intensity in the nucleus. RNA foci were identified using FIJI 3D Objects Counter plugin, with an intensity threshold as 1.6x the mean fluorescence intensity in the nucleus of the cell, and a size cut-off of >50 adjoining pixels (pixel size, 83 nm x 83 nm). This algorithm faithfully identified the foci as depicted in (c). (d–h) We compared the extent of foci formation in 47xCAG and 5xCAG expressing cells. (d) The mean nuclear fluorescence intensity is similar between the 47xCAG and 5xCAG expressing cells. The cells are compared via various metrics: (e) number of foci per cell, (f) total volume of foci per cell, (g) integrated fluorescence intensity of the foci per cell and, (h) normalized variance in the fluorescence intensity in the nucleus per cell. Scale bar represents 5 μm. Error bars depict median and interquartile range.
Extended Data Figure 5. CAG RNA foci co-localize with nuclear speckles.

Representative immunofluorescence micrographs depicting that the 47xCAG RNA foci co-localize with the marker for nuclear speckles (SC-35) but not with other nuclear bodies such as PML bodies (PML), paraspeckles (nmt55), nucleoli (Fib1) or Cajal bodies (coilin). RNA foci are stained using an antibody against GFP. Nuclei are stained with DAPI. Data are representative of 3 or more independent experiments. Scale bars are 5 μm.
Extended Data Figure 6. RNA foci are disrupted by treatments that prevent RNA gelation in vitro.

(a) RNA FISH using a probe directed against MS2 hairpin loops confirms that 47xCAG RNA foci are disrupted by treatment with 100 mM NH4OAc, thus precluding the possibility that the observed disruption of RNA foci in live cells is due to dissociation of MS2CP-YFP from the MS2 hairpins. Representative images and corresponding quantification. (b) Transfection of an 8xCTG oligonucleotide disrupted 47xCAG RNA foci while control oligonucleotides (3xC4G2 or Control) did not. Representative images and quantification of the number of RNA foci per cell. Sequences of the oligonucleotides are provided in Supplementary Table 2. (c) Doxorubicin disrupts 47xCAG RNA clustering in vitro in a dose-dependent manner. Representative micrographs and the quantification of the inhomogeneity in the solution at indicated RNA and doxorubicin concentrations. (d) RNA FISH using a probe directed against MS2 hairpin loops confirms that 47xCAG RNA foci are disrupted by treatment with 2.5 μM doxorubicin, suggesting that the observed disruption of RNA foci in live cells is likely not an artefact of MS2CP-YFP dissociation from MS2 hairpins. Scale bars represent 5 μm. Error bars depict median and interquartile range. Data are representative of 3 or more independent experiments.
Extended Data Figure 7. Doxorubicin disrupts RNA foci but not nuclear speckles.

(a) Representative immunofluorescence micrographs of U-2OS cells expressing 47xCAG stained with antibodies against GFP (MS2-YFP) and SC-35, as a marker for nuclear speckles. Treatment with 2 μM tautomycin for 4 h (b), or 100 mM NH4OAc for 10 min (c) disrupts both the 47xCAG RNA foci as well as the nuclear speckles. (d) Treatment with 2.5 μM doxorubicin for 2 h specifically abrogated the 47xCAG RNA foci but the nuclear speckles are not disrupted. Nuclei are counterstained with DAPI (blue). Scale bars represent 5 μm. (e) Quantification of the total volume occupied by nuclear speckles (left) and the integrated intensity of the SC-35 immunofluorescence per cell under various treatments (right). Error bars depict median and interquartile range. Data are representative of 3 or more independent experiments.
Extended Data Figure 8. Doxorubicin disrupts RNA foci in DM1 fibroblasts.

(a) Fibroblasts derived from DM1 patients (DM1a, DM1b) or control fibroblasts (Hs27) were stained using a FISH probe directed against expanded CUG-repeats (8xCAG labeled with Atto647N). Representative fluorescence images showing RNA foci in DM1 cell lines but not in control. Nuclei are counterstained with DAPI (blue). (b) Quantification of the number of RNA foci per cell across various cell lines. (c) Single molecule FISH using probes designed against the wild-type DMPK allele shows isolated diffraction limited spots in control fibroblasts (Hs27), indicated by white arrows, likely arising due to single mRNA. In the patient derived fibroblasts (DM1a, DM1b), isolated spots (white arrows) as well as several bright puncta (yellow arrows) are observed. Since both the wild-type as well as the mutant transcript with expanded CUG repeats can each accommodate the same number of fluorescent probes (48 probes per transcript), the higher brightness indicates that each punctum in DM1 patient cells (yellow arrows) contains multiple DMPK mRNAs. (d) Treatment with 2 μM doxorubicin for 24 h reduced the average number of RNA foci per cell by 66% and the total volume of foci per cell by 87%. Representative images with or without doxorubicin treatment and corresponding quantification. Data are aggregated from 2 independent experiments, and are representative of 4 or more independent experiments. Scale bars represent 5 μm. Error bars depict median and interquartile range.
Extended Data Figure 9. GGGGCC repeat containing RNAs form clusters in vitro and foci in cells.

(a) Binary phase diagram for 23xGGGGCC RNA clustering in vitro as a function of NaCl and MgCl2 concentrations. RNA concentration was 1.5 μM. Blue dots represent two-phase regime while the red dots indicate a homogenous single phase regime. (b) Representative fluorescence micrographs for 3xGGGGCC RNA clusters before and after photobleaching at indicated time points. The lack of fluorescence recovery indicates that the RNA in the clusters is immobile or in a solid-like state. Scale bar is 1 μm. (c) Quantification of the number of RNA foci per cell for U-2OS cells expressing 29xGGGGCC or 29xCCCCGG RNA. (d) Number of 29xGGGGCC RNA foci increases with the increasing level of RNA expression. The expression levels were controlled by increasing the virus titer. (e) Representative fluorescence micrographs and corresponding quantification of the total volume of foci per cell in U-2OS cells transduced with 12xMS2 tagged RNA with the indicated number of GGGGCC repeats. (f) GGGGCC RNA foci exhibit incomplete recovery upon fluorescence photobleaching. Representative fluorescence micrographs for 29xGGGGCC RNA foci at indicated time points. (g) Fluorescence recovery plots for GGGGCC RNA foci with indicated number of repeats. Data are average of n = 10 foci at each repeat number. (h) Percentage of 29xGGGGCC RNA retained in the nucleus compared against a control RNA encoding for mCherry. (i) Effect of flanking sequences on the formation of GGGGCC RNA foci. Construct G1 has 29xGGGGCC repeats with 12xMS2 hairpins (~0.7 kb) downstream of the repeats for RNA visualization. Incorporation of a ~1 kb long sequence (G2, sequence in Supplementary Table 1) between the 29xGGGGCC repeats and 12xMS2 repeats did not inhibit foci formation. Similarly, RNA foci were observed in construct G3 which has the same 5′ flanking sequence as found in the endogenous locus in intron 1 of c9orf72. However, incorporation of a longer ~1 kb 5′ flanking sequence (G4) inhibited the formation of RNA foci. (j) Transfection of U-2OS cells with a 3xCCCCGG ASO disrupted the 29xGGGGCC RNA foci while a control ASO did not. Representative micrographs and the quantification of the number of RNA foci per cell. (k) Representative fluorescence micrographs and corresponding quantification of inhomogeneity for 23xGGGGCC RNA in vitro with or without 1 mM doxorubicin (l) Same as (k) with or without 100 mM NH4OAc. Scale bars in (e, f, j, k, l) are 5 μm. Error bars in (c, d, e, i, j, k, l) represent median and interquartile range, and in (g, h) represent mean and s.d. Data are representative of 3 or more independent experiments.
Extended Data Figure 10. GGGGCC RNA foci co-localize with nuclear speckles.

Representative immunofluorescence images illustrating that the 29xGGGGCC RNA foci co-localize with the marker for nuclear speckles (SC-35) but not to Cajal bodies (coilin). The GGGGCC RNA foci also recruited endogenous hnRNP H and MBNL1. Scale bars are 5 μm. Data are representative of 3 or more independent experiments.
Supplementary Material
Acknowledgments
We thank M. K. Rosen, W. W. Seeley and the members of Vale lab for helpful discussions. A.J. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-2181-14).
Footnotes
Author contributions:
A.J. and R.V. designed research and wrote the paper. A.J. performed experiments and analyzed data.
The authors declare that they have no competing financial interests.
References
- 1.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 2.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krzyzosiak WJ, et al. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res. 2012;40:11–26. doi: 10.1093/nar/gkr729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeJesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renton AE, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee DY, McMurray CT. Trinucleotide expansion in disease: why is there a length threshold? Curr Opin Genet Dev. 2014;26:131–140. doi: 10.1016/j.gde.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chew J, et al. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348:1151–1154. doi: 10.1126/science.aaa9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mankodi A, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 9.Jin P, et al. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39:739–747. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- 10.Zu T, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cleary JD, Ranum LPW. Repeat-associated non-ATG (RAN) translation in neurological disease. Hum Mol Genet. 2013;22:R45–51. doi: 10.1093/hmg/ddt371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wojciechowska M, Krzyzosiak WJ. Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet. 2011;20:3811–3821. doi: 10.1093/hmg/ddr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller JW, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conlon EG, et al. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife. 2016;5:345. doi: 10.7554/eLife.17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timchenko NA, et al. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem. 2001;276:7820–7826. doi: 10.1074/jbc.M005960200. [DOI] [PubMed] [Google Scholar]
- 17.Todd PK, Paulson HL. RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol. 2010;67:291–300. doi: 10.1002/ana.21948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belzil VV, Gendron TF, Petrucelli L. RNA-mediated toxicity in neurodegenerative disease. Mol Cell Neurosci. 2013;56:406–419. doi: 10.1016/j.mcn.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flory PJ. Principles of Polymer Chemistry. Cornell University Press; 1953. [Google Scholar]
- 20.Li P, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–340. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stockmayer WH. Theory of Molecular Size Distribution and Gel Formation in Branched Polymers II. General Cross Linking. The Journal of Chemical Physics. 1944;12:125–131. [Google Scholar]
- 22.Semenov AN, Rubinstein M. Thermoreversible gelation in solutions of associative polymers. 1. Statics. Macromolecules. 1998;31:1373–1385. [Google Scholar]
- 23.Falkenberg CV, Blinov ML, Loew LM. Pleomorphic ensembles: formation of large clusters composed of weakly interacting multivalent molecules. Biophys J. 2013;105:2451–2460. doi: 10.1016/j.bpj.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aumiller WM, Keating CD. Phosphorylation-mediated RNA/peptide complex coacervation as a model for intracellular liquid organelles. Nat Chem. 2016;8:129–137. doi: 10.1038/nchem.2414. [DOI] [PubMed] [Google Scholar]
- 25.Larson RG. The Structure and Rheology of Complex Fluids. Oxford University Press; 1999. [Google Scholar]
- 26.Bertrand E, et al. Localization of ASH1 mRNA particles in living yeast. Molecular Cell. 1998;2:437–445. doi: 10.1016/s1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- 27.Kearse MG, et al. CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Molecular Cell. 2016;62:314–322. doi: 10.1016/j.molcel.2016.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brangwynne CP, et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- 29.Urbanek MO, Jazurek M, Switonski PM, Figura G, Krzyzosiak WJ. Nuclear speckles are detention centers for transcripts containing expanded CAG repeats. Biochim Biophys Acta. 2016;1862:1513–1520. doi: 10.1016/j.bbadis.2016.05.015. [DOI] [PubMed] [Google Scholar]
- 30.Spector DL, Lamond AI. Nuclear speckles. Cold Spring Harb Perspect Biol. 2011;3:a000646–a000646. doi: 10.1101/cshperspect.a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li LB, Yu Z, Teng X, Bonini NM. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453:1107–1111. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamaguchi MS, Watanabe K, Hamaguchi Y. Regulation of intracellular pH in sea urchin eggs by medium containing both weak acid and base. Cell Struct Funct. 1997;22:387–398. doi: 10.1247/csf.22.387. [DOI] [PubMed] [Google Scholar]
- 34.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 35.Frederick CA, et al. Structural comparison of anticancer drug-DNA complexes: adriamycin and daunomycin. Biochemistry. 1990;29:2538–2549. [PubMed] [Google Scholar]
- 36.Reddy K, Zamiri B, Stanley SYR, Macgregor RB, Pearson CE. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem. 2013;288:9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo JU, Bartel DP. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science. 2016;353:aaf5371–aaf5371. doi: 10.1126/science.aaf5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hyman AA, Weber CA, Jülicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. 2014;30:39–58. doi: 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- 39.Holmes SE, et al. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet. 2001;29:377–378. doi: 10.1038/ng760. [DOI] [PubMed] [Google Scholar]
- 40.Elden AC, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsoi H, Lau TCK, Tsang SY, Lau KF, Chan HYE. CAG expansion induces nucleolar stress in polyglutamine diseases. Proc Natl Acad Sci USA. 2012;109:13428–13433. doi: 10.1073/pnas.1204089109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freibaum BD, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang K, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scior A, Preissler S, Koch M, Deuerling E. Directed PCR-free engineering of highly repetitive DNA sequences. BMC Biotechnol. 2011;11:87. doi: 10.1186/1472-6750-11-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jain A, et al. Probing cellular protein complexes using single-molecule pull-down. Nature. 2011;473:484–488. doi: 10.1038/nature10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shav-Tal Y, et al. Dynamics of single mRNPs in nuclei of living cells. Science. 2004;304:1797–1800. doi: 10.1126/science.1099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phair RD, Gorski SA, Misteli T. Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Meth Enzymol. 2004;375:393–414. doi: 10.1016/s0076-6879(03)75025-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data that support the findings of this study are available from the corresponding author upon reasonable request.