

Abstract
Rate-limiting dissociation of the tetrameric protein transthyretin (TTR), followed by monomer misfolding and misassembly, appears to cause degenerative diseases in humans known as the transthyretin amyloidoses, based on human genetic, biochemical and pharmacologic evidence. Small molecules that bind to the generally unoccupied thyroxine binding pockets in the native TTR tetramer kinetically stabilize the tetramer, slowing subunit dissociation proportional to the extent that the molecules stabilize the native state over the dissociative transition state—thereby inhibiting amyloidogenesis. Herein, we use previously reported structure-activity relationship data to develop two semi-quantitative algorithms for identifying the structures of potent and selective transthyretin kinetic stabilizers/amyloidogenesis inhibitors. The viability of these prediction algorithms, in particular the more robust in silico docking model, is perhaps best validated by the clinical success of tafamidis, the first-in-class drug approved in Europe, Japan, South America, and elsewhere for treating transthyretin aggregation-associated familial amyloid polyneuropathy. Tafamidis is also being evaluated in a fully-enrolled placebo-controlled clinical trial for its efficacy against TTR cardiomyopathy. These prediction algorithms will be useful for identifying second generation TTR kinetic stabilizers, should these be needed to ameliorate the central nervous system or ophthalmologic pathology caused by TTR aggregation in organs not accessed by oral tafamidis administration.
TOC Graphic
Human genetic, biochemical and pharmacologic evidence implicates rate-limiting transthyretin (TTR) tetramer dissociation, followed by rapid monomer misfolding and misassembly, as the cause of several degenerative diseases exhibiting overlapping phenotypes, collectively referred to as the transthyretin amyloidoses.1–16 The amyloidogenic TTR monomer misassembles into a variety of aggregate structures during amyloidogenesis, including cross-β-sheet amyloid fibrils, for which these diseases are named.17–19 Amyloidogenesis of wild-type (WT) TTR or aggregation of certain mutants along with WT-TTR in heterozygotes leads to cardiomyopathies, affecting up to 500,000 individuals (disorders historically called senile systemic amyloidosis (SSA) and familial amyloid cardiomyopathy (FAC), respectively).14, 20 Amyloidogenesis of distinct TTR mutants along with WT-TTR in heterozygotes results in a primary autonomic and peripheral neuropathy, often called familial amyloid polyneuropathy (FAP). The latter disease has historically been treated by liver transplant-mediated gene therapy, wherein the mutant-TTR/WT-TTR liver (which secretes destabilized TTR heterotetramers) is replaced by a WT-TTR/WT-TTR liver (which secretes a more stable WT-TTR homotetramer). Interestingly, slowing the course of peripheral disease progression by liver transplantation has led to the appearance of TTR aggregation in the central nervous system (CNS) and eyes, which manifests as a consequence of treatment-associated lifespan extension.21–26
Another strategy to prevent TTR amyloidogenesis is to fashion small molecules that bind selectively in human blood to one or both of the thyroxine (T4) binding sites comprising the tetramer made up of WT or mutant and WT subunits. Selective binding to the native tetrameric ground state of TTR over the dissociative transition state raises the kinetic barrier for subunit dissociation, substantially slowing TTR aggregation. The extent of kinetic stabilization of tetrameric TTR determines the extent to which amyloidogenesis is inhibited.27–31 A placebo-controlled clinical trial in V30M FAP patients (a prominent mutation causing tetramer destabilization), along with a 12-month extension study, demonstrates the efficacy of this strategy in slowing the progression of autonomic and peripheral neuropathy.32, 33
Our studies carried out over the last two decades to develop small molecule TTR amyloidogenesis inhibitors have revealed that optimal TTR kinetic stabilizers are typically composed of two aryl rings joined by linkers of variable chemical composition.28, 29, 34–55 Figure S1 and Table S1 in the Supporting Information contain compilations of the structures and experimental results for the majority of the inhibitors procured or synthesized by the Kelly laboratory during this period. Binding of these small molecules to one or both of the generally unoccupied, funnel-shaped, T4 binding pockets strengthens the weaker dimer-dimer interface of TTR by non-covalently bridging adjacent monomeric subunits through specific hydrophobic and electrostatic interactions, as exemplified in the TTR•(201)2 crystal structure (Figure 1). To gauge the efficacy of candidate molecules to bind to the T4 pockets and kinetically stabilize the TTR tetramer from dissociating and aggregating in complex biological environments, we rely on two primary assays: 1) an in vitro acid-mediated TTR aggregation assay carried out with recombinant TTR in buffer; and 2) an ex vivo TTR immunoprecipitation/HPLC assay to quantify the stoichiometry of a candidate kinetic stabilizer bound to TTR in blood plasma. These two assays are briefly explained below, with complete experimental details presented in the Supporting Information).56, 57
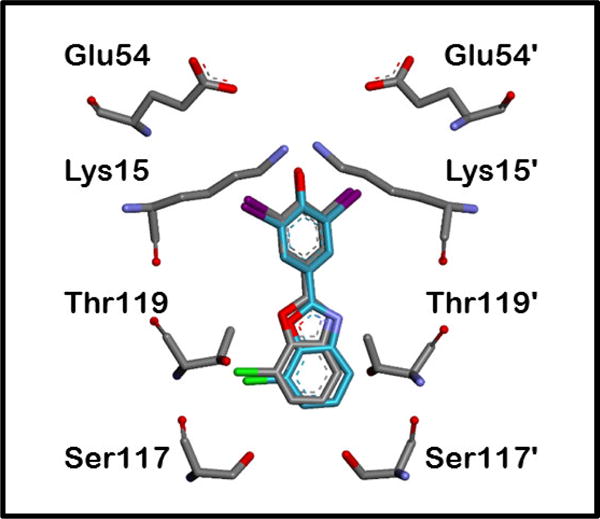
Figure 1.
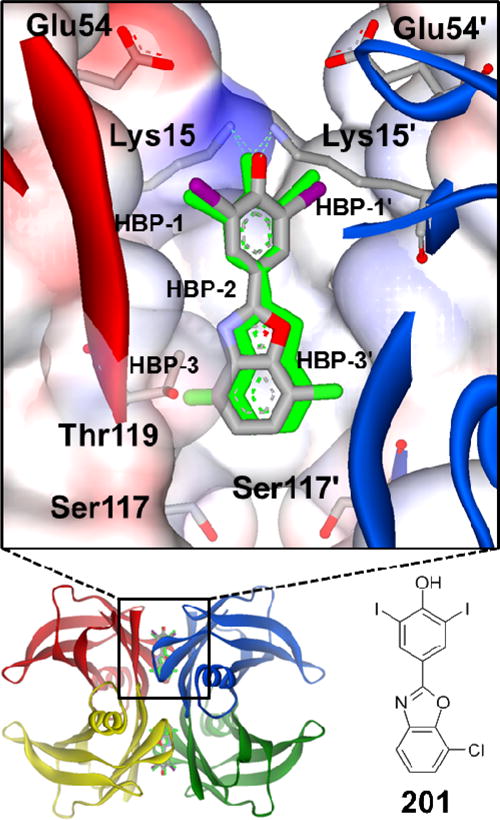
X-ray structure of the TTR•(201)2 complex (PDB ID 5TZL) highlights the interactions known to be important for tight binding to TTR. Compound 201 is bound in its equivalent symmetry-related binding modes (grey and green, respectively), which results from ligand binding along the crystallographic 2-fold axis. The omit FO-FC density (contoured at +/− 3.5σ) for 201 is shown in Figure S3 of the Supporting Information. The binding pocket is characterized by a smaller inner cavity and a larger outer cavity, throughout which are distributed three pairs of symmetric hydrophobic depressions, referred to as the halogen binding pockets (HBPs). The iodine and chlorine atoms of 201 reside within HBPs 1 and 3. Primed amino acids or HBPs refer to symmetry-related monomers of TTR comprising each T4 binding pocket. The phenolate of 201 makes charged interactions with the Lys 15 and 15′ residues in the outer cavity; however, it is known that other kinetic stabilizers composed of phenols exhibiting a higher pKa preferentially bind in the opposite orientation so that the phenols can hydrogen bond with the Ser-117 and 117′ residues in the inner binding cavity (details of this phenomenon have been previously reported).46–50
The acid-mediated TTR aggregation assay (pH 4.4; 100 mM acetate buffer) probes the ability of a candidate small molecule kinetic stabilizer to bind tetrameric TTR in vitro and prevent amyloidogenesis under denaturing conditions that destabilize the tetramer, and largely convert the dissociated TTR monomers to aggregates within 3 days in the absence of a kinetic stabilizer. The best inhibitors prevent >90% of TTR aggregation over a 72 h time course when incubated at a concentration twice that of tetrameric TTR (i.e., 7.2 μM candidate inhibitor versus 3.6 μM tetrameric TTR). We have traditionally presented results as % fibril formation (% FF), with complete arrest of TTR aggregation (i.e., 100% inhibition) corresponding to 0% FF.
Since plasma TTR is the form that undergoes amyloidogenesis and causes degenerative phenotypes in the periphery, a kinetic stabilizer candidate must bind selectively to TTR over all the other proteins in blood plasma to be efficacious. The ex vivo TTR immunoprecipitation/HPLC quantification assay probes the ability of candidate kinetic stabilizers to bind selectively to TTR over the 4000+ other proteins that are present in human blood plasma. Selectively binding to TTR over albumin, which is the primary T4 transport protein in the blood, can be particularly challenging: TTR represents <0.5% by mass of the total plasma proteome, whereas albumin comprises ∼57%.58 Potent TTR kinetic stabilizers can display average plasma TTR binding stoichiometry values, also referred to as the Plasma Binding Selectivity (PS) values, between zero (i.e., the candidate small molecule binds predominantly to plasma proteins other than TTR) and the theoretical maximum of two (i.e., the candidate kinetic stabilizer binds very selectively to TTR in blood plasma and exhibits a slow dissociation rate), owing to the presence of the two T4 binding sites per TTR tetramer. A PS value of 0 renders a compound useless as a TTR amyloidogenesis inhibitor in vivo, whereas a PS value of 1 or higher is acceptable as our previous studies show that only one inhibitor bound per TTR tetramer is necessary to kinetically stabilize the WT-TTR tetramer against amyloidogenesis.29 In addition to mitigating off-target toxicity during the envisioned life-long use of a kinetic stabilizer to ameliorate a TTR amyloid disease, achieving a high PS value may also allow administration of lower amounts of drug while still maintaining efficacy.
Our early studies looked at the potential of repurposing approved non-steroidal anti-inflammatory drugs (NSAIDs) to serve as TTR kinetic stabilizers, and thus inhibit the process of TTR amyloidogenesis.38–41 During these investigations, we discovered that diflunisal (compound 389) is a viable TTR kinetic stabilizer and inhibitor of TTR aggregation in vitro. Diflunisal is an effective TTR kinetic stabilizer in humans because it can achieve extremely high plasma concentrations, which makes up for its modest binding constant and mediocre binding selectivity. Since diflunisal is already an approved drug in many countries around the world, this discovery was instrumental in setting up an international academic clinical trial to test the efficacy of TTR kinetic stabilizers as a strategy to treat FAP. While the clinical trial of tafamidis to ameliorate V30M FAP (see below) was completed first, the diflunisal FAP trial (all mutations) ultimately showed that treatment with diflunisal reduces the rate of progression of neurological impairment and preserves the quality of life in polyneuropathy patients.59 However, the long term therapeutic use of diflunisal for the treatment of the transthyretin amyloidoses is limited by the fact that, as an NSAID, it can trigger gastrointestinal bleeds and impair renal flow,59–64 which is already diminished in cardiomyopathy patients. Hence, diflunisal is contraindicated for TTR cardiomyopathy patients. Thus, we focused on developing lead TTR kinetic stabilizers/amyloidogenesis inhibitors that did not inhibit cyclooxygenase activity.
Early on in our TTR kinetic stabilizer development program, it became clear that some of the most potent TTR kinetic stabilizers under evaluation also exhibited binding to the thyroid hormone receptors, which would likely produce undesirable metabolism and heart rate side-effects in the envisioned long-term use of TTR amyloidogenesis inhibitors. Thus, we also sought TTR kinetic stabilizers that were neither thyroid agonists nor antagonists.
Through extensive medicinal chemistry optimization efforts and counter-screens to avoid NSAID or thyroid hormone receptor activity, one TTR kinetic stabilizer emerged as a lead drug candidate, tafamidis (compound 250), which has since been approved by regulatory agencies in >30 countries for treating FAP, causatively linked to over 100 TTR mutations. The 18-month-long clinical trial, the 12-month extension study, and observations of more than 1000 patients, some for longer than 8 years, indicate a very favorable safety profile and clearly demonstrate the therapeutic efficacy of tafamidis at delaying neurologic impairment and preserving nutritional status in polyneuropathy patients, while enhancing quality of life and likely lifespan.32, 33, 64, 65 Unfortunately, a drawback to current therapies is that a subset of liver transplant patients with FAP, and some tafamidis-treated patients, present with continued ocular and CNS deposition of TTR aggregates (tafamidis does not enter the eye or the brain upon oral administration). It seems that the majority of FAP patients treated by liver transplant-mediated gene therapy develop CNS amyloid pathology after more than a decade of treatment. Thus, it may be necessary to develop a second generation, tafamidis-like molecule that enters the eye and the brain while still maintaining TTR kinetic stabilization in the periphery.
To increase the success of developing second generation transthyretin kinetic stabilizers/amyloidosis inhibitors, in the present study, we have developed two semi-quantitative models to predict the structures of potent TTR kinetic stabilizers/amyloidogenesis inhibitors that bind with high selectivity to TTR in the blood, and that exhibit minimal off-target effects, such as binding to the thyroid hormone receptor or the cyclooxygenase enzymes. The development of these two prediction models will allow us to more effectively focus future research efforts on optimizing the CNS and ocular permeability of TTR kinetic stabilizers, since we can be confident that, even before any synthesis commences, proposed lead candidates will have a high probability of selectively targeting transthyretin.
Previously, we synthesized and evaluated three libraries of TTR kinetic stabilizer candidates in an effort to optimize the three substructures of a typical TTR amyloidogenesis inhibitor — the two aromatic rings and the linker joining them (Figure S1, compounds 1–134).46–48 The structure-activity relationship (SAR) data from these three mini-libraries (detailed in the previous studies and summarized in Figure 2)46–48 were envisioned to contain the information enabling one to predict the potency and selectivity of a larger combinatorial library of “theoretical” TTR amyloidogenesis inhibitors. For the envisioned predictive algorithm, Equation 1 was developed, incorporating an equal weighting of inhibitor potency (based on % FF) and plasma TTR binding selectivity (PS), to semi-quantitatively rank compounds by what we call an Experimental Efficacy Score (EES).
| Equation 1 |
EESs range from 0 (least potent and selective inhibitors) to a maximum of 1, which corresponds to a compound displaying maximal amyloidogenesis inhibition (0% FF) and a maximum binding selectivity value of 2 (i.e., both T4 binding sites are occupied by a kinetic stabilizer in blood plasma). The EES values for the Aryl-X, Aryl-Z, and Linker-Y substructures can then be used to calculate what we refer to as a Predicted Efficacy Score (PES) for a theoretical candidate molecule (Equation 2).
| Equation 2 |
In this algorithm, the Aryl-X substructure EESs have been further subdivided into Xs and Xp EESs (values are presented in Figure 2), to differentiate contributions from substituents and the substitution patterns, respectively. The Aryl-Z substructure EESs have been similarly subdivided into Zs and Zp EESs (values are presented in Figure 2). A detailed description of how to calculate PES values for a given molecule is presented in the Supporting Information (page 16). Briefly, to calculate a molecule’s PES, the EES values for the Xs, Xp, Y, Zs, and Zp substructures that characterize the molecule (as defined in Figure 2) are simply added together (Equation 2). With EESs for the Xs, Xp, Y, Zs, and Zp parameters (9, 6, 10, 10, and 8 substructures, respectively) derived from the three small libraries consisting of 134 molecules,46–48 we are able to calculate PESs for 43,200 theoretical TTR kinetic stabilizer small molecules, although some structural overlap occurs owing to molecular symmetry, which reduces the total number of distinct chemical entities. In theory, PES values can range from 0 (least potent and selective inhibitors) to a maximum of 5 (most potent and selective inhibitors); however, the current practical range spans between ∼0.5–3.0 as EESs never reach the limits of 0 or 1 for any of the individual molecular substructures.
Figure 2.
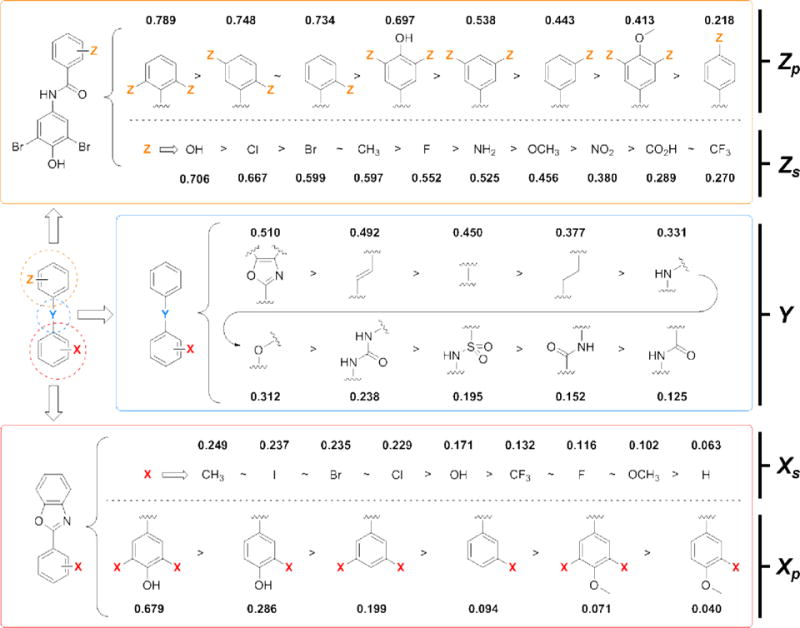
Summary of the structure-activity relationships (data derived) from a trio of previous libraries designed to screen the three substructures of a typical small molecule TTR amyloidosis inhibitor (i.e., Aryl-X, Linker-Y, and Aryl-Z).46–48 Substructures are quantitatively ranked according to the average experimental efficacy scores (EES) as determined using Equation 1, which evaluates the ability of a substructure to afford potent aggregation inhibitors in vitro that bind selectively to TTR in human blood plasma ex vivo.
Next, we scrutinized whether the PES algorithm could enhance our ability to chemically synthesize a high proportion of potent and selective TTR amyloidogenesis inhibitors. As our test case, we employed a library of stilbene and dihydrostilbene compounds reported previously49 with calculated PESs in the 1.9–2.9 range (i.e., at the higher end of the practical PES range). In this test library, 93% of the stilbenes (52/56, compounds 135–174) and 100% of the dihydrostilbenes (34/34, compounds 175–199) displayed EESs greater than 0.667, an important cutoff that represents a scenario of 0% FF and a binding stoichiometry of ≥1 molecule per TTR tetramer. A complete tabulation of % FF, PS, EES, and PES values for the stilbene and dihydrostilbene compounds can be found in Table S1 in the Supporting Information.
When EESs are plotted vs. PESs for those compounds containing substructures encompassed within this prediction model (i.e., substructures presented in Figure 2) — from the four aforementioned small molecule libraries (1–199)46–49 along with 19 new compounds reported here (200–218) and previously evaluated compounds (243–314)28, 29, 35–41, 43–45, 51–55 — it becomes evident that enrichment of compounds with EESs above the 0.667 cutoff occurs for compounds with PESs >1.8 (Figure 3; shaded area). Solid black data points (see rightmost Y-axis in Figure 3) represent the percentage of compounds exceeding the EES = 0.667 cutoff (PES data plotted in increments of 0.2). Thus, focusing synthetic efforts on compounds with PESs ≥1.8 should enhance the development of a higher proportion of potent and selective TTR amyloidogenesis inhibitors. While this PES approach appears to have value in terms of directing synthetic efforts towards developing potent TTR kinetic stabilizers that display high binding selectivity in blood plasma, and presumably in cerebrospinal fluid (to be determined), experimental evidence suggests that these types of predictions may not directly translate into developing the most promising clinical candidates, as exemplified by the situation for compound 201.
Figure 3.
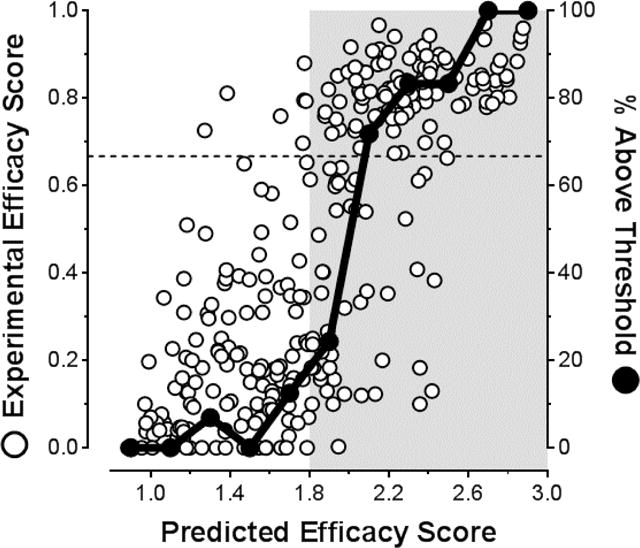
Examination of the correlation between Predicted Efficacy Scores (PES) of kinetic stabilizers and their Experimental Efficacy Scores (EES). EES and PES values for individual compounds were calculated using Equations 1 and 2, respectively. The dashed line at EES = 0.667 represents a scenario where an inhibitor binds with a stoichiometry of 1 molecule per TTR tetramer, which has been shown sufficient to completely inhibit TTR aggregation (i.e., 0% fibril formation).29 Solid black data points represent the percentage of compounds within the 0.2 PES bins that exceed the EES = 0.667 cutoff. The area shaded grey represents the PES region where significant enrichment of molecules above this cutoff occurs: that is, where the greatest proportion of highly desirable, potent and selective TTR aggregation inhibitors are predicted (PES > 1.8). Please refer to Table S1 in the Supporting Information for a complete tabulation of all values.
Compound 201 (PES = 2.682) was one of the first candidates to undergo further evaluation as a result of this substructure optimization study and embodies the pinnacle of TTR aggregation inhibition and blood plasma TTR binding selectivity (EES = 0.97). Examination of the TTR•(201)2 co-crystal structure (Figure 1) reveals that 201 complements the T4 binding site nearly perfectly, justifying its binding affinity and plasma selectivity. However, it fails as a promising drug candidate because it binds significantly to the thyroid hormone (TH) nuclear receptor (relative TH receptor binding = 0.93 — i.e., it displaces 93% of radiolabled T3 [the endogenous thyroid hormone] from the receptor at a concentration of 10 μM), which is an undesirable characteristic for a molecule that would be used daily for the remainder of a patient’s life. Forewarning of this off-target binding was not possible using the PES approach, since most of the compounds in the three libraries from which the Xs, Xp, Y, Zs, and Zp parameters were derived lacked any appreciable TH receptor binding.46–48 Of the 70 highly potent and selective TTR kinetic stabilizers evaluated for TH receptor binding efficacy in the aforementioned 3 libraries, only 3 compounds displaced T3 by >20% (compounds 46, 110, and 111, with relative TH receptor activities of 0.28, 0.21, and 0.28, respectively).
Prior to the creation of the fourth library, the stilbene and dihydrostilbene library,49 emerging evidence from a panel of compounds designed to validate this lead candidate predictive algorithm (compounds 200–218 and others that were later incorporated into the stilbene and dihydrostilbene scaffold) suggested that molecules at the high end of the PES range stood a greater chance of binding to the TH nuclear receptor. However, it appeared that investigating compounds with sub-optimal PES values could alleviate this issue. To probe what appeared to be the transition zone from low-to-high TH receptor binding, we investigated the stilbene and dihydrostilbene compound series.49 Of the 21 stilbenes tested, 15 (68%) display relative TH receptor binding activities exceeding the 0.2 threshold; however, only 17% of the dihydrostilbene compounds tested (2/12) exceeded the 0.2 threshold. A correlation is evident from plotting the accumulated relative TH receptor binding results as a function of PES for all compounds evaluated to date (Figure 4). TH receptor binding increases significantly at PES values >2.5. As evidenced by the stilbene/dihydrostilbene results (ΔPES∼0.115), decreasing the PES by a small amount can have a profound effect on the likelihood that a candidate TTR kinetic stabilizer will bind the thyroid hormone receptor.
Figure 4.
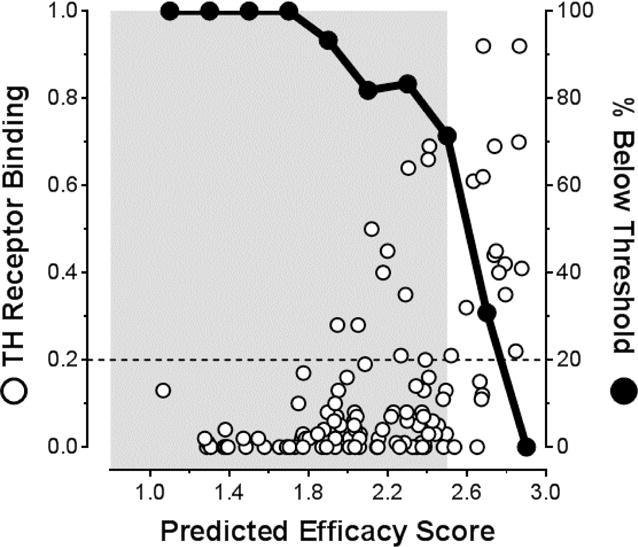
Examination of the correlation between kinetic stabilizer Predicted Efficacy Scores (PES) of the candidate kinetic stabilizers and their Relative Thyroid Hormone (TH) receptor binding. A cutoff for undesirable TH receptor binding has been selected at 0.2, represented by the dashed line. Solid black data points represent the percentage of compounds within the 0.2 PES bins that are below this threshold. The area shaded grey represents the PES region where significant enrichment of molecules below this cutoff occurs: that is, where the greatest proportion of highly desirable molecules that do not bind to the TH receptor are predicted (PES < 2.5). Please refer to Table S1 in the Supporting Information for a complete tabulation of all values.
Given that the most promising clinical candidates are not necessarily those with the highest Predicted Efficacy Scores, we reassessed the scoring algorithm to incorporate a “negative design” component, i.e., to select against compounds exhibiting TH receptor binding capacity. Toward this end, the Experimental Efficacy Scoring function of Equation 1 was modified to integrate relative TH receptor binding results and compute what we refer to as a Thyroid Hormone Receptor Experimental Efficacy Score, or THREE Score (Equation 3).
| Equation 3 |
Consistent with data in Figures 3 and 4, plotting THREE Scores as a function of PES (Figure 5) demonstrates an enrichment of compounds that exceed the 0.667 threshold within the 1.8–2.5 PES range (shaded area), with a maximum occurring at a PES of ∼2.2 (refer to Table S1 in the Supporting Information for a tabulation of all THREE Score results).
Figure 5.
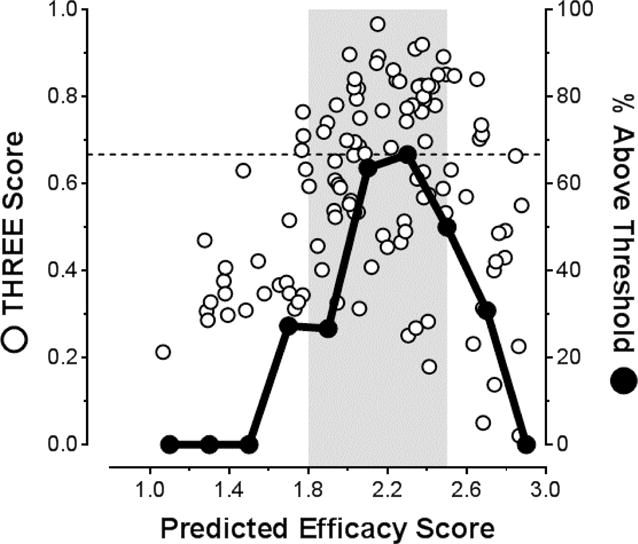
Examination of the correlation between the Predicted Efficacy Scores (PES) of the candidate kinetic stabilizers and their Thyroid Hormone Receptor Experimental Efficacy Scores (THREE Scores). THREE Score values for individual compounds were calculated using Equation 3. The cutoff line at 0.667 represents 1 inhibitor bound per TTR tetramer as described in Figure 3. Solid black data points represent the percentage of compounds within the 0.2 PES bins that exceed the 0.667 cutoff. As in Figure 3, the highest proportion of potent and selective TTR aggregation inhibitors occurs at PES values >1.8; however, at PES values above 2.5, binding to the TH receptor decreases the THREE score values significantly. Thus, the most promising potent and selective TTR amyloidogenesis inhibitors that display minimal binding to the TH receptor are predicted in the PES = 1.8–2.5 range (i.e., the area shaded grey). Please refer to Table S1 in the Supporting Information for a complete tabulation of all values.
While these Predicted Efficacy Scores are useful for identifying candidate TTR kinetic stabilizers, this semi-quantitative optimization algorithm has several deficiencies that limit its effectiveness. For one, this algorithm is limited to predicting the efficacies of compounds encompassed within the scope of the scaffold substructures included in the Aryl-X, Aryl-Z, and Linker-Y optimization studies (i.e., limited to the substructures presented in Figure 2).46–48 Furthermore, PES calculations can be ambiguous owing to molecular symmetry that does not permit unambiguous assignment of Aryl-X and Aryl-Z rings; thus, scoring in both directions is possible, which may not give the same PES values (this issue is described in more detail with compound 88 in Figure S2B of the Supporting Information). Scaffolds such as the benzoxazoles, featuring substituent patterns that do not fall within the general classification of ortho, meta, and para, also complicate PES calculations. PES comparisons are further complicated by the differences in linker lengths and angles between the aryl rings (e.g., biphenyls vs. a urea linker vs. a biphenyl ether linker, etc.), distinctions in substituent orientation on different scaffolds, and the orientation differences of the candidate kinetic stabilizers within the TTR T4 binding sites, which this algorithm assumes are equivalent. We also note that carboxylates were not highly represented in the three mini-libraries that serve as the basis for this strategy.46–48 In prior SAR studies, carboxylate-bearing aryls strongly favor binding to the outer cavity of the T4 binding site. However, in one of the mini-libraries,48 a 3,5-Br2-4-OH aryl-X group was employed that preferred binding in the outer cavity; thus, the carboxylates on the aryl-Z ring were unfavorably positioned into the inner binding cavity, leading to their assignment of low Zs scores. We know that for many inhibitors, carboxylates are not as much of a liability as this scoring algorithm would suggest. This deficiency is highlighted by the results of tafamidis (compound 250), which, while proving to be an excellent drug for treating FAP, would not have been predicted as a lead candidate using this algorithm: PES = 1.558, EES = 0.597, and THREE Score = 0.597. Thus, if we had evaluated a larger data set, carboxylate substituents might have been suggested as more favorable and tafamidis would likely have fallen within the 1.8–2.5 PES range for optimal candidates to select for further pre-clinical and clinical evaluation.
To counter the deficiencies inherent in the PES algorithm described above, we evaluated the viability of an in silico docking model for predicting the efficacy of TTR kinetic stabilizers. In this approach, we obtained docking energies for all of the compounds listed in Figure S1 and Table S1 by docking them to the unliganded TTR derived from the TTR•(201)2 complex structure using AutoDock 4.66–68 We used the apo-TTR from the TTR•(201)2 crystal structure because 201 exhibits one of the highest experimental efficacy scores that we have ever observed (EES = 0.97) — the idea being that TTR adopts this structure because of complementarity to an ideal kinetic stabilizer from a TTR binding perspective. The detailed protocol employed for compound docking to TTR is outlined in the Supporting Information. Briefly, compound structures were created using ChemDraw, then their conformational energy was minimized using Accelrys Discovery Studio 4. The compound docking files, the receptor docking files, and docking parameter files were prepared using AutoDockTools 1.5.6. The receptor grid maps were established using the AutoGrid4 program, with a grid box large enough to encompass both of the thyroxine binding sites so that the molecules could dock to either site in a single simulation. Compounds were then docked in the receptor employing triplicate simulations and the docking conformations and energies were evaluated in AutoDockTools. If the docked conformations were considered reasonable (i.e., bound in conformations that we would expect based on our prior experience observing numerous TTR•(Inhibitor)2 co-crystal structures, but in general with RMSDs < 8Å — see validation discussion below), the docking energies were recorded in Table S1 (representing the average of the three docking energies). In 92.2% of the dockings, the lowest energy conformations were considered reasonable and bound in conformations we would expect. For the remaining 7.8% of the dockings, the lowest energy conformations had the molecules either not positioned within the binding site or in conformations that we have not previously observed; however, in all cases, we were able to observe reasonable binding conformations for higher energy docked structures. Therefore, we recorded these higher energy docking conformations in Table S1, rather than the lowest docking energies for the conformations that were deemed unreasonable.
We validated that the docking of inhibitors provided reasonable binding conformations by comparing the docked conformations of 30 inhibitors with their established TTR•(kinetic stabilizer)2 co-crystal structures. Of the 30 docked compounds, 23 of them (77%) had RMSD values that differed by <2.5 Å compared to their X-ray co-crystal structures (refer to Figure S4 in the Supporting Information). Compound 201 docked with an RMSD of 0.44 Å compared to the established TTR•(201)2 crystal structure (Figure 6). The remaining compounds had higher RMSD values (but still < 8Å) owing to binding in the opposite orientation in the T4 binding site relative to that observed in the crystal structures (i.e., binding backwards in the TTR thyroid hormone binding pocket). While this indicates a failure of AutoDock to re-capitulate the conformations observed in these seven TTR•(Inhibitor)2 co-crystal structures, we still considered these docking conformations reasonable as we have often seen compounds binding in orientations opposite to what we might have predicted based on historical SAR. Furthermore, we have also observed compounds binding in mixed forward and reversed orientations in the same structures, indicating the energetic differences between the two orientations can be small, with both likely sampled in the solution state. Based on these results, we had high confidence that AutoDock 4 was effective at predicting reasonable binding conformations and that the docking model would provide robust and physiologically relevant binding rankings.
Figure 6.
The lowest energy docked conformation of inhibitor 201 calculated using Autodock 4 superimposes nearly identically with the TTR•(201)2 co-crystal structure (RMSD = 0.44 Å; calculated using the Accelrys Discovery Studio 4 crystallographic visualization program).
The initial docking scores for each of the compounds (i.e., raw docking scores as computed by AutoDock 4, presented in Table S1) were plotted against in vitro biochemical results to evaluate potential correlations (Figure S5, left panels, in the Supporting Information). Autodock 4 generates calculated docking scores as binding energies (in kcal/mol units); thus, lower docking scores (i.e., more negative) correspond to tighter binding kinetic stabilizers. Upon closer examination of the correlations, it appears that the scoring algorithm that AutoDock 4 uses to calculate docking energies was over-estimating the contributions that −CO2H and −NO2 substituents made to the docking energies (at least in this context of binding to TTR). Thus, we decided to assign “correction factors” to the AutoDock 4 calculated docking energies for compounds bearing −CO2H and −NO2 substituents, which came to be +1.03 and +2.37 kcal/mol for the carboxyl and nitro substituents, respectively. Since we are perturbing these docking energies using correction factors for the −CO2H and −NO2 substituents, we feel that representing the corrected values as energies is not entirely valid, and thus we represent the results as docking “scores” omitting the units (kcal/mol). A detailed description of how we determined these “correction factors” and how they are used to compute corrected docking scores is presented in the Supporting Information.
As with the Predicted Efficacy Scores, we compared the corrected in silico docking scores for all the inhibitors against the various biochemical results and scoring functions (Figure 7). As shown in Figure 7A, the most potent TTR kinetic stabilizers (aggregation inhibitors) in vitro are those with corrected docking scores lower than −7. The corrected docking scores also correlated with the candidate kinetic stabilizer plasma binding stoichiometries (Figure 7B) and, consequently, the Experimental Efficacy Scores (Figure 7C). There was also a trend noted that the lower the corrected docking score, the greater the chance a molecule had for binding to the thyroid hormone receptors (Figure 7D). From these correlations, it appears that the “sweet spot” for predicting the most promising lead candidate kinetic stabilizers is associated with a docking score ranging from −7.5 to −9.0 (Figure 7E). Perhaps not surprisingly, the corrected docking score for tafamidis is −7.83, which falls within this range. Thus, this in silico optimization algorithm further supports that tafamidis is an excellent transthyretin kinetic stabilizer, a prediction borne out by its clinical efficacy.
Figure 7.
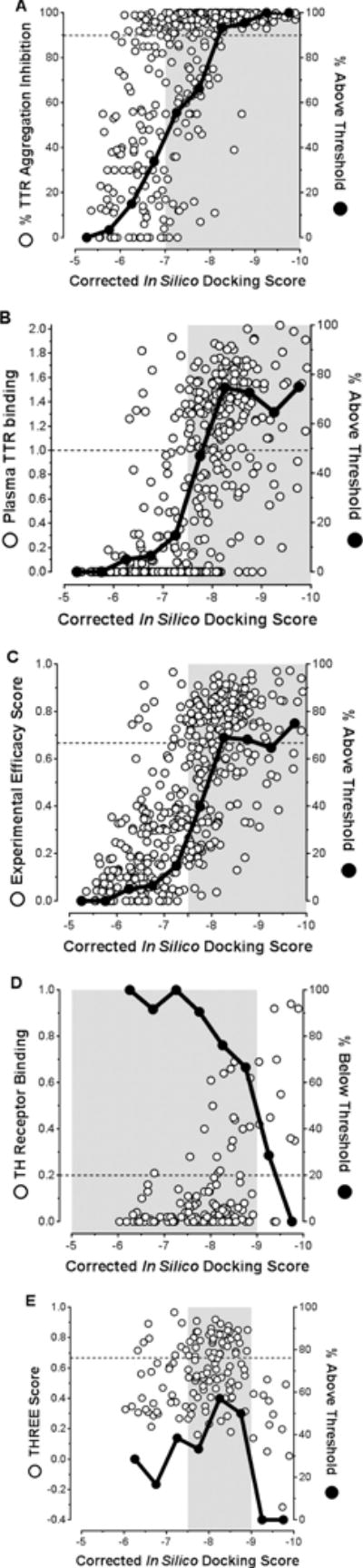
Examination of the correlations between corrected in silico docking scores of candidate kinetic stabilizers and their in vitro % inhibition of TTR aggregation (A); ex vivo plasma TTR binding stoichiometry (B); Experimental Efficacy Scores (C); Relative Thyroid Hormone receptor binding (D); and Thyroid Hormone Receptor Experimental Efficacy Scores (E). Please refer to Table S1 in the Supporting Information for a complete tabulation of all values. In panels A, B, C, and E, solid black data points represent the percentage of compounds within the 0.5 docking score bins that are above the respective thresholds (90% for inhibition of TTR aggregation for panel A; 1 molecule bound per TTR tetramer for panel B; and 0.667 for C and E, representing 100% inhibition of TTR aggregation and 1 molecule bound per tetramer). In panel D, solid black data points represent the percentage of compounds within the 0.5 docking score bins that are below the 0.2 threshold for undesirable thyroid hormone receptor binding. Areas shaded grey represent the docking score regions where significant enrichment of molecules above the respective cutoffs occur (or below with respect to thyroid hormone receptor binding). In panel E, the greatest proportion of highly desirable, potent, and selective TTR aggregation inhibitors are predicted within the −7.5 to −9.0 docking score region.
While we would like to include a “negative design” component to this in silico algorithm that incorporates scores from docking to the thyroid hormone receptors and cyclooxygenases, the scale of this endeavor renders it beyond the scope of what can be included in the initial paper. Ideally, we would like more than ten X-ray crystal structures of candidate TTR kinetic stabilizers bound to the thyroid hormone receptors and cyclooxygenases, and these data are lacking. While other thyroid hormone receptor and cyclooxygenase crystal structures are available (i.e. those without TTR kinetic stabilizer ligands), we fear that docking results using these structures may not prove reliable. Future studies linking thyroid hormone receptors and cyclooxygenase co-crystal structures and docking experiments with TTR kinetic stabilizer candidates are warranted to potentially add this negative design component.
In conclusion, we have developed two semi-quantitative models for predicting the optimal structures of potent and selective transthyretin kinetic stabilizers/amyloidogenesis inhibitors. While in our initial Predicted Efficacy Scoring model, tafamidis fell slightly out of the range for predicting the best lead candidates to pursue for further pre-clinical development, it did fall within the optimized inhibitor zone using our in silico docking model, which we believe is the more robust of the two predictive algorithms. Thus, not only is tafamidis predicted to be a bona fide lead candidate, but the clinical success of tafamidis also supports this algorithm as a useful tool for predicting promising lead transthyretin amyloidogenesis inhibitors for treating the transthyretin amyloidoses. These prediction algorithms should be useful for identifying second generation TTR kinetic stabilizers, should these be needed to ameliorate CNS or ophthalmologic challenges caused by TTR aggregation from TTR synthesized by the choroid plexus and the retinal pigment epithelial cells, respectively.
Supplementary Material
Acknowledgments
We are grateful for the support of the NIH (DK 46335), the Skaggs Institute for Chemical Biology and the Lita Annenberg Hazen Foundation (JWK), as well as startup funding from the Indiana University School of Medicine, Department of Biochemistry and Molecular Biology (SMJ). David Mortenson is supported by a postdoctoral fellowship from the Hewitt Foundation. While tafamidis was discovered at The Scripps Research Institute, it was developed at FoldRx Pharmaceuticals by the substantial expertise of Richard Labaudinière and his colleagues. FoldRx Pharmaceuticals is now owned by Pfizer. The technical expertise of Ted Foss, Maria Dendle, and Mike Saure from the Kelly lab is greatly appreciated. Crystallographic data were collected at beamline 11-1 at the Stanford Synchrotron Radiation Lightsource (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. The authors would also like to thank Drs. Xiaoping Dai, Andre Schiefner and Xueyong Zhu from the Wilson laboratory for assistance in data collection.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information
Supporting information associated with this article can be found in the online version, which includes a compilation of compounds developed in the Kelly lab for which % fibril formation and Plasma Selectivity values are available; tabulated values of experimental results for these compounds; guidelines for calculating compound Predicted Efficacy Scores (PESs); synthesis and evaluation procedures for new molecules presented in this manuscript; X-ray crystallography procedures and structure data for the TTR•(201)2 X-ray co-crystal structure; procedures for docking of molecules to the unliganded TTR from the TTR•(201)2 crystal structure using AutoDock 4; comparison of the lowest energy docked conformations of molecules with their TTR•(molecule)2 co-crystal structures; and a description of the procedure for determining the −CO2H and −NO2 substituent correction factors and calculation of corrected docking scores.
References
- 1.Buxbaum JN. The systemic amyloidoses. Curr Opin Rheumatol. 2004;16:67–75. doi: 10.1097/00002281-200401000-00013. [DOI] [PubMed] [Google Scholar]
- 2.Buxbaum JN, Tagoe CE. The genetics of the amyloidoses. Annu Rev Med. 2000;51:543–69. doi: 10.1146/annurev.med.51.1.543. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, Buxbaum JN. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336:466–73. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 4.Jiang X, Buxbaum JN, Kelly JW. The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc Natl Acad Sci USA. 2001;98:14943–8. doi: 10.1073/pnas.261419998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gambetti P, Russo C. Human brain amyloidoses. Nephrol Dial Transplant. 1998;13(Suppl 7):33–40. doi: 10.1093/ndt/13.suppl_7.33. [DOI] [PubMed] [Google Scholar]
- 6.Westermark P, Sletten K, Johansson B, Cornwell GG., 3rd Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci USA. 1990;87:2843–5. doi: 10.1073/pnas.87.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikeda S, Nakazato M, Ando Y, Sobue G. Familial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneit. Neurology. 2002;58:1001–7. doi: 10.1212/wnl.58.7.1001. [DOI] [PubMed] [Google Scholar]
- 8.Sekijima Y, Hammarstrom P, Matsumura M, Shimizu Y, Iwata M, Tokuda T, Ikeda S, Kelly JW. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab Invest. 2003;83:409–17. doi: 10.1097/01.lab.0000059937.11023.1f. [DOI] [PubMed] [Google Scholar]
- 9.Sousa A, Coelho T, Barros J, Sequeiros J. Genetic epidemiology of familial amyloidotic polyneuropathy (FAP)-type I in Povoa do Varzim and Vila do Conde (north of Portugal) Am J Med Genet. 1995;60:512–21. doi: 10.1002/ajmg.1320600606. [DOI] [PubMed] [Google Scholar]
- 10.McCutchen SL, Lai Z, Miroy GJ, Kelly JW, Colon W. Comparison of lethal and nonlethal transthyretin variants and their relationship to amyloid disease. Biochemistry. 1995;34:13527–36. doi: 10.1021/bi00041a032. [DOI] [PubMed] [Google Scholar]
- 11.Hammarstrom P, Sekijima Y, White JT, Wiseman RL, Lim A, Costello CE, Altland K, Garzuly F, Budka H, Kelly JW. D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: a prescription for central nervous system amyloidosis? Biochemistry. 2003;42:6656–63. doi: 10.1021/bi027319b. [DOI] [PubMed] [Google Scholar]
- 12.Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 13.Sipe JD. Amyloidosis. Crit Rev Clin Lab Sci. 1994;31:325–54. doi: 10.3109/10408369409084679. [DOI] [PubMed] [Google Scholar]
- 14.Westermark P, Bergstrom J, Solomon A, Murphy C, Sletten K. Transthyretin-derived senile systemic amyloidosis: clinicopathologic and structural considerations. Amyloid. 2003;10(Suppl 1):48–54. [PubMed] [Google Scholar]
- 15.Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012;421:185–203. doi: 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson SM, Wiseman RL, Reixach N, Paulsson J, Choi S, Powers ET, Buxbaum JN, Kelly JW. Understanding and Ameliorating the Transthyretin Amyloidoses. In: Ramirez-Alvarado M, Kelly JW, Dobson CM, editors. Protein Misfolding Diseases: Current and Emerging Principles and Therapies. John Wiley & Sons; 2010. pp. 967–1003. [Google Scholar]
- 17.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–90. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 18.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–4. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 19.Kelly JW. Alternative conformations of amyloidogenic proteins govern their behavior. Curr Opin Struct Biol. 1996;6:11–7. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 20.Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60:765–74. doi: 10.1111/j.1532-5415.2011.03868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandgren O, Kjellgren D, Suhr OB. Ocular manifestations in liver transplant recipients with familial amyloid polyneuropathy. Acta Ophthalmol. 2008;86:520–4. doi: 10.1111/j.1600-0420.2007.01098.x. [DOI] [PubMed] [Google Scholar]
- 22.Ohya Y, Okamoto S, Tasaki M, Ueda M, Jono H, Obayashi K, Takeda K, Okajima H, Asonuma K, Hara R, Tanihara H, Ando Y, Inomata Y. Manifestations of transthyretin-related familial amyloidotic polyneuropathy: long-term follow-up of Japanese patients after liver transplantation. Surg Today. 2011;41:1211–8. doi: 10.1007/s00595-010-4488-5. [DOI] [PubMed] [Google Scholar]
- 23.Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86:1036–43. doi: 10.1136/jnnp-2014-308724. [DOI] [PubMed] [Google Scholar]
- 24.Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S, Lewis WD, Obici L, Plante-Bordeneuve V, Rapezzi C, Said G, Salvi F. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31. doi: 10.1186/1750-1172-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sekijima Y, Yazaki M, Oguchi K, Ezawa N, Yoshinaga T, Yamada M, Yahikozawa H, Watanabe M, Kametani F, Ikeda S. Cerebral amyloid angiopathy in posttransplant patients with hereditary ATTR amyloidosis. Neurology. 2016;87:773–81. doi: 10.1212/WNL.0000000000003001. [DOI] [PubMed] [Google Scholar]
- 26.Maia LF, Magalhaes R, Freitas J, Taipa R, Pires MM, Osorio H, Dias D, Pessegueiro H, Correia M, Coelho T. CNS involvement in V30M transthyretin amyloidosis: clinical, neuropathological and biochemical findings. J Neurol Neurosurg Psychiatry. 2015;86:159–67. doi: 10.1136/jnnp-2014-308107. [DOI] [PubMed] [Google Scholar]
- 27.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–6. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 28.Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski-Werner SL, Kelly JW. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Acc Chem Res. 2005;38:911–21. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 29.Wiseman RL, Johnson SM, Kelker MS, Foss T, Wilson IA, Kelly JW. Kinetic stabilization of an oligomeric protein by a single ligand binding event. J Am Chem Soc. 2005;127:5540–51. doi: 10.1021/ja042929f. [DOI] [PubMed] [Google Scholar]
- 30.Foss TR, Kelker MS, Wiseman RL, Wilson IA, Kelly JW. Kinetic stabilization of the native state by protein engineering: Implications for inhibition of transthyretin amyloidogenesis. J Mol Biol. 2005;347:841–54. doi: 10.1016/j.jmb.2005.01.050. [DOI] [PubMed] [Google Scholar]
- 31.Foss TR, Wiseman RL, Kelly JW. The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry. 2005;44:15525–33. doi: 10.1021/bi051608t. [DOI] [PubMed] [Google Scholar]
- 32.Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, Suhr OB, Campistol JM, Conceicao IM, Schmidt HH, Trigo P, Kelly JW, Labaudiniere R, Chan J, Packman J, Wilson A, Grogan DR. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79:785–92. doi: 10.1212/WNL.0b013e3182661eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coelho T, Maia LF, da Silva AM, Cruz MW, Plante-Bordeneuve V, Suhr OB, Conceicao I, Schmidt HH, Trigo P, Kelly JW, Labaudiniere R, Chan J, Packman J, Grogan DR. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260:2802–14. doi: 10.1007/s00415-013-7051-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, Sacchettini JC. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 2000;7:312–21. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 35.Miroy GJ, Lai Z, Lashuel HA, Peterson SA, Strang C, Kelly JW. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc Natl Acad Sci USA. 1996;93:15051–6. doi: 10.1073/pnas.93.26.15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oza VB, Petrassi HM, Purkey HE, Kelly JW. Synthesis and evaluation of anthranilic acid-based transthyretin amyloid fibril inhibitors. Bioorg Med Chem Lett. 1999;9:1–6. doi: 10.1016/s0960-894x(98)00696-9. [DOI] [PubMed] [Google Scholar]
- 37.Oza VB, Smith C, Raman P, Koepf EK, Lashuel HA, Petrassi HM, Chiang KP, Powers ET, Sachettini J, Kelly JW. Discovering transthyretin amyloid fibril inhibitors by limited screening. J Med Chem. 2002;45:321–32. doi: 10.1021/jm010257n. [DOI] [PubMed] [Google Scholar]
- 38.Baures PW, Peterson SA, Kelly JW. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg Med Chem. 1998;6:1389–401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 39.Baures PW, Oza VB, Peterson SA, Kelly JW. Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the non-steroidal anti-inflammatory drug, flufenamic acid. Bioorg Med Chem. 1999;7:1339–47. doi: 10.1016/s0968-0896(99)00066-8. [DOI] [PubMed] [Google Scholar]
- 40.Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem. 2004;47:355–74. doi: 10.1021/jm030347n. [DOI] [PubMed] [Google Scholar]
- 41.Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest. 2004;84:545–52. doi: 10.1038/labinvest.3700059. [DOI] [PubMed] [Google Scholar]
- 42.Purkey HE, Palaninathan SK, Kent KC, Smith C, Safe SH, Sacchettini JC, Kelly JW. Hydroxylated polychlorinated biphenyls selectively bind transthyretin in blood and inhibit amyloidogenesis: rationalizing rodent PCB toxicity. Chem Biol. 2004;11:1719–28. doi: 10.1016/j.chembiol.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 43.Razavi H, Palaninathan SK, Powers ET, Wiseman RL, Purkey HE, Mohamedmohaideen NN, Deechongkit S, Chiang KP, Dendle MT, Sacchettini JC, Kelly JW. Benzoxazoles as transthyretin amyloid fibril inhibitors: synthesis, evaluation, and mechanism of action. Angew Chem Int Ed Engl. 2003;42:2758–61. doi: 10.1002/anie.200351179. [DOI] [PubMed] [Google Scholar]
- 44.Razavi H, Powers ET, Purkey HE, Adamski-Werner SL, Chiang KP, Dendle MT, Kelly JW. Structure-based design of N-phenyl phenoxazine transthyretin amyloid fibril inhibitors. Bioorg Med Chem Lett. 2005;15:1075–8. doi: 10.1016/j.bmcl.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 45.Petrassi HM, Klabunde T, Sacchettini JC, Kelly JW. Structure-based design of N-phenyl phenoxazine transthyretin amyloid fibril inhibitors. J Am Chem Soc. 2000;122:2178–2192. [Google Scholar]
- 46.Johnson SM, Connelly S, Wilson IA, Kelly JW. Biochemical and structural evaluation of highly selective 2-arylbenzoxazole-based transthyretin amyloidogenesis inhibitors. J Med Chem. 2008;51:260–70. doi: 10.1021/jm0708735. [DOI] [PubMed] [Google Scholar]
- 47.Johnson SM, Connelly S, Wilson IA, Kelly JW. Toward optimization of the linker substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem. 2008;51:6348–58. doi: 10.1021/jm800435s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson SM, Connelly S, Wilson IA, Kelly JW. Toward optimization of the second aryl substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem. 2009;52:1115–25. doi: 10.1021/jm801347s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi S, Reixach N, Connelly S, Johnson SM, Wilson IA, Kelly JW. A substructure combination strategy to create potent and selective transthyretin kinetic stabilizers that prevent amyloidogenesis and cytotoxicity. J Am Chem Soc. 2010;132:1359–70. doi: 10.1021/ja908562q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Connelly S, Choi S, Johnson SM, Kelly JW, Wilson IA. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr Opin Struct Biol. 2010;20:54–62. doi: 10.1016/j.sbi.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson SM, Petrassi HM, Palaninathan SK, Mohamedmohaideen NN, Purkey HE, Nichols C, Chiang KP, Walkup T, Sacchettini JC, Sharpless KB, Kelly JW. Bisaryloxime ethers as potent inhibitors of transthyretin amyloid fibril formation. J Med Chem. 2005;48:1576–87. doi: 10.1021/jm049274d. [DOI] [PubMed] [Google Scholar]
- 52.Green NS, Foss TR, Kelly JW. Genistein, a natural product from soy, is a potent inhibitor of transthyretin amyloidosis. Proc Natl Acad Sci USA. 2005;102:14545–50. doi: 10.1073/pnas.0501609102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Green NS, Palaninathan SK, Sacchettini JC, Kelly JW. Synthesis and characterization of potent bivalent amyloidosis inhibitors that bind prior to transthyretin tetramerization. J Am Chem Soc. 2003;125:13404–14. doi: 10.1021/ja030294z. [DOI] [PubMed] [Google Scholar]
- 54.Petrassi HM, Johnson SM, Purkey HE, Chiang KP, Walkup T, Jiang X, Powers ET, Kelly JW. Potent and selective structure-based dibenzofuran inhibitors of transthyretin amyloidogenesis: kinetic stabilization of the native state. J Am Chem Soc. 2005;127:6662–71. doi: 10.1021/ja044351f. [DOI] [PubMed] [Google Scholar]
- 55.Peterson SA, Klabunde T, Lashuel HA, Purkey H, Sacchettini JC, Kelly JW. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc Natl Acad Sci USA. 1998;95:12956–60. doi: 10.1073/pnas.95.22.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lai Z, Colon W, Kelly JW. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry. 1996;35:6470–82. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 57.Purkey HE, Dorrell MI, Kelly JW. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc Natl Acad Sci USA. 2001;98:5566–71. doi: 10.1073/pnas.091431798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joven J, Cliville X, Camps J, Espinel E, Simo J, Vilella E, Oliver A. Plasma protein abnormalities in nephrotic syndrome: effect on plasma colloid osmotic pressure and viscosity. Clin Chem. 1997;43:1223–31. [PubMed] [Google Scholar]
- 59.Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, Heneghan MA, Gorevic PD, Litchy WJ, Wiesman JF, Nordh E, Corato M, Lozza A, Cortese A, Robinson-Papp J, Colton T, Rybin DV, Bisbee AB, Ando Y, Ikeda S, Seldin DC, Merlini G, Skinner M, Kelly JW, Dyck PJ, Diflunisal Trial C. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658–67. doi: 10.1001/jama.2013.283815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13:236–49. doi: 10.1080/13506120600960882. [DOI] [PubMed] [Google Scholar]
- 61.Tojo K, Sekijima Y, Kelly JW, Ikeda S. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res. 2006;56:441–9. doi: 10.1016/j.neures.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 62.Glenner GG. Amyloid deposits and amyloidosis: the beta-fibrilloses (second of two parts) N Engl J Med. 1980;302:1333–43. doi: 10.1056/NEJM198006123022403. [DOI] [PubMed] [Google Scholar]
- 63.Sekijima Y, Tojo K, Morita H, Koyama J, Ikeda S. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid. 2015;22:79–83. doi: 10.3109/13506129.2014.997872. [DOI] [PubMed] [Google Scholar]
- 64.Obici L, Merlini G. An overview of drugs currently under investigation for the treatment of transthyretin-related hereditary amyloidosis. Expert Opin Investig Drugs. 2014;23:1239–51. doi: 10.1517/13543784.2014.922541. [DOI] [PubMed] [Google Scholar]
- 65.Waddington Cruz M, Benson MD. A Review of tafamidis for the treatment of transthyretin-related amyloidosis. Neurol Ther. 2015;4:61–79. doi: 10.1007/s40120-015-0031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 67.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.