

Abstract
Background
Campylobacter species are a leading cause of bacterial foodborne illness worldwide. Despite the global efforts to curb them, Campylobacter infections have increased continuously in both developed and developing countries. The development of effective strategies to control the infection by this pathogen is warranted. The essential genes of bacteria are the most prominent targets for this purpose. In this study, we used transposon sequencing (Tn-seq) of a genome-saturating library of Tn5 insertion mutants to define the essential genome of C. jejuni at a high resolution.
Result
We constructed a Tn5 mutant library of unprecedented complexity in C. jejuni NCTC 11168 with 95,929 unique insertions throughout the genome and used the genomic DNA of the library for the reconstruction of Tn5 libraries in the same (C. jejuni NCTC 11168) and different strain background (C. jejuni 81–176) through natural transformation. We identified 166 essential protein-coding genes and 20 essential transfer RNAs (tRNA) in C. jejuni NCTC 11168 which were intolerant to Tn5 insertions during in vitro growth. The reconstructed C. jejuni 81–176 library had 384 protein coding genes with no Tn5 insertions. Essential genes in both strain backgrounds were highly enriched in the cluster of orthologous group (COG) categories of ‘Translation, ribosomal structure and biogenesis (J)’, ‘Energy production and conversion (C)’, and ‘Coenzyme transport and metabolism (H)’.
Conclusion
Comparative analysis among this and previous studies identified 50 core essential genes of C. jejuni, which can be further investigated for the development of novel strategies to control the spread of this notorious foodborne bacterial pathogen.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4032-8) contains supplementary material, which is available to authorized users.
Keywords: Campylobacter, Transposon sequencing (Tn-seq), Essential genes
Background
Campylobacter species are a leading cause of bacterial foodborne illness worldwide and one of the most common infectious agents of the last century. Despite a reduction in the incidence of infections by a number of major foodborne pathogens due to global efforts, Campylobacter infections have continued to increase in both developed and developing regions across the globe including the U.S., Europe, Australia, Africa, Asia and the Middle East. Additionally, Campylobacter harbors antimicrobial resistance genes with the capability of horizontal transfer between pathogenic and commensal microorganisms, which could possibly lead to the emergence of multidrug resistant microorganisms. Hence, researchers speculate that Campylobacter will continue to remain a threat to global public health for years to come [1, 2]. The current issue warrants a multifaceted approach to intervene and control Campylobacter infections, including identification of indispensable essential genes that are related to basic cellular functions or metabolic pathways/processes. These genes have the potential of becoming novel targets for the development of new antibiotics or vaccines.
Essential genes are defined as those that are absolutely required for the viability of cellular life [3, 4]. Experimental techniques, such as single-gene knockouts [5–7], transposon mutagenesis [8, 9], and antisense RNA and RNA interference [10, 11] have been used to identify essential genes. In addition, computational approaches to track down essential genes involve comparative genomics, supervised machine learning, constraint-based methods, and integrative genomics approaches based on orthology and phylogeny [12–16]. However, the most reliable method used to define the essential genome is transposon mutagenesis via transposon sequencing (Tn-seq). The basic principle of this approach involves the creation of a transposon insertion library in the bacteria of interest and identification of individual transposon-genome junction sites on a global scale by the Tn-seq method. This process helps identify virtually all dispensable genes, which allows for the identification of the entire set of essential genes in the genome simultaneously in a single experiment by negative selection of transposon mutants [17]. Methods with minor variations are known as InSeq [18], TraDIS [19], HITS [20], Tn-seq circle [21], Tn-seq [22], and RB-TnSeq [23]. Recently, Hutchison III et al. (2016) has used improved transposon mutagenesis methods for the identification of quasi-essential genes, which were then used as a basis to create a minimal synthetic bacterial genome, Mycoplasma mycoides JCVI-syn3.0, smaller than the genomes of any autonomously replicating cell found in nature [24].
Stahl and Stintzi (2011) identified 195 essential genes of Campylobacter jejuni (C. jejuni) NCTC 11168 required for growth at 37 °C under a microaerophilic atmosphere on a rich Muller-Hinton medium with 7201 individual mutants (Tn5) using a microarray-based approach for tracking transposon insertions [25]. Furthermore, Metris et al. (2011) also identified 233 essential genes of C. jejuni strain NCTC 11168 strain based on a total of 9550 transposon insertions in the genome using two different transposons (Mariner and Tn7) on Blood Agar Base no.2 (Oxoid) plates supplemented with 5% v/v defibrinated horse blood at 42 °C under microaerophilic conditions [26]. More recently, Gao et al. (2014) identified 175 essential genes of C. jejuni 81–176 based on ~50,000 transposon insertion mutants of C. jejuni 87–176 on brucella agar plates at 37 °C in 10% CO2 atmosphere [27]. However, these studies have had only a limited overlap in their lists of essential genes, probably because of the different culture conditions for recovery of the mutants, different strain backgrounds, varying levels of saturation of transposon insertions, or different analytical approaches. Since these approaches for essential gene discovery are based on the identification of the genomic regions that do not tolerate transposon insertions, the accuracy of essential gene discovery should be critically dependent on the saturation level of the transposon insertion library.
In this study, we created a highly complex Tn5 mutant library of C. jejuni NCTC 11168 (seed library) with more than 95,000 unique insertions in the genome. C. jejuni NCTC 11168 required 166 essential protein coding genes for the growth on Muller-Hinton (MH) agar at 37 °C under microaerophilic conditions. Additionally, we reconstructed a Tn5 mutant library in the same (C. jejuni NCTC 11168) and a different strain background (C. jejuni 81–176) by transferring the insertions in the seed library to the recipient cells via natural transformation to develop and validate a powerful approach for comparative functional genomics of C. jejuni.
The reconstruction technique of a Tn5 library in Campylobacter developed in this study has several advantages. First, the viability of Campylobacter strains normally reduces significantly over time even during storage at −80 °C for both the wild type and mutant strains. Additionally, this reduction in viability can be more severe depending on the mutations the mutant strains carry. This presents the challenge of storing the mutant library without altering the representativeness of the viable mutant strains. However, if we could reconstruct the mutant library in a wild type background without significant changes in the representativeness of the mutations present, it would be an important advancement in functional genomics. Second, the ability to reproduce the mutant library by natural transformation with genomic DNA of a complex library would facilitate the use of the DNA as an important resource for functional genomic analysis of C. jejuni among different labs. Third, the reconstruction of the library will facilitate comparative genomic analysis across different strains, including different deletion mutant backgrounds. For example, this reconstruction method can be employed to perform genetic interaction mapping by introducing the same library into wild type and different deletion mutants of the same strain background, followed by Tn-seq analysis.
Finally, we combined all existing data from the previous and current studies to define a core set of essential genes of C. jejuni.
Results and discussion
Evaluation and comparison of the libraries based on Illumina sequencing data
We generated complex library of C. jejuni NCTC 11168 following natural transformation of in vitro mutagenized genomic DNA with commercially available Tn5 transposome complex kit (EZ-Tn5™ < KAN-2 > Tnp Transposome™ Kit). Fourteen natural transformations were performed, each producing ~100,000 mutants and combined to create a complex library that contains a total of 1.4 million mutants. Previously, various strategies have been attempted for efficient transposon mutagenesis of C. jejuni. For in vivo mutagenesis, the preformed Tn5 transposome complex was introduced into the various C. jejuni strains via electroporation [28]. This strategy yielded ~3000 random mutants per electroporation for C. jejuni strain 81–176, but the efficiency was extremely low for other strains tested, limiting the application of the approach. On the contrary, for in vitro mutagenesis, transposition reactions were conducted using genomic DNA of C. jejuni, purified transposon sequence plus purified transposase enzyme of either Tn5 [29] mariner [30]or Tn552 [31]. Then the in vitro mutagenized DNA was used to transform C. jejuni cells through natural transformation, yielding 3000–7000 transposon mutants per reaction. In the current study, we achieved an efficiency of transposon mutagenesis far higher than previously reported (100,000 vs. 3000–7000 transposon mutants per transformation). We speculate that the high efficiency in our study was due to the use of a preformed transposome complex for in vitro mutagenesis of genomic DNA as compared to adding transposase and transposon sequences separately into the reaction, as has been done in all previous studies for in vitro transposon mutagenesis in Campylobacter [29–31].
Equal volumes of the mutant pools were combined to make a seed library (S-CJ11168). Genomic DNA from the seed library was used for reconstruction of the Tn5 mutant library. We collected 281,000 mutants from the natural transformation of the seed library in the same strain background (C. jejuni NCTC 11168: R-CJ11168-D) and 82,000 mutants in a different strain background (C. jejuni 81–176: R-CJ81176-D) as shown in Fig. 1 and Table 1.
Fig. 1.
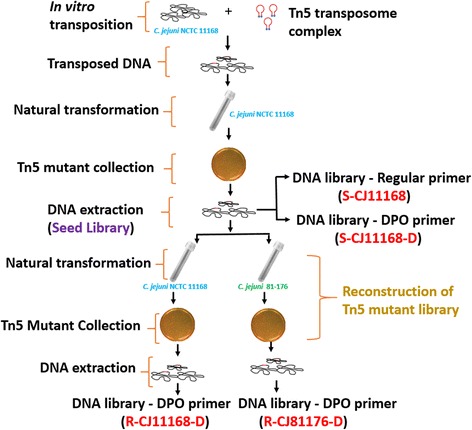
Design of experiment. EZ-Tn5™ < KAN-2 > Tnp Transposome™ Kit was used for in vitro transposition of genomic DNA of C. jejuni NCTC 11168. The transposed DNA was then naturally transformed into C. jejuni NCTC 11168 and mutants were cultured on MH agar with TMP and Km (Seed library: S-CJ11168). Seed library DNA was extracted and used for the reconstruction of the Tn5 library in the same (C. jejuni NCTC 11168: R-CJ11168-D) and different strain backgrounds (C. jejuni 81–176: R-CJ81176-D). Regular and DPO primers were used for linear extension to make DNA libraries for Illumina sequencing. [S: the seed library, R: the reconstructed library, D: dual priming oligonucleotide (DPO), CJ11168: C. jejuni NCTC 11168; and CJ81176: C. jejuni 81–176]
Table 1.
Overview of C. jejuni Tn5 mutant libraries
| Library | # Tn5 Mutants | Total Reads | Mapped Reads (%) | # Unique Insertions | Mean (± SE) |
|---|---|---|---|---|---|
| S-CJ11168 | 1,400,000 | 9,040,241 | 6,812,731 (75.36) | 95,929 | 71.02 ± 0.90 |
| S-CJ11168-D | 1,400,000 | 6,920,934 | 6,448,244 (93.1) | 79,178 | 81.44 ± 11.54 |
| R-CJ11168-D | 281,000 | 6,052,446 | 5,685,107 (93.93) | 52,607 | 108.07 ± 13.89 |
| R-CJ81176-D (a) | 82,000 | 1,638,463 | 1,303,248 (79.54) | 29,565 | 44.08 ± 5.89 |
| *a- > CJ11168 | 1,493,694 (91.16) | 32,623 | 45.79 ± 5.46 |
Mean is the average reads per unique insertion in the Tn5 library with standard error (SE)
S: Seed library, R: Reconstruction, D: DPO, CJ11168: C. jejuni NCTC 11168, CJ81116: C. jejuni 81–176
*Tn5 library (R-CJ81176-D, a) when mapped against the donor strain C. jejuni NCTC 11168 genome (a- > CJ11168), a higher alignment rate was achieved (91.6%)
Transposon junction sequences were amplified using two different primers i.e. regular and dual priming oligonucleotide (DPO) in the linear extension step with downstream processes remaining the same for all DNA libraries. The regular primer was used for linear extension of the seed library in C. jejuni NCTC 11168 (S-CJ11168) while the DPO primer was used for the seed library in C. jejuni NCTC 11168 (S-CJ11168-D), the reconstructed library in same strain background C. jejuni NCTC 11168 (R-CJ11168-D), and the other strain background C. jejuni 81–176 (R-CJ81176-D). The DPO primer is believed to block mismatched priming, thereby accomplishing higher PCR specificity [32]. The site of Tn5 insertion in each Tn5 library was determined through next-generation sequencing on the HiSeq platform. Demultiplexing Illumina sequencing reads without any mismatch in the barcode sequence and Tn5 mosaic end produced 9,040,241 reads for S-CJ11168; 6,920,934 reads for S-CJ11168-D; 6,052,446 reads for R-CJ11168-D and 1,638,463 reads for R-CJ81176-D. Similarly, R-CJ1168-D had the highest number of reads per unique insertion site and R-CJ81176-D had the lowest reads per unique insertion site with S-CJ11168 having the highest and R-CJ81176-D having the lowest median reads per unique insertion site (Table 1).
Next, the 22 bp transposon genome junction sequence reads were mapped to their respective genome using the default parameter of Bowtie2.2.8, which reports the best alignment. It is interesting to note that C. jejuni NCTC 11168 has 30.6% GC content [33], however, the GC content of the 22 bp genomic sequence across all the Tn5 libraries was 40.25% (SE ± 2.22). This might reflect the preference of Tn5 transposition into guanosine (G) and cytidine (C) rich sequences [34]. The overall alignment rate was 85.50% (SE ± 5.46). The regular primer had a significantly lower alignment rate when compared to the DPO primer library (75.36% vs. 92.87% respectively), clearly indicating a higher specificity in binding the target DNA by the DPO primer. It was also observed that the regular primer produced a lower standard error (SE) with mean reads per unique insertion when compared to the DPO primer (0.90 vs. 11.54, respectively, for S-CJ11168 library). Thus, from this data, we can conclude that the DPO primer produced the Tn-seq amplicon libraries are more representative of the Tn5 mutant pools than those produced using the regular primer based on the higher alignment rate (Table 1).
Furthermore, the seed libraries S-CJ11168 and S-CJ11168-D had the most unique insertions throughout the genome, with 95,920 and 79,178 unique insertions respectively; followed by the reconstructed library in same strain background (R-CJ11168-D: 52,607) and the fewest in the other strain background (R-CJ81176-D: 29,565) (Table 1). C. jejuni NCTC 11168 libraries had 47,090 shared unique insertions genome-wide. Importantly, only 2218 (2.1%) of the Tn5 insertion sites were unique to the reconstructed library of C. jejuni NCTC 11168 (R-CJ11168-D). Similarly, the seed library amplified using the regular and the DPO primer had 73,649 (71%) unique insertions in common (Additional file 1: Figure S1). The fact that a significantly lower number of unique insertions was detected in R-CJ11168-D in comparison to S-CJ11168-D (52,607 vs. 79,178) may be due to the insufficient number of Tn5 mutants (281,000 mutant colonies) collected during the reconstruction experiment. The considerably lower number of unique insertions in R-CJ81176-D in comparison to R-CJ11168-D (29,565 vs. 52,607) is likely due to the genomic differences in the two strains but may also be due to the insufficient number (82,000 mutants) of Tn5 mutants collected to form the reconstructed library R-CJ81176-D. The saturation level of reconstructed libraries could possibly be increased by increasing the number of Tn5 mutants by carrying out more natural transformations of the seed library.
Identification of essential gene in Campylobacter jejuni NCTC 11168
The EL-ARTIST pipeline was used for the identification of essential genes of C. jejuni required for optimal growth on MH agar plates under microaerophilic conditions at 37 °C. The Campylobacter Tn5 libraries had no noticeable replication bias in the reads distribution throughout the genome (Fig. 2a, b, c, and d). Replication bias is usually suggested by a ‘V’ shaped reads distribution with a higher number of reads at the origin of replication. Absence of a replication bias in C. jejuni NCTC 11168 might be due to the higher doubling time (112 min) when grown in MH broth as opposed to lower doubling times of some bacteria that have a replication bias, such as V. cholera which has a doubling time of 16–20 min when grown in rich media [35, 36]. Read counts of Tn5 libraries were mapped to 400 bp genomic windows against the C. jejuni NCTC 11168 genome. A high Spearman correlation was observed between the seed library prepared with the regular (S-CJ11168) and the DPO primer (S-CJ11168-D) based on read counts binned using a 400 bp window size (R2 = 0.95, p < 0.0001) as shown in Fig. 2e. A slightly lower Spearman correlation (R2 = 0.92, p < 0.0001) was observed between the reconstructed library in the same genetic background (R-CJ11168-D) with the seed libraries prepared with the regular (S-CJ11168) and the DPO primer (S-CJ11168-D) as shown in Fig. 2f and g, respectively.
Fig. 2.
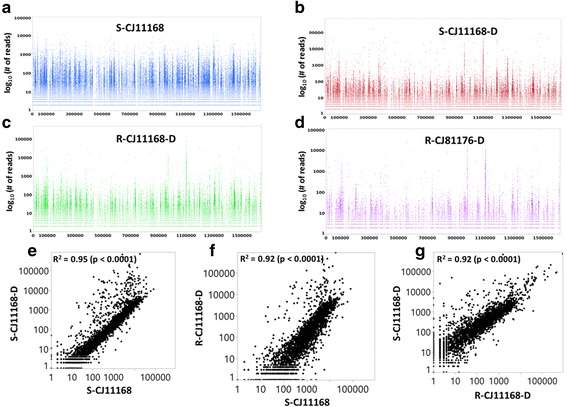
Overview of Illumina sequencing of C. jejuni Tn5 libraries. Read distribution of Tn5 mutant libraries of C. jejuni: (a) S-CJ1118, (b) S-CJ11168-D, (c) R-CJ11168-D and (d) R-CJ81176 (X-axis: Genomic coordinate of Campylobacter; Y-axis: log10 transformed read counts). Spearman correlation (R2) of Tn5 reads distribution based on a 400 bp window size between different libraries of C. jejuni NCTC 11168: (e) S-CJ11168 vs S-CJ11168-D, (f) S-CJ11168 vs R-CJ11168-D and (g) R-Cj11168-D vs S-CJ11168-D. Tn5 libraries are named as previously
Tn5 sequencing analysis using the EL-ARTIST pipeline revealed ~250 genes essential in C. jejuni NCTC 11168 required for optimal growth on rich MH agar under microaerophilic conditions at 37 °C. The seed library C. jejuni NCTC 11168 (S-CJ11168) identified 280 essential genes (15 domain essential and 265 entirely essential genes) for optimal growth. Likewise, the same seed library prepared with DPO primer (S-CJ11168-D) identified 278 essential genes (9 domain essential and 269 entirely essential genes) and the reconstructed library in the same genetic background (R-CJ11168-D) revealed 284 essential genes (18 domain essential and 266 entirely essential genes) using the same conditions. All three of these libraries shared 200 common essential genes (Additional file 1: Figure S2).
However, when the Tn5 insertions were examined closely at the gene level, multiple genes identified as essential by EL-ARTIST had significant sequence reads corresponding to the insertions in the genes. For example, spoT had 64 Tn5 unique insertions with 358 reads (in S-CJ11168 library) that was called as essential for optimal growth by EL-ARTIST. Furthermore, the EL-ARTIST pipeline was not sensitive enough to pick up the genes with less than a 400 bp window size. In addition, windows far smaller than 400 bp, for instance 100 bp, can give false positive results due to a lack of insertions in many 100 bp windows.
Bioinformatics pipeline analysis results are subject to variation depending upon the algorithm and statistical power. To overcome the noticed limitation of EL-ARTIST in analyzing our dataset, the same dataset was re-analyzed to identify the essential genes according to the definition of essential genes as those that cannot tolerate any insertions. The genes meeting this condition for the internal 80% of the coding region in all three Tn5 libraries of C. jejuni NCTC 11168 were considered to be essential for growth or survival in this study. We identified 166 essential coding sequences (CJ-11168) of C. jejuni NCTC 11168 with no Tn5 insertions (Additional file 2: SH2). 52.4% of the essential genes of C. jejuni NCTC 11168 were on the negative strand while 47.5% were on the positive strand. Genes that contain even one insertion can be labeled as non-essential, but genes lacking insertions cannot be necessarily classified as essential due to sequence bias of Tn5 insertions and the smaller genes having a lower chance of transposon insertion [26]. However, in this study, there was a significantly low correlation between the Tn5 insertion read counts and gene length (Spearman correlation = 0.1852, p < 0.0001) (Fig. 3a). Also, Tn5 transposons are inserted randomly throughout the entire genome with some preference towards GC rich DNA sequences [34]. Nevertheless, we did not observe any correlation between the Tn5 read insertion in the central 80% of genes (CDS) and GC content (%) of the entire gene (CDS) as shown in Fig. 3b (Spearman correlation = 0.0488, p > 0.0531).
Fig. 3.
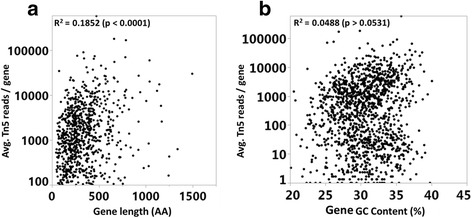
Tn5 insertion bias analysis in Tn5 libraries of C. jejuni NCTC 11168. Spearman correlation (R2) between (a) gene length (amino acid length) and average Tn5 insertion reads and (b) GC content (%) of all genes and average Tn5 insertion reads. Average Tn5 reads is the average of Tn5 reads in the central 80% of gene in all the 3 libraries of C. jejuni NCTC 11168 (S-CJ11168, S-CJ11168-D, and R-CJ11168-D)
Furthermore, to back up our analysis at the gene level, we looked for the Tn5 insertion in pseudogenes of C. jejuni NCTC 11168. Out of a total of 38 pseudogenes in the entire genome of C. jejuni NCTC 11168, all of the pseudogenes had Tn5 insertions in at least one of the seed libraries of C. jejuni NCTC 11168 (Additional file 2: SH4). This also indicates the high coverage of Tn5 mutagenesis in the seed library. Nonetheless, the seed library S-CJ11168 (sequenced with the regular primer) and S-CJ11168-D (sequenced with the DPO primer) missed Tn5 insertions in only one pseudogene each, Cj0740 and Cj0742, respectively. However, the reconstructed library in the same background (R-CJ11168-D) had no Tn5 insertions in 11 pseudogenes. This is most likely due to the low saturation level in the reconstructed libraries, which was likely caused by the limited number of transposon mutants collected to form the reconstructed libraries. This explanation is supported by the significantly low level of unique insertions in the reconstructed libraries in comparison to the seed libraries (Table 1). This shortcoming may be able to be mitigated by increasing the number of Tn5 mutants recovered to form reconstructed libraries and ensuring a similar level of sequencing depth across the libraries that are compared.
Next, we assigned essential genes to Cluster of Orthologous Groups (COG) using the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/archive/old_refseq/Bacteria/). The COG categories highly enriched among the 166 essential genes were: J- Translation, ribosomal structure and biogenesis (20.48%), Not in COG (18.07%), M- Cell wall/membrane/envelope biogenesis (10.24%), H- Coenzyme transport and metabolism (8.43%), C- Energy production and conversion (7.83%) and I- Lipid transport and metabolism (6.02%). The COG moderately enriched were: U- Intracellular trafficking, secretion, and vesicular transport (4.22%), P– Inorganic ion transport and metabolism (3.01%), F- Nucleotide transport and metabolism (2.41%), O- Post-translational modification, protein turnover, and chaperones (2.41%), and R- General function prediction only (2.41%). Also, the low abundant COG categories with only one gene each were CP, HR, JO, JT, Q (Secondary metabolites biosynthesis, transport, and catabolism) and TK (Fig. 4). Similar to our findings, the most commonly enriched COG in essential genes of other bacteria like Porphyromonas gingivalis, Herbaspirillum seropedicae, Vibrio cholera, Rhodopseudomonas palustris, Burkholderia cenocepacia, and the synthetic bacteria Mycoplasma mycoides JCVI-syn3.0 were related to translation, ribosomal structure and biogenesis (J), cell wall/membrane/envelope biogenesis (M), and coenzyme transport and metabolism (H) [8, 9, 24, 37–39].
Fig. 4.
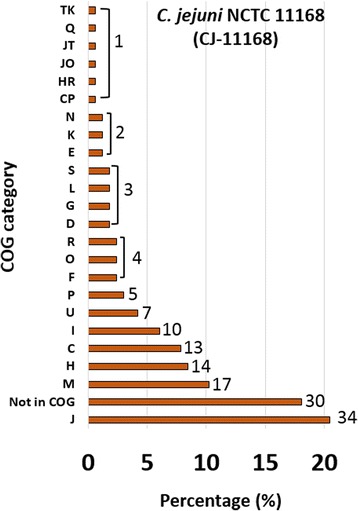
Cluster of orthologous group (COG) categories of essential genes of C. jejuni NCTC 11168 (CJ-11168). Essential genes were defined as genes without any Tn5 insertions in central 80% of genes among all the three libraries of C. jejuni NCTC 11168 (S-CJ11168, S-CJ11168-D, and R-CJ11168-D). The number at the top of bar is the number of essential genes in that COG category. J- Translation, ribosomal structure and biogenesis; X– Not in COG; M- Cell wall/membrane/envelope biogenesis; H- Coenzyme transport and metabolism; C- Energy production and conversion; I- Lipid transport and metabolism; U- Intracellular trafficking, secretion, and vesicular transport; P– Inorganic ion transport and metabolism; F- Nucleotide transport and metabolism; O- Post-translational modification, protein turnover, and chaperones; R- General function prediction only; D– Cell cycle control, cell division, chromosome partitioning; G- Carbohydrate metabolism and transport; L- Replication, recombination and repair; S- Function unknown; E- Amino acid transport and metabolism; K- Transcription; N- Cell motility; V- Defense mechanisms; Q- Secondary metabolites biosynthesis, transport, and catabolism; and T- Signal transduction mechanisms
Interestingly, we identified 20 essential transfer RNA (tRNA) out of a total of 43 tRNA genes in the entire genome of C. jejuni NCTC 11168 and none of the ribosomal RNA genes encoding 16S ribosomal RNA, 23S ribosomal RNA, and 5S ribosomal RNA were identified as essential (Additional file 2: SH3). The 20 essential tRNA genes are those that are required for the transfer of arginine, asparagine, aspartic acid, glutamic acid, glycine, leucine, lysine, methionine, selenocysteine, serine, tryptophan, tyrosine, and valine during protein synthesis. Another essential ncRNA was rnpB which is a component RNA of the ribonuclease P enzyme (RNase P) together with the RnpA protein (also identified as an essential gene in our study) functions in the processing of 4.5S RNA and tRNA precursor molecules in E. coli (https://ecocyc.org/gene?orgid=ECOLI&id=EG30069). Similar to our findings, in a recent study, Rosconi et al. (2016) found 22 tRNA genes and one 23S ribosomal RNA gene to be essential in Herbaspirillum seropedicae, an endophyte that colonizes crops like rice and maize, during in vitro growth in TY medium [38].
Reconstruction of insertion libraries in a different strain background (C. jejuni 81–176)
We transformed the seed library genomic DNA through natural transformation into a different strain background (C. jejuni 81–176). Homologous recombination of the seed library genomic DNA in a different strain requires significant homology between the incoming DNA fragments and the target regions at the DNA level. C. jejuni strains have a high level of genomic diversity, and Poly et al. (2005) previously reported that C. jejuni 81–176 had 63 kb of new chromosomal DNA sequences unique to this strain, which contains 86 open reading frames, when compared to C. jejuni NCTC 11168, based on a shotgun DNA microarray analysis [40]. To identify the genomic region in the donor strain with significant homology to the recipient genome, DNA sequences that consist of 1 kb flanking upstream and downstream of each coding sequence (CDS) in C. jejuni NCTC 11168 along with the CDS were BLASTed against C. jejuni 81–176. This analysis produced 1535 best BLAST hits based on the highest bit score for each query sequence. Further filtering (described in the Materials and Methods section) resulted in 904 query genes with substantial similarity of which 895 are orthologous genes commonly present in both C. jejuni 11,168 and C. jejuni 81–176 genomes (Additional file 3: SH1). This process was designed to eliminate the genes that are unique to the C. jejuni 81–176 genome from the downstream analysis so that we can prevent or minimize the false identification of the genes with zero Tn5 insertions as essential genes, where the absence of insertion is in fact due to the lack of identity.
Out of 895 orthologous genes with significant similarity including 1 kbp flanking sequences, 384 genes had zero Tn5 insertions (Additional file 3: SH2) and are thus considered to be essential genes required for the in vitro growth of C. jejuni 81–176 on rich MH agar at 37 °C under microaerophilic conditions. These 384 essential genes (CJ-81176) of C. jejuni 81–176 were categorized into cluster of orthologous groups (COGs). The highly enriched COGs were: J- Translation, ribosomal structure and biogenesis (16.67%), Not in COG (13.28%), C- Energy production and conversion (7.55%), H- Coenzyme transport and metabolism (6.25%), R- General function prediction only (5%), E- Amino acid transport and metabolism (5.73%), S- Function unknown (4.95%), M- Cell wall/membrane/envelope biogenesis (4.43%), and F- Nucleotide transport and metabolism (4.17%). Other moderately enriched COGs ranging from 3.5% to 2.5% in abundance were: posttranslational modification, protein turnover, chaperones (O), inorganic ion transport and metabolism (P), replication, recombination and repair (L), intracellular trafficking, secretion, and vesicular transport (U), cell cycle control, cell division, chromosome partitioning (D), carbohydrate transport and metabolism (G), lipid transport and metabolism (I); and transcription (K). The other COGs with 1 or 2 genes were 12.50% of the total number of genes (Additional file 1: Figure S3).
Both strains, C. jejuni NCTC 11168 and C. jejuni 81–176, had similar levels of enrichment in the COGs including: nucleotide transport and metabolism (F), transcription (K), replication, recombination and repair (L), posttranslational modification, protein turnover, chaperones (O), cell cycle control, cell division, chromosome partitioning (D), carbohydrate transport and metabolism (G), inorganic ion transport and metabolism (P), energy production and conversion (C), and intracellular trafficking, secretion, and vesicular transport (U) with differences in relative abundance ranging from −1.76% to +1.62%. The COG category such as ‘Amino acid transport and metabolism’ (E) was relatively higher in C. jejuni 81–176 by 4.53% than in C. jejuni NCTC 11168, and ‘Lipid transport and metabolism’ (I) and ‘Cell wall/membrane/envelope biogenesis’ (M) were higher in C. jejuni NCTC 11168 by 3.68% and 5.81% respectively than in C. jejuni NCTC 81–176 (Fig. 5). These differences may have been due to the considerable variation in the genomic backgrounds of the two strains, which in turn likely reflects differences in the regulatory networks and their responses to environmental stimuli such as the availability of nutrients and temperature. In addition, the fact that the essential genes in C. jejuni 81–176 were identified only from the genomic regions common in both strains could have resulted in some bias in the overall enrichment procedure.
Fig. 5.
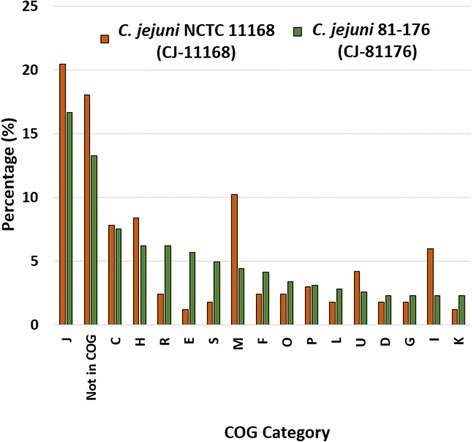
Comparison of major COG categories enriched in the essential genome of C. jejuni NCTC 11168 (CJ11168) and C. jejuni 81–176 (CJ81176). COG categories annotations are the same as in Fig. 4
Comparative study
Two research groups have previously reported on the essential genes in C. jejuni NCTC 11168 on a genome-wide scale. Surprisingly, there is only a limited correlation between the essential genes compliment identified in the reports by Metris et al. (2011) and Stahl and Stintzi (2011). We compared the essential genes of C. jejuni previously identified by Metris et al. (2011) and Stahl and Stintzi (2011) with the findings in our current study. Since the level of library saturation is very much higher in our study, we expected that our result would show significantly higher overlaps with each of these previous reports. However, there were limited overlaps in the essential genes of C. jejuni NCTC 11168 among the two previous studies and our current study as shown in Fig. 6a. Likely explanations are: 1) a limited number of transposon mutants (~ 10,000 mutants) in previous studies, 2) differences in growth conditions: 37 °C vs. 42 °C, and MH agar vs. Blood agar, and 3) techniques used for transposon insertion site mapping: microarray used in previous studies vs. next-generation sequencing in this study. These arguments are also substantiated by Gao et al. (2014).
Fig. 6.
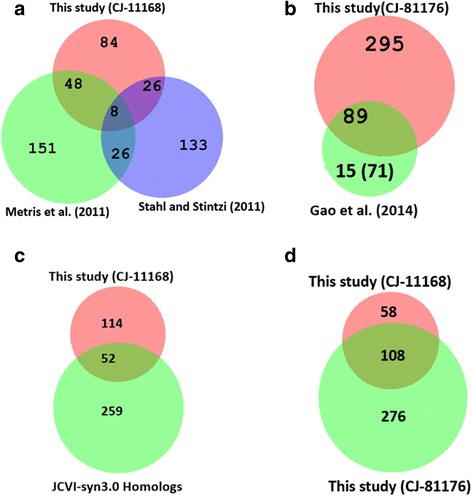
Venn diagrams indicating the numbers of shared essential genes of C. jejuni found between this and previous studies. a Essential genes of C. jejuni NCTC 11168 common between Metris et al. (2011), Stahl and Stintzi (2011) and this study (CJ-11168). b Essential genes of C. jejuni 81–176 common between Gao et al. (2014) and this study (CJ-81176). Number of genes inside small bracket did not have highly homologous sequences in the C. jejuni NCTC 11168 background according to our criteria. c Common genes shared between this study (CJ-11168) and homologous proteins of JCVI-syn3.0 against C. jejuni NCTC 11168. d Common essential genes between the two Campylobacter strains identified in this study (CJ-11168 - C. jejuni NCTC 11168 and CJ-81176 - C. jejuni 81–176)
Interestingly, substantial overlapping was observed between the essential genes of C. jejuni 81–176 identified in this study and the previous report by Gao et al. (2014) as shown in Fig. 6b, despite the considerable differences in experimental procedures. Twenty-three percent of essential genes from this study were common to Gao et al. (2014) and more than 50% of the genes identified by Gao et al. (2014) were identified in our study. The primary source of variation with the previous study (Gao et al. 2014) was the method of transposon library construction. In our study, genomic DNA from the C. jejuni NCTC 11168 Tn5 library was naturally transformed into C. jejuni 81–176 to create the Tn5 mutant library. Thus, Tn5 mutagenesis occurred only at the genomic loci in C. jejuni 81–176 with a significant amount of homology between the two strains.
Additionally, we were interested in comparing our results with the essential genes used for the creation of JCVI-syn3.0, the first synthesized minimal bacterium with the smallest genome (473 genes) capable of self-replication in laboratory media. C. jejuni NCTC 11168 protein coding genes were searched for homologous proteins against the JCVI-syn3.0 genome using BLASTp with a similarity score cutoff of 1e−5 and 311 homologous proteins were found in C. jejuni NCTC 11168. Among the 311 homologous proteins of C. jejuni, 52 genes encoding these proteins overlapped with the essential genes of C. jejuni NCTC 11168 identified in our study (Fig. 6c). As expected, little overlap was seen between the two distantly related bacteria probably due to the significant differences in their genetic networks and physiology.
However, there was substantial overlap between essential genes of C. jejuni NCTC 11168 and C. jejuni 81–176 (Fig. 6d). Approximately, 65% of the essential genes of C. jejuni NCTC 11168 were common to C. jejuni 81–176. In contrast, fewer essential genes of C. jejuni 81–176 were shared (~28%) with C. jejuni NCTC 11168. This could be partly due to the assumption that the genes with no Tn5 insertion are essential genes, which may have led to the inclusion of some nonessential genes in the list of essential genes for C. jejuni 81–176. This difference may be also due to a lack of sufficient nucleotide sequence identity between genes in the two different strain backgrounds of C. jejuni, despite extensive filtering for homologous sequences to reduce the noise in the data analysis. Another important factor that may have contributed to the identification of false essential genes in C. jejuni 81–176, may be the insufficient number of Tn5 mutants collected during the reconstruction of the library.
Notably, the essential genes of C. jejuni NCTC 11168 identified in this study had an extensive homolog hits in the Database of Essential Genes (DEG, http://www.essentialgene.org. DNA sequences of 166 essential genes of C. jejuni NCTC 11168 were found using BLASTx among the essential genes from 46 bacterial species in the database with default parameters (Expect - 1E-05, Score - 100, and Matrix –BLOSUM62). Out of 166 essential protein-coding genes of C. jejuni NCTC 11168, 135 genes had homologs among the DEG and 2879 DEG genes had homology with our essential genes of C. jejuni NCTC 11168. Most of the essential genes with no hits in the DEG were hypothetical proteins (15 genes). However, six integral membrane proteins (Cj0369c, Cj0423, Cj0430, Cj0544, Cj0564, and Cj0851c), three periplasmic proteins (Cj0659c, Cj0854c, and Cj0114), and other genes (pseH and rnpA) were also essential genes in this study with no hit in the DEG.
Next, we studied the core essential genes of C. jejuni through comparative analysis of all essential genes identified in this study (C. jejuni NCTC 11168 and C. jejuni 81–176), previous studies by Metris et al. (2011), Stahl and Stintzi (2011), Gao et al. (2014), and those in the synthetic bacterium, JCVI-syn3.0 (Fig. 7 and Additional file 4). Essential genes of C. jejuni 81–176 orthologous to C. jejuni NCTC 11168 strain and proteins of JCVI-syn3.0 homologous to C. jejuni NCTC 11168 were considered. There were 50 genes common to the six studies with each of these genes being shared amongst at least four of the studies (Fig. 7). Most of these genes belonged to COG category ‘translation, ribosomal structure and biogenesis’ (J; 34%), ‘carbohydrate metabolism and transport’ (G; 10%), followed by genes ‘Not in COG’ (8%), ‘coenzyme transport and metabolism’ (H; 6%) and ‘intracellular trafficking, secretion, and vesicular transport’ (U; 6%). The importance of category J being a large portion of the core essential genes is further supported by the fact that ribosomal proteins are the most prominent drug targets in bacteria that have been used to control infections [41].
Fig. 7.
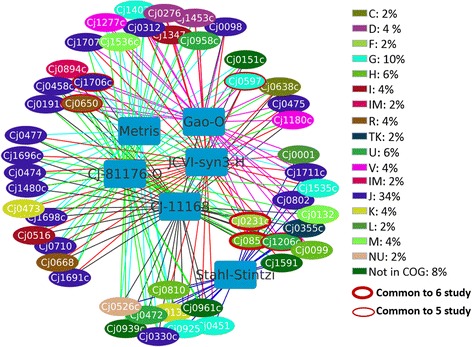
Core essential genes of C. jejuni NCTC 11168. Genes are colored to indicate the COG category. Numbers after the legend indicate percentages of the COG category enriched in the core essential gene list. Soft edge rectangle- various studies: Metris [26], Stahl-Stintzi [25], Gao-O [27], JCVI-syn3-H [24], CJ-81176-O: C. jejuni 81–176 (this study), and CJ-11168: C. jejuni NCTC 11168 (this study). O and H in the study name indicate orthologs and homologs respectively. Genes circled in thick red and thin red are common in six and five studies, respectively. All the genes in this network were identified as essential in at least four of the studies
NrdF and folD were identified as essential in all of the above 6 studies while ftsY, fba, engB and rplD were identified as essential in 5 of the studies (Fig. 7 and see Additional file 4 for corresponding gene name and locus_tag). A live attenuated Salmonella Typhimurium aroA vector expressing the Mycoplasma hyopneumoniae ribonucleotide reductase R2 subunit (NrdF) gene was shown to induce a cell-mediated immune response [42]. A deletion mutant of ftsY, the gene encoding a signal recognition particle protein, in Streptococcus pneumonia induced potent serotype-independent protection against otitis media, sinusitis, pneumonia and invasive pneumococcal disease [43]. fba encodes the class II fructose 1,6-bisphosphate aldolase enzyme, which is important for bacterial, fungal and protozoan glycolysis and gluconeogenesis and is considered as a putative drug target against Mycobacterium tuberculosis, the causative agent of tuberculosis [44].
Many other essential Campylobacter genes shared among at least 4 of the above studies have been used as drug targets and vaccine constructions to mitigate several bacterial infections. BirA, a biotin protein ligase, is an emerging drug target against E. coli and other pathogens such as Staphylococcus aureus and M. tuberculosis. Inhibition of lpxC, an enzyme that has a role in lipid A biosynthesis, by a small-molecule antibiotic in Acinetobacter baumannii, protected mice from infection by modulation of inflammation and enhancing opsonophagocytic killing [45]. MreB, a rod shape-determining protein, when blocked by MreB-specific antibiotics inhibited the growth of Chlamydia [46]. IspH (4-hydroxy-3-methylbut-2-enyl diphosphate reductase) satisfied all criteria of being a putative drug target against Corynebacterium pseudotuberculosis, a pathogenic bacterium that causes caseous lymphadenitis (CLA), ulcerative lymphangitis, mastitis, and edematous in a broad spectrum of hosts [47]. KsgA (rRNA small subunit methyltransferase A) has been associated with clarithromycin resistance in M. tuberculosis [48]. A GalU (UTP-glucose-1-phosphate uridylyltransferase) mutation in Francisella tularensis, the causative agent of tularemia, was protective against homologous challenge in mice [49]. GapA (glyceraldehyde 3-phosphate dehydrogenase) was also used in the construction of an effective DNA vaccine against Haemophilus parasuis, the causative agent of swine polyserositis, polyarthritis, and meningitis [50].
Conclusion
We constructed an unprecedentedly complex Tn5 library of C. jejuni NCTC 11168 with more than 95,000 unique insertions in the genome. The genomic DNA of the seed library was effectively used for reconstruction of a Tn5 library in the same strain background (C. jejuni NCTC 11168) and with limited success in a different strain background (C. jejuni 81–176). Despite discrepancies among studies, comparative analysis conducted in this study showed the essential genes of C. jejuni were also found to be essential in other bacterial species and many of the genes have been exploited as drug target and in vaccine development against a wide range of bacterial diseases. By definition, the absence of essential genes have the potential to severely affect the survival of a bacterium, therefore, these genes can be further exploited to develop novel strategies to curb this important foodborne pathogen.
Methods
Bacteria strains and growth conditions
C. jejuni NCTC11168 and C. jejuni 81–176 were grown on Muller-Hinton (MH) agar plates at 37 °C under microaerophilic conditions (O2–5%, CO2–10%, and N2- Balance). Trimethoprim (TMP, 10 μg/ml,) and Kanamycin (Km, 50 μg/ml) were added to the MH agar when required. Bacterial pellets and extracted DNA were stored at −20 °C. C. jejuni frozen stocks were stored at −80 °C in 50% glycerol.
Construction of the Tn5 seed library
The Tn5 transposon mutant library of C. jejuni NCTC11168 was generated using EZ-Tn5™ < KAN-2 > Tnp Transposome™ Kit (Cat. No. TSM99K2, Epicentre Biotechnologies, Madison, WI, USA) following the manufacturer’s protocol. Briefly, in vitro transposition reactions consisted of 2 μl 10× EZ-Tn5 reaction buffer, 1 μl of transposome complex, 2 μg of chromosomal DNA, and 15 μl of distilled deionized H2O that were incubated for 4 h at 37 °C. The transposed DNA was purified after adding 60 μl of distilled deionized H2O followed by phenol-chloroform extraction and then ethanol precipitation of DNA. DNA was recovered in 40 μl TE buffer (pH 8.0). Next, in vitro transposed DNA was repaired by adding 40 μl of transposed DNA, 6 μl of T4 DNA polymerase buffer (New England Biolabs, NEB), 4.8 μl of dNTPs mix (2.5 mM), 7.7 μl distilled H2O, and 1.5 μl T4 DNA polymerase (1 U/ μl, NEB) and incubating at 11 °C for 20 min in a thermal cycler. The reaction was inactivated by incubating at 75 °C for 15 min. The second repair reaction consisted of a 60 μl reaction mixture (previous reaction), 12 μl T4 DNA ligase buffer (NEB), 1.5 μl T4 DNA ligase (NEB) and 46.5 μl dH2O that was incubated overnight at 16 °C. This was followed by DNA dialysis on top of a nitrocellulose membrane floating on 10–20 ml distilled deionized water for 20 min. All of the reaction was used for one transformation of C. jejuni NCTC11168 following the natural transformation method described by Davis et al. 2008 (briefly explained in the next section) [25]. Naturally transformed C. jejuni NCTC11168 were selected on MH agar plates with TMP and Km. The mutants were scrapped off the plate into 1× PBS, centrifuged, and the pellet was stored at −80 °C. We performed 14 transformations with each producing ~100,000 mutants. An equal number of mutants from each transformation was combined to create Tn5 seed mutant library (the seed library) as shown in Fig. 1.
Reconstruction of Tn5 libraries
Genomic DNA extracted from the complex Tn5 seed library (Tn5 seed library DNA) was used for reconstruction of Tn5 libraries in the same and different strain backgrounds of C. jejuni. The Tn5 seed library was naturally transformed into C. jejuni NCTC11168 and C. jejuni 81–176 backgrounds following the natural transformation protocol for C. jejuni as described by Davis et al. 2008 [51]. Briefly, C. jejuni strains from frozen stock were streaked on MH agar containing TMP and incubated for 16 h under microaerophilic conditions at 37 °C. Next day, a heavy inoculum from the plate was streaked on MH agar with TMP and incubated for 16 h. The bacterial growth from the 16 h growth plate was resuspended in 1 ml MH broth without antibiotics and the OD600 was adjusted to 0.5 in MH broth. One ml fresh melted MH agar (without antibiotics) was pipetted into a 5 ml plastic test-tube and was allowed to solidify. An aliquot of 0.5 ml bacteria adjusted to OD600 was added into a test-tube containing 1 ml of solidified MH agar and mixed gently and incubated for 3 h at 37 °C in microaerophilic conditions (bi-phasic medium). Following this, 500 μg of seed library DNA was added to the biphasic medium and it was incubated for 4 h in the above conditions. The transformants were collected in a microcentrifuge tube, centrifuged for 2 min and resuspended in MH broth. Finally, the transformants were plated directly or after serial dilutions on MH agar plates supplemented with TMP and Km and incubated for 2 days. The colonies were counted from dilution plates and collected from direct plates in 1X PBS, and centrifuged. The supernatant was discarded and the bacterial pellet was stored at −20 °C (Fig. 1).
Transposon junction amplification and sequencing
Genomic DNA was extracted from the bacterial pellets of complex Tn5 libraries using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol. Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA) was used to check the concentration and purity of extracted DNA. Tn-seq DNA libraries for Illumina sequencing were prepared using the previously developed protocol in our laboratory [52] with minor modifications as described in detail in Additional file 1 (Protocol S1). The first step was a single primer extension step using a primer specific to one end of the transposon. In this study, for each library DNA, the linear extension step was performed either a regular primer, dual priming oligonucleotide (DPO) primer or both. DPO primers were designed as described by Chun et al. (2007) [32] and used to increase the specificity of PCR amplification of the Tn5-chromosome junction sequences. The regular primer (5′-GATCCTCTAGAGTCGACCTGCAGGCATGCA-5′) and DPO primer (5′-ACCGTGGCGGGGATCCTCTAGAGTIIIIITGCAGGCAT-3′) were located 32 bp and 35 bp upstream of the Tn5-genome junction, respectively (Additional file 1: Fig. S4). Briefly, either regular or DPO primers were used for linear extension PCR using GoTaq G2 hot start colorless master mix (Promega Corporation, Madison, USA). The linear extension was followed by addition of a C tail and then exponential PCR to amplify Tn5-genome junction sequences. Then the product was heated at 65 °C for 15 min, mixed with loading buffer and the PCR products were run on 1% agarose gel. DNA from 300 to 500 bp were gel-purified using Zymoclean™ Gel DNA Recovery Kit following the manufacturer’s protocol (Irvine, Ca, USA). The oligonucleotide used in this study are shown in Additional file 1: Table S1. Equal quantities of DNA (10 ng) were mixed for each library and sent for Illumina HiSeq 4000 single end read sequencing with 90 cycles (DNA Technologies Core, UC Davis Genome Center, Davis, CA 95616).
Data analysis
Illumina sequencing reads were demultiplexed allowing a perfect match of barcode and transposon mosaic end using a custom Perl script (Additional file 1: Script S1). The 22 bp genomic junction sequences were extracted and used for downstream analysis in different manners for (1) the seed library and the library reconstructed in the same C. jejuni NCTC11168 strain background, and (2) the library reconstructed in a different background (C. jejuni 81–176). For the libraries in the C. jejuni NCTC11168 background, the junction sequences were aligned to the complete sequence of the same genome using Bowtie version 2.2.8 [53]. The sequence alignment map (SAM) files were then inputted into EL-ARTIST for the analysis of the essential genome following the user manual [54]. Briefly, the C. jejuni NCTC11168 Tn5 library mapped to 400 bp genomic windows and the C. jejuni 81–176 Tn5 library to 500 bp genomic windows. Insertion sites were linked to their associated annotated genomic features. There was no obvious insertion bias according to the insertion sites with respect to the replication of origin (Fig. 2a, b, c, and d). Thus, raw data were used for downstream analysis. Then, sliding window analysis was used to define regions with lower read counts, which were used to train a hidden Markov model to predict each window to be essential or non-essential for growth. Because of the differences in read numbers and complexity of the Tn5 library, we used sliding windows of different sizes and different p-value thresholds for calling a region significantly underrepresented in reads, appropriate for each Tn5 library (Additional file 2: SH1).
Additionally, we used Tn-seq Explorer to assign the number of unique Tn5 insertions sites and read counts to each gene using Bowtie aligned SAM files [55]. Tn5 insertion read counts and unique insertion sites were only considered in the central 80% of the gene (CDS) excluding insertions from the beginning and end 10% of the gene. While, for transfer RNA (tRNA), ribosomal (rRNA) and pseudogenes, unique insertion and read counts were considered for the whole gene length.
In contrast, to the libraries in the C. jejuni 11,168 background, we had to employ a different strategy for the downstream analysis using the 22 bp junction sequences for the library reconstructed in the C. jejuni 81–176 background due to the previously known differences in the genomic regions [40]. For this library, genomic DNA from the seed library (C. jejuni NCTC 11168) was transferred to C. jejuni 81–176 through natural transformation. For the incoming genomic DNA fragments containing Tn5 insertions to integrate into the recipient genome, there must be sufficient homology between the two strains at the DNA sequence level. To determine the homology levels in C. jejuni 81–176, DNA sequences flanking 1000 bp upstream and downstream sequences of the coding sequences (CDS) were extracted along with the CDS for all genes in C. jejuni NCTC 11168 using a custom Python script (Additional file 1: Script S2) and these NCTC 11168 sequences were BLASTed against C. jejuni 81–176 genome using the BLASTn from the command-line interface to display result in tabular output format 6. Single best BLAST hits were kept for each query sequence based on the highest bit score. Then, BLAST output tables were filtered so that genes having the highest homology with flanking sequences were retained. We arbitrarily used the following combination of condition to filter the BLAST tabular output file: alignment over >55% of query length, percent identity ≥98%, mismatches <50 nucleotide and gaps <5 with were kept. Next, the genes of C. jejuni NCTC 11168 having the highest probability of homologous recombination in C. jejuni 81–176 were searched again for orthologous genes present in C. jejuni 81–176. Only the orthologous genes with flanking sequences of C. jejuni 81–176 having high homology in C. jejuni NCTC 11168 were considered for the analysis of the essential genome (Additional file 3: SH1).
Additional files
This pdf file contains supplementary figures (Figures S1-S6), a detailed protocol for DNA library preparation (Protocol S1), Table S1, and Perl and Python scripts (Script S1 and S2). Figure legends are available within file. (PDF 662 kb)
Essential genes of C. jejuni NCTC 11168. This xlsx file contains Microsoft Excel spreadsheet: SH1-SH4. (XLSX 342 kb)
Essential genes of C. jejuni 81–176. This xlsx file contains Microsoft Excel spreadsheet: SH1 and SH2. (XLSX 236 kb)
Comparative analysis of the essential genome of C. jejuni with previously identified essential genes. (XLSX 68 kb)
Acknowledgements
We would like to thank Arkansas Biosciences Institute (ABI) for funding support and Arkansas High Performance Computing Center (AHPCC) - University of Arkansas for their computational support.
Funding
The research was supported by Arkansas Bioscience Institute (ABI) grant.
Availability of data and materials
The supplementary information is provided in the form of Additional files 1, 2, 3, and 4. The Tn-seq raw Illumina sequencing data is deposited in the NCBI Sequence Read Archive (SRA) under accession number SRR5571033 and SRR5572448.
Authors’ contributions
YMK and RKM- Conceived the study; RKM and TJ- performed the experiments; RKM- analyzed the data and drafted the manuscript; YMK- revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4032-8) contains supplementary material, which is available to authorized users.
Contributor Information
Rabindra K. Mandal, Phone: (479) 276-2203, Email: rabindra.mandal@louisville.edu
Tieshan Jiang, Email: txj001@uark.edu.
Young Min Kwon, Email: ykwon@uark.edu.
References
- 1.Kaakoush NO, Castano-Rodriguez N, Mitchell HM, Man SM. Global epidemiology of Campylobacter infection. Clin Microbiol Rev. 2015;28(3):687–720. doi: 10.1128/CMR.00006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Epps SV, Harvey RB, Hume ME, Phillips TD, Anderson RC, Nisbet DJ. Foodborne Campylobacter: infections, metabolism, pathogenesis and reservoirs. Int J Environ Res Public Health. 2013;10(12):6292–6304. doi: 10.3390/ijerph10126292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Juhas M, Eberl L, Church GM. Essential genes as antimicrobial targets and cornerstones of synthetic biology. Trends Biotechnol. 2012;30(11):601–607. doi: 10.1016/j.tibtech.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Xu P, Ge X, Chen L, Wang X, Dou Y, Xu JZ, Patel JR, Stone V, Evans K, Kitten T. Genome-wide essential gene identification in Streptococcus sanguinis. Sci Rep. 2011;1:125. doi: 10.1038/srep00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Berardinis V, Vallenet D, Castelli V, Besnard M, Pinet A, Cruaud C, Samair S, Lechaplais C, Gyapay G, Richez C, Durot M, Kreimeyer A, Le Fevre F, Schachter V, Pezo V, Doring V, Scarpelli C, Medigue C, Cohen GN, Marliere P, Salanoubat M, Weissenbach J. A complete collection of single-gene deletion mutants of Acinetobacter baylyi ADP1. Mol Syst Biol. 2008;4:174. doi: 10.1038/msb.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porwollik S, Santiviago CA, Cheng P, Long F, Desai P, Fredlund J, Srikumar S, Silva CA, Chu W, Chen X. Defined single-gene and multi-gene deletion mutant collections in Salmonella enterica sv Typhimurium. PLoS One. 2014;9(7):e99820. doi: 10.1371/journal.pone.0099820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pechter KB, Gallagher L, Pyles H, Manoil CS, Harwood CS. Essential genome of the metabolically versatile Alphaproteobacterium Rhodopseudomonas palustris. J Bacteriol. 2015;198(5):867–876. doi: 10.1128/JB.00771-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong Y, El Ghany MA, Naeem R, Lee K, Tan Y, Pain A, Nathan S. Candidate essential genes in Burkholderia cenocepacia J2315 identified by genome-wide TraDIS. Front Microbiol. 2016;7:1288. doi: 10.3389/fmicb.2016.01288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ji Y, Zhang B, Van SF. Horn, Warren P, Woodnutt G, Burnham MK, Rosenberg M: identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science. 2001;293(5538):2266–2269. doi: 10.1126/science.1063566. [DOI] [PubMed] [Google Scholar]
- 11.Juhas M, Eberl L, Glass JI. Essence of life: essential genes of minimal genomes. Trends Cell Biol. 2011;21(10):562–568. doi: 10.1016/j.tcb.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Mushegian AR, Koonin EV. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci U S A. 1996;93(19):10268–10273. doi: 10.1073/pnas.93.19.10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng J. An integrated machine-learning model to predict prokaryotic essential genes. Gene Essent Methods Protoc. 2015:137–51. https://www.ncbi.nlm.nih.gov/pubmed/25636617. [DOI] [PubMed]
- 14.Perumal D, Samal A, Sakharkar KR, Sakharkar MK. Targeting multiple targets in Pseudomonas aeruginosa PAO1 using flux balance analysis of a reconstructed genome-scale metabolic network. J Drug Target. 2011;19(1):1–13. doi: 10.3109/10611861003649753. [DOI] [PubMed] [Google Scholar]
- 15.Wei W, Ning L, Ye Y, Guo F. Geptop: a gene essentiality prediction tool for sequenced bacterial genomes based on orthology and phylogeny. PLoS One. 2013;8(8):e72343. doi: 10.1371/journal.pone.0072343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long JL. Gene essentiality: methods and protocols: Humana press. 2015. [Google Scholar]
- 17.Kwon YM, Ricke SC, Mandal RK. Transposon sequencing: methods and expanding applications. Appl Microbiol Biotechnol. 2016;100(1):31–43. doi: 10.1007/s00253-015-7037-8. [DOI] [PubMed] [Google Scholar]
- 18.Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, Gordon JI. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe. 2009;6(3):279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langridge GC, Phan MD, Turner DJ, Perkins TT, Parts L, Haase J, Charles I, Maskell DJ, Peters SE, Dougan G, Wain J, Parkhill J, Turner AK. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 2009;19(12):2308–2316. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gawronski JD, Wong SM, Giannoukos G, Ward DV, Akerley BJ. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc Natl Acad Sci U S A. 2009;106(38):16422–16427. doi: 10.1073/pnas.0906627106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallagher LA, Shendure J, Manoil C. Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. MBio. 2011;2(1):e00315–e00310. doi: 10.1128/mBio.00315-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods. 2009;6(10):767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wetmore KM, Price MN, Waters RJ, Lamson JS, He J, Hoover CA, Blow MJ, Bristow J, Butland G, Arkin AP, Deutschbauer A. Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. MBio. 2015;6(3):e00306–e00315. doi: 10.1128/mBio.00306-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hutchison CA, Chuang R, Noskov VN, Assad-Garcia N, Deerinck TJ, Ellisman MH, Gill J, Kannan K, Karas BJ, Ma L. Design and synthesis of a minimal bacterial genome. Science. 2016;351(6280):aad6253. doi: 10.1126/science.aad6253. [DOI] [PubMed] [Google Scholar]
- 25.Stahl M, Stintzi A. Identification of essential genes in C. jejuni genome highlights hyper-variable plasticity regions. Funct Integr Genomics. 2011;11(2):241–257. doi: 10.1007/s10142-011-0214-7. [DOI] [PubMed] [Google Scholar]
- 26.Metris A, Reuter M, Gaskin DJ, Baranyi J, van Vliet AH. In vivo and in silico determination of essential genes of Campylobacter jejuni. BMC Genomics. 2011;12(1):1. doi: 10.1186/1471-2164-12-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao B, Lara-Tejero M, Lefebre M, Goodman AL, Galan JE. Novel components of the flagellar system in Epsilonproteobacteria. MBio. 2014;5(3):e01349–e01314. doi: 10.1128/mBio.01349-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin J, Wang Y, Van Hoang K. Systematic identification of genetic loci required for polymyxin resistance in Campylobacter jejuni using an efficient in vivo transposon mutagenesis system. Foodborne Pathog Dis. 2009;6(2):173–185. doi: 10.1089/fpd.2008.0177. [DOI] [PubMed] [Google Scholar]
- 29.Reid AN, Pandey R, Palyada K, Whitworth L, Doukhanine E, Stintzi A. Identification of Campylobacter jejuni genes contributing to acid adaptation by transcriptional profiling and genome-wide mutagenesis. Appl Environ Microbiol. 2008;74(5):1598–1612. doi: 10.1128/AEM.01508-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hendrixson DR, Akerley BJ, DiRita VJ. Transposon mutagenesis of Campylobacter jejuni identifies a bipartite energy taxis system required for motility. Mol Microbiol. 2001;40(1):214–224. doi: 10.1046/j.1365-2958.2001.02376.x. [DOI] [PubMed] [Google Scholar]
- 31.Colegio OR. Griffin TJ,4th, Grindley ND, Galan JE: in vitro transposition system for efficient generation of random mutants of Campylobacter jejuni. J Bacteriol. 2001;183(7):2384–2388. doi: 10.1128/JB.183.7.2384-2388.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chun JY, Kim KJ, Hwang IT, Kim YJ, Lee DH, Lee IK, Kim JK. Dual priming oligonucleotide system for the multiplex detection of respiratory viruses and SNP genotyping of CYP2C19 gene. Nucleic Acids Res. 2007;35(6):e40. doi: 10.1093/nar/gkm051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parkhill J, Wren B, Mungall K, Ketley J, Churcher C, Basham D, Chillingworth T, Davies R, Feltwell T, Holroyd S. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403(6770):665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 34.Green B, Bouchier C, Fairhead C, Craig NL, Cormack BP. Insertion site preference of mu, Tn5, and Tn7 transposons. Mob DNA. 2012;3(1):1. doi: 10.1186/1759-8753-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han F, Pu S, Wang F, Meng J, Ge B. Fitness cost of macrolide resistance in Campylobacter jejuni. Int J Antimicrob Agents. 2009;34(5):462–466. doi: 10.1016/j.ijantimicag.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 36.Dryselius R, Izutsu K, Honda T, Iida T. Differential replication dynamics for large and small Vibrio chromosomes affect gene dosage, expression and location. BMC Genomics. 2008;9(1):1. doi: 10.1186/1471-2164-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein BA, Tenorio EL, Lazinski DW, Camilli A, Duncan MJ, Hu LT. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics. 2012;13:578. doi: 10.1186/1471-2164-13-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosconi F, de Vries SP, Baig A, Fabiano E, Grant AJ. Essential genes for in vitro growth of the endophyte Herbaspirillum seropedicae SmR1 revealed by transposon insertion sequencing. Appl Environ Microbiol. 2016;82(22):6664–6671. doi: 10.1128/AEM.02281-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chao MC, Pritchard JR, Zhang YJ, Rubin EJ, Livny J, Davis BM, Waldor MK. High-resolution definition of the Vibrio cholerae essential gene set with hidden Markov model-based analyses of transposon-insertion sequencing data. Nucleic Acids Res. 2013;41(19):9033–9048. doi: 10.1093/nar/gkt654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poly F, Threadgill D, Stintzi A. Genomic diversity in Campylobacter jejuni: identification of C. jejuni 81-176-specific genes. J Clin Microbiol. 2005;43(5):2330–2338. doi: 10.1128/JCM.43.5.2330-2338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gladki A, Kaczanowski S, Szczesny P, Zielenkiewicz P. The evolutionary rate of antibacterial drug targets. BMC Bioinformatics. 2013;14(1):1. doi: 10.1186/1471-2105-14-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen AY, Fry SR, Forbes-Faulkner J, Daggard GE, Mukkur T. Comparative immunogenicity of M. hyopneumoniae NrdF encoded in different expression systems delivered orally via attenuated S. Typhimurium aroA in mice. Vet Microbiol. 2006;114(3):252–259. doi: 10.1016/j.vetmic.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 43.Rosch JW, Iverson AR, Humann J, Mann B, Gao G, Vogel P, Mina M, Murrah KA, Perez AC, Edward Swords W, Tuomanen EI, McCullers JA. A live-attenuated pneumococcal vaccine elicits CD4+ T-cell dependent class switching and provides serotype independent protection against acute otitis media. EMBO Mol Med. 2014;6(1):141–154. doi: 10.1002/emmm.201202150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Capodagli GC, Sedhom WG, Jackson M, Ahrendt KA, Pegan SD. A noncompetitive inhibitor for Mycobacterium tuberculosis’s class IIa fructose 1, 6-bisphosphate aldolase. Biochem. 2013;53(1):202–213. doi: 10.1021/bi401022b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin L, Tan B, Pantapalangkoor P, Ho T, Baquir B, Tomaras A, Montgomery JI, Reilly U, Barbacci EG, Hujer K, Bonomo RA, Fernandez L, Hancock RE, Adams MD, French SW, Buslon VS, Spellberg B. Inhibition of LpxC protects mice from resistant Acinetobacter baumannii by modulating inflammation and enhancing phagocytosis. MBio. 2012;3(5) doi:10.1128/mBio.00312-12. Print 2012 [DOI] [PMC free article] [PubMed]
- 46.Ouellette SP, Karimova G, Subtil A, Ladant D. Chlamydia co-opts the rod shape-determining proteins MreB and Pbp2 for cell division. Mol Microbiol. 2012;85(1):164–178. doi: 10.1111/j.1365-2958.2012.08100.x. [DOI] [PubMed] [Google Scholar]
- 47.Hassan SS, Tiwari S, Guimarães LC, Jamal SB, Folador E, Sharma NB, de Castro SS, Almeida S, Ali A, Islam A. Proteome scale comparative modeling for conserved drug and vaccine targets identification in Corynebacterium pseudotuberculosis. BMC Genomics. 2014;15(7):1. doi: 10.1186/1471-2164-15-S7-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phunpruch S, Warit S, Suksamran R, Billamas P, Jaitrong S, Palittapongarnpim P, Prammananan T. A role for 16S rRNA dimethyltransferase (ksgA) in intrinsic clarithromycin resistance in Mycobacterium tuberculosis. Int J Antimicrob Agents. 2013;41(6):548–551. doi: 10.1016/j.ijantimicag.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 49.Jayakar HR, Parvathareddy J, Fitzpatrick EA, Bina XR, Bina JE, Re F, Emery FD, Miller MA. A galU mutant of Francisella tularensis is attenuated for virulence in a murine pulmonary model of tularemia. BMC Microbiol. 2011;11:179. doi: 10.1186/1471-2180-11-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu S, Zhang M, Ou J, Liu H, Tan C, Liu J, Chen H, Bei W. Construction and immune effect of Haemophilus parasuis DNA vaccine encoding glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in mice. Vaccine. 2012;30(48):6839–6844. doi: 10.1016/j.vaccine.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 51.Davis L, Young K, DiRita V. Genetic manipulation of Campylobacter jejuni. Curr Protoc Microbiol. 2008;8A:2.1-8A–2.2.17. doi: 10.1002/9780471729259.mc08a02s10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dawoud TM, Jiang T, Mandal RK, Ricke SC, Kwon YM. Improving the efficiency of transposon mutagenesis in Salmonella Enteritidis by overcoming host-restriction barriers. Mol Biotechnol. 2014;56(11):1004–1010. doi: 10.1007/s12033-014-9779-4. [DOI] [PubMed] [Google Scholar]
- 53.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pritchard JR, Chao MC, Abel S, Davis BM, Baranowski C, Zhang YJ, Rubin EJ, Waldor MK. ARTIST: high-resolution genome-wide assessment of fitness using transposon-insertion sequencing. PLoS Genet. 2014;10(11):e1004782. doi: 10.1371/journal.pgen.1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Solaimanpour S, Sarmiento F, Mrázek J. Tn-seq explorer: a tool for analysis of high-throughput sequencing data of transposon mutant libraries. PLoS One. 2015;10(5):e0126070. doi: 10.1371/journal.pone.0126070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This pdf file contains supplementary figures (Figures S1-S6), a detailed protocol for DNA library preparation (Protocol S1), Table S1, and Perl and Python scripts (Script S1 and S2). Figure legends are available within file. (PDF 662 kb)
Essential genes of C. jejuni NCTC 11168. This xlsx file contains Microsoft Excel spreadsheet: SH1-SH4. (XLSX 342 kb)
Essential genes of C. jejuni 81–176. This xlsx file contains Microsoft Excel spreadsheet: SH1 and SH2. (XLSX 236 kb)
Comparative analysis of the essential genome of C. jejuni with previously identified essential genes. (XLSX 68 kb)
Data Availability Statement
The supplementary information is provided in the form of Additional files 1, 2, 3, and 4. The Tn-seq raw Illumina sequencing data is deposited in the NCBI Sequence Read Archive (SRA) under accession number SRR5571033 and SRR5572448.