

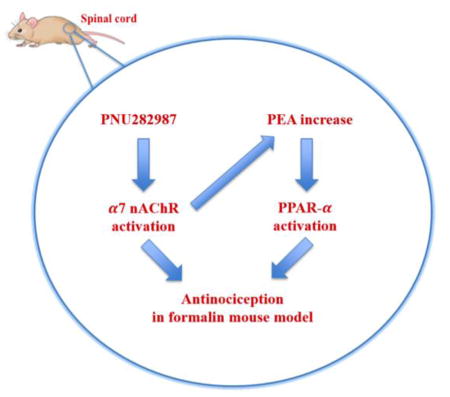
Abstract
Recently, α7 nicotinic acetylcholine receptors (nAChRs), primarily activated by binding of orthosteric agonists, represent a target for anti-inflammatory and analgesic drug development. These receptors may also be modulated by positive allosteric modulators (PAMs), ago-allosteric ligands (ago-PAMs), and α7-silent agonists. Activation of α7 nAChRs has been reported to increase the brain levels of endogenous ligands for nuclear peroxisome proliferator-activated receptors type-α (PPAR-α), palmitoylethanolamide (PEA) and oleoylethanolamide (OEA), in a Ca2+-dependent manner. Here, we investigated potential crosstalk between α7 nAChR and PPAR-α, using the formalin test, a mouse model of tonic pain. Using pharmacological and genetic approaches, we found that PNU282987, a full α7 agonist, attenuated formalin-induced nociceptive behavior in α7-dependent manner. Interestingly, the selective PPAR-α antagonist GW6471 blocked the antinociceptive effects of PNU282987, but did not alter the antinociceptive responses evoked by the α7 nAChR PAM PNU120596, ago-PAM GAT107, and silent agonist NS6740. Moreover, GW6471 administered systemically or spinally, but not via the intraplantar surface of the formalin-injected paw blocked PNU282987-induced antinociception. Conversely, exogenous administration of the naturally occurring PPAR-α agonist PEA potentiated the antinociceptive effects of PNU282987. In contrast, the cannabinoid CB1 antagonist rimonabant and the CB2 antagonist SR144528 failed to reverse the antinociceptive effects of PNU282987. These findings suggest that PPAR-α plays a key role in a putative antinociceptive α7 nicotinic signaling pathway.
Keywords: alpha7, nicotinic acetylcholine receptors, nuclear peroxisome proliferator-activated receptor type-α, palmitoylethanolamide, tonic pain, mice
Graphical abstract
1. Introduction
Nicotinic acetylcholine receptors (nAChRs) play an active role in modulating pain transmission pathways (Khan et al., 2003), in which their stimulation produces antinociception in pre-clinical and clinical pain models (Umana et al., 2013). Functional nAChRs are pentameric structures that may be homomeric, containing only α subunits, or heteromeric, containing α and β subunits (Jensen et al., 2005). Homomeric α7 nAChRs are abundantly expressed in the central and peripheral nervous systems, including neuronal and non-neuronal cells (Girod et al., 1999). These receptors represent viable drug targets for cognitive and neurodegenerative disorders, and may have potential to treat inflammatory and pain disorders.
Activation of the α7 nAChR ion channel is primarily controlled by the binding of agonists at orthosteric sites, and may also be regulated by allosteric conformational stabilization (Horenstein et al., 2016). Full and partial α7 nAChR agonists elicit significant anti-inflammatory and antinociceptive effects in several experimental models of tonic and chronic pain (Damaj et al., 2000; Feuerbach et al., 2009; Wang et al., 2005). Moreover, previous studies demonstrated that α7 nAChR selective positive allosteric modulators (PAMs) were also active in rodent models of chronic and inflammatory pain (Freitas et al., 2013; Freitas et al., 2013c; Munro et al., 2012). PAMs facilitate endogenous neurotransmission and enhance efficacy and potency of α7 nAChR agonists without directly stimulating the orthosteric binding site (Bertrand and Gopalakrishnan, 2007; Faghih et al., 2007). PAMs lack intrinsic agonist activation, but ago-allosteric ligands (ago-PAMs) exhibit dual activity, via allosteric and orthosteric interactions (Gill et al., 2011; Horenstein et al., 2016; Papke et al., 2014; Thakur et al., 2013), eliciting antinociceptive and anti-inflammatory effects in mice (Bagdas et al., 2016). A new class of modulators, α7 nAChR silent agonists, are unique in that they bind the receptor but preferentially induce non-conducting states, which modulate inflammation and nociception in rodents (Papke et al., 2015) via an unknown signaling mechanism.
Several mechanisms may mediate the anti-inflammatory properties of α7 nAChR agonists. In the central nervous system (CNS), α7 nAChRs exhibit rapid activation and desensitization, as well as high calcium permeability, leading to activation of calcium-dependent intracellular phosphatases and kinases (Feuerbach et al., 2009; Williams et al., 2011). While the intracellular pathways following α7 nAChR activation in non-neuronal cells may involve calcium influx through the channel, signaling pathways independent of ion flux were also reported (Papke et al., 2014). Recently, α7 nAChRs pharmacological activation have been found to increase the brain levels of palmitoylethanolamide (PEA) and oleoylethanolamide (OEA), endogenous agonists for nuclear peroxisome proliferator-activated receptor type-α (PPAR-α), in a Ca2+-dependent manner (Melis et al., 2013). These findings suggest a pharmacological crosstalk between PPAR-α and α7 nAChRs in the CNS.
Based on the research outlined above, we hypothesized that PPAR-α activation might represent a novel pathway that mediates analgesic effects of α7 nAChR. To test this hypothesis, we manipulated both receptors in mice using pharmacological and genetic approaches, and then evaluated the mice in the formalin test, a tonic model of pain.
2. Material and methods
2.1 Animals
Male ICR mice (8–10 weeks of age) were obtained from ENVIGO (Indianapolis, IN). Mice null for the α7 (The Jackson Laboratory, Bar Harbor ME) or β2 subunits (Pasteur Institute, Paris, France) on a C57BL/6J background were bred with wild-type (WT) littermates in an Association for Assessment and Accreditation of Laboratory Animal Care approved animal care facility at Virginia Commonwealth University. For all experiments, mice were backcrossed for ≥ 8 generations. Knockout (KO) and WT mice were obtained by crossing heterozygote mice. Mice were housed in groups of four at 21°C in a humidity-controlled environment. Animals had ad libitum access to food and water. The rooms were on a 12-hour light/dark cycle (lights on at 7:00 AM) with all experiments performed during the light cycle. Unless otherwise noted, animals were promptly euthanized after experiments so as to minimize suffering. The study was approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University. All studies were carried out in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
2.2 Drugs
PNU282987 [N-(3R)-1-Azabicyclo[2.2.2]oct-3-yl-4-chlorobenzamide] (selective α7 full agonist), PNU120596 [N-(5-chloro-2,4-dimethoxyphenyl)-N′-(5-methyl-3-isoxazolyl)-urea] (α7 nAChR PAM), PHA543613 (selective α7 full agonist), SR144528 [5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]heptyl-2-yl]-1H-pyrazole-3-carboxamide] (CB2 antagonist) and rimonabant (CB1 antagonist) were obtained from the National Institute on Drug Abuse (NIDA) supply program (Rockville, MD). GW6471 [N-((2S)-2-(((1Z)-1-Methyl-3-oxo-3-(4-(trifluoromethyl)phenyl)prop-1-enyl)amino)-3-(4-(2-(5-methyl-2-phenyl-1,3-oxazol-4-yl)ethoxy)phenyl)propyl)propanamide] (selective PPAR-α antagonist), and N-palmitoylethanolamine (PEA) were purchased from Tocris Biosciences (Minneapolis, MN). Methyllycaconitine citrate (MLA) (selective α7 antagonist) was purchased from RBI (Natick, MA). GAT107 ((3aR,4S,9bS)-4-(4-bromophenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide) (α7 ago-PAM) was synthesized as described previously (Kulkarni et al., 2013; Thakur et al., 2013). NS6740 ((1,4-diazabicyclo[3.2.2]nonan-4-yl(5-(3-(trifluoromethyl) phenyl) furan-2-yl) methanone) (α7 silent agonist) was prepared as previously described (Peters et al., 2004; Papke et al., 2015).
GW6471, PNU120596, GAT107, PEA, rimonabant, and SR144528 were dissolved in a mixture of 1:1:18 [1 volume ethanol/1 volume Emulphor-620 (Rhone-Poulenc, Inc., Princeton, NJ)/18 volumes distilled water] and administered intraperitoneally (i.p.) for systemic injections. In addition to i.p. route, GW6471 was also administered intraplantar (i.pl.) and intrathecal (i.t.). PNU282987, PHA54613, NS6740 were dissolved in physiological saline (0.9% sodium chloride) and injected subcutaneously (s.c.), with the exception of NS6740, which was administered i.p. All drugs were injected at a total volume of 1 ml/100 g body weight, unless noted otherwise. All doses are expressed as the free base of the drug.
2.3 Formalin test
The formalin test was carried out in an open, empty Plexiglas cage (29 × 19 × 13 cm). Mice were allowed to acclimate for 15 min in the test cage prior to injection. Each animal was injected with 20 μL of (2.5%) formalin to the right hind paw (i.pl.). Mice were observed from 0 to 5 min (phase I) and 20 to 45 min (phase II) post-formalin injection. The amount of time spent attending to (i.e., licking) the injected paw was recorded with a digital stopwatch. Unless otherwise noted, all experiments were performed on ICR mice. PNU282987 (0.1, 1, 10 and 20 mg/kg, s.c.) or vehicle was administered 15 min prior to formalin injection. In a separate cohort, PNU282987 (10 mg/kg, s.c.) effects in the formalin test were measured in α7 and β2 WT and KO mice. In order to test the involvement of PPAR-α and its site of action in PNU282987-evoked antinociception, i.p., i.t., and i.pl. injections of varying doses of the PPAR-α antagonist GW6471 or vehicle, were injected before PNU282987 (10 mg/kg, s.c.) or vehicle. For systemic experiments, GW6471 (0.2 and 2 mg/kg, i.p.) or vehicle was administered 30 min prior to PNU282987. For CNS experiments, GW6471 (0.2 or 1 μg/5μL/mouse, i.t.) or its vehicle were administered 5 min before PNU282987 or its vehicle. The i.t. injections were performed free-hand between the fifth and sixth lumbar vertebra in unanesthetized mice according to the method of Hylden and Wilcox (1980). For local experiments, GW6471 (1 μg/20μL/mouse) or vehicle was administered i.pl. 5 min before PNU282987 or its vehicle. Formalin test was performed 15 min after PNU282987 injection.
To test the effects of GW6471 on various α7 nAChR modulators in the formalin test, we assessed several compounds which preferentially induce different α7 nAChR conformational states (Bagdas et al., 2016; Freitas et al., 2013a; Papke et al., 2015). For these experiments, the α7 nAChR full agonist PHA-543613, silent agonist NS6740, PAM PNU120596, and ago-PAM GAT107 were used. PHA-543613 (6 mg/kg, s.c.), NS6740 (9 mg/kg, i.p.), PNU120596 (10 mg/kg, i.p.), and GAT107 (10 mg/kg, i.p.) or their vehicles were administered 15 min after GW6471 (2 mg/kg, i.p.) or its vehicle. The formalin test began 15 min later.
Additionally, we assessed the pharmacological interaction between the PPAR-α agonist PEA and PNU282987 in the formalin test. We determined the antinociceptive effects of PEA pretreatment (0, 1, 3, 10 and 30 mg/kg, i.p.). Following the determination of PEA dose-response curve, the inactive doses of PEA (1 mg/kg and 3 mg/kg) and PNU282987 (0.1 mg/kg, s.c.) were co-administered to evaluate the possible interaction. For these experiments, PEA was injected 45 min before PNU282987 and the formalin test was conducted 15 min later.
Finally, we investigated the possible role of cannabinoid (CB) receptors in the antinociceptive effect of PNU282987 in the formalin test. To perform this part of the study, physiologically active doses of the CB1 antagonist rimonabant (3 mg/kg) or CB2 antagonist SR144528 (3 mg/kg) or vehicle were injected i.p. 10 min before PNU282987 (10 mg/kg, s.c.) or vehicle. Animals were administered formalin 15 min later.
2.4 Statistical Analysis
The data obtained were analyzed using GraphPad Prism software, version 6.0 (GraphPad Software, Inc., La Jolla, CA) and expressed as the mean ± S.E.M. Statistical analysis was conducted using one-way or two-way analysis of variance (ANOVA), followed by Tukey’s post hoc comparison. Student’s unpaired t-test was used to analyze differences between treatment groups if further analysis needed. P values < 0.05 were considered significant.
3. Results
3.1 PNU282987 attenuates formalin-induced pain responses
In the first experiment, we investigated the antinociceptive effects of a s.c. injection of vehicle or PNU282987 (0.1, 1, 10 or 20 mg/kg) administered 15 min before i.pl. formalin injection. In phase I of the formalin test, ANOVA revealed a significant antinociceptive effect [F (4, 30) = 11.75, P < 0.001, Fig. 1A], with PNU282987 (20 mg/kg) significantly reducing paw licking behavior (Tukey post hoc, P < 0.05). In phase II of the test, PNU282987 dose-dependently attenuated formalin-induced licking behavior [F (4, 30) = 18.75, P < 0.001, Fig. 1B], with 1, 10, and 20 mg/kg doses of PNU282987 significantly attenuating licking behaviors (P < 0.05).
Fig. 1.

PNU282987 elicits antinociceptive effects in formalin-injected mice in a dose-dependent fashion. Mice were treated with s.c. administration of PNU282987 (0.1, 1, 10, and 20 mg/kg) 15 min prior to formalin (2.5%, 20 μl) injection into the plantar region of the right hind paw. The cumulative pain response of time of licking was measured during the period of (A) 0–5 min (phase I), and (B) 20–45 min (phase II). Data reflect the mean ± S.E.M. of 7 animals for each group. * P < 0.05, significantly different from its vehicle group.
3.2 The PNU282987 effects are α7- but not β2-mediated
To evaluate the in vivo receptor selectivity of PNU282987 between α7 and β2 nicotinic subunits, we tested the antinociceptive effects of the drug in α7 and β2 KO mice. Two-way ANOVA revealed there was a significant effect of PNU282987 administration. Consistent with ICR results, PNU282987 (10 mg/kg, s.c.) significantly reduced phase I [Ftreatment(1,24) = 11.84, P = 0.0021, Fig. 2A and phase II [Ftreatment (1, 24) = 52.25, P < 0.0001, Fig. 2B] nociceptive responses in α7 WT mice. However, a significant effect of genotype was observed. The antinociceptive effects of PNU282987 in α7 KO mice was abolished in phase I [Fgenotype (1,24) = 5.116, P= 0.0330, Fig. 2B], and phase II [Fgenotype (1,24) = 40.17, P= 0.0330, Fig. 2B] of the formalin test. In contrast, PNU282987 (10 mg/kg, s.c.) maintained its antinociceptive effects in phase I [Ftreatment (1, 24) = 34.97, P < 0.0001, Fig. 2C] and phase II [Ftreatment (1, 24) = 209.2, P < 0.0001, Fig. 2D] of the formalin test. There was no effect of difference in antinociception in the β2 WT and β2 KO mice in phase I [Fgenotype (1,24) = 0.4265, P= 0.5199, Fig. 2C] or phase II [Fgenotype (1,24) = 1.4, P= 0.2483, Fig. 2D]. To confirm the data obtained in α7 null mice, we administered MLA (10 mg/kg, s.c.), a selective α7 nicotinic antagonist in naïve mice, 15 minutes prior PNU282987 (10 mg/kg, s.c.) or vehicle injection. MLA was able to block the PNU282987 antinociceptive effects in formalin-injected mice in both phase I and II (Supplemental, Fig. 1).
Fig. 2.

The antinociceptive effects of PNU282987 in the formalin test requires α7 nAChR subunit but not β2. WT and KO mice for α7 and β2 nAChRs subunits were treated with s.c. administration of PNU282987 (10 mg/kg) 15 min prior to formalin (2.5%, 20 μl) injection into the right hind paw. The cumulative pain response of time of licking was measured during the period of 0–5 min (phase I) and 20–45 min (phase II). (A) PNU282987 reduced nociceptive responses in WT mice but not in α7 KO mice in both phase I and (B) phase II. (C) PNU282987 evoked a decrease in licking behavior in WT mice as well as in β2 KO mice in both phase I and (D) phase II. Data reflect the mean ± S.E.M. of 7 animals for each group. * P < 0.05, significantly different from its vehicle group; # P < 0.05, significantly different from its corresponding control group (WT group).
3.3 PPAR-α mediates the antinociceptive effects of PNU282987
To test whether PPAR-α plays a role in the antinociceptive effects of subcutaneously administered PNU282987 in the formalin test, we used the selective PPAR-α antagonist GW6471. Figure 3A and B show that systemically administered GW6471 completely blocked the antinociceptive effects of PNU282987 (One-way ANOVA for phase I [Ftreatments (4, 25) = 11.32, P < 0.001, Fig. 3A] and phase II [Ftreatments (4, 25) = 10.53, P < 0.001, Fig. 3B]), as well as PHA-543613 (Supplemental, Fig. 2), but did not attenuate formalin-induced pain responses when paired with vehicle control of PNU282987 (P > 0.05). We next tested gave mice i.t. injections of GW6471 in order to assess whether spinal PPAR-α receptors mediate the antinociceptive effects of systemically administered PNU282987. GW6471 (1 μg) blocked the antinociceptive effects of PNU282987 (10 mg/kg, s.c.) without altering formalin-induced nociceptive responses when given alone (at phase I [Ftreatments(4,25) = 8.379, P < 0.001, Fig. 3C] or phase II [Ftreatments(4,25) = 4.934, P < 0.01, Fig. 3D]). In contrast to i.p. and i.t. routes of administration, i.pl. GW6471 (1 μg/20μl/mouse) failed to reverse the antinociceptive effects of PNU282987 (10 mg/kg, s.c.; Fig. 3E) in phase I [F (3, 20) = 5.564, P < 0.01, Fig. 3E] or phase II [F (3, 20) = 17.96, P < 0.001, Fig. 3F] of the formalin test (P > 0.05). These results suggest that PPAR-α receptors in the spinal cord and possibly elsewhere, but not in the formalin-injected paw, mediate PNU282987-induced antinociception.
Fig. 3.

The systemic and spinal, but not intraplantar, injection of PPAR-α antagonist blocks the antinociceptive effects of PNU282987 in the formalin test. GW6471 (2 mg/kg, i.p.) blocks the antinociceptive effects of PNU282987 (10 mg/kg, s.c.) when administrated systemically in both (A) phase I and (B) phase II. Intrathecal injection of GW6471 (1 μg/5μl) was able to block the antinociceptive effects of PNU282987 (10 mg/kg, s.c.) in both (C) phase I and (D) phase II. Intraplantar injection of GW6471 (1 μg/20μl) failed to block the effects of PNU282987 (10 mg/kg, s.c.) in (E) phase I and (F) phase II, respectively. Data reflect the mean ± S.E.M. of 6 animals for each group. * P < 0.05, significantly different from its vehicle group; # P < 0.05, significantly different from its corresponding control group (PNU282987 treated group).
3.4 PPAR-α receptor involvement in the antinociceptive effects of other types of α7 nAChR modulators
The next series of experiments investigated the involvement of PPAR-α receptors in the antinociceptive effects of other types of α7 receptor ligands, including the α7 nAChR silent agonist NS6740, the α7 nAChR PAM PNU120596, and α7 nAChR ago-PAM GAT107 (Bagdas et al., 2016; Kelen Freitas et al., 2013a; Papke et al., 2015) in the formalin test (Fig. 4). Two-way ANOVA revealed significant effects of treatments at phase I [Ftreatments (3, 40) = 14.04, P < 0.0001, Fig. 4A] and at phase II [Ftreatments (3, 40) = 12.58, P < 0.0001, Fig. 4B] in the formalin test.
Fig. 4.

PPAR-α is differentially involved in the ligand-induced α7 nAChR-mediated antinociception in the formalin test. The α7 nAChR silent agonist NS6740 (9 mg/kg, i.p.), α7 nAChR positive allosteric modulator (PAM) PNU120596 (10 mg/kg, i.p.), and dual functional α7 nAChR allosteric agonist and PAM (ago-PAM) GAT107 (10 mg/kg, i.p.) evoked antinociceptive effects in formalin-injected mice in (A) phase I and (B) phase II, respectively. The PPAR-α antagonist GW6471 (2 mg/kg, i.p.) was not able to block their antinociceptive effects in both (A) phase I and (B) phase II. Data reflect the mean ± S.E.M. of 6 animals for each group. * P < 0.05, significantly different from its vehicle group.
We tested the PPAR-α antagonist GW6471 (2 mg/kg, i.p.) in the presence of the α7 nAChR silent agonist NS6740 (9 mg/kg, i.p.) (Papke et al., 2015) in the formalin test (Fig. 4). Consistent with our previous study (Papke et al., 2015), NS6740 was able to attenuate the formalin-induced nociceptive behavior in both phases of the formalin test (Tukey post hoc, P < 0.05). However, GW6471 (2 mg/kg, i.p.) pretreatment could not inhibit the effects of NS6740 in either phase of the test (P > 0.05). We also investigated whether the antinociceptive effects of the α7 nAChR PAM PNU120596 (Freitas et al., 2013b) would be blocked by GW6471. As shown in Figure 4, PNU120596 (10 mg/kg, i.p.) produced significant antinociceptive effects in phase I and phase II (Tukey post hoc, P < 0.05). However, GW6471 (2 mg/kg, i.p.) failed to attenuate the effects of PNU120596 in either phase (P > 0.05). Additionally, the α7 nAChR ago-PAM GAT107 (10 mg/kg i.p.) reduced formalin-induced nociceptive behavior in phase II (Tukey post hoc, P < 0.05) (Fig. 4B), but not phase I (P > 0.05) (Fig. 4A), which is consistent with our previous study (Bagdas et al., 2016). GW6471 (2 mg/kg, i.p.) failed to block the effects of GAT107 in Phase II (P > 0.05) (Fig. 4B).
3.5 Potentiation of the antinociceptive effects of the PNU282987 by PEA
Given that a PPAR-α antagonist blocked the antinociceptive effects PNU282987-induced antinociception in the formalin test, we next examined whether combination of PNU282987 and the PPAR-α PEA would produce enhanced antinociceptive effects. One-way ANOVA analysis revealed significant effects for phase I [FPEA treatment (4, 30) = 9.690, P < 0.001, Fig. 5A] and phase II [FPEA treatment (4, 30) = 13.34, P < 0.001, Fig. 5B]. PEA significantly reduced paw licking behavior in a dose-dependent manner. Particularly, higher doses (10 and 30 mg/kg, i.p.) significantly inhibited licking (P < 0.05) in phase II, whereas lower doses (1 and 3 mg/kg, i.p.) did not (P > 0.05) (Fig. 5A, B). Based on these results, we chose subthreshold doses of PEA (1 and 3 mg/kg) in combination with an ineffective dose of PNU282987 (0.1 mg/kg, s.c.). As shown in Figure 5C and D, treatment with a low dose of PEA (1 and 3 mg/kg, i.p.) or PNU282987 (0.1 mg/kg, s.c) failed to attenuate formalin-induced pain responses in both phases when given alone (P > 0.05). However, two-way ANOVA showed that PEA significantly reversed pain behavior in the presence of PNU282987 in phase I [Ftreatment(2,36) = 17.37, P < 0.0001, Fig. 5C] and phase II [Ftreatment(2,36) = 23.84, P < 0.001, Fig. 5D]. Tukey’s post hoc analysis showed that combination evoked significant antinociceptive effects at doses of 3 mg/kg of PEA in phase I (P < 0.05) and at doses of 1 and 3 mg/kg of PEA in phase II (P < 0.05).
Fig. 5.

The systemic administration of PEA potentiates the antinociceptive effects of PNU282987. Intraperitoneal injection of PEA (1, 3, 10 and 30 mg/kg) dose-dependently reduced the licking behavior in (A) phase I and (B) phase II, respectively. The combination of ineffective doses of PEA (1, 3 mg/kg, i.p.) and PNU282987 (0.1 mg/kg, s.c.) elicited antinociceptive effects in both (C) phase I and (D) phase II compare to the effects of drugs given alone. Data reflect the mean ± S.E.M. of 7 animals for each group. * P < 0.05, significantly different from its corresponding control group.
3.6 CB receptors do not mediate the effects of PNU282987
In order to test whether the structurally-related endogenous cannabinoids anandamide (AEA) and 2-arachidonoylglycerol (2-AG) contribute to the antinociceptive effects of PNU282987, we investigated the possible involvement of CB1 receptors using the CB1 antagonist rimonabant (3 mg/kg) or CB2 ntagonist SR144528 (3 mg/kg) (Kinsey et al., 2009) injected i.p. 10 min before the PNU282987 (10 mg/kg, s.c.). Two-way ANOVA revealed significant effects for treatment of PNU282987 at phase I [FPNU282987 treatment (1, 34) = 25.32, P < 0.0001, Fig. 6A] and at phase II [FPNU282987 treatment(1, 34) = 55.23, P < 0.0001, Fig. 6B] of the formalin test. As shown in Figure 6, CB1 receptor antagonist rimonabant and CB2 receptor antagonist SR144S28 [at phase I, Fantagonist treatments (2, 34) = 1.658, P = 0.2055, Fig. 6A and at phase II [Fantagonist treatments (2, 34) = 0.6588, P = 0.5239, Fig. 6B] did not have antinociceptive effects by themselves at phase I and II of formalin test (P > 0.05) and did not significantly alter antinociceptive behavior induced by PNU282987 (P > 0.05). Although there was a trend toward reversal of PNU282987’s effect in phase I of the formalin test by CB1 and CB2 antagonists, there was no statistically significant differences between group means as determined by Tukey’s post hoc. Furthermore, we went a step further and tested group means by using t-test. The comparisons between rimonabant-vehicle vs rimonabant-PNU282987, and SR144528-vehicle vs SR144528-PNU282987 were significant in simple t-test analysis (t=2.675, df=12, P < 0.05 and t=2.157, df=11, P = 0.05; respectively) indicating the persistence of antinociceptive effects of PNU282987 in the presence of CB antagonists. In addition, the t-test analysis confirmed that rimonabant and SR144528 had no effect on PNU282987-induced antinociception (t=1.964, df=13, P > 0.05 and t=1.478, df=11, P > 0.05; respectively).
Fig. 6.

The systemic injection of CB1 and CB2 receptor antagonists do not block the antinociceptive effects of PNU282987 in the formalin test. CB1 receptor antagonist rimonabant (3 mg/kg, i.p.) and CB2 receptor antagonist SR144S28 (3 mg/kg, i.p.) do not block the antinociceptive effects of PNU282987 (10 mg/kg, s.c.) in both (A) phase I and (B) phase II. Data reflect the mean ± S.E.M. of 6–8 animals for each group. * P < 0.05, significantly different from its corresponding control group.
4. Discussion
α7-nAChRs represent viable therapeutic targets for cognitive and neuro-degenerative disorders as well as potential anti-inflammatory and analgesic candidates (Damaj et al., 2000; Feuerbach et al., 2009; Wang et al., 2005). Additionally, it has been demonstrated that the selective activation of α7 nAChR subtype may be involved in a rat model of oxaliplatin-induced neuropathy (Di Cesare Mannelli et al., 2014), and induce neuroprotection associated with an increase in astrocyte density (Di Cesare Mannelli et al., 2015). As we mentioned in the introduction, several possible mechanisms were proposed to mediate these disparate pharmacological and behavioral responses (Bagdas et al., 2016; Damaj et al., 2000; Egea et al., 2015; Feuerbach et al., 2009; Liu et al., 2012; Wang et al., 2005). The present study identified in vivo crosstalk between α7 nAChR and PPAR-α, suggesting a novel signaling pathway in reducing nociception in the mouse formalin model of tonic pain.
Consistent with the anti-inflammatory and antinociceptive effects of α7 nAChR full and partial agonists reported in several rodent models of tonic and chronic pain (Bagdas et al., 2011; Damaj et al., 2000; Feuerbach et al., 2009; Wang et al., 2005), we show that PNU282987, an α7-nAChRs orthosteric agonist, dose-dependently attenuated pain-related behaviors in both phases of the formalin test in male mice. The antinociceptive effects of PNU282987 were selectively mediated by α7 nAChRs since PNU282987’s effects were abolished in α7 KO mice, and spared in β2 KO mice.
Although there has been mounting evidence that α7 nAChRs have a large intracellular (Paulo et al., 2009) interactome that may function both as metabotropic as well as ionotropic receptors (de Jonge and Ulloa, 2007; Kabbani et al., 2013; King and Kabbani, 2016), specific signaling intermediaries remain to be identified. The conformational changes induced by the binding of orthosteric ligands may modulate the Jak/Stat cascade, which is implicated in the regulation of inflammation (de Jonge et al., 2005). The main objective of the present study was to elucidate the pharmacological interaction between α7 nAChRs and PPAR-α signaling in the formalin test. Our findings demonstrate that the PPAR-α antagonist GW6471 blocked the antinociceptive effects of PNU282987. Moreover, in order to identify the locus of action of this α7 nAChRs/PPAR-α interaction, we tested multiple routes of administration, including i.p., i.t., and i.pl. Our results show that GW6471 blocked the effects of PNU282987 when given i.t., but not i.pl., which supports a spinal site of action. This pharmacological interaction between α7 nAChRs and PPAR-α signaling in reducing formalin-induced nociception are consistent with the previous findings of Melis et al. (2013), who showed an interaction between PPAR-α and α7 nAChRs in the ventral tegmental area (Melis et al., 2013).
Conventional orthosteric α7 nAChR agonists control conformational changes of this ion channel binding sites. However, ligands binding allosteric modulatory sites or conformations also regulate α7 nAChR. Accordingly, it has been reported that not only orthosteric agonists, but also other α7 ligands elicit antinociceptive effects in preclinical models of pain. In particular, α7-selective positive allosteric modulators (PAMs) showed activity in rodent models of chronic and inflammatory pain (Bagdas et al., 2015; Freitas et al., 2013; Freitas et al., 2013b; Munro et al., 2012). α7 ago-PAMs elicit antinociceptive and anti-inflammatory effects in mice (Bagdas et al., 2016), as well. Furthermore, a new class of α7 ligands, α7-silent agonists, acting through non-conducting conformations of the α7 nAChR channel, also elicit anti-inflammatory and antinociceptive properties in mice (Papke et al., 2015). Given this plethora of evidence, we investigated the interaction between α7 nAChR and PPAR-α using a variety of classes of α7 nAChR modulators. In particular, we evaluated antagonism of PPAR-α by GW6471 would block the antinociceptive effects elicited by the α7 PAM PNU120596, ago-PAM GAT107, silent agonist NS6740, and full agonists PHA-543613 and PNU282987 in the formalin test. Interestingly, among the different classes of α7 ligands tested, GW6471 only inhibited the antinociceptive effects evoked by the orthosteric agonists PHA-543613 and PNU282987. These findings suggest that α7 nAChR/PPAR-α signaling in this system may be limited to the conformational state induced by orthosteric agonists. One possibility is that the coupling of α7 nAChR to PPAR-α requires α7 channel activation per se, as previously suggested for agents active in assays of cognition (Briggs et al., 2009). Also, it should be noted that PAMs and silent agonists can induce distinct conformational states in both the conducting and non-conducting forms of the receptor, as determined by their sensitivity to select antagonists. This suggests that coupling of α7 to PPAR-α signaling may be selective for a specific conformational state of the α7 nAChR.
To examine further a possible in vivo crosstalk between α7 nAChR and PPAR-α, we tested whether subthreshold doses of PEA, an endogenous PPAR-α agonist, and PNU282987 would produce enhanced antinociceptive effects in the formalin test. Strikingly, combined administration of inactive doses of PNU282987 and PEA significantly reduced the formalin-induced pain compared with single administration of these drugs, further supporting pharmacological crosstalk between the antinociceptive effects of α7 nAChR and PPAR-α agonists.
As PEA has been demonstrated to potentiate the effects of anandamide on CB1 and CB2 receptors by competing for their common hydrolytic enzyme fatty acid amide hydrolase, a phenomenon known as “the entourage effect” (Jonsson et al., 2001). Given this possible involvement of cannabinoid receptor signaling, we investigated whether CB1 or CB2 antagonists would reverse PNU282987’s antinociceptive effects in formalin-injected mice. However, the fact that neither CB1 nor CB2 receptor antagonist blocked these antinociceptive effects further supports the idea of a direct α7 nAChR/PPAR-α interaction in this model of tonic pain.
In conclusion, the results in the present study support the main hypothesis that the selective orthosteric nAChR full agonist (i.e. PNU282987) may lead to increases of endogenous neuronal PPAR-α tone mediating the observed antinociceptive effects. This in vivo crosstalk between α7 nAChR and PPAR-α represents a novel signaling pathway mediating antinociceptive effects in the mouse formalin model of tonic pain.
Supplementary Material
Highlights.
The α7 nAChR and PPAR-α crosstalk is investigated using a mouse model of tonic pain
The PPAR-α antagonist blocks the antinociceptive effects of α7 nAChR full agonist
The cannabinoid receptors are not involved in the effects of α7 nAChR full agonist
Palmitoylethanolamide potentiates the antinociceptive effects of α7 nAChR full agonist
PPAR-α plays a key role in a putative antinociceptive α7 nicotinic signaling pathway
Acknowledgments
This study was supported by NIH grants GM57481 (RLP), and R01 CA206028 (MID).
Abbreviations
- nAChRs
α7 nicotinic acetylcholine receptors
- PPAR-α
nuclear peroxisome proliferator-activated receptors type-α
- PAMs
positive allosteric modulators
- ago-PAMs
ago-allosteric ligands
- PEA
palmitoylethanolamide
- OEA
oleoylethanolamide
- CB1 and CB2
cannabinoid receptors 1 and 2
- CNS
central nervous system
- AEA
anandamide
- 2-AG
2-arachidonoylglycerol
Footnotes
Conflict of interest
The authors have declared no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bagdas D, Sonat FA, Hamurtekin E, Sonal S, Gurun MS. The antihyperalgesic effect of cytidine-5′-diphosphate-choline in neuropathic and inflammatory pain models. Behav Pharmacol. 2011;22:589–98. doi: 10.1097/FBP.0b013e32834a1efb. [DOI] [PubMed] [Google Scholar]
- Bagdas D, Targowska-Duda KM, López JJ, Perez EG, Arias HR, Damaj MI. The Antinociceptive and Antiinflammatory Properties of 3-furan-2-yl-N-p-tolyl-acrylamide, a Positive Allosteric Modulator of α7 Nicotinic Acetylcholine Receptors in Mice. Anesth Analg. 2015;121:1369–1377. doi: 10.1213/ANE.0000000000000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdas D, Wilkerson JL, Kulkarni A, Toma W, AlSharari S, Gul Z, Lichtman AH, Papke RL, Thakur GA, Damaj MI. The α7 nicotinic receptor dual allosteric agonist and positive allosteric modulator GAT107 reverses nociception in mouse models of inflammatory and neuropathic pain. Br J Pharmacol. 2016;173:2506–2520. doi: 10.1111/bph.13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Gopalakrishnan M. Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol. 2007;74:1155–63. doi: 10.1016/j.bcp.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Briggs CA, Grønlien JH, Curzon P, Timmermann DB, Ween H, Thorin-Hagene K, Kerr P, Anderson DJ, Malysz J, Dyhring T, Olsen GM, Peters D, Bunnelle WH, Gopalakrishnan M. Role of channel activation in cognitive enhancement mediated by α7 nicotinic acetylcholine receptors. Br J Pharmacol. 2009;158:1486–1494. doi: 10.1111/j.1476-5381.2009.00426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damaj MI, Meyer EM, Martin BR. The antinociceptive effects of alpha7 nicotinic agonists in an acute pain model. Neuropharmacology. 2000;39:2785–91. doi: 10.1016/s0028-3908(00)00139-8. [DOI] [PubMed] [Google Scholar]
- de Jonge WJ, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol. 2007;151:915–29. doi: 10.1038/sj.bjp.0707264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6:844–51. doi: 10.1038/ni1229. [DOI] [PubMed] [Google Scholar]
- Di Cesare Mannelli L, Pacini A, Matera C, Zanardelli M, Mello T, De Amici M, Dallanoce C, Ghelardini C. Involvement of α7 nAChR subtype in rat oxaliplatin-induced neuropathy: Effects of selective activation. Neuropharmacology. 2014;79:37–48. doi: 10.1016/j.neuropharm.2013.10.034. [DOI] [PubMed] [Google Scholar]
- Di Cesare Mannelli L, Tenci B, Zanardelli M, Failli P, Ghelardini C. α7 Nicotinic Receptor Promotes the Neuroprotective Functions of Astrocytes against Oxaliplatin Neurotoxicity. Neural Plast. 2015;2015:1–10. doi: 10.1155/2015/396908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea J, Buendia I, Parada E, Navarro E, León R, Lopez MG. Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol. 2015;97:463–72. doi: 10.1016/j.bcp.2015.07.032. [DOI] [PubMed] [Google Scholar]
- Faghih R, Gfesser GA, Gopalakrishnan M. Advances in the discovery of novel positive allosteric modulators of the alpha7 nicotinic acetylcholine receptor. Recent Pat CNS Drug Discov. 2007;2:99–106. doi: 10.2174/157488907780832751. [DOI] [PubMed] [Google Scholar]
- Feuerbach D, Lingenhoehl K, Olpe HR, Vassout A, Gentsch C, Chaperon F, Nozulak J, Enz A, Bilbe G, McAllister K, Hoyer D. The selective nicotinic acetylcholine receptor alpha7 agonist JN403 is active in animal models of cognition, sensory gating, epilepsy and pain. Neuropharmacology. 2009;56:254–63. doi: 10.1016/j.neuropharm.2008.08.025. [DOI] [PubMed] [Google Scholar]
- Freitas K, Carroll FI, Damaj MI. The antinociceptive effects of nicotinic receptors α7-positive allosteric modulators in murine acute and tonic pain models. J Pharmacol Exp Ther. 2013a;344:264–75. doi: 10.1124/jpet.112.197871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas K, Carroll FI, Damaj MI. The antinociceptive effects of nicotinic receptors α7-positive allosteric modulators in murine acute and tonic pain models. J Pharmacol Exp Ther. 2013b;344:264–75. doi: 10.1124/jpet.112.197871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas K, Ghosh S, Ivy Carroll F, Lichtman AH, Imad Damaj M. Effects of α7 positive allosteric modulators in murine inflammatory and chronic neuropathic pain models. Neuropharmacology. 2013c;65:156–64. doi: 10.1016/j.neuropharm.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas K, Negus SS, Carroll FI, Damaj MI. In vivo pharmacological interactions between a type II positive allosteric modulator of α7 nicotinic ACh receptors and nicotinic agonists in a murine tonic pain model. Br J Pharmacol. 2013;169:567–79. doi: 10.1111/j.1476-5381.2012.02226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill JK, Savolainen M, Young GT, Zwart R, Sher E, Millar NS. Agonist activation of alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci U S A. 2011;108:5867–72. doi: 10.1073/pnas.1017975108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girod R, Crabtree G, Ernstrom G, Ramirez-Latorre J, McGehee D, Turner J, Role L. Heteromeric complexes of alpha 5 and/or alpha 7 subunits. Effects of calcium and potential role in nicotine-induced presynaptic facilitation. Ann N Y Acad Sci. 1999;868:578–90. doi: 10.1111/j.1749-6632.1999.tb11331.x. [DOI] [PubMed] [Google Scholar]
- Horenstein NA, Papke RL, Kulkarni AR, Chaturbhuj GU, Stokes C, Manther K, Thakur GA. Critical Molecular Determinants of α7 Nicotinic Acetylcholine Receptor Allosteric Activation: SEPARATION OF DIRECT ALLOSTERIC ACTIVATION AND POSITIVE ALLOSTERIC MODULATION. J Biol Chem. 2016;291:5049–67. doi: 10.1074/jbc.M115.692392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AA, Frølund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem. 2005;48:4705–45. doi: 10.1021/jm040219e. [DOI] [PubMed] [Google Scholar]
- Jonsson KO, Vandevoorde S, Lambert DM, Tiger G, Fowler CJ. Effects of homologues and analogues of palmitoylethanolamide upon the inactivation of the endocannabinoid anandamide. Br J Pharmacol. 2001;133:1263–75. doi: 10.1038/sj.bjp.0704199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabbani N, Nordman JC, Corgiat BA, Veltri DP, Shehu A, Seymour VA, Adams DJ. Are nicotinic acetylcholine receptors coupled to G proteins? BioEssays. 2013;35:1025–1034. doi: 10.1002/bies.201300082. [DOI] [PubMed] [Google Scholar]
- Khan I, Osaka H, Stanislaus S, Calvo RM, Deerinck T, Yaksh TL, Taylor P. Nicotinic acetylcholine receptor distribution in relation to spinal neurotransmission pathways. J Comp Neurol. 2003;467:44–59. doi: 10.1002/cne.10913. [DOI] [PubMed] [Google Scholar]
- King JR, Kabbani N. Alpha 7 nicotinic receptor coupling to heterotrimeric G proteins modulates RhoA activation, cytoskeletal motility, and structural growth. J Neurochem. 2016;138:532–545. doi: 10.1111/jnc.13660. [DOI] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O’Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330:902–10. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni AR, Thakur GA. Microwave-assisted Expeditious and Efficient Synthesis of Cyclopentene Ring-fused Tetrahydroquinoline Derivatives Using Three-component Povarov Reaction. Tetrahedron Lett. 2013;54:6592–6595. doi: 10.1016/j.tetlet.2013.09.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Hu J, Wu J, Zhu C, Hui Y, Han Y, Huang Z, Ellsworth K, Fan W. α7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J Neuroinflammation. 2012;9:617. doi: 10.1186/1742-2094-9-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Scheggi S, Carta G, Madeddu C, Lecca S, Luchicchi A, Cadeddu F, Frau R, Fattore L, Fadda P, Ennas MG, Castelli MP, Fratta W, Schilstrom B, Banni S, De Montis MG, Pistis M. PPARα regulates cholinergic-driven activity of midbrain dopamine neurons via a novel mechanism involving α7 nicotinic acetylcholine receptors. J Neurosci. 2013;33:6203–11. doi: 10.1523/JNEUROSCI.4647-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro G, Hansen R, Erichsen H, Timmermann D, Christensen J, Hansen H. The α7 nicotinic ACh receptor agonist compound B and positive allosteric modulator PNU-120596 both alleviate inflammatory hyperalgesia and cytokine release in the rat. Br J Pharmacol. 2012;167:421–35. doi: 10.1111/j.1476-5381.2012.02003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Bagdas D, Kulkarni AR, Gould T, AlSharari SD, Thakur GA, Damaj MI. The analgesic-like properties of the alpha7 nAChR silent agonist NS6740 is associated with non-conducting conformations of the receptor. Neuropharmacology. 2015;91:34–42. doi: 10.1016/j.neuropharm.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Chojnacka K, Horenstein NA. The Minimal Pharmacophore for Silent Agonism of the 7 Nicotinic Acetylcholine Receptor. J Pharmacol Exp Ther. 2014;350:665–680. doi: 10.1124/jpet.114.215236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Horenstein NA, Kulkarni AR, Stokes C, Corrie LW, Maeng CY, Thakur GA. The activity of GAT107, an allosteric activator and positive modulator of α7 nicotinic acetylcholine receptors (nAChR), is regulated by aromatic amino acids that span the subunit interface. J Biol Chem. 2014;289:4515–31. doi: 10.1074/jbc.M113.524603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, Brucker WJ, Hawrot E. Proteomic analysis of an alpha7 nicotinic acetylcholine receptor interactome. J Proteome Res. 2009;8:1849–58. doi: 10.1021/pr800731z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur GA, Kulkarni AR, Deschamps PR., JR Expeditious synthesis, enantiomeric resolution. J Med Chem. 2013;56:8943–7. doi: 10.1021/jm401267t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umana IC, Daniele CA, McGehee DS. Neuronal nicotinic receptors as analgesic targets: it’s a winding road. Biochem Pharmacol. 2013;86:1208–14. doi: 10.1016/j.bcp.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Su DM, Wang RH, Liu Y, Wang H. Antinociceptive effects of choline against acute and inflammatory pain. Neuroscience. 2005;132:49–56. doi: 10.1016/j.neuroscience.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Williams DK, Wang J, Papke RL. Investigation of the molecular mechanism of the α7 nicotinic acetylcholine receptor positive allosteric modulator PNU-120596 provides evidence for two distinct desensitized states. Mol Pharmacol. 2011;80:1013–32. doi: 10.1124/mol.111.074302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.