

Abstract
GTP cyclohydrolase I catalyzes the first step in folic acid biosynthesis in bacteria and plants, biopterin biosynthesis in mammals, and the biosynthesis of 7-deazaguanosine modified tRNA nucleosides in bacteria and archaea. The type IB GTP cyclohydrolase (GCYH-IB) is a prokaryotic-specific enzyme found in a number of pathogens. GCYH-IB is structurally distinct from the canonical type IA GTP cyclohydrolase involved in biopterin biosynthesis in humans and animals, and thus is of interest as a potential antibacterial drug target. We report kinetic and inhibition data of Neisseria gonorrhoeae GCYH-IB, and two high-resolution crystal structures of the enzyme; one in complex with the reaction intermediate analog and competitive inhibitor 8-oxo-GTP, and one with a TRIS molecule bound in the active site and mimicking another reaction intermediate. Comparison with the type IA enzyme bound to 8-oxo-GTP reveals an inverted mode of binding of the inhibitor ribosyl moiety and, together with site-directed mutagenesis data, shows that the two enzymes utilize different strategies for catalysis. Notably, the inhibitor interacts with a conserved active site Cys149, and this residue is S-nitrosylated in the structures. This is the first structural characterization of a biologically S-nitrosylated bacterial protein. Mutagenesis and biochemical analyses demonstrate that Cys149 is essential for the cyclohydrolase reaction, and S-nitrosylation maintains enzyme activity, suggesting a potential role of the S-nitrosothiol in catalysis.
Keywords: Transfer RNA (tRNA), bacterial metabolism, metalloenzyme, 7-deazaguanosine, modified nucleoside, Post-Translational Modification (PTM)
Introduction
GTP cyclohydrolase-I (GCYH-I; FolE) is a Zn2+-dependent enzyme that catalyzes the conversion of GTP to 7,8-dihydroneopterin triphosphate (H2NTP), the first intermediate in the de novo tetrahydrofolate (THF, folate) biosynthesis pathway in bacteria, fungi and plants (Fig. 1) [1]. THF is an essential cofactor involved in one carbon transfer reactions in the biosynthesis of purines, thymidylate, panthothenate, glycine, serine and methionine in all domains of life [1], and formylmethionyl-tRNA in bacteria [2]. GCYH-I also catalyzes the first step in the biosynthesis of the 7-deazaguanosine modified tRNA nucleosides queuosine and archaeosine in bacteria and archaea [3], the 7-deazapurine natural products in some bacteria [4], and the 7-deazaguanosine modified nucleosides recently discovered in the genomes of some organisms [5]. GCYH-I is also found in mammals and other higher eukaryotes where it catalyzes the first and rate-limiting step of the tetrahydrobiopterin (BH4) pathway (Fig. 1) which functions in mammals as a cofactor of aromatic amino acid hydroxylases, nitric oxide synthase, and glyceryl ether monooxygenase [6].
Figure 1.

The reaction catalyzed by GTP cyclohydrolase I, and the metabolic pathways in which it is found.
GCYH-I has been studied in some detail both biochemically and structurally. It is part of the tunneling-fold (T-fold) structural superfamily, a small but catalytically diverse group of pterin/purine binding proteins that utilize a common oligomerization of the T-fold domain, the core structural component of the superfamily. The T-fold domain is comprised of a 4-stranded antiparallel β-sheet and two helices, that upon assembly form a β2nαn barrel [7]. The functional enzymes are comprised of two barrels that join in a head-to-head fashion, forming a tunnel-like center (the origin of the name).
The GCYH-I catalyzed conversion of GTP to H2NTP is a mechanistically complex transformation that involves hydrolytic opening of the purine ring at C-8 of GTP to generate an N-formyl intermediate, followed by deformylation and subsequent rearrangement and cyclization of the ribosyl moiety to generate the pterin system. This mechanistic complexity coupled with the biological importance of GCYH-I has made the enzyme the subject of interest since its discovery by Burg and Brown 48 years ago [8]. While the outlines for the chemical mechanism of the reaction were proposed shortly after the enzyme discovery [9], only relatively recently has an understanding of the molecular basis for GCYH-I catalysis begun to emerge.
Despite the essential role of GCYH-I as the initiating step in the bacterial folate pathway, its role in the biopterin pathway in humans, coupled with the high homology between the E. coli and human GCYH-I enzymes, precluded GCYH-I as a viable target for new antibacterials. However, a significant number of bacteria possess a second class of GCYH-I enzymes (COG1469) that display virtually no sequence homology to the canonical GCYH-I despite exhibiting the high topological homology characteristic of this superfamily. The new subfamily of GCYH-I is prokaryotic specific and was named GCYH-IB, while the canonical enzymes were renamed GCYH-IA [10]. Notably, GCYH-IB is found in a number of pathogens, including the clinically important Staphylococcus and Neisseria where it is essential [11, 12].
Crystal structures have been determined for GCYH-IA from Escherichia coli [13], humans [14], rat [15], and Thermus thermophilus [16], and for Zn2+- and Mn2+-metallated forms of GCYH-IB from Neisseria gonorrhoeae [12]. Remarkably, while both subfamilies catalyze the same reaction and belong to the same structural superfamily (T-fold), they share only 2 residues in common and exhibit major differences in global structure and active site architecture [12]. While GCYH-IA is a unimodular, homodecameric, Zn2+-dependent enzyme, GCYH-IB is a bimodular, homotetrameric enzyme activated by a variety of divalent cations [12], with maximal activity observed with Mn2+. In both enzymes, as in all T-fold members, catalysis occurs at the interfaces between the T-fold subunits [12, 17]. However, several helical insertions and deletions near the interface render the active sites of the two enzymes significantly different. These differences, coupled with the occurrence of the latter enzyme in clinically important human pathogens that lack GCYH-IA, and thus depend solely on GCYH-IB for folate biosynthesis, have positioned GCYH-IB as a potential new antibacterial target in the folate pathway [12, 18]. These differences also led to the proposal that the two GCYH-I subfamilies may employ different catalytic strategies [12].
While details of the mechanism have been proposed for the type IA enzyme [14, 16, 19], the mechanism of the type IB enzyme has not been explicitly addressed. Here, we present kinetic parameters and competitive inhibition data of N. gonnorrhoae GCYH-IB, and describe crystal structures of two enzyme-ligand complexes that represent two states of the enzyme catalytic cycle: one structure is of the complex with 8-oxo-GTP, an analog of a reaction intermediate, and the other structure is of the complex with Tris(hydroxymethyl)aminomethane (TRIS), representing another intermediate previously postulated for the GCYH-I reaction. Additionally, we provide data on the consequences of site-directed mutagenesis of putative catalytic residues implicated from the crystal structures. Finally, we present crystallographic and biochemical evidence of specific, posttranslational S-nitrosylation of a conserved, catalytic cysteine residue, suggesting a role of the S-nitrosothiol in catalysis. The results provide insight into the catalytic strategy of GCYH-IB, and offer a starting point for the design of specific inhibitors against GCYH-IB.
Experimental Procedures
General
Reagents, salts and buffers were of the highest quality grade, and were purchased from Sigma unless otherwise noted. Millipore ultrapure water (18.2 MΩ) was used for all solutions. [8-14C]-GTP was purchased from Moravek. Ultra pure grade 2′-dGTP and GTP were from Ameresco or Sigma. 8-oxo-GTP was purchased from Jena Bioscience. 7-deaza-GTP was purchased from Trilink Biotechnologies. Neopterin and glutathione were purchased from Sigma.
Preparation of wild-type protein
Wild-type N. gonorrhoeae GCYH-IB was overexpressed as an N-terminally His6 tagged protein in Bl21(DE3) cells (New England Biolabs Inc.) grown in Luria–Bertani (LB) medium with vigorous aeration as described previously [10], and purified by Ni-NTA chromatography. The His6 tag was removed by cleavage with factor-Xa (New England BioLabs, product number P8010L) according to manufacturer's protocol, followed by capture of the affinity tag on Ni-NTA resin. For enzyme used for biochemical assays, the His6 tag-free protein was subjected to a final purification step by gel filtration chromatography using a Enrich SEC 650 column (Bio-Rad, Hercules, CA) pre-equilibrated with TRIS-free buffer containing 100 mM HEPES (pH 7.4), 100 mM KCl, and 1 mM dithiothreitol (DTT) or β-mercaptoethanol (BME). For enzyme used for crystallization, all purification steps were conducted in buffer containing 50 mM TRIS-acetate (pH 8.0), 100 mM KCl, 5 mM ZnCl2 and 1 mM BME, and the final gel filtration step was skipped. For all preparations, reducing agent (DTT or BME) was kept at a concentration of 1 mM in all purification steps, and protein purity was confirmed to be greater than 95% by SDS-PAGE.
Enzyme activity assays
Enzymatic activities were measured using either of three different enzyme assays, the standard fluorescence based GTP cyclohydrolase assay that relies on post-reaction oxidation of the enzymatic product H2NTP to the fluorescent neopterin [10], a continuous UV-vis assay that measures H2NTP directly [10], and a radiochemical assay that uses [8-14C]-GTP and measures the co-product formic acid (as [14C]-HCO2H) [10]. The fluorescence assay were performed in 100 mM HEPES (pH 8.0), 100 mM KCl, 0.5 mM MnCl2, 1 mM MgCl2, and 0.5 – 5 μM of N. gonorrhoeae GCYH-IB in a total volume of 100 μL. Reactions were initiated by the addition of GTP (final concentration 100 μM) and incubated at 37 °C for 50-60 minutes. Reactions were terminated by addition of 12 μL of a 1% I2, 2% KI solution in 1.0 M HCl to oxidize the H2NTP to neopterin. Following a 15-minute incubation at room temperature in the dark, excess iodine was quenched with the addition of 6 μL of 2% ascorbic acid, and the samples were analyzed by fluorescence spectroscopy using either a FluoroMax-3 instrument (Horiba Jobin Yvon Scientific), a PTI Time-Master fluorometer, or a Gemini XPS microplate spectrofluorometer (Molecular Devices) at λEx=365 nm. Emission spectra were recorded between 400 and 500 nm, and the neopterin peak at 446 nm was monitored. Neopterin was quantified using a linear calibration curve of the fluorescence signal from a pure neopterin standard.
The UV-vis assays were carried out in 100 mM HEPES (pH 8.0), 100 mM KCl, 0.5 mM MnCl2, 1 mM DTT, 0.5 - 5 μM enzyme, and 50 μM GTP in a final volume of 100 μL. The reaction was held at 37 °C and monitored over 40 minutes at a wavelength of 330 nm with a CARY 100 Bio UV-visible spectrophotometer (Varian). Radiochemical assays were performed in 100 mM HEPES (pH 8.0), 100 mM KCl, 0.5 mM MnCl2, 1 mM DTT, 0.5 - 5 μM enzyme, and 50 μM of [8-14C]GTP (10 μCi/μmol). The reactions were incubated at 37 °C and 50 μL aliquots were taken at 5, 10, 20, and 40 minutes. Aliquots were immediately quenched with 62.5 μL of 0.5 M formic acid, and loaded onto columns packed with 100 mg of activated charcoal pre-equilibrated with 0.1 M formic acid (pH 7.0). The flowthrough and a 3 mL elution of 0.1 M formic acid (pH 7.0) were collected together in a scintillation vial, combined with Econo-Safe scintillation cocktail (RPI), and counted on a LS6000 Series Scintillation Counter (Beckman).
Determination of pH optima
The optimal pH of N. gonorrhoeae GCYH-IB was determined in a 3 component buffer system composed of Tris-MES-acetic acid (100-50-50 mM) over a pH range of 4.0-9.0. Buffers also contained 100 mM KCl, 0.5 mM MnCl2, 2 mM DTT, 50 μM GTP and 1 μM protein in a final volume of 100 μL. After incubating at 37 °C in the dark for 30 min, the reactions were terminated and analyzed by fluorescence with a Gemini XPS microplate spectrofluorometer (Molecular Devices) as described above.
Metal activation studies
The ability of various metal ions to support catalytic activity was examined by incubating the dematellated apoenzyme (2 μM), prepared as described previously [12], in the presence of varying concentration (0.1 μM - 4 mM) of metal chlorides (MnCl2, ZnCl2, MgCl2, NiCl2, CaCl2, CdCl2, CoCl2, CuCl2, CoCl3, FeCl3) in 100 mM HEPES (pH 8.0) and 100 mM KCl for 10 min at 37 oC. Since iron(II) readily oxidizes in the presence of oxygen, all assays involving iron were conducted with Fe(SO4)2 under anaerobic conditions with buffers that had been degassed and sparged with nitrogen prior to use. Assays were initiated upon the addition of GTP to a final concentration of 0.1 mM and the reactions were allowed to proceed at 37 °C for 30 min in the dark. Reactions were terminated and analyzed by fluorescence with either a PTI Time-Master fluorometer or a Gemini XPS microplate spectrofluorometer (Molecular Devices) as described above.
Steady state kinetic analysis of wild-type N. gonorrhoeae GCYH-IB
Steady-state kinetic measurements were conducted under initial velocity conditions with varying concentrations of GTP (3-50 μM) as substrate. Assays were carried out in 100 mM HEPES (pH 8.0), 100 mM KCl, 0.5 mM MnCl2, 1 mM DTT and 0.5 μM of protein in a final volume of 100 μL at 37 oC. Reactions were terminated at various time points and analyzed by fluorescence with either a PTI Time-Master fluorometer or a Gemini XPS microplate spectrofluorometer (Molecular Devices) as described above. Kinetic parameters were calculated from the average of minimally 4 triplicates with the Michaelis-Menten equation using the software Kaleidagraph 4.0 (Synergy Software, Reading, PA).
Inhibition analysis was conducted by assessing enzyme activity in the presence of various fixed concentrations of 2′-deoxy-GTP (0, 50, 100, 200 μM), 7-deaza-GTP (0, 15, 30, 60 μM), or 8-oxo-GTP (0.1, 0.25, 0.5 μM). Reactions were carried out as described above in the presence of 1 mM DTT, 0.5 μM protein, 0.5 mM MnCl2, variable GTP concentration (3-50 μM), and analyzed by fluorescence. The data (average of minimally 4 triplicates) were fitted to the equation for competitive inhibition (eq. 1)
| (1) |
where ν is the initial rate of reaction, S is the varied substrate concentration, I is the inhibitor concentration, Ki is the inhibition constant, and V is the maximum reaction velocity.
Substrate specificity analysis
Substrate specificity was determined with various nucleotides in 100-μL reactions performed in the presence of 100 mM HEPES (pH 8.0), 100 mM KCl, 0.5 mM MnCl2, 1 mM DTT and 5.0 μM of protein and incubated at 37 °C for 60 minutes. Activity with GTP was assessed by both fluorescence and reversed-phase HPLC, while activities with 2′-dGTP, 7-deaza-GTP or 8-oxo-GTP as substrates were assessed by reversed-phase HPLC, as described previously [10, 12].
Crystallization
Crystals of the enzyme complex with TRIS were generated using the enzyme prepared in buffer solutions containing 50 mM TRIS-acetate (pH 8.0), 100 mM KCl, 5 mM ZnCl2 and 1 mM BME, and concentrated to 9 mg/mL in an Amicon device. Crystals were grown at 20 °C by vapor diffusion in sitting drops prepared by mixing 1 μL enzyme solution and 1 μL reservoir solution containing 10-16% (w/v) polyethylene glycol 6000, 1-1.4 M LiCl, 50 mM TRIS (pH 9.0), and 50 mM TRIS-HCl (pH 7.0), and equilibrated against reservoir solution for 1 week.
For preparation of the complex with 8-oxo-GTP, first TRIS was removed from a sample containing 9 mg/mL N. gonorrhoeae GCYH-IB by dialysis against 2 L of buffer solution containing 50 mM HEPES (pH 7.4), 100 mM KCl, 5 mM ZnCl2, and 1 mM BME for 12 hours. 8-oxo-GTP was then added directly to the dialyzed protein to a final concentration of 1 mM and incubated for 30 minutes. Crystals were grown by vapor diffusion in sitting drops prepared by mixing 1 μL enzyme-inhibitor complex and 1 μL reservoir solution containing 10-16% (w/v) polyethylene glycol 6000, 0.6-1.4 M LiCl, 60 mM HEPES (pH 8.2), and 40 mM HEPES (pH 6.8). For both enzyme-ligand complexes, rhomb shaped crystals grew in 1 week to dimensions of 0.2 × 0.2 × 0.1 mm3 and were harvested and cryo-protected in mother liquor containing additional 25% ethylene glycol as cryo-protecting agent, and flash-cooled in liquid nitrogen.
X-ray data collection, structure determination and refinement
For the complex with TRIS, single-wavelength X-ray diffraction data were collected at the Stanford Synchrotron Research Laboratory beam line 7-1. For the complex with 8-oxo-GTP, data were collected at the Advanced Light Source beam line 5.0.1. All data were processed in the HKL2000 program suite [20]. The structures were determined using the difference Fourier method, and the previously determined structure of the enzyme (PDB ID 3D1T, [12]) as a start model after removing all heteroatoms. Initial rigid-body refinement, followed by restrained crystallographic refinement and solvent fitting were done using refmac-5 [21] and Coot [22]. Clear density corresponding to bound ligands and zinc ions could be seen in the active sites in both structures after rigid-body refinement. Zn2+ and ligands were modeled in the density and the structures were subjected to several rounds of restrained maximum-likelihood refinement of coordinates and temperature factors, iterated with solvent modeling.
Preparation of site-directed mutants
Point mutagenesis was carried out on pSAB-8-142 (N. gonorrhoeae GCYH-IB gene cloned in pET-30 Xa/LIC vector [10]) using the Q5® Site-Directed Mutagenesis Kit (NEW ENGLAND Biolabs Inc.) or the QuikChange™ site-directed mutagenesis kit (Stratagene). The Glu152 residue was mutated to Ala (GAG→GCT). Glu243 was mutated to Ala (GAG→GCT). His246 was mutated to Ala (CAC→GCT), Asp (CAC→GAC), Gln (CAC→CAG), Asn (CAC→AAC), and Lys (CAC→AAG). Cys149 was mutated to Ser (TGT→TCT) and Ala (TGT→GCC). The primers used for the mutations are listed in Supplementary Table 1. The resulting DNA constructs were confirmed to be correct and in frame by sequencing (at Genewiz, Inc, South Plainfield, NJ, or at the Portland State University-Keck Genomic Facility, Portland, OR). The His246Ala and Glu243Ala mutants were overexpressed in E. coli DH5α competent cells, and all other mutants were overexpressed in E. coli BL21(DE3) as described for the wild-type enzyme. All mutant proteins were purified by Ni-NTA and size-exclusion chromatography as described for the wild-type enzyme. Purity was verified to be greater than 95% by SDS-PAGE.
Enzymatic assays of all GCYH-IB mutants and wild-type enzyme were monitored via both UV-vis and radiochemical assays as described above. Initial velocities were calculated from non-linear fits of the reaction progress curves.
Test for in vitro formylation of GCYH-IB
To test for formylation of GCYH-IB 50 μL reactions containing 100 mM HEPES (pH 8.0), 100 mM KCl, 0.5 mM MnCl2, 1 mM DTT, and 100 μM [8-14C]GTP (10 μCi/μmol) were prepared. Reactions were initiated with either 25 μM recombinant His6-tagged N. gonorrhoeae GCYH-IB that possessed a modified Cys149 residue, or 25 μM enzyme that had been subjected to the GSH treatment described below. The reactions were incubated in 1.5 mL Eppendorf tubes at 37°C for 40 minutes before the addition of 20 μL of Ni-NTA resin and a further 30 minute room-temperature incubation with gentle mixing. The reactions were centrifuged for 5 minutes at 500g and the supernatant saved. The Ni-NTA resin pellets were subjected to 10 washes with 200 μL of buffer (100 mM HEPES (pH 8.0), 100 mM KCl) with centrifuging for 2 minutes at 500g between each wash. The supernatant, the ten washes, and the 1.5 mL reaction tube holding the Ni-NTA resin pellet (but with cap removed) were collected in 5 mL scintillation vials and combined with Econo-Safe scintillation cocktail (RPI). Counting was performed on a Hidex 300 SL scintillation counter (Hidex) using MikroWin software (Mikrotek Laborsysteme GmbH).
In vitro de-nitrosylation of GCYH-IB
To prepare de-nitrosylated GCYH-IB, a sample containing 600 μg of freshly purified, active enzyme was made at a concentration of 28 μM in buffer containing 100 mM HEPES, 100 mM KCl, and 0.5 mM MnCl2 (pH 8.0). Glutathione (GSH) was freshly dissolved in water and added to the protein sample to a final concentration of 280 μM and was incubated for 30 minutes in the dark. Excess GSH was removed in two buffer exchange steps by loading onto Zeba Spin Desalting Columns (MWCO 7,000 Da, ThermoFisher Scientific, Waltham, MA) pre-equilibrated in buffer containing 100 mM HEPES (pH 7.4), and 100 mM KCl; and centrifuging at 1000 g for 2 minutes. The resulting GSH-treated protein was immediately assayed for activity using the fluorescence assay described above, and for its state of nitrosylation using the iodoTMT-switch assay (vida infra).
IodoTMT switch assay
This assay was carried out using the Pierce™ S-nitrosylation Western Blot Kit (ThermoFisher Scientific, Waltham, MA) following a modified version of the manufacturer's protocol. Briefly, a 100 μL sample containing 1 μg/μL protein in HENS buffer was prepared, and 5 μL of a 100 mM solution of the irreversible blocking agent N-ethylmaleimide (NEM, prepared in ultrapure water) were added to block the protein free thiols. After vortexing vigorously and incubating for 30 minutes at 50 °C, excess NEM was removed in two buffer exchange steps using Zeba Spin Desalting Columns (MWCO 7,000 Da). The protein was then precipitated by adding 6 volumes of cold acetone (pre-equilibrated at -20 °C) and incubating for one hour at -20 °C. The precipitated protein was pelleted by centrifuging at 10,000 g for 10 minutes, and the pellet was re-suspended in 100 μL of HENS buffer and divided into two 50-μL batches. To one batch, 4 μL of 1.0 M sodium ascorbate (prepared in ultrapure water) and 1 μL of 20 mM iodoTMT6-126 labeling reagent (iodoTMT) were added to reduce and label the nitrosylated thiols, respectively. (The final protein, ascorbate, and iodoTMT concentrations were 30 μM, 75 mM and 0.4 mM, respectively). For a negative control, the second batch was treated similarly except that the ascorbate was replaced with ultrapure water. Following incubation for 2 hours at room temperature, excess labeling reagent and ascorbate were removed in two buffer exchange steps using Zeba Spin Desalting Columns. The proteins were then precipitated by adding 6 volumes of cold acetone and incubating over night at -20 °C, followed by centrifugation at 10,000 g for 10 minutes. The precipitated proteins were each re-suspended in 50 μL of HENS buffer and analyzed by Western blotting (see below). All steps were performed in the dark and the protein concentration after each precipitation/resuspension step was determined using a Bradford assay, absorption at 280 nm, and BSA-calibrated SDS-PAGE on an Any kD Mini-Protean TGX gel (Bio-Rad, Hercules, CA).
Western blot analysis of iodoTMT-labeled proteins
Equal amounts of labeled proteins were loaded on a denaturing Mini-Protean TGX precast gel (Bio-Rad, Hercules, CA) and electrophoresed without any reducing agents for 15 minutes at 300 V. The proteins were then transferred to a PVDF membrane (0.45 μm, ThermoFisher Scientific) by electroblotting for 48 mins at 60 V in a wet transfer apparatus (Bio-Rad, Hercules, CA). The membrane was blocked by incubating for 5 hours in 5 mL TRIS-Buffered Saline and Tween (TBST buffer: 20 mM TRIS, 150 mM NaCl and 0.1% Tween 20, pH 7.5) containing 2% BSA, and immunolabeled by incubation overnight in TBST solution containing mouse anti-TMT monoclonal antibody (IgG2b, ThermoFisher Scientific, product number 90075) at a dilution of 1:500. Following 5 washes in TBST, the membrane was probed with goat anti-mouse IgG-horseradish peroxidase conjugate antibody (ThermoFisher Scientific, product number Ab97040) at 1:10,000 dilution for one hour. Subsequently, the membrane was washed 5 times with TBST and developed with SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher Scientific). All steps were carried out at room temperature. To verify equal loading of proteins, a second, identical SDS-PAGE was performed in parallel on the same samples and that gel was stained with Coomassie to visualize total protein.
Oxidation of GCYH-IB by hydrogen peroxide (H2O2)
A 20-μL sample of each of the freshly purified GCYH-IB (verified to be S-nitrosylated by the iodoTMT assay as described above), and the GSH-treated GCYH-IB was prepared in assay buffer (100 mM HEPES (pH 8.0), 100 mM KCl, 0.5 mM MnCl2, 1 mM MgCl2) at a final protein concentration of 25 μM. H2O2 was added to a final concentration of 0.25 mM and the reactions were incubated in the dark for 5, 10, 15, 30 and 60 seconds before addition of catalase (from bovine liver, Sigma-Aldrich) to a final concentration of 600 U/mL to quench excess H2O2. The samples were immediately assayed for H2NTP production using the fluorescence assay.
Results
Biochemical characterization of GYCH-IB from N. gonorrhoeae
We previously reported the biochemical properties of GCYH-IB from B. subtilis [12], and the N. gonorrhoeae enzyme exhibited similar characteristics. In particular, the N. gonorrhoeae GCYH-IB shows optimal activity broadly between pH 8.0 – 8.5 in HEPES buffer (data not shown), and is able to utilize a variety of metals for catalysis (Supplementary Table 2), exhibiting maximal activity in the presence of 0.5 mM Mn2+.
Kinetic parameters and competitive inhibition of N. gonorrhoeae GCYH-IB with substrate analogs
Steady-state kinetic analysis of recombinant N. gonorrhoeae GCYH-IB, overexpressed in E. coli and freshly purified, provided a Km of 9.15 ± 0.67 μM and kcat of 0.0159 ± 0.0013 min-1 with GTP as substrate, similar to the previously reported kinetic parameters for the Bacillus subtilis enzyme (Km of 9.87 μM and kcat of 0.0011 min-1 [12]). The substrate analogs 2′-dGTP, 7-deaza-GTP and 8-oxo-GTP were investigated for their effects as inhibitors, and all three analogs gave competitive inhibition patterns with respect to GTP, but only 8-oxo-GTP functioned as a potent inhibitor with a Ki/Km of 0.022, which is 300 and 1000 fold lower than the Ki/Km for 7-deaza-GTP and 2′-dGTP, respectively (Table 1, Fig. 2). These analogs were also investigated as alternate substrates, but reversed-phase HPLC analysis of incubations with the enzyme failed to detect any products. Indeed, in each case only a single peak corresponding to the starting nucleotide was observed in the HPLC chromatogram (data not shown), indicating that they were not turned over by the enzyme.
Table 1.
Competitive inhibition constants for N. gonorrhoeae GCYH-IB with substrate analogs.
| Inhibitor | Ki (μM)a | Ki/Km |
|---|---|---|
| 2′-deoxyGTP | 164.2 ± 3.1 | 24.3 |
| 7-deaza-GTP | 45.7 ± 1.5 | 6.8 |
| 8-oxo-GTP | 0.149 ± 0.0049 | 0.022 |
Values are presented with respect to their standard errors.
Figure 2.

Competitive inhibition of N. gonorrhoeae GCYH-IB with GTP analogs. Reactions were performed with varied GTP concentration (3-50 μM) and in the presence of various fixed concentrations of inhibitor. Activity was monitored by fluorescence with excitation at 365 nm and emission at 446 nm. (A) 2′-dGTP: (•) 0, (□) 50, (◊) 100, and (○) 200 μM; (B) 7-deaza-GTP: (•) 0, (□) 15, (◊) 30, and (○) 60 μM; and (C) 8-oxo-GTP: (•) 0, (□) 0.1, (◊) 0.25, and (○) 0.5 μM. Data were fit to equation 1 using KaleidaGraph 4.0.
Crystal structure of N. gonnorrhoae GCYH-IB in complex with 8-oxo-GTP
The enzyme complex with the inhibitor nucleotide 8-oxo-GTP crystallized in space group C2221. The structure was determined by difference Fourier calculation using model phases calculated from the previously determined crystal structure of the inhibitor-free enzyme (PDB ID 3D1T [12]), and was refined at a resolution of 2.77 Å. Data collection and structure refinement statistics are listed in Table 2. The structure reveals the GCYH-IB homotetramer [12] with half a tetramer (subunits A and B) comprising the asymmetric unit (Fig. 3). Each subunit (257 amino acids) is composed of two tunnel-fold (T-fold) modules with the architecture characteristic of bimodular T-fold enzymes. In each subunit, an eight-stranded tightly twisted antiparallel β-sheet is backed by four antiparallel α-helices. The β-sheets from the two adjacent subunits form a continuous 16-stranded β-barrel, and two barrels join together head-to-head to form a central tunnel. The enzyme active sites are located at the interfaces between subunits.
Table 2.
X-ray data collection parameters and structure refinement statistics.
| Parameter | Value | Value |
|---|---|---|
| Data collection | ||
| Structure | TRIS complex | 8-oxo-GTP complex |
| Beamline | ALS-5.0.1 | ALS-5.0.1 |
| Space group | C 2 2 21 | C 2 2 21 |
| Unit cell (Å) | 92.71, 100.56, 114.02 | 91.88, 100.56, 115.04 |
| Resolution (Å) | 68.16-1.9 (1.95-1.9) | 67.83-2.77 (2.90-2.77) |
| Wavelength (Å) | 0.9774 | 0.9774 |
| Unique Reflections | 41,799 (2,2068) | 13,898 (1,297) |
| Completeness (%)a | 99.2 (91.6) | 99.7 (96.8) |
| Redundancy | 4.4 (2.7) | 8.4 (6.0) |
| Rsym b | 0.053 (0.45) | 0.076 (0.67) |
| Average I/σ(I) | 36.4 (2.1) | 16.4 (1.2) |
| Refinement statistics | ||
| Resolution range (Å) | 68.1-1.9 | 67.8-2.8 |
| Rcrsyt c | 0.172 | 0.19 |
| Rfreed | 0.208 | 0.25 |
| R.m.s. deviation from ideality | ||
| Bond length (Å) | 0.019 | 0.010 |
| Bond angle (°) | 1.89 | 1.49 |
| Average B factor (Å2) | 42.4 | 61.6 |
| Ramachandran plot: | ||
| Preferred (%) | 94.4 | 90.8 |
| Allowed (%) | 5.6 | 6.8 |
| Outlier (%) | 0.0 | 2.4 |
Values in parentheses are for the highest-resolution shell.
Rsym = Σ|Iobs − <I>|/Σ Iobs.
Crystallographic R-factor = Σ||Fobs| − |Fcalc||/Σ|Fobs|.
Rfree is monitored with 5% of the reflections excluded from the refinement.
Figure 3.

Crystal structure of N. gonnorrhoae GCYH-IB in complex with 8-oxo-GTP. A) Side (left) and top (right) views of the enzyme homotetramer with subunits shown in different colors. Bound Zn2+ ions are shown as red spheres, and bound 8-oxo-GTP molecules in two of the four active sites in the homotetramer are shown in red stick representation.
Difference electron density maps reveal a bound zinc ion and 8-oxo-GTP molecule in two of the four active sites in the biological tetramer (Fig. 3 and Fig. 4A). The other two active sites are more exposed to solvent in the crystal lattice and are unoccupied. The inhibitor nucleotide is in the syn conformation and its binding pocket lies at the interface between three subunits, two from the same β-barrel (subunits A and B), and one from the opposite β-barrel (subunit B′, Fig. 4). The purine moiety of the inhibitor is inserted into the pocket, while the ribosyl and triphosphate moieties lie on the surface near the pocket entrance.
Figure 4.
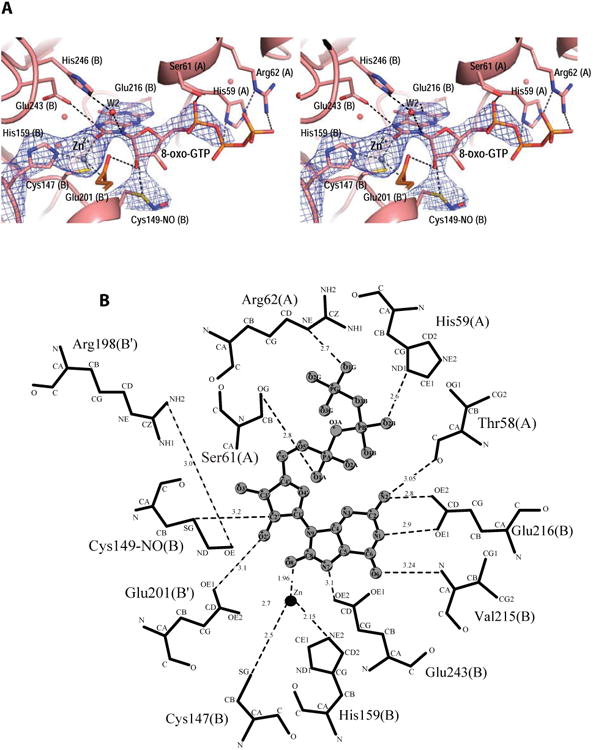
View of the active site of N. gonnorrhoae GCYH-IB with bound 8-oxo-GTP. A) Stereoview of the 2Fo-Fc electron density map (contour level 1.1 σ, resolution 2.77 Å), contoured around the bound Zinc ion, 8-oxo-GTP, and the nitrosylated Cys149, and superposed on the refined model. Zn2+ is shown as purple sphere, and water molecules as red spheres. Side chains interacting with 8-oxo-GTP are labeled and the subunits from which they originate are indicated in parentheses. B) Schematic of enzyme-inhibitor interactions. The ligand is shown in ball-and-stick representation. Dotted lines indicate hydrogen bonds or interactions with the metal ion. Residues are labeled as in panel A. For clarity, His246 and the water molecule W2 are not shown in panel B. The figure was made with the program LIG-PLOT [54].
The Zn2+ ion binds at the interface between opposite β-barrels and is coordinated in a distorted tetrahedral geometry by the Sγ atom of Cys147, the Nε2 of His159, the Oε2 of Glu201 (all invariant residues in the type IB enzymes), and the O8 atom of 8-oxo-GTP (Fig. 4). The purine moiety of the inhibitor is sandwiched between Phe214 (from subunit B) and Met60 (from subunit A), both conserved hydrophobic residues in the GCYH-IB family. The purine ring is anchored to the pocket by side chain and backbone interactions with Glu216, Val215, and Thr58; while the purine N7, which is protonated in 8-oxo-GTP, donates a hydrogen bond to the side chain of the invariant residue Glu243 (Fig. 4). The latter interaction indicates a role for Glu243 in the protonation of N7 of the GTP substrate, an event necessary for the first hydration step of the GCYH-I reaction. Consistent with this interpretation, replacement of N7 with a carbon in the 7-deaza-GTP analog results in compromised binding (Table 1).
The ribose ring adopts a C3′-endo pucker conformation, a common conformation in nucleosides and nucleotides in general [23]. Two key interactions are seen with the ribosyl moiety: 1) The ribosyl 2′-hydroxyl group is coordinated via a hydrogen bond (H-bond) by the carboxylate Oε1 of Glu201 from the adjacent β-barrel (thus, Glu201 bridges between the metal ion and the inhibitor). The importance of this interaction for substrate binding is demonstrated by the 24-fold higher Ki value of 2′-deoxyGTP relative to the Km of GTP (Table 2). 2) The C2′ atom is within contact distance from the Sγ atom of the conserved residue Cys149. Close inspection of the difference electron density maps revealed extra density extending from the Sγ atom of Cys149, corresponding to a nitrosyl group (Fig. 4A). Although the resolution of this structure does not permit distinguishing a nitrosyl from a formyl group, the latter was ruled out based on the higher resolution Tris-bound structure and a biochemical test (vida infra). The S-nitrosothiol adopts a staggered anti conformation with a Cβ-Sy-N-O dihedral angle of 103°. As shown by Zhao and Houk [24], this conformation suggests a stable reduced thionitroxide radical form in which a hydrogen is bonded to the nitrogen atom (Cβ-Sy-NH-O). Consistent with this interpretation, the putative radical is stabilized in the structure by interaction with the conserved Arg198 side chain protruding into the active site from the opposite β-barrel (Fig. 4B). Notably, the nitrosyl group is present even though the protein was purified and crystallized in the presence of 1 mM DTT/βME, consistent with a highly stable thionitroxide form. S-nitrosylation of Cys149 is also seen in the unoccupied active sites, and with a similar staggered conformation (dihedral angle ∼ 122°). The triphosphate tail of 8-oxo-GTP is stabilized by H-bonds with conserved residues His59, Ser61 and Arg62.
Bound TRIS molecule mimics a reaction intermediate
We also determined a high resolution (1.9 Å) crystal structure of the enzyme prepared and crystallized in the presence of TRIS buffer. In this structure, a bound TRIS molecule is seen coordinating the Zn2+ ion in two of the four active sites in the homotetramer. The Zn2+ ion forms a trigonal bipyramidal coordination shell with its five ligands: the Sγ atom of Cys147, the Oε2 atom of Glu201 from subunit B′, and the amino group of bound TRIS in the equatorial plane; and the O3 hydroxyl group of TRIS, and the Nε2 atom of His159 as axial ligands (Fig. 5). This rather unusual zinc coordination geometry is common in proposed transition states of metalloenzymes [25]. Cys149 is also found S-nitrosylated in this structure. To rule out the possibility that the modification may be S-formylation associated with the formate-releasing activity of the enzyme, we modeled a formyl group in the electron density and observed the resulting temperature factors after structure refinement. The refined temperature factor for the formyl carbon atom was 8 Å3 lower than the temperature factor of the Sγ atom, indicating that the electron density is under-represented by the model in this case. In contrast, when modeled as a nitrosyl group, the refined temperature factor of the nitrosyl nitrogen atom is within the expected range for optimal fit to the density (< 3 Å3 lower than the temperature factor of the Sγ atom). In this structure, the S-nitrosyl group is in a syn conformation and exhibits a planar geometry with a small Cβ-Sγ-N-O dihedral angle of ∼24°, close to the geometry of small-molecule S-nitrosothiols [26-31], indicating that it is the oxidized form Cβ-Sγ-N=O [24].
Figure 5.
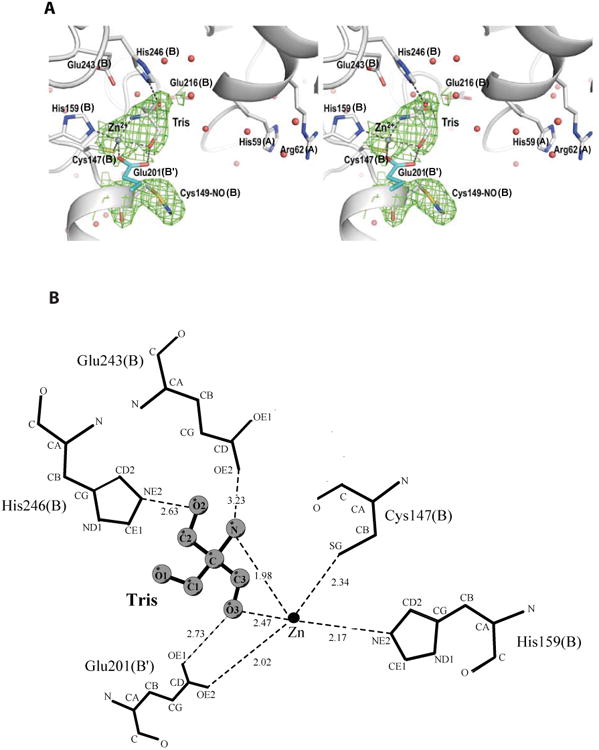
The TRIS occupied active site in the crystal structure of N. gonnorrhoae GCYH-IB in complex with TRIS. A) Stereoview of the 2Fo-Fc electron density map (contour level 1.3 σ, resolution 1.9 Å), contoured around the bound Zinc ion, TRIS, and the nitrosylated Cys149; and superposed on the refined model. Zn2+ is shown as purple sphere, and water molecules as red spheres. Side chains interacting with TRIS are labeled and the subunits from which they originate are indicated in parentheses. B) Schematic of enzyme-TRIS interactions. The ligand is shown in ball-and-stick representation. Dotted lines indicate hydrogen bonds or interactions within the metal coordination sphere. The figure was made with the program LIG-PLOT [54].
Superposition of the TRIS-bound structure with the 8-oxo-GTP-bound structure (r.m.s.d. of 0.27 Å over 237 of subunit B Cα atoms, subunit A is much less ordered) reveals that the bound Tris molecule is reminiscent of a tetrahedral transition-state intermediate proposed to occur in the GCYH-I-catalyzed reaction prior to breakage of the C8-N7 bond and release of formate from GTP (Fig. 6) [16]. In this intermediate, C8 is the tetrahedral center that is imitated by the tetrahedral carbon atom of TRIS. In the superposition, the N atom of TRIS occupies the same site as the O8 atom of 8-oxo-GTP, thus mimicking the state of the metal-coordinated hydroxyl ion after it performs the first nucleophilic attack on the C8 atom of the substrate. The C2 carbon of TRIS mimics the state of the second water molecule after it performs the second nucleophilic attack on C8. Notably, the O2 atom of TRIS makes a hydrogen bond with the conserved side chain His246 (Fig. 5 and Fig 6), and the position of O2 may represent the position of the second nucleophilic water molecule just before its attack on C8 and formation of the tetrahedral center. Further, the metal-coordinating O3 atom of TRIS occupies an analogous position to the 2′-OH of 8-oxo-GTP, thus may represent the position of the 2′-OH of the substrate during a late step in the reaction after opening of the ribose ring. Finally, a 6 Å-wide water-filled cavity located behind the purine binding pocket is seen in both structures and may serve as the putative binding site for the formate molecule released in the early steps of the GTP cyclohydrolase reaction (Fig. 6).
Figure 6.

View in the active site region of the superposition of the 8-oxo-GTP-bound structure (pink) and the TRIS-bound structure (grey). Zn2+ is shown as purple sphere, and water molecules as red spheres. For clarity, Ser61(A) is not shown. The ligand-coordinating side chains are shown and labeled as in figure 4. TRIS and 8-oxo-GTP atoms discussed in the text are labeled in black and pink, respectively. The water filled cavity serving as the putative formate binding site is marked with an asterisk.
The crystal of the complex with TRIS was grown under the same crystallization conditions and using the same sample preparation conditions as the previously described structure with PDB ID 3D1T [12] and a resolution of 2.2 Å. Re-refinement of PDB ID 3D1T also revealed a bound TRIS molecule in the active site and S-nitrosylated Cys149, and was therefore replaced with the higher resolution structure presented here.
S-nitrosylation of GCYH-IB is site specific and preserves enzyme activity
The structural observation of S-nitrosylation of a catalytic active-site cysteine residue prompted us to investigate this post-translational modification biochemically. First, we confirmed its presence in the freshly purified enzyme in solution using the iodoTMT-switch assay, and investigated the effect of the SNO on enzyme activity. In the iodoTMT-switch assay, the free thiols in the protein are first blocked using the irreversible blocking agent NEM. The S-nitrosylated cysteines are then reduced with ascorbate, and the newly formed free thiols are labeled with iodoTMT. The labeled thiols are then detected and quantified by Western blot analysis using an anti-TMT antibody. To elucidate the effect of S-nitrosylation of Cys149 on enzyme activity, we denitrosylated the freshly purified wild-type enzyme by treating it with reduced glutathione (GSH, a widely used method for denitrosylation of proteins [32]) and, after removal of excess GSH, assayed the resulting denitrosylated form for H2NTP production activity using the fluorescence assay. To eliminate the effect of possible glutathione chelation of the catalytic metal, the GSH-treated enzyme was incubated in assay buffer containing 0.5 mM metal before adding substrate. While the GSH-treated enzyme was fully denistrosylated, as confirmed by the iodoTMT-switch assay (Fig. 7A), its activity was found to be comparable to that of the untreated enzyme, with only 20% reduction (Fig. 7B), indicating that Cys149 supports catalysis in both the nitrosylated and reduced states.
Figure 7.

S-nitrosylation of N. gonnorrhoae GCYH-IB. A) iodoTMT-switch assay of freshly purified wild-type GCYH-IB, of the enzyme after treatment with 0.5 mM GSH, and of the Cys149Ala mutant. Top: Western blot of TMT-labeled proteins visualized using an anti-TMT antibody. Bottom: Coomassie Brilliant Blue staining of the same gel showing equal loading in all lanes. B) Relative H2NTP producing activities of the enzyme samples in panel A as measured using the fluorescence assay. The error bars indicate the standard error of triplicate measurements.
As further proof that the Cys149 modification occurred as a consequence of nitrosylation, and not formylation in the course of enzymatic turnover, we probed for the possibility of formyl transfer from GTP with reactions containing [8-14C]GTP followed by capture of the protein on Ni2+-NTA resin and liquid scintillation counting. To test for formylation that might occur during each turnover (with consequent loss of starting formyl group) as well as advantageous formylation that might occur to unmodified Cys149 and then be retained in subsequent turnovers, we tested enzyme that possessed modification of Cys149 and enzyme that had been treated with GSH as described above, and thus lacked modification. In no case did we see radioactivity above background associated with protein fractions (data not shown), allowing us to conclude that no transfer of a formyl group to the protein occurred in any of the reactions. In these experiments the background radioactivity corresponded to the calculated radioactivity that would be observed for ∼0.5% labeling.
In addition to Cys149, N. gonnorrhoae GCYH-IB contains two other cysteine residues in each subunit: the metal coordinating Cys147 in the active site, and Cys190 located in a partially solvated hydrophobic pocket 12 Å away from the active site. In the crystal structures, only Cys149 is seen S-nitrsoylated. To confirm this observation, we assessed the nitrosylation state of the Cys149Ala mutant using the iodoTMT-switch assay. The results show that while the fully active, wild-type protein in solution and as purified from E. coli is S-nitrosylated, the Cys149Ala mutant protein purified under the same conditions lacks any S-nitrosylated thiols (Fig. 7A). These results demonstrate that the observed S-nitrsoylation of GCYH-IB occurs posttranslationally during heterologous expression in E. coli and that it is specific to Cys149. Although the physiological relevance of the observed S-nitrosylation of GCYH-IB in a heterologous system is unclear, the specificity of the nitrosylation to a conserved and essential active-site cysteine, and the direct interaction of the thionitroxide group with the substrate analog seen in the crystal structure suggest that it may participate directly in catalysis (see details in the discussion).
Role of Cys149, Glu152, Glu243, and His246 in catalysis
Of the active site residues that interact with 8-oxo-GTP and TRIS in the structures, three residues make contacts that suggest a direct role in catalysis. These residues are Glu243 and Cys149, which interact with the guanine imidazole ring and ribosyl moiety of 8-oxo-GTP, respectively, and His246, which coordinates the O2 atom of the transition state mimic TRIS. Additionally Glu152 is positioned where it might serve as an acid/base residue to modulate the protonation state of Cys149. Glu243, and His246 are conserved residues in all GCYH-IB sequences, whereas Cys149 is only conserved in bacterial GCYH-IB sequences and Glu152 is present in only a subset of bacterial sequences (see Supplementary Fig. 1). To investigate the role of these residues in catalysis, we prepared site-directed mutants of Cys149, Glu152, Glu243, and His246, and measured the abilities of the mutant proteins to catalyze formation of the reaction product H2NTP, as well as their abilities to catalyze the early steps in the reaction leading to formate release from GTP. For the former activity we monitored the formation of H2NTP using a fluorescence assay as well as a UV-vis continuous assay measuring the absorbance of H2NTP at 330 nm. For the latter activity, we monitored release of [14C]-formate from [8-14C]GTP using a radiochemical assay. Notably, studies with the IA enzyme have demonstrated the ability to uncouple the early steps in the reaction from the late steps by independently measuring formate and H2NTP formation, and showing that some mutants compromised in H2NTP formation are able to efficiently catalyze the early steps in the reaction and release formate along with the intermediate 2,5-diamino-(6β)-ribosylamino-4(3H)-pyrimidinone 5′-triphosphate (DRPT) [33]. All mutants were overexpressed in E. coli and freshly purified under the same conditions used to purify the wild-type enzyme. The structural integrity of the mutant proteins was checked by size exclusion chromatography and their elution profiles were identical to that of the wild-type enzyme, showing a single peak corresponding to an intact homotetramer with apparent molecular weight of 135,000 Da (Supplementary Fig. 2).
With the exception of the Glu152 mutant, which exhibited ∼20% of the wild-type activity, all of the alanine mutants exhibited impaired ability to carry out full catalytic turnover to produce H2NTP, with activities <1% that of the wild-type enzyme (Table 3, Supplementary Fig. 3). In contrast, when assaying for production of [14C]-formate only the Glu152Ala and Glu243Ala mutants showed a similarly impaired ability in the production of formate, while the His246Ala mutant exhibited an almost 3x higher rate of formate production than H2NTP, and the Cys149Ala mutant, for which we were unable to detect H2NTP production, exhibited a robust rate of formate production roughly 10% that of the wild-type enzyme. Consistent with this pattern, the Cys149Ser mutant displayed activity for H2NTP formation ∼3% that of wild-type, and for formate production almost 20% that of wild-type.
Table 3.
Relative catalytic activities (%) of GCYH-IB mutants.
| Protein | Radiochemicala | UV-visa |
|---|---|---|
| Wild-type | 100 | 100 |
| E152A | 24.7 | 21.4 |
| E243A | 0.36 | 0.37 |
| C149A | 11.0 | NAb |
| C149S | 17 | 2.7 |
| H246A | 2.0 | 0.74 |
| H246D | 72 | 3.0 |
| H246Q | 67 | 2.4 |
| H246N | 24 | NA |
| H246K | 5.0 | NA |
Activity reflects the relative initial velocities determined via the radiochemical and UV-vis time course assays, which quantify the formation of [14C]-HCO2H and H2NTP, respectively. Standard errors for the initial velocity measurements calculated from the radiochemical and UV-vis data varied from 2-8% and 2.5-10%, respectively.
NA indicates that no activity was detected. Detection limits were 0.25% of wild-type activity.
The observed catalytic activities of the Glu243 and Cys149 mutants suggest roles in the early and late stages of the chemical mechanism, respectively, conclusions consistent with the crystallographic information from the GCYH-IB structures containing 8-oxo-GTP and TRIS. In contrast, mutation of His246, which interacts with the O2 atom of TRIS in the TRIS-bound structure, an interaction suggestive of the transition state leading to formate release (vide supra) in an early step in the reaction, results in enhanced production of formate relative to H2NTP.
To further probe the role of His246, we constructed His246 mutants substituted with Asn, Gln, Asp and Lys and measured their ability to catalyze the formation of formate and H2NTP. All of these mutants were compromised in their ability to catalyze the formation of H2NTP (Table 3), with only the Asp and Gln mutants capable of complete turnover. Remarkably, all of the mutants were capable of modest to robust production of formate, with the Asp and Gln mutants capable of formate production at a rate >65% that of the wild-type enzyme (Table 3).
S-nitrosylation of GCYH-IB does not provide protection against oxidation
Since the nitrosylated cysteine is a catalytic residue, we interrogated the possibility of the SNO modification serving a protective role against oxidative inactivation of the enzyme. In this experiment, the activities of the freshly purified (S-nitrosylated) GCYH-IB and the GSH-treated (denitrosylated) enzyme were monitored following incubation with 0.5 mM hydrogen peroxide for 30 seconds. The activities of both the nitrsoylated and denitrosylated enzyme dropped by the same extent and rate with exposure time to H2O2 (data not shown), indicating no observable protection by the SNO against oxidative inactivation.
Discussion
Comparison with GCYH-IA
8-oxo-GTP is the most potent inhibitor of GCYH-IB identified to date, exhibiting a Ki = 150 nM with the N. gonorrhoeae enzyme. Notably, however, inhibition is more modest than observed with GCYH-IA, which exhibits a Ki = 5.4 nM with the enzyme from T. thermophilus [16], almost 28-fold lower than the observed Ki for GCYH-IB. Comparison with the crystal structure of the ternary complex of T. thermophilus GCYH-IA with Zn2+ and 8-oxo-GTP (PDB ID 1WUQ) [16] reveals that the two enzymes bind 8-oxo-GTP in the same general orientation and provide the same distorted tetrahedral geometry for coordinating the Zn2+, with the O8 atom of 8-oxo-GTP acting as the fourth and axial ligand (Fig. 8A), consistent with a similar role for the metal ion in their catalytic mechanisms.
Figure 8.

Comparison between GCYH-IB (left) and GCYH-IA (right) ternary complexes with zinc and 8-oxo-GTP showing interactions in the active site. Cα chains and active site residues are shown as ribbons and sticks, respectively. Subunits A, B and B′ are colored orange, cyan and magenta, respectively. Hydrogen bonds and Zinc ion interactions are shown as dashed yellow lines. A) The Zinc-binding site and common distorted tetrahedral coordination geometry. Two subunits contribute ligands to the metal site in the -IB enzyme, whereas in the -IA enzyme, all ligands originate from one subunit. B) The purine and ribose binding pocket showing similar interactions and mode of binding for the purine ring and rotation of the ribosyl group (the triphosphate tail was removed from this view for clarity). C) Hydrogen-bond interactions for the triphosphate moiety. One subunit contributes residues to stabilization of the triphosphate in the –IB enzyme, whereas in the –IA enzyme, all three interface subunits participate.
Both enzymes make the same number of H-bonds (five direct and one water-mediated H-bonds) and similar hydrophobic interactions with the guanidine ring of 8-oxo-GTP. However, their recognition of the ribosyl and triphosphate moieties is different (Fig. 8B). First, the two enzymes favor different orientations of the ribosyl moiety relative to the base, as achieved through rotation around the β-N9-glycosidic bond. In GCYH-IB, the bound nucleotide's chi (χ) torsion angle (defined by O4′-C1′-N9-C4) is 34°, whereas in GCYH-IA it is -106° (averaged over all active sites), amounting to a counterclockwise rotation around the glycosidic bond of 140° in the GCYH-IB-bound conformer relative to the GCYH-IA-bound conformer. This rotation is stabilized by enzyme-specific interactions with the 2′- and 3′-hydroxyl groups. In the GCYH-IB-bound conformer, O2′ and O3′ point toward the B and B′ subunits and O2′ interacts with the -IB specific and invariant side chain Glu201, whereas in the GCYH-IA-bound conformer these hydroxyl groups point toward subunit A and are coordinated by the -IA specific and invariant Ser133 (in T. thermophilus numbers, Fig. 8B). Second, the two enzymes accommodate different conformers of the ribose ring in their active sites; in GCYH-IB the ribose adopts the common C3′-endo conformation, while in GCYH-IA it adopts a rare C1′-exo conformation [16]. Third, in GCYH-IB, the triphosphate tail binds in a shallow pocket formed by residues from one subunit, whereas in GCYH-IA this pocket is deeper and is formed by basic side chains from all three interfacing subunits (Fig. 8C).
Overall, the inhibitor makes only 15 H-bonds with the GCYH-IB active site (14 direct and one water-mediated) versus 20 H-bonds with GCYH-IA (17 direct and 3 water-mediated), and 4 of the 5 additional bonds are with triphosphate atoms. Thus, the apparent difference in Ki exhibited by 8-oxo-GTP to the two enzymes appears to be mostly due to differences in recognition of the triphosphate tail.
In summary, GCYH-IA and GCYH-IB exhibit similarities in their binding to the purine moiety of the substrate analog, but differ in binding the ribosyl and triphosphate groups. These differences may be employed in the design of improved inhibitors specific for GCYH-IB.
S-nitrosylation of GCYH-IB
S-nitrosylation is a nitric oxide (NO) mediated posttranslational modification that regulates protein function [34-38]. Our structural and biochemical data show that GCYH-IB is S-nitrosylated at an essential, active-site catalytic cysteine and that the nitrosylated enzyme is active, if not more active than the denitrosylated form. Although many enzymes are known to be reversibly inactivated by S-nitrosylation of an active-site cysteine (e.g., the human mitochondrial caspases [39], protein tyrosine phosphatases [40], mammalian methionine adenosyltransferases [41], and dimethylarginine dimethylaminohydrolases [42], for partial review see [43]), to our knowledge, there are no reported examples of enzymes in which S-nitrosylation of a catalytic cysteine residue maintains (or enhances) activity.
S-nitrosylation of active site cysteines has also been shown to protect some enzymes from oxidative inactivation (e.g., protein tyrosine phosphatase IB [44] and galectin-2 [45]), however we did not detect such protection of the S-nitrosylated N. gonorrhoeae GCYH-IB from oxidative inactivation by H2O2. This could be due to oxidation of one or both of the other two cysteine thiols in the protein, especially the metal ligand Cys147, thus compromising the structural integrity of the enzyme tetramer. Consistent with this interpretation, purification of the enzyme in the absence of DTT results in a destabilized tetramer (as detected by gel filtration chromatography) and subsequent loss of activity (Supplementary Figure 4). Additionally, GCYH-IB is transcriptionally upregulated in N. gonorrhoeae when the organism is subjected to oxidative stress by exposure to peroxide [46].
The precise source of the NO that posttranslationally modifies GCYH-IB during heterologous expression in E. coli remains unclear. S-nitrosylation in E. coli has been shown to be a prominent feature of anaerobic respiration on nitrate where the nitrate reductase NarG acts as the major source of S-nitrosylating activity [37]. However, in the present study, we did not provide the E. coli cultures with exogenous nitrate, and the cultures were grown aerobically. In general, biological S-nitrosylation of proteins occurs through 1) an autocatalytic mechanism in which the protein hydrophobic core accumulates NO and accelerates its oxidation to the more reactive N2O3 causing S-nitrosylation of a nearby cysteine thiol within the protein interior [47], or 2) oxidation of NO to the nitrosonium cation (NO+) by reaction with a transition metal or transfer from another S-nitrosylated protein or small S-nitroso compounds (e.g., S-nitrosoglutathione) to a solvent-exposed thiol (transnitrosylation) [48]. The nitrosylated cysteine in GCYH-IB resides in a short helix on the surface of the protein, completely exposed to solvent, consistent with transnitrosylation, not autocatalysis.
Proposed catalytic mechanism of GCYH-IB
A chemical mechanism has been proposed for the GCYH-IA enzyme based on crystallography and a variety of biochemical studies, including mutagenesis, NMR analysis and transient state kinetics [14, 16, 19, 33, 49-52]. In the early steps in the mechanism, the reaction is initiated with reversible hydrolysis of GTP at C8 via nucleophilic attack of a zinc activated water molecule and cleavage of the C8-N9 bond to form an N-formyl intermediate, which subsequently undergoes a second hydrolysis reaction at C8 resulting in the cleavage of the C8-N7 bond and elimination of C8 as formate. In the second half of the reaction the ribosyl moiety undergoes ring opening and an Amadori rearrangement followed by cyclization and loss of the 2′-oxygen as H2O to give H2NTP.
Our biochemical and structural data are consistent with GCYH-IB facilitating the same chemical steps as GCYH-IA. 8-oxo-GTP, 7-deaza-GTP and 2′-deoxyGTP do not act as substrates for GCYH-IB, consistent with the proposed GCYH-IA chemical mechanism in which oxidation of C8 to the carbonyl state and protonation of N7 (i.e. 8-oxo-GTP), although formally analogous to the N-formyl intermediate (III, Fig. 9A), precludes the second nucleophilic attack of water due to the reduced electrophilicity of C8 as a consequence of electron delocalization in the 8-oxo-guanine ring. Likewise, replacing N7 with a carbon (i.e. 7-deaza-GTP) prevents the initial hydrolysis step due to reduced electrophilicity of C8. Finally, the absence of the 2′-OH (i.e. 2′-deoxyGTP) prevents the Amadori rearrangement necessary for the formation of the pterin ring system (VIII to X, Fig. 9B).
Figure 9.


Proposed mechanism for the GCYH-IB catalyzed reaction. A) The early hydrolytic steps in the reaction leading to the formation of formic acid and the intermediate 2,5-diamino-(6β)-ribosylamino-4(3H)-pyrimidinone 5′-triphosphate (DRPT). B) The later steps in the reaction involving the conversion of DRPT to H2NTP.
However, differences in the active-site architecture and the different modes of recognition of the ribosyl moiety by the two enzymes suggest that they utilize different catalytic strategies. Our proposal of the GCYH-IB catalyzed mechanism is shown in Figure 9. In this proposal, a water molecule coordinated to the metal is first deprotonated by Glu243 to produce a metal-coordinated hydroxyl ion (I, Fig. 9A) that carries out nucleophilic attack at C8, accompanied by protonation of N7 by Glu243 (II, Fig. 9A). In contrast, in GCYH-IA, protonation of N7 is carried out by a water molecule [16] coordinated to a His residue. Decomposition of the tetrahedral intermediate and breakage of the C8-N9 bond (II, Fig. 9A) is mediated by a bound water molecule (W2 in 8-oxo-GTP bound structure, Fig 4A), yielding the intermediate 2-amino-5-formylamino-(6β)-ribosylamino-4(3H)-pyrimidinone 5′-triphosphate (AFRPT, III, Fig. 9A). This intermediate has been experimentally verified in the reaction catalyzed by GCYH-IA [33, 50] and is mimicked by 8-oxo-GTP except for the out of plane geometry of the CHO group relative to the base.
The water molecule (W2), H-bonded to His246, is deprotonated by Glu243 to yield a second nucleophilic hydroxyl ion that attacks the out of plane electrophilic carbonyl C8 to produce a second tetrahedral intermediate (IV, Fig. 9A). This intermediate is mimicked by TRIS in the TRIS-bound crystal structure of GCYH-IB (Fig. 5). Collapse of this intermediate coupled with protonation of N7 by Glu243 and bond cleavage between N7-C8 produces formic acid and the intermediate 2,5-diamino-(6β)-ribosylamino-4(3H)-pyrimidinone 5′-triphosphate (DRPT, V, Fig. 9A) [8].
Ring opening of the ribose moiety is facilitated by protonation of the ring oxygen by a water molecule coordinated to His246 coupled with Schiff base formation at C1′-N9 (VI, Fig. 9B). Ring opening facilitates a conformational rearrangement that allows the 2′-hydroxyl group to displace formate from the metal, a process facilitated by Glu201, which is seen H-bonded to the 2′-hydroxyl in the 8-oxo-GTP-bound structure (Fig. 4). The fate of the formate is unclear; the presence of a water-filled cavity behind 8-oxo-GTP (Fig. 6) suggests that formate may be trapped there until the end of the catalytic cycle. Deprotonation of C2′ by Cys149 (VII, Fig. 9B) yields the enol-enamine and the protonated Cys149 thiol (VIII, Fig. 9B). The proximity of the carboxyl group of Glu152 suggested that the this step might be facilitated by proton transfer from Cys149 to Glu152, but the mutagenesis data, in which activity for the Glu152Ala mutant was decreased by only ∼75% in both assays, does not support such a role in catalysis. Glu201, then deprotonates the 2′-OH with protonation at C1′ by Cys149 (VIII, Fig. 9B) to give a C2′ keto group (IX, Fig. 9B). Subsequent nucleophilic attack by N7 on the electrophilic keto carbonyl at C2′ is facilitated by deprotonation at N7 by Glu243 and protonation of the carbonyl by Glu201, giving the bicyclic pterin ring system (X, Fig. 9B). Dehydration coupled with imine formation then occurs with protonation of the OH group by Glu243, forming dihydroneopterin triphosphate (H2NTP).
The proposed mechanism is consistent with all of the mutagenesis and crystallographic data. Specifically, mutation of Glu243 essentially abolishes both the formate releasing activity as well as complete turnover to H2NTP, as is expected for a residue involved in the early steps of the reaction. Replacement of Cys149 with either Ala or Ser removes a base necessary for initiation of the Amadori rearrangement, thus precluding the formation of the pterin ring. However, mutation does not prevent the hydrolytic steps in the reaction as these occur prior to the direct participation of Cys149. Nevertheless, there is a decrease in the rate of hydrolytic activity (Table 3) for the two mutants, indicating that Cys149 does play some role in this process, potentially in substrate binding as suggested by the 8-oxo-GTP bound structure. The role of His246 is more complex; while the structural data suggests a role in the second hydrolytic step (vide supra), the mutagenesis data instead implicate a more direct role in the late steps of the reaction and only a minor role in the steps involved in formate release. Thus, in our mechanistic proposal His246, while involved in H-bonding in the early steps of the reaction, does not participate directly in any chemical steps (Fig. 9A), but does participate in protonation of the ribose ring via an intervening water molecule in the second half of the reaction (Fig. 9B).
Notably, the proposal is also consistent with the observation that Cys149 can be nitrosylated (Fig. 10). S-nitroso groups on cysteine side chains can exist in several oxidation and protonation states, and computational quantum mechanical studies have shown that the dihedral angle and the S-N bond length is a function of its chemical state [24]. Accordingly, the staggered conformers seen in the 8-oxo-GTP-occupied sites and in the empty active sites (dihedral angle ∼103° and ∼122°, respectively) are consistent with the radical thionitroxide form C-S-NH-O•, while the almost planar CSNO conformer (dihedral angle ∼24°) seen in the TRIS-occupied active site represents the oxidized C-S-N=O form. The latter species, constituting a minority of states, may be an artifact of oxidation by O2 or X-ray exposure during data acquisition. Although detailed acid/base properties for these species have not been reported, they are observed to be involved in H-bonding interactions (e.g., Fig. 4) and presumed to readily undergo protonation/deprotonation reactions. It is not clear which atom of C-S-NH-O• serves as the site for deprotonating C2′, but we show this occurring on the oxygen (Fig. 10) since this position has been explicitly highlighted as a putative basic site [53].
Figure 10.

Proposed mechanism for the participation of the thionitroxide of Cys149 in catalysis.
Since its discovery GCYH-I has come to exemplify the chemical complexity possible for enzymatic catalysis, and it continues to astonish some five decades later. Remarkably, this complex reaction is now known to be catalyzed by two enzymes that, despite their common origin, share only two amino acids in common and bind GTP in markedly distinct ways. As a further surprise, an essential catalytic cysteine in a GCYH-IB family member undergoes nitrosylation to form a thionitroxide species, a post-translational modification heretofore associated only with metabolic regulation, here utilized directly in catalysis. While the formation of this modification of GCYH-IB in N. gonorrhoeae awaits confirmation, the observation that the enzyme undergoes site-specific nitrosylation in E. coli, and that nitrosylation enhances catalytic activity, offers an exciting new insight into the chemistry of GCYH-I, and potentially new roles for nitrosylation in biology.
Supplementary Material
Supplementary Figure S1: Multisequence alignments of bacterial and archaeal GCYH-IB proteins. A) 990 non-redundant bacterial GCYH-IB sequences sourced from SWISS-PROT plus the N. gonorrhoeae GCYH-IB sequence (top sequence) were aligned using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/). The alignment shows strict conservation of Cys149 (yellow) in all 990 sequences, but a less conserved Glu152 (cyan). B) 337 non-redundant archaeal GCYH-IB sequences obtained from SWISS-PROT were aligned using Clustal Omega. Unlike the bacterial sequences, the archaeal sequences exhibit low sequence similarity and 43 of them contained His at the position homologous to Cys149 of N. gonorrhoeae GCYH-IB. None of the 43 sequences has been experimentally validated for function, but 3D structural models generated in Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) from a sample of these sequences revealed a tunnel fold architecture, consistent with GYCH-IB proteins. The strictly conserved Glu243, and His246 in all GCYH-IB sequences are highlighted in red and green, respectively. For the interest of space, only 95 bacterial sequences and 37 archaeal sequences are shown.
Supplementary Figure 2. Examples of gel filtration analyses of wild-type N. gonorrhoeae GCYH-IB and some of the mutants under study demonstrating the integrity of the biological tetramer. Size-exclusion chromatography elution profiles of wild-type (WT) protein, and the mutant proteins His246Ala, Glu243Ala, and Cysl49Ala purified from E. coli in the presence of 1 mM DTT show a single peak for each protein, corresponding to a homotetramer of an apparent molecular weight of 135,000 Da.
Supplementary Figure 3. Sample activity data of wild-type GCYH-IB and several mutants. A) Sample data from radiochemical activity assays. ■ Wild-type GCYH-IB; O Glul52Ala; □ His246Ala; ● Glu243Ala. Each time point is the average of 3 independent duplicates, and error bars represent the standard error. B) Sample data from UV-vis activity assays. Solid line: wild-type GCYH-IB; dashed line: Glul52Ala; dotted line: His246Ala; dash-dot line: Glu243Ala. Each trace is the average of 3 independent acquisitions with background subtraction.
Supplementary Figure 4. The structural integrity and activity of N. gonorrhoeae GCYH-IB require DTT. A) Size exclusion chromatography analysis of N. gonorrhoeae GCYH-IB in the presence (dashed line) and absence (solid line) of 1 mM DTT.The inset shows the molecular weight standard graph all peaks were resolved against. In the presence of 1 mM DTT, a homogeneous tetramer elutes with an apparent Mr of 135,000 Da (Peaks 1 and 2). In the absence of DTT, the enzyme elutes as a mixture with a lower apparent molecular weight species of Mr 70,000 Da, indicating a destabilized tetramer. B) Effect of DTT on enzyme relative activity as measured by the UV-Vis assay.
Supplementary Table 1. Primers used for the preparation of N. gonorrhoeae GCYH-IB mutants.
Supplementary Table 2. Effect of metal ions on N. gonorrhoeae apoenzyme GCYH-IB Activity
Acknowledgments
Portions of this research were conducted at the Advanced Light Source, a national user facility operated by Lawrence Berkeley National Laboratory, on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health (NIGMS). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. Coordinates and structure factors for the enzyme/TRIS complex and the enzyme/8-oxo-GTP complex have been deposited into the Protein Data Bank (PDB) under accession number 5K9G and 5K95, respectively.
Funding: This project was supported by NSF grant CHE-1309323 to D. Iwata-Reuyl and M.A. Swairjo, NIH grant GM110588 to M.A. Swairjo and D. Iwata-Reuyl, NIH grant GM70641 to D. Iwata-Reuyl, and an intramural grant from Western University of Health Sciences to M.A. Swairjo.
Abbreviations
- ATP
adenosine 5′-triphosphate
- 2′-dGTP
2′-deoxyguanosine triphosphate
- 7-deazaGTP
7-deazaguanosine 5′-triphosphate
- DTT
DL-dithiothreitol
- BME
β-mercaptoethanol
- EDTA
ethylenediaminetetraacetate
- GCYH-I
GTP cyclohydrolase I
- GCYH-IA
Type IA GTP cyclohydrolase
- GCYH-IB
Type IB GTP cyclohydrolase
- GTP
guanosine 5′-triphosphate
- HEPES
N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)
- TRIS
2-Amino-2-hydroxymethyl-propane-1,3-diol
- H2NTP
7,8-dihydroneopterin triphosphate
- HPLC
high-pressure liquid chromatography
- MES
4-morpholineethanesulfonic acid
- 8-oxo-GTP
8-oxoguanosine 5′-triphosphate
- PEG
polyethylene glycol
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- T-fold
tunnel fold
- R.m.s.d
root mean square deviation
- GSH
reduced glutathione
- TBST
TRIS-buffered saline and Tween 20
- MWCO
molecular weight cut-off
- BSA
bovine serum albumin
Footnotes
Author contribution: NP purified recombinant wild-type and mutant proteins, conducted the S-nitrosylation detection assays, de-nitrosylation, peroxide inactivation, fluorescence based activity assays of mutant and wild-type proteins, and proposed the enzyme mechanism. JA and BS processed the X-ray data and refined and analyzed the structures. AL designed and constructed vectors for expression of mutant proteins and supervised protein purification. SAB carried out the full characterization of the recombinant wild-type enzyme, constructed and assayed mutants, and carried out the kinetic and inhibition studies. AG carried out enzyme activity assays on the mutant proteins and carried out formylation experiments. DIR supervised the kinetic and inhibition studies, construction of some mutants and their assay, and assisted in deriving the enzyme mechanism. MAS collected the X-ray data, supervised structural work and nitrosylation analysis, and drafted the manuscript. MAS and DIR conceived and coordinated the study. All authors reviewed the results, contributed to the writing of the manuscript, and approved the final version of the manuscript.
Declarations of interest: The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Nichol CA, Smith GK, Duch DS. Biosynthesis and metabolism of tetrahydrobiopterin and molybdopterin. Annu Rev Biochem. 1985;54:729–764. doi: 10.1146/annurev.bi.54.070185.003501. [DOI] [PubMed] [Google Scholar]
- 2.Clark BF, Marcker KA. The role of N-formyl-methionyl-sRNA in protein biosynthesis. J Mol Biol. 1966;17:394–406. doi: 10.1016/s0022-2836(66)80150-x. [DOI] [PubMed] [Google Scholar]
- 3.Phillips G, El Yacoubi B, Lyons B, Alvarez S, Iwata-Reuyl D, de Crécy-Lagard V. Biosynthesis of 7-deazaguanosine-modified tRNA nucleosides: a new role forGTP cyclohydrolase I. J Bacteriol. 2008;190:7876–7884. doi: 10.1128/JB.00874-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCarty RM, Bandarian V. Deciphering deazapurine biosynthesis: pathway for pyrrolopyrimidine nucleosides toyocamycin and sangivamycin. Chem Biol. 2008;15:790–798. doi: 10.1016/j.chembiol.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thiaville JJ, Kellner SM, Yuan Y, Hutinet G, Thiaville PC, Jumpathong W, et al. Novel genomic island modifies DNA with 7-deazaguanine derivatives. Proc Natl Acad Sci U S A. 2016;113:E1452–1459. doi: 10.1073/pnas.1518570113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thony B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347:1–16. [PMC free article] [PubMed] [Google Scholar]
- 7.Colloc'h N, Poupon A, Mornon JP. Sequence and structural features of the T-fold, an original tunnelling building unit. Proteins. 2000;39:142–154. doi: 10.1002/(sici)1097-0134(20000501)39:2<142::aid-prot4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 8.Burg AW, Brown GM. The biosynthesis of folic acid. 8 Purification and properties of the enzyme that catalyzes the production of formate from carbon atom 8 of guanosine triphosphate J Biol Chem. 1968;243:2349–2358. [PubMed] [Google Scholar]
- 9.Wolf WA, Brown GM. The biosynthesis of folic acid. X Evidence for an Amadori rearrangement in the enzymatic formation of dihydroneopterin triphosphate from GTP Biochim Biophys Acta. 1969;192:468–478. doi: 10.1016/0304-4165(69)90396-1. [DOI] [PubMed] [Google Scholar]
- 10.El Yacoubi B, Bonnett S, Anderson JN, Swairjo MA, Iwata-Reuyl D, de Crecy-Lagard V. Discovery of a new prokaryotic type I GTP cyclohydrolase family. J Biol Chem. 2006;281:37586–37593. doi: 10.1074/jbc.M607114200. [DOI] [PubMed] [Google Scholar]
- 11.Chaudhuri RR, Allen AG, Owen PJ, Shalom G, Stone K, Harrison M, et al. Comprehensive identification of essential Staphylococcus aureus genes usingTransposon-Mediated Differential Hybridisation (TMDH) BMC Genomics. 2009;10:291–309. doi: 10.1186/1471-2164-10-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sankaran B, Bonnett SA, Shah K, Gabriel S, Reddy R, Schimmel P, et al. Zinc-independent folate biosynthesis: genetic, biochemical, and structural investigations reveal new metal dependence for GTP cyclohydrolase IB. J Bacteriol. 2009;191:6936–6949. doi: 10.1128/JB.00287-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nar H, Huber R, Meining W, Schmid C, Weinkauf S, Bacher A. Atomic structure of GTP cyclohydrolase. Structure (Camb) 1995;3:459–466. doi: 10.1016/s0969-2126(01)00179-4. [DOI] [PubMed] [Google Scholar]
- 14.Auerbach G, Herrmann A, Bracher A, Bader G, Gutlich M, Fischer M, et al. Zinc plays a key role in human and bacterial GTP cyclohydrolase I. Proc. Natl Acad Sci U S A. 2000;97:13567–13572. doi: 10.1073/pnas.240463497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maita N, Hatakeyama K, Okada K, Hakoshima T. Structural basis of biopterin-induced inhibition of GTP cyclohydrolase I by GFRP, its feedback regulatory protein. J Biol Chem. 2004;279:51534–51540. doi: 10.1074/jbc.M409440200. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka Y, Nakagawa N, Kuramitsu S, Yokoyama S, Masui R. Novel reaction mechanism of GTP cyclohydrolase I. High-resolution X-ray crystallography of Thermus thermophilus HB8 enzyme complexed with a transition state analogue, the 8-oxoguanine derivative. J Biochem. 2005;138:263–275. doi: 10.1093/jb/mvi120. [DOI] [PubMed] [Google Scholar]
- 17.Rebelo J, Auerbach G, Bader G, Bracher A, Nar H, Hosl C, et al. Biosynthesis of pteridines. Reaction mechanism of GTP cyclohydrolase I J Mol Biol. 2003;326:503–516. doi: 10.1016/s0022-2836(02)01303-7. [DOI] [PubMed] [Google Scholar]
- 18.Bourne CR. Utility of the biosynthetic folate pathway for targets in antimicrobial discovery. Antibiotics. 2014;3:1–28. doi: 10.3390/antibiotics3010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nar H, Huber R, Auerbach G, Fischer M, Hösl C, Ritz H, et al. Active site topology and reaction mechanism of GTP cyclohydrolase I. Proc. Natl Acad Sci U S A. 1995;92:12120–12125. doi: 10.1073/pnas.92.26.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr,Sect D: Biol Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 22.Emsley P, Cowtan K. COOT: Model-building tools for molecular graphics. Acta Crystallogr,Sect D: Biol Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 23.Sundaralingam M. Stereochemistry of nucleic acids and their constituents. IV Allowed and preferred conformations of nucleosides, nucleoside mono-, di-, tri-, tetraphosphates, nucleic acids and polynucleotide Biopolymers. 1969;7:821–860. [Google Scholar]
- 24.Zhao YL, Houk KN. Thionitroxides, RSNHO*: the structure of the SNO moiety in “S-Nitrosohemoglobin”, a possible NO reservoir and transporter. J Am Chem Soc. 2006;128:1422–1423. doi: 10.1021/ja057097f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purich DL. Catalysis and Control: A reference of theory and best practice methods. Chapter 2. Academic Press 2010; London, Burlington, San Diego: 2010. Enzyme Kinetics; pp. 85–86. [Google Scholar]
- 26.Field L, Dilts RV, Ravichandran R, Lenhert PG, Carnahan GE. An unusually stable thionitrite from N-acetyl-d, l-penicillamine; X-ray crystal and molecular structure of 2-(acetylamino)-2-carboxy-1,1-dimethylethyl thionitrite. J. Chem. Soc., Chem. Commun. 1978:249–250. [Google Scholar]
- 27.Arulsamy N, Bohle DS, Butt JA, Irvine GJ, Jordan PA, Sagan E. Interrelationships between conformational dynamics and the redox chemistry of S-nitrosothiols. J Am Chem Soc. 1999;121:7115–7123. [Google Scholar]
- 28.Carnahan GE, Lenhert PG, Ravichandran R. S-Nitroso-N-acetyl-DL-penicillamine. Acta Crystallogr B. 1978;34:2645–2648. [Google Scholar]
- 29.Lee J, Yi GB, Powell DR, Khan MA, Richter-Addo GB. Synthesis, characterization, and protonation of octaethylporphyrin osmium nitrosyl complexes containing axial thiolate ligands. X-ray structures of an alkyl thionitrite (RSNO) and its (OEP)Os(NO)(SR) addition product Can J Chem. 2001;79:830–840. [Google Scholar]
- 30.Bartberger MD, Houk KN, Powell SC, Mannion JD, Lo KY, Stamler JS, et al. Theory, spectroscopy, and crystallographic analysis of S-nitrosothiols: conformational distribution dictates spectroscopic behavior. J Am Chem Soc. 2000;122:5889–5890. [Google Scholar]
- 31.Baciu C, Gauld JW. An assessment of theoretical methods for the calculation of accurate structures and SN bond dissociation energies of S-nitrosothiols (RSNOs) The Journal of Physical Chemistry A. 2003;107:9946–9952. doi: 10.1021/jp035205j. [DOI] [Google Scholar]
- 32.Raju K, Doulias PT, Tenopoulou M, Greene JL, Ischiropoulos H. Strategies and tools to explore protein S-nitrosylation. Biochim Biophys Acta. 2012;1820:684–688. doi: 10.1016/j.bbagen.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bracher A, Fischer M, Eisenreich W, Ritz H, Schramek N, Boyle P, et al. Histidine 179 mutants of GTP cyclohydrolase I catalyze the formation of 2-amino-5-formylamino-6-ribofuranosylamino-4(3H)-pyrimidinone triphosphate. J Biol Chem. 1999;274:16727–16735. doi: 10.1074/jbc.274.24.16727. [DOI] [PubMed] [Google Scholar]
- 34.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: A physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 35.Anand P, Stamler JS. Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J Mol Med (Berl) 2012;90:233–244. doi: 10.1007/s00109-012-0878-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gusarov I, Nudler E. S-nitrosylation signaling in Escherichia coli. Sci Signal. 2012;5:pe26. doi: 10.1126/scisignal.2003181. [DOI] [PubMed] [Google Scholar]
- 37.Seth D, Hausladen A, Wang YJ, Stamler JS. Endogenous protein S-Nitrosylation in E. coli: regulation by OxyR. Science. 2012;336:470–473. doi: 10.1126/science.1215643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haldar SM, Stamler JS. S-nitrosylation: integrator of cardiovascular performance and oxygen delivery. J Clin Invest. 2013;123:101–110. doi: 10.1172/jci62854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, et al. S-Nitrosylation of mitochondrial caspases. J Cell Biol. 2001;154:1111–1116. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S, Whorton AR. Regulation of protein tyrosine phosphatase 1B in intact cells by S-nitrosothiols. Arch Biochem Biophys. 2003;410:269–279. doi: 10.1016/s0003-9861(02)00696-3. [DOI] [PubMed] [Google Scholar]
- 41.Perez-Mato I, Castro C, Ruiz FA, Corrales FJ, Mato JM. Methionine adenosyltransferase S-nitrosylation is regulated by the basic and acidic amino acids surrounding the target thiol. J Biol Chem. 1999;274:17075–17079. doi: 10.1074/jbc.274.24.17075. [DOI] [PubMed] [Google Scholar]
- 42.Leiper J, Murray-Rust J, McDonald N, Vallance P. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc Natl Acad Sci U S A. 2002;99:13527–13532. doi: 10.1073/pnas.212269799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 44.Chen YY, Chu HM, Pan KT, Teng CH, Wang DL, Wang AH, et al. Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J Biol Chem. 2008;283:35265–35272. doi: 10.1074/jbc.M805287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tamura M, Saito M, Yamamoto K, Takeuchi T, Ohtake K, Tateno H, et al. S-nitrosylation of mouse galectin-2 prevents oxidative inactivation by hydrogen peroxide. Biochem Biophys Res Commun. 2015;457:712–717. doi: 10.1016/j.bbrc.2015.01.055. [DOI] [PubMed] [Google Scholar]
- 46.Stohl EA, Criss AK, Seifert HS. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol Microbiol. 2005;58:520–532. doi: 10.1111/j.1365-2958.2005.04839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nedospasov A, Rafikov R, Beda N, Nudler E. An autocatalytic mechanism of protein nitrosylation. Proc Natl Acad Sci U S A. 2000;97:13543–13548. doi: 10.1073/pnas.250398197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 49.Schramek N, Bracher A, Fischer M, Auerbach G, Nar H, Huber R, et al. Reaction mechanism of GTP cyclohydrolase I: single turnover experiments using a kinetically competent reaction intermediate. J Mol Biol. 2002;316:829–837. doi: 10.1006/jmbi.2001.5339. [DOI] [PubMed] [Google Scholar]
- 50.Shiota T, Baugh CM, Myrick J. The assignment of structure to the formamidopyrimidine nucleoside triphosphate precursor of pteridines. Biochim Biophys Acta. 1969;192:205–210. doi: 10.1016/0304-4165(69)90357-2. [DOI] [PubMed] [Google Scholar]
- 51.Bracher A, Schramek N, Bacher A. Biosynthesis of pteridines. Stopped-flow kinetic analysis of GTP cyclohydrolase I Biochemistry. 2001;40:7896–7902. doi: 10.1021/bi010322v. [DOI] [PubMed] [Google Scholar]
- 52.Nar H, Huber R, Meining W, Schmid C, Weinkauf S, Bacher A. Atomic structure of GTP cyclohydrolase I. Structure. 1995;3:459–466. doi: 10.1016/s0969-2126(01)00179-4. [DOI] [PubMed] [Google Scholar]
- 53.Liang J, Cheng S, Hou J, Xu Z, Zhao YL. Car-Parinello molecular dynamics simulations of thionitroxide and S-nitrosothiol in the gas phase, methanol, and water—A theoretical study of S-nitrosylation. Sci China Chem. 2012;55:2081. doi: 10.1007/s11426-012-4712-9. [DOI] [Google Scholar]
- 54.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to gernerate schematic diagrams of protein-ligan interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1: Multisequence alignments of bacterial and archaeal GCYH-IB proteins. A) 990 non-redundant bacterial GCYH-IB sequences sourced from SWISS-PROT plus the N. gonorrhoeae GCYH-IB sequence (top sequence) were aligned using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/). The alignment shows strict conservation of Cys149 (yellow) in all 990 sequences, but a less conserved Glu152 (cyan). B) 337 non-redundant archaeal GCYH-IB sequences obtained from SWISS-PROT were aligned using Clustal Omega. Unlike the bacterial sequences, the archaeal sequences exhibit low sequence similarity and 43 of them contained His at the position homologous to Cys149 of N. gonorrhoeae GCYH-IB. None of the 43 sequences has been experimentally validated for function, but 3D structural models generated in Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) from a sample of these sequences revealed a tunnel fold architecture, consistent with GYCH-IB proteins. The strictly conserved Glu243, and His246 in all GCYH-IB sequences are highlighted in red and green, respectively. For the interest of space, only 95 bacterial sequences and 37 archaeal sequences are shown.
Supplementary Figure 2. Examples of gel filtration analyses of wild-type N. gonorrhoeae GCYH-IB and some of the mutants under study demonstrating the integrity of the biological tetramer. Size-exclusion chromatography elution profiles of wild-type (WT) protein, and the mutant proteins His246Ala, Glu243Ala, and Cysl49Ala purified from E. coli in the presence of 1 mM DTT show a single peak for each protein, corresponding to a homotetramer of an apparent molecular weight of 135,000 Da.
Supplementary Figure 3. Sample activity data of wild-type GCYH-IB and several mutants. A) Sample data from radiochemical activity assays. ■ Wild-type GCYH-IB; O Glul52Ala; □ His246Ala; ● Glu243Ala. Each time point is the average of 3 independent duplicates, and error bars represent the standard error. B) Sample data from UV-vis activity assays. Solid line: wild-type GCYH-IB; dashed line: Glul52Ala; dotted line: His246Ala; dash-dot line: Glu243Ala. Each trace is the average of 3 independent acquisitions with background subtraction.
Supplementary Figure 4. The structural integrity and activity of N. gonorrhoeae GCYH-IB require DTT. A) Size exclusion chromatography analysis of N. gonorrhoeae GCYH-IB in the presence (dashed line) and absence (solid line) of 1 mM DTT.The inset shows the molecular weight standard graph all peaks were resolved against. In the presence of 1 mM DTT, a homogeneous tetramer elutes with an apparent Mr of 135,000 Da (Peaks 1 and 2). In the absence of DTT, the enzyme elutes as a mixture with a lower apparent molecular weight species of Mr 70,000 Da, indicating a destabilized tetramer. B) Effect of DTT on enzyme relative activity as measured by the UV-Vis assay.
Supplementary Table 1. Primers used for the preparation of N. gonorrhoeae GCYH-IB mutants.
Supplementary Table 2. Effect of metal ions on N. gonorrhoeae apoenzyme GCYH-IB Activity