

Abstract
Aging is associated with impaired renal artery function, which is partly characterized by arterial stiffening and a reduced vasodilatory capacity due to excessive generation of reactive oxygen species by NADPH oxidases (Nox). The abundance and activity of Nox depends on constitutive activity of the heptahelical transmembrane receptor GPER; however, whether GPER contributes to age-dependent functional changes in renal arteries is unknown. This study investigated the effect of aging and Nox activity on renal artery tone in wild-type and GPER-deficient (Gper−/−) mice (4 and 24 months old). In wild-type mice, aging markedly impaired endothelium-dependent, nitric oxide-mediated relaxations to acetylcholine, which were largely preserved in renal arteries of aged Gper−/− mice. The Nox inhibitor gp91ds-tat abolished this difference by greatly enhancing relaxations in wild-type mice, while having no effect in Gper−/− mice. Contractions to angiotensin II and phenylephrine in wild-type mice were partly sensitive to gp91ds-tat but unaffected by aging. Again, deletion of GPER abolished effects of Nox inhibition on contractile responses. In conclusion, constitutive expression of GPER is required for the age-dependent impairment in endothelium-dependent, nitric oxide-mediated relaxation in the renal artery. Restoration of relaxation by a Nox inhibitor in aged wild-type but not GPER-deficient mice strongly supports a role for Nox-dependent ROS as the underlying cause of the impaired relaxation. Pharmacological blockers of GPER signaling may thus be suitable to inhibit functional endothelial aging of renal arteries by reducing Nox-derived oxidative stress, and possibly the associated age-dependent deterioration of kidney function.
Keywords: acetylcholine, angiotensin, endothelium, GPR30, NADPH oxidase, nitric oxide, phenylephrine, vascular
Introduction
Chronic kidney disease affects more than half of adults older than 70 years of age, and is strongly associated with an increased risk of cardiovascular disease and mortality [1]. Kidney function critically depends on adequate perfusion, which is provided by the renal artery. Advanced age is an independent risk factor for the development of both vascular and kidney disease [2], indicating a strong relationship between arterial and renal pathophysiolgy [3, 4]. In fact, age-dependent stiffening of renal arteries due to vascular calcification, fibrosis, inflammation, and a reduced vasodilatory capacity is centrally involved in detrimental renal changes, such as the development of glomerulosclerosis and tubulointerstitial fibrosis [3–5].
A common mediator of age-dependent structural and functional organ damage is excessive production of reactive oxygen species (ROS), commonly known as “oxidative stress” [6]. Among several cellular sources, ROS are mainly generated by NADPH oxidase (Nox) enzymes, of which 7 mammalian isoforms have been identified [7]. Nox-dependent regulation of redox-sensitive signaling pathways is particularly important in vascular and renal pathophysiology mainly involving Nox1, Nox2 and Nox4 [7, 8]. In arteries, ROS reduce the bioavailability of endothelial nitric oxide (NO) with subsequent loss of its vasodilatory, anti-inflammatory and anti-thrombotic properties [9]. At the same time, oxidative stress promotes arterial constriction through activators of G protein-coupled receptors (GPCRs), such as angiotensin and α-adrenergic receptors [2, 7]. Furthermore, ROS-induced alterations in cellular signaling pathways have been implicated in arterial remodeling and hypertrophy, as well as glomerulosclerosis, tubulointerstitial fibrosis and proteinuria [7, 8]. Thus, therapeutic approaches interfering with increased ROS formation will likely reduce the progression both vascular and renal diseases associated with aging [7, 8, 10].
We have recently identified an unexpected role of a constitutively active GPCR termed GPER, a heptahelical transmembrane receptor that also signals via estrogen-dependent mechanisms [11], which facilitates Nox1 expression and activity in the aorta and hearts of aged mice, as well as in human aortic smooth muscle cells [12]. Ligand-independent GPER activity was also found to mediate Nox1-dependent impaired vascular function in arterial hypertension [12], however, whether GPER contributes to age-dependent functional changes of renal arteries is unknown.
We hypothesized that constitutive GPER signaling contributes to the Nox-dependent deterioration of renal artery function with age. To this end, we studied the effects of GPER deletion and Nox activity on endothelium-dependent, NO-mediated vasodilation and vasoconstrictor responses mediated by angiotensin AT1 and α-adrenergic receptors in the renal artery from young and aged healthy mice.
Methods
Transgenic mice and aging model
Gper−/− mice were generated and backcrossed as described [12]. Wild-type C57BL/6 and Gper−/− mice were housed at the Animal Resource Facility of the University of New Mexico Health Sciences Center under controlled temperature (22 °C) on a 12-hour light-dark cycle with unrestricted access to water and a rodent diet devoid of alfalfa or soybean meal to minimize the presence of natural phytoestrogens (Teklad 2020SX, Harlan Laboratories, Madison, WI). Animals were killed by intraperitoneal injection of sodium pentobarbital (2.2 mg/g body weight) at 4 and 24 months of age, as age-dependent functional and structural changes in 24 month-old mice closely resemble human vascular aging [13]. All procedures were approved by and carried out in accordance with institutional policies and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Preparation of renal arteries
Immediately after sacrifice, renal arteries were carefully excised and dissected free from adherent connective tissue and fat in cold (4 °C) physiological saline solution (PSS, composition in mmol/L: 129.8 NaCl, 5.4 KCl, 0.83 MgSO4, 0.43 NaH2PO4, 19 NaHCO3, 1.8 CaCl2, and 5.5 glucose; pH 7.4). Using two 25 μm tungsten wires, vessels were mounted in organ chambers of a Mulvany-Halpern myograph (620M Multi-Channel Myograph System, Danish Myo Technology, Aarhus, Denmark) containing PSS (37 °C, pH 7.4, bubbled with 21% O2, 5% CO2, and balanced N2).
Vascular reactivity studies
Recordings of isometric tension were performed as described [14] using a PowerLab 8/35 data acquisition system and LabChart Pro software (AD Instruments, Colorado Springs, CO, USA). Briefly, arteries were stretched stepwise to the optimal level of passive tension for force generation, and the functional integrity of vascular smooth muscle was confirmed by repeated exposure to KCl (PSS with substitution of 60 mmol/L potassium for sodium). In subsequent experiments, the role of NADPH oxidase was studied by randomly treating the left or right renal artery with the Nox1/2-selective inhibitor gp91ds-tat (3 μmol/L for 30 minutes) [15]. This peptide is derived from a gp91phox (now termed Nox2) sequence in the region that interacts with the organizer protein p47phox, thus disrupting p47phox binding to and activation of the homologous Nox2 and Nox1 catalytic subunits [7, 12]. To study endothelium-dependent, NO-mediated relaxations, arteries were precontracted with phenylephrine to ~70% of KCl-induced contractions, and responses to acetylcholine (0.1 nmol/L – 10 μmol/L) were recorded. Precontraction did not differ between groups. Similarly, responses to the NO donor sodium nitroprusside (10 μmol/L) were determined. Contractions to agonists of angiotensin AT1 and α-adrenergic receptors, angiotensin II (100 nmol/L) and phenylephrine (0.3 μmol/L), respectively, were studied in separate experiments.
Calculations and statistical analyses
Relaxation is expressed as the percentage of precontraction, and contraction is given as the percentage of contraction compared to KCl. EC50 values (given as negative logarithm: pD2), area under the curve (AUC, given as arbitrary units (AU)), and maximal responses (Emax) were calculated by non-linear regression analysis [16]. Data were analyzed by one-way or two-way ANOVA, as appropriate, followed by the Bonferroni post hoc test to correct for multiple comparisons (GraphPad Prism version 5.0 for Macintosh, GraphPad Software, San Diego, CA, USA). Values are expressed as means±SEM; n equals the number of animals per group. Statistical significance was accepted at a p value <0.05.
Drugs
Gp91ds-tat was from Anaspec (Fremont, CA, USA), and sodium nitroprusside was from MP Biomedicals (Solon, OH, USA). All other drugs were from Sigma-Aldrich (St. Louis, MO, USA). Stock solutions were prepared according to the manufacturer’s instructions, and diluted in PSS to the required concentrations before use.
Results
GPER deletion preserves renal artery NO bioactivity with aging
In renal arteries of young wild-type and Gper−/− mice, endothelium-dependent, NO-mediated relaxation induced by acetylcholine was similar and unaffected by the Nox1/2 inhibitor gp91ds-tat (fig. 1). By contrast, in wild-type mice, aging markedly impaired endothelium-dependent relaxation (Emax, −27±9% vs. −75±10%, n=4–5, p<0.001 vs. young, fig. 1), also indicated by reduced sensitivity (pD2, 6.4±0.2 vs. 7.4±0.2, n=4–5, p<0.01 vs. young) and AUC values (0.4±0.2 vs. 1.9±0.4, n=4–5, p<0.01 vs. young). This age-dependent deterioration in NO bioactivity was largely absent in mice lacking Gper (−61±8% vs. −27±9%, n=4–5, p<0.01 vs. wild-type, fig. 1). As a result, vasodilatory responses were 2.3-fold greater in aged Gper−/− mice. The Nox1/2 inhibitor gp91ds-tat abolished this difference by greatly enhancing relaxation in aged wild-type mice (2.3-fold, from −27±9% to −63±11%, n=5, p<0.01), to the level observed in Gper−/− mice (−61±8%). Furthermore, gp91ds-tat had no effect in Gper−/− mice (fig 1). Vasodilation in response to the exogenous NO donor sodium nitroprusside was unaffected by aging or Gper deletion, indicating that the differences observed in acetylcholine-mediated relaxation are not due to alterations in the sensitivity to NO (not shown). Thus, aging impairs endothelium-dependent, NO-mediated relaxation in renal arteries largely through increased Nox activity, an effect that is absent in mice lacking Gper.
Fig. 1.
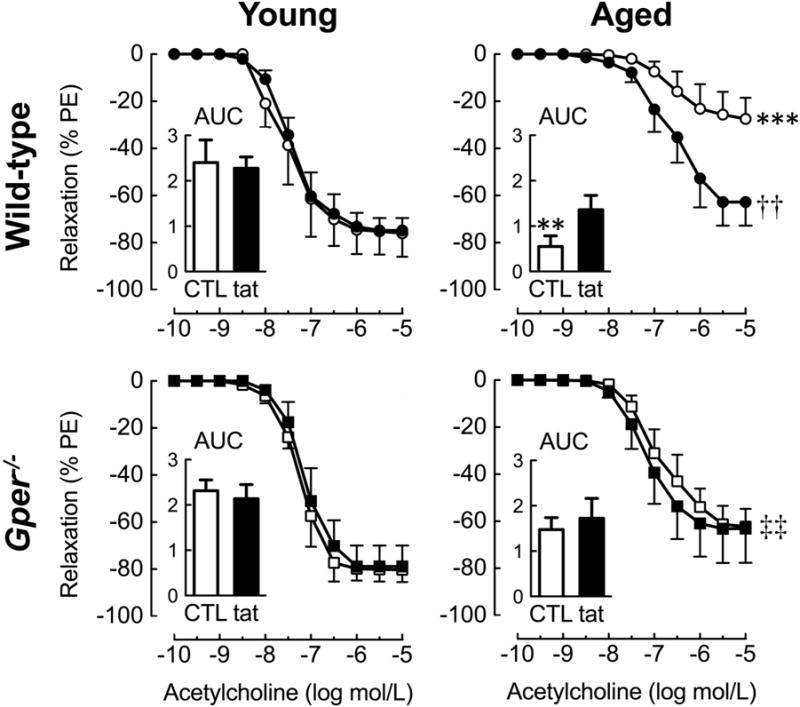
Effect of aging and GPER on endothelium-dependent relaxation to acetylcholine in renal arteries. The role of Nox-derived oxidative stress was assessed using the Nox1/2-inhibitor gp91ds-tat (tat, 3 μmol/L) in wild-type and Gper−/− mice at 4 (young) or 24 (aged) months of age. Responses are expressed as percent of precontraction to phenylephrine (PE). Insets: area under the curve (AUC) of the concentration-response curves given as arbitrary units. Data (n=4–5 per group) are means±SEM. **p<0.01, ***p<0.001 vs. young; ††p<0.01 vs. untreated control (CTL); ‡‡p<0.01 vs. wild-type.
GPER mediates Nox-dependent signaling to GPCR agonists
Given that Gper mediates age-dependent, Nox-mediated changes in endothelium-dependent relaxation of renal arteries, we next studied whether similar alterations also exist in contractile responses induced by agonists of other GPCRs. In wild-type mice, contractions to angiotensin II were unaffected by age (fig 2A); the Nox1/2 inhibitor gp91ds-tat reduced contractions to angiotensin II in young (1.9-fold) and aged (1.3-fold) animals (both n=5–7, p<0.05, fig 2A). Deletion of Gper reduced contractions to Ang II in young mice only (~50% reduction, n=5–7, p<0.01, fig 2A). Notably, in the absence of Gper, Nox1/2 inhibition had no effect on angiotensin II-induced contractions regardless of age (fig 2B).
Fig. 2.
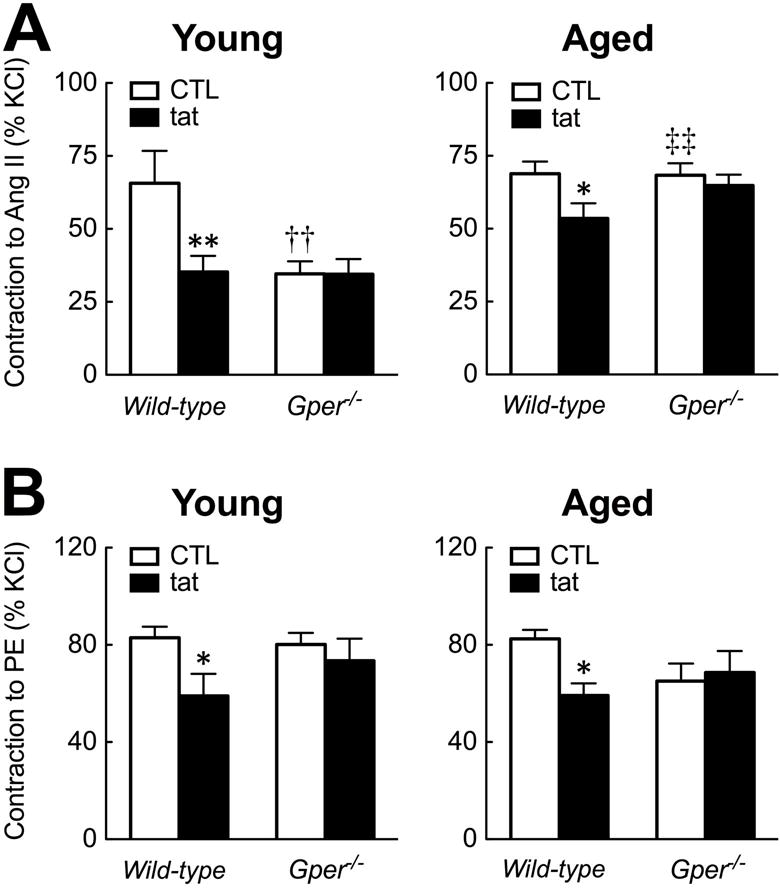
Effect of aging and GPER on Nox-mediated contractions to GPCR agonists in renal arteries. Responses were recorded in the presence and absence of the Nox1/2-inhibitor gp91ds-tat (3 μmol/L) in young (4 months old) and aged (24 months old) wild-type and Gper−/− mice. A: Contractions to the angiotensin AT1 receptor agonist angiotensin II (Ang II, 100 nmol/L). B: Contractions to the α-adrenergic agonist phenylephrine (PE, 0.3 μmol/L). Responses are expressed as percent of contractions to KCl (60 mmol/L). Data (n=5–7 per group) are means±SEM. *p<0.05, **p<0.01 vs. untreated control (CTL); ††p<0.01 vs. wild-type; ‡‡p<0.01 vs. young.
Aging did not affect contractions to the α-adrenergic agonist phenylephrine independent of genotype. However, responses were partially blocked by gp91ds-tat in young and aged wild-type (both ~30%, n=4–7, p<0.05), but not in Gper−/− mice (fig 2). Together, given that the Nox1/2 inhibitor gp91ds-tat is ineffective in mice lacking the Gper gene, these findings indicate that constitutive Gper activity is required for Nox-mediated contractions to GPCR agonists in addition to the age-dependent impaired NO bioactivity in renal arteries.
Discussion
In the present study, we have sought to characterize functional changes in the renal artery during the physiological aging process and to assess whether and how GPER, a GPCR recently found to regulate Nox1 protein and function [12], contributes to ROS-dependent modulation of endothelium-dependent and -independent vascular tone. The results show that endothelium-dependent, NO-mediated vasodilation in the renal artery is markedly reduced with aging, a functional change that largely depends on the activity of both Gper and Nox. These results are compatible with our previous report showing that Nox-derived ROS formation is largely absent in the heart and aorta of aged Gper−/− mice [12]. In both studies, we used the Nox1/2-selective inhibitor gp91ds-tat [15], a peptide derived from a gp91phox (now termed Nox2) sequence in the region that interacts with the organizer protein p47phox, thus disrupting p47phox binding to and activation of the homologous Nox2 and Nox1 catalytic subunits [7, 12]. Detailed mechanistic studies in murine and human vascular smooth muscle cells revealed that GPER in fact selectively regulates Nox1 protein abundance and function, while having no effect on Nox2 [12]. It is thus highly likely that Nox1, facilitated by constitutive GPER expression, is centrally involved in the age-dependent regulation of renal artery tone.
In contrast to the murine aorta or carotid arteries from young animals [12, 17], the endothelium-dependent, NO-mediated relaxant response induced by acetylcholine was about 20% less potent in renal arteries, and impaired to an even greater extent with age. Although this age-dependent reduction was largely reversed by inhibiting Nox, several mechanisms may also play a role. Acetylcholine-dependent generation of cyclooxygenase-derived vasoconstrictor prostanoids counteracts the vasodilatory activity of NO [18], and their formation and activity increases with aging [19], an effect that may become insensitive to Nox1/2 inhibition by gp91ds-tat [17]. Of note, compared to other arterial beds, the renal artery produces higher levels of endothelium-derived vasoconstrictor prostanoids [20], which are co-stimulated with NO in response to acetylcholine. Second, the renal artery may be more sensitive to uncoupling of endothelial NO synthase compared to other vascular beds, an effect that can be induced by Nox1-derived ROS [21]. As a result, uncoupled endothelial NO synthase increasingly produces ROS instead of vasodilatory NO, which is further reduced by direct interaction with superoxide and hydrogen peroxide [22].
We also studied whether and how aging and GPER deficiency affect renal artery contractility to GPCR agonists, including angiotensin II and phyenylephrine. Previously, angiotensin II was used to stimulate Nox activity in vascular smooth muscle cells [23] and subsequent cloning of Nox1 [24]. Subsequently, angiotensin II has emerged as the prototypic inducer of Nox1 both at the molecular and functional level [7]. We recently found that, in the murine aorta of wild-type mice, contraction to angiotensin II (and the ROS formation associated with it) is partially sensitive to inhibition by gp91ds-tat; of note, mice lacking GPER showed an equal reduction in contraction to angiotensin II, with gp91ds-tat having no additional effect, indicating that Nox-dependent contractions to angiotensin II require functional GPER [12]. The present study extends these findings, showing a similar regulation of angiotensin II-dependent contractions by GPER in the renal artery, suggesting a common mechanism in different vascular beds that persists independent of age. Furthermore, contractions to the α-adrenergic agonist phenylephrine were partly mediated by GPER-dependent Nox activity, indicating that the ROS-dependent regulation of vascular tone by GPER is not limited to a specific agonist, but may be involved in a broader number of signaling pathways governed by Nox1.
The present study was conducted in male animals to exclude any effects of endogenous (ovarian) estrogen, the prototypical physiological GPER agonist [11]. The data reinforce that ligand-independent or basal activity of constitutively expressed GPER affects regulation of vascular tone, consistent with our previous studies [12, 25, 26]. Indeed, many GPCRs exhibit basal or intrinsic activity sufficient to regulate cell signaling [27]. Furthermore, effects reported in the present study are independent of blood pressure, as both wild-type and GPER-deficient mice at 3 and 24 months of age, respectively, remain normotensive [12, 17].
In conclusion, this study shows that aging substantially attenuates endothelium-dependent, NO-mediated relaxation in the renal artery, which is largely preserved in GPER-deficient mice due to a lack of Nox activity. These findings indicate a novel role for GPER in ROS-dependent functional aging of the renal artery. With the availability of small molecule GPER blockers (GRBs) such as G36 [28], which represent a new drug class capable of selectively reducing the increased expression and activity of Nox1, future therapeutic applications in disease conditions characterized by oxidative stress may emerge [29]. Inhibition of GPER signaling may thus provide a novel approach to inhibit the age-dependent impairment of renal artery function, and possibly the associated detrimental structural and functional renal changes.
Acknowledgments
This study was supported by NIH R01 grants (CA127731, CA163890 and CA194496 to E.R.P.), the UNM Comprehensive Cancer Center (NIH grant P30 CA118100), Dedicated Health Research Funds from the UNM School of Medicine allocated to the Signature Program in Cardiovascular and Metabolic Diseases (to E.R.P.), a grant from Dialysis Clinic, Inc (to E.R.P.), and the Swiss National Science Foundation (grants 135874 and 141501 to M.R.M. as well as grants 108258 and 122504 to M.B.). The authors thank Jan S. Rosenbaum (Proctor & Gamble, Cincinnati) for providing GPER-deficient mice.
Footnotes
This work was presented in part at the 12. International Symposium on Mechanisms of Vasodilation (MOVD) - Celebrating the 30th Anniversary of the Discovery of Nitric Oxide as an Intracellular Messenger, November 7–9, 2016, Mayo Clinic, Rochester, MN, and published in abstract form (J Vasc Res 2016; 53 (suppl. 1): 15)
Competing interests
M.R.M, M.B. & E.R.P. are inventors on a U.S. patent application for the therapeutic use of compounds targeting GPER. E.R.P. is an inventor on U.S. patent nos. 7,875,721 and 8,487,100 for GPER-selective ligands and imaging agents.
References
- 1.Levey AS, Inker LA, Coresh J. Chronic kidney disease in older people. JAMA. 2015;314(6):557–558. doi: 10.1001/jama.2015.6753. [DOI] [PubMed] [Google Scholar]
- 2.Barton M, Husmann M, Meyer MR. Accelerated vascular aging as a paradigm for hypertensive vascular disease: prevention and therapy. Can J Cardiol. 2016;32(5):680–686 e684. doi: 10.1016/j.cjca.2016.02.062. [DOI] [PubMed] [Google Scholar]
- 3.Jia G, Aroor AR, Sowers JR. Arterial Stiffness: A Nexus between Cardiac and Renal Disease. Cardiorenal Med. 2014;4(1):60–71. doi: 10.1159/000360867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang N, Foster MC, Mitchell GF, Andresdottir MB, Eiriksdottir G, Gudmundsdottir H, Harris TB, Launer LJ, Palsson R, Gudnason V, Levey AS, Inker LA. Aortic stiffness and change in glomerular filtration rate and albuminuria in older people. Nephrol Dial Transplant. 2017;32(4):677–684. doi: 10.1093/ndt/gfw050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolignano D, Mattace-Raso F, Sijbrands EJ, Zoccali C. The aging kidney revisited: a systematic review. Ageing Res Rev. 2014;14:65–80. doi: 10.1016/j.arr.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 7.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110(10):1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holterman CE, Read NC, Kennedy CR. Nox and renal disease. Clin Sci (Lond) 2015;128(8):465–481. doi: 10.1042/CS20140361. [DOI] [PubMed] [Google Scholar]
- 9.Munzel T, Sinning C, Post F, Warnholtz A, Schulz E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann Med. 2008;40(3):180–196. doi: 10.1080/07853890701854702. [DOI] [PubMed] [Google Scholar]
- 10.Altenhofer S, Radermacher KA, Kleikers PW, Wingler K, Schmidt HH. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid Redox Signal. 2015;23(5):406–427. doi: 10.1089/ars.2013.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 12.Meyer MR, Fredette NC, Daniel C, Sharma G, Amann K, Arterburn JB, Barton M, Prossnitz ER. Obligatory role for GPER in cardiovascular aging and disease. Sci Signal. 2016;9(452):ra105. doi: 10.1126/scisignal.aag0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107(3):490–497. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- 14.Meyer MR, Barton M, Prossnitz ER. Functional heterogeneity of NADPH oxidase-mediated contractions to endothelin with vascular aging. Life Sci. 2014;118:226–231. doi: 10.1016/j.lfs.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res. 2001;89(5):408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 16.DeLean A, Munson PJ, Rodbard D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am J Physiol. 1978;235(2):E97–102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- 17.Meyer MR, Fredette NC, Barton M, Prossnitz ER. Prostanoid-mediated contractions of the carotid artery become Nox2-independent with aging. Age (Dordr) 2015;37(4):9806. doi: 10.1007/s11357-015-9806-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial Dysfunction and Vascular Disease - A Thirthieth Anniversary Update. Acta Physiol (Oxf) 2015 doi: 10.1111/apha.12646. [DOI] [PubMed] [Google Scholar]
- 19.Heymes C, Habib A, Yang D, Mathieu E, Marotte F, Samuel J, Boulanger CM. Cyclooxygenase-1 and -2 contribution to endothelial dysfunction in ageing. Br J Pharmacol. 2000;131(4):804–810. doi: 10.1038/sj.bjp.0703632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barton M, Vos I, Shaw S, Boer P, D’Uscio LV, Grone HJ, Rabelink TJ, Lattmann T, Moreau P, Luscher TF. Dysfunctional renal nitric oxide synthase as a determinant of salt-sensitive hypertension: mechanisms of renal artery endothelial dysfunction and role of endothelin for vascular hypertrophy and Glomerulosclerosis. J Am Soc Nephrol. 2000;11(5):835–845. doi: 10.1681/ASN.V115835. [DOI] [PubMed] [Google Scholar]
- 21.Dikalova AE, Gongora MC, Harrison DG, Lambeth JD, Dikalov S, Griendling KK. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am J Physiol Heart Circ Physiol. 2010;299(3):H673–679. doi: 10.1152/ajpheart.00242.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Forstermann U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr Opin Pharmacol. 2013;13(2):161–167. doi: 10.1016/j.coph.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74(6):1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 24.Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401(6748):79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 25.Meyer MR, Amann K, Field AS, Hu C, Hathaway HJ, Kanagy NL, Walker MK, Barton M, Prossnitz ER. Deletion of G protein-coupled estrogen receptor increases endothelial vasoconstriction. Hypertension. 2012;59(2):507–512. doi: 10.1161/HYPERTENSIONAHA.111.184606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer MR, Field AS, Kanagy NL, Barton M, Prossnitz ER. GPER regulates endothelin-dependent vascular tone and intracellular calcium. Life Sci. 2012;91:623–627. doi: 10.1016/j.lfs.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin AL, Steurer MA, Aronstam RS. Constitutive Activity among Orphan Class-A G Protein Coupled Receptors. PLoS One. 2015;10(9):e0138463. doi: 10.1371/journal.pone.0138463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, Oprea TI, Yamaguchi Y, Hayashi SI, Sklar LA, Hathaway HJ, Arterburn JB, Prossnitz ER. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol. 2011;127:358–366. doi: 10.1016/j.jsbmb.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer MR, Barton M. GPER blockers as Nox downregulators: A new drug class to target chronic non-communicable diseases. J Steroid Biochem Mol Biol. 2017 doi: 10.1016/j.jsbmb.2017.03.019. [DOI] [PubMed] [Google Scholar]