

Abstract
The 3′X domain is a 98-nt region located at the 3′ end of hepatitis C virus genomic RNA that plays essential functions in the viral life cycle. It contains an absolutely conserved, 16-base palindromic sequence that promotes viral RNA dimerization, overlapped with a 7-nt tract implicated in a distal contact with a nearby functional sequence. Using small angle X-ray scattering measurements combined with model building guided by NMR spectroscopy, we have studied the stoichiometry, structure, and flexibility of domain 3′X and two smaller subdomain sequences as a function of ionic strength, and obtained a three-dimensional view of the full-length domain in its monomeric and dimeric states. In the monomeric form, the 3′X domain adopted an elongated conformation containing two SL1′ and SL2′ double-helical stems stabilized by coaxial stacking. This structure was significantly less flexible than that of isolated subdomain SL2′ monomers. At higher ionic strength, the 3′X scattering envelope nearly doubled its size, reflecting the formation of extended homodimers containing an antiparallel SL2′ duplex flanked by coaxially stacked SL1′ helices. Formation of these dimers could initialize and/or regulate the packaging of viral RNA genomes into virions.
Keywords: 3′X domain, dimer, hepatitis C virus, RNA, small-angle X-ray scattering, structure
INTRODUCTION
The Hepatitis C virus (HCV) represents a major global health problem. The genome of HCV is formed by a positive sense RNA molecule comprising a single open reading frame (ORF) flanked by 5′ and 3′ untranslated regions (UTR). Upon cell entry, the ORF is translated and then the virus replicates in the cytoplasm through a negative-sense intermediate to produce multiple copies of positive-sense RNA molecules, which are ultimately encapsidated into new virions. Due to the low fidelity of the virus-encoded RNA polymerase and a high rate of virion production, HCV has substantial genetic diversity: Seven genotypes have been identified around the world, along with several subtypes and many quasispecies (Bartenschlager 2013).
The UTRs contain structured regions that play essential functions and are well conserved relative to the ORF (Fig. 1A). The 5′UTR comprises the internal ribosome entry site (IRES), a 340-nucleotide (nt) structure that is capable of directly recognizing the small ribosomal subunit. With this mechanism, the virus advantageously triggers translation of its ORF, skipping the intensely regulated steps involving mRNA cap recognition (Lukavsky 2009; Piñeiro and Martinez-Salas 2012). On the other side of the ORF, the 3′UTR comprises a variable region, a pyrimidine-rich segment, and a strongly conserved 98-nt terminal domain called 3′X (Fig. 1A; Tanaka et al. 1995; Kolykhalov et al. 1996). This domain is essential for replication (Kolykhalov et al. 2000; Friebe and Bartenschlager 2002; Yi and Lemon 2003a,b; Masante et al. 2015), as it is necessarily recognized by the viral replication complex to start synthesis of the negative polarity strand (Shi and Lai 2006; Lohmann 2013). A distal interaction between a 7-nt k sequence in the 3′X domain and a complementary k′ sequence in stem–loop 5BSL3.2 of the ORF influences the replication and translation processes (Friebe et al. 2005; You and Rice 2008; Murayama et al. 2010; Tuplin et al. 2012, 2015). This contact is part of a complex network of distal RNA–RNA connections that likely includes interactions between an internal loop of 5BSL3.2 and upstream sequences located in the ORF and the IRES (Fig. 1A; Diviney et al. 2008; Romero-López and Berzal-Herranz 2009; Tuplin et al. 2012; Shetty et al. 2013; Romero-López et al. 2014). In addition, the 3′X region contains a 16-nt palindromic sequence that has been shown to promote viral RNA dimerization in vitro (Cristofari et al. 2004; Ivanyi-Nagy et al. 2006; Shetty et al. 2010). This tract partially overlaps the k sequence and is termed the dimer linkage sequence (DLS) by analogy to the dimerization sequences of retroviruses (Fig. 1B). The presence of such long DLS in the 3′UTR of this virus is intriguing, as HCV virions are widely thought to contain a single copy of genomic RNA. A dimeric form of domain 3′X may be involved in genomic RNA packaging through interactions with the core protein, which has been shown to favor 3′X dimer formation. Alternatively, dimer formation might function as a mechanism for diminishing replication (Cristofari et al. 2004; Ivanyi-Nagy et al. 2006; Shetty et al. 2010). In any case, the overlapping k and DLS sequences are absolutely invariable among the different HCV genotypes (Yamada et al. 1996), attesting to the functional importance of this region.
FIGURE 1.

HCV RNA and 3′X terminal domain. (A) Functional structures identified in the HCV genomic RNA; distal RNA–RNA interactions proposed to have functional relevance are identified. (B) Secondary structure of two alternative conformations of full-length domain 3′X. These structures expose differently the absolutely conserved, overlapping sequences k (indicated with black circles) and DLS (red nt). Sequence k has been proposed to establish a distal kissing-loop interaction with a complementary k′ sequence located in the apical loop of ORF hairpin 5BSL3.2, and DLS is a 16-nt palindromic sequence likely involved in HCV RNA dimerization. (C) Secondary structure of 3′X subdomains SL2′ (also identified as X55 in the literature) and SL1.
Despite this functional relevance and high sequence conservation among HCV genotypes, the three-dimensional structure adopted by the 3′X sequence remains elusive. This contrasts with the HCV IRES, whose atomic structure has been determined through a variety of techniques (for review, see Lukavsky 2009; Piñeiro and Martinez-Salas 2012). So far, all analyses of the 3′X domain have focused on secondary structure, and the majority of them have relied on chemical modification experiments. Most reports proposed one of two possible folds, containing either two (SL1′ and SL2′) or three (SL1, SL2, and SL3) helical stems (Fig. 1B; Blight and Rice 1997; Ito and Lai 1997; Dutkiewicz and Ciesiolka 2005; Ivanyi-Nagy et al. 2006; Romero-López et al. 2014). These folds expose the overlapping k and DLS sequences differently. In the two-stem fold, the k tract is almost completely buried in the upper double-helical stem of SL2′, whereas palindromic DLS nt occupy the apical loop and would be available to initiate dimerization through the formation of a kissing loop with another SL2′ hairpin. In contrast, the three-stem structure would expose the k sequence in the apical loop of the SL2 hairpin for pairing with the k′ nt of 5BSL3.2, and initiation of dimer formation would imply the disruption of the SL2 stem (Fig. 1B). It has been proposed that these two folds are part of an RNA-based switch signaling the transition between the replication, translation, and possibly genome packaging processes of the virus (Fig. 1B; Dutkiewicz and Ciesiolka 2005; Shetty et al. 2010, 2013; Tuplin et al. 2012; Palau et al. 2013; Romero-López et al. 2014). Based on NMR spectroscopy analyses under different solution conditions, we recently reported that the monomeric form of the isolated 3′X domain comprised two stems, SL2′ and SL1′ (Cantero-Camacho and Gallego 2015). This fold exposed three unpaired terminal nt after the SL1′ stem, a configuration that has been shown to favor primer-independent initiation of RNA synthesis by the viral NS5B polymerase (Kao et al. 2000). At higher ionic strength, the domain formed extended homodimers comprising an intermolecular SL2′ double-helical stem. The three-stem conformation of the domain was not detected under our experimental conditions, leading us to suggest that it would require additional RNA or protein factors to be stabilized (Cantero-Camacho and Gallego 2015).
Here we have applied small-angle X-ray scattering (SAXS) and a structure prediction algorithm guided by NMR-based secondary structure restraints to build three-dimensional models of domain 3′X and two smaller subdomains in conditions of low and higher ionic strength. The scattering data have also allowed us to study the dimerization of these sequences as a function of ionic strength, as well as their flexibility in monomeric and dimeric states. In the monomeric form, the structure adopted by the full-length domain was elongated and relatively inflexible, and the best atomic models comprised two SL1′ and SL2′ double-helical stems stabilized by coaxial stacking. In conditions of higher ionic strength, the 3′X envelope almost doubled in length, reflecting the formation of extended dimers containing an intermolecular SL2′ stem flanked by SL1′ stem–loops. These results provide a three-dimensional view of the domain in its monomeric and dimeric states, as well as a stepping stone for further analyses of the structure of the HCV 3′UTR and its complexes.
RESULTS
Strategy for SAXS analyses
Large RNA molecules can be difficult to crystallize, and their characterization by NMR is challenging due to spectral overlap and fast relaxation. The SAXS methodology is particularly well suited to study these systems, as the electron-rich phosphate backbone increases the overall contrast between nucleic acid and buffer, and this facilitates outlining the shape of the RNA under study (Lipfert et al. 2008; Ali et al. 2010; Burke and Butcher 2012; Fang et al. 2015). To tackle the study of the 3′X domain and verify whether it formed two or three helical stems, we separately studied by SAXS two smaller subdomains in addition to the full-length sequence: a 46-nt sequence containing the extended SL1 subdomain present in the three-stem domain conformation, and a 55-nt construct adopting the extended SL2′ stem–loop structure contained in the two-stem domain fold (Fig. 1C). We used a modular approach based on the same sequences in our previous NMR spectroscopy study (Cantero-Camacho and Gallego 2015).
Each of the sequences was analyzed by SAXS under low and higher ionic strength conditions. At lower salt concentration, all SL1, SL2′, and 3′X systems were formerly shown by NMR spectroscopy and gel electrophoresis to form monomers, whereas at higher ionic strength subdomain SL2′ and the full-length sequence, containing the DLS, dimerized (Cantero-Camacho and Gallego 2015). This was supported by gel electrophoresis experiments and by the appearance of diagnostic NMR signals indicating dimerization at 2 mM MgCl2 or 100 mM NaCl (Supplemental Fig. S2; Cantero-Camacho and Gallego 2015). Three-dimensional envelopes were obtained from the scattering data of domain 3′X and subdomains SL1 and SL2′ sequences dissolved in similar low and higher ionic strength solutions. All-atom models giving rise to the best fits with the experimental SAXS curves were selected from ensembles generated with a structure prediction algorithm and NMR-based secondary structure restraints, and overlaid on the ab initio shapes. The results attained with the scattering data of the 3′X sequence were further supported by reconstructing the envelopes of the full-length domain with the shapes of subdomains SL1 and SL2′ at both ionic strength conditions. The stoichiometry of the RNA sequences at each ionic strength condition was evaluated with molecular weight calculations based on the scattering data, and by comparing the theoretical SAXS profiles of monomer, dimer, and mixture models with the experimental curves.
The experimental SAXS curves of subdomains SL1 and SL2′ and domain 3′X at low (red) and higher (blue) ionic strength are shown in Figure 2. The Guinier region of the scattering curves (insets in Fig. 2A,D,G) was linear in all cases, indicating that the SL1, SL2′, and 3′X samples were not aggregated in the two solution conditions.
FIGURE 2.

Small-angle X-ray scattering data of subdomains SL1 and SL2′ and full-length domain 3′X of hepatitis C virus RNA. (A,D,G) Scattering intensities versus momentum transfer q, obtained at low (red) and higher (blue) ionic strength. The insets show, for each ionic strength condition, the Guinier region of the scattering curve with a linear fit line. (B,E,H) Porod–Debye plots at low (red) and higher (blue) ionic strength. (C,F,I) Pair distance distribution functions (PDDF) at low (red) and higher (blue) ionic strength. Experimental conditions: 0.5–2 mg mL−1 (subdomains SL1 and SL2′) or 0.375–1.5 mg mL−1 (full-length domain 3′X); 10 mM Tris–HCl pH 7.0, 0 mM NaCl/MgCl2 (all sequences, low ionic strength); 10 mM Tris–HCl pH 7.0 and 2 mM MgCl2 (subdomains SL1 and SL2′, higher ionic strength); or 10 mM Tris–HCl pH 7.0, 2 mM MgCl2 and 50 mM NaCl (domain 3′X, higher ionic strength).
Solution structure of subdomains SL1 and SL2′
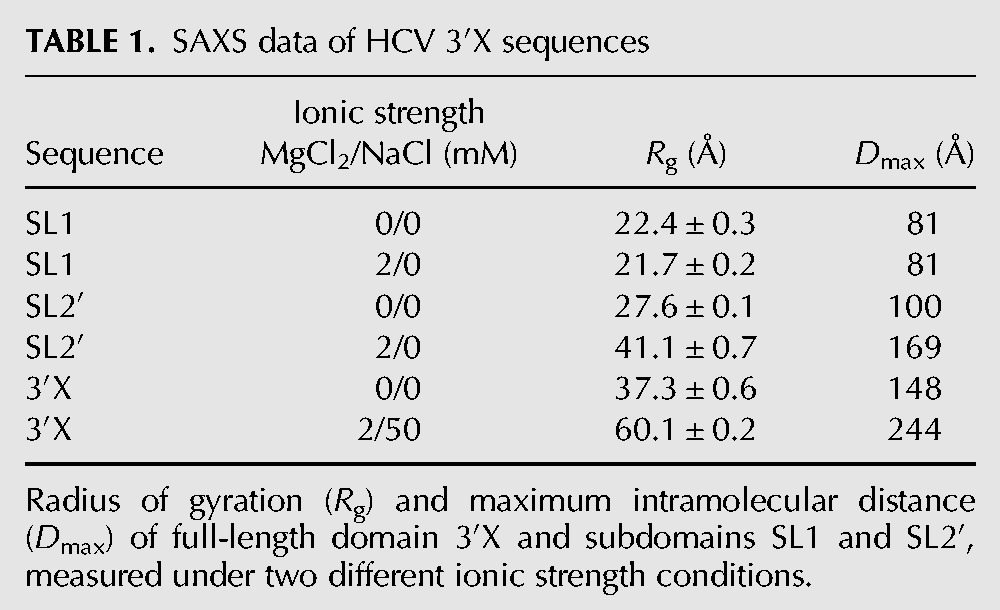
For construct SL1, the Porod–Debye plot (Rambo and Tainer 2011) showed a distinct plateau at higher salt that was somewhat less pronounced at low ionic strength (Fig. 2B). Together with the shape of the Kratky plots (Supplemental Fig. S3A), this suggested that SL1 was structured under both ionic strength conditions and exhibited slightly enhanced flexibility at low ionic strength. The pair distance distribution functions (PDDF; Fig. 2C) showed that SL1 had one main distance population at ∼20 Å, which approximately coincided with the diameter of an A-form RNA helix, and a maximum intramolecular distance (Dmax) of ∼81 Å in both conditions (Table 1).
TABLE 1.
SAXS data of HCV 3′X sequences
An ensemble of 10,000 SL1 atomic models was generated using as restraints the base pairs (Fig. 3A) previously detected for the same sequence by NMR spectroscopy under similar solution conditions (Cantero-Camacho and Gallego 2015), and the models giving rise to the best fit with the experimental scattering curve were selected and energy-minimized. The best energy-minimized atomic model obtained for this subdomain is shown in Figure 3B, superposed with the three-dimensional envelope of SL1 determined from the SAXS data at low ionic strength. The SL1 envelope calculated from the higher ionic strength scattering data was very similar (data not shown). Figure 3C shows the fit of the theoretical profile of the best energy-minimized model to the experimental SAXS curve. Altogether, the scattering data indicated that SL1 adopted a monomeric stem–loop structure in both ionic strength conditions, confirming the previous observations (Cantero-Camacho and Gallego 2015). The stoichiometry of the SL1 molecules was also supported by SAXS-based molecular weight calculations using three different methods (Supplemental Table S1). The SL1 atomic model comprised three A-form double-helical segments separated by two 1-nt bulges (U69 and A92), and was closed by a U72GAUACUG79 apical octaloop comprising a wobble U72:G79 pair at its base (Fig. 3B).
FIGURE 3.
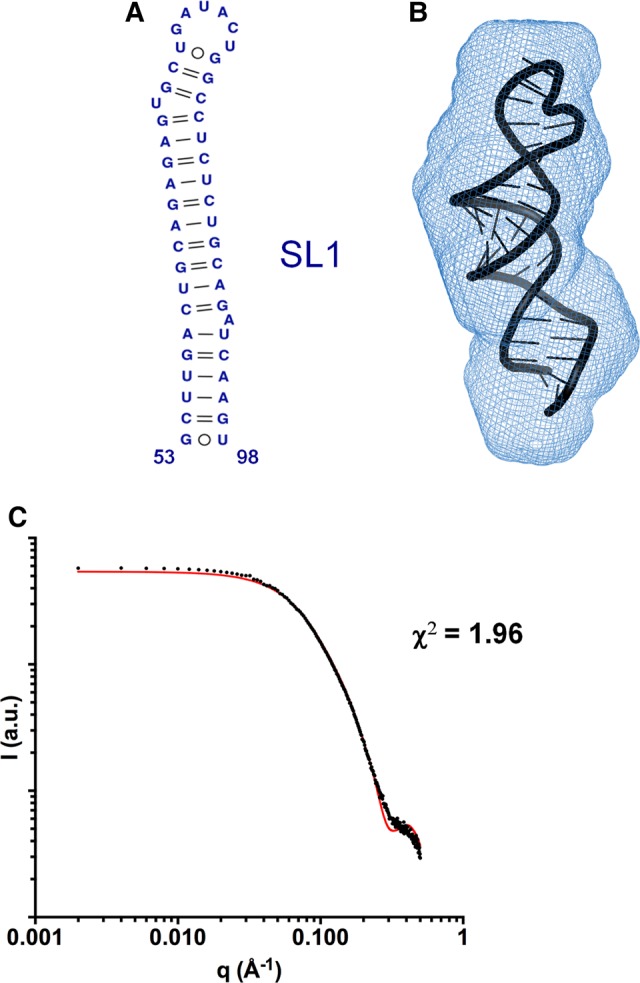
Three-dimensional structure of subdomain SL1 determined by SAXS. (A) Secondary structure of SL1 detected by NMR spectroscopy at low and higher salt concentration (Cantero-Camacho and Gallego 2015). (B) Average ab initio envelope of SL1 calculated from the SAXS profile at low ionic strength, superposed with the best energy-minimized SL1 atomic model, selected by fitting the theoretical SAXS profiles to the experimental curve. The normalized spatial discrepancy (NSD) between scattering envelopes was 0.624. (C) Theoretical SAXS profile calculated from the best energy-minimized SL1 atomic model (red line), overlaid with the experimental profile obtained at low ionic concentration (black dots).
In contrast to SL1, subdomain SL2′ contains the 16-nt palindromic DLS tract (Fig. 1C), and was previously shown by NMR spectroscopy and gel electrophoresis to form monomeric hairpins at low ionic strength and extended dimers at higher ionic strength (Fig. 4A,B; Supplemental Fig. S2A; Cantero-Camacho and Gallego 2015). The experimental SAXS profiles collected for this construct were consistent with these observations. The PDDFs obtained at low and higher ionic strength of salts were very different: Both had the main distance population of 20 Å detected for SL1, but at higher ionic strength Dmax increased by a factor of 1.7 relative to the low ionic strength value (169 versus 100 Å) (Table 1; Fig. 2F). Likewise, molecular weight calculations based on the scattering data indicated the presence of monomeric particles at low ionic strength and dimeric molecules at higher ionic strength (Supplemental Table S1). The Porod–Debye and Kratky graphs indicated that the structure was more flexible at low ionic strength relative to the higher ionic strength condition (Fig. 2E; Supplemental Fig. S3B).
FIGURE 4.

SAXS structural analysis of subdomain SL2′. (A,B) Secondary structures of SL2′ determined by NMR spectroscopy. This sequence formed monomeric hairpins at low ionic strength (A) and symmetric dimers in the presence of salts (B) (Cantero-Camacho and Gallego 2015). (C,D) Average ab initio envelopes of SL2′ obtained by SAXS analyses in conditions of low (C) and higher (D) ionic strength, superposed with the best energy-minimized respective atomic models, selected by fitting the theoretical scattering profiles to the experimental curves. The NSD values between SAXS envelopes were 0.560 and 0.555, respectively. (E,F) Experimental SAXS curves (black dots) of subdomain SL2′ obtained at low (E) and higher (F) salt concentration, overlaid with the theoretical profiles calculated from the best energy-minimized monomer (red lines) and dimer (blue lines) models, and from a mixture of monomer and dimer models (green lines).
At higher ionic strength, the SL2′ ab initio envelope almost doubled the size of that obtained at low ionic strength (Fig. 4), reflecting the differences detected in the PDDF plots at low and high salt concentration. In these conditions, previous NMR spectroscopy and gel electrophoresis experiments detected the formation of extended, 110-nt symmetric dimers by this sequence (Fig. 4B; Supplemental Fig. S2A; Cantero-Camacho and Gallego 2015). Atomic models generated using these secondary structure restraints gave rise to a very good fit with the experimental SAXS profile (Fig. 4F), indicating the agreement between both methodologies. The fits of monomeric SL2′ atomic models were significantly worse in these conditions (Fig. 4F; Supplemental Table S2). Likewise, the fitting of a mixture of monomer and dimer models indicated a 95% proportion of dimer in the mixture (Supplemental Table S2), and there was no visible difference in the fitting between the dimer-only model and the dimer/monomer model mixture (Fig. 4F).
At low ionic strength, the initial all-atom monomer models generated with base-pair restraints based on NMR observations under similar solution conditions (Fig. 4A; Cantero-Camacho and Gallego 2015) gave rise to a relatively poor fit with the experimental SAXS curve (χ2 = 8.58). Since the Porod–Debye and Kratky graphs indicated that the SL2′ structure was more flexible in these conditions (Fig. 2E; Supplemental Fig. S3B), we explored the flexibility of the best SL2′ monomer model with SREFLEX (Panjkovich and Svergun 2016) and obtained an energy-minimized model with significantly better fit to the scattering curve (χ2 = 2.04) (Fig. 4E; Supplemental Fig. S4). The fit worsened significantly with an atomic dimer model, and the fitting of a monomer/dimer model mixture indicated a 92% presence of monomer (Fig. 4E; Supplemental Table S2). This agreed with the SAXS-based molecular weight calculations (Supplemental Table S1) and with the observation by NMR and gel electrophoresis experiments of monomeric SL2′ structures at low ionic strength (Supplemental Fig. S2A; Cantero-Camacho and Gallego 2015). The atomic models indicated that the SL2′ sequence formed in these conditions a monomeric hairpin structure involving all 55 nt and closed by an apical tetraloop containing the palindromic C29UAG32 segment of the DLS (Fig. 4A,C). This structure contained two large internal loops separating two upper and lower double-helical stems. The presence of these loops may be responsible for the increased flexibility detected in the Porod–Debye and Kratky plots, as these regions are likely more dynamic and/or less compact than canonical helices. Structurally, the SL2′ dimers were similar relative to SL2′ monomers, except that the continuous 16–base-pair duplex formed by the self-complementary DLS nt in the middle of the dimer replaced the 6–base-pair DLS hairpin located at the apex of the monomeric structure. The presence of this long DLS duplex probably accounts for the more compact profile detected in the Porod–Debye and Kratky plots of SL2′ under higher ionic strength (Fig. 2E; Supplemental Fig. S3B).
Structure of the full-length 3′X domain
The Porod–Debye plots of the 3′X sequence tended asymptotically to a constant value in the absence and presence of salts (Fig. 2H). Together with the corresponding Kratky plots (Supplemental Fig. S3C), this suggested that the full-length domain formed extended structures under both ionic strength conditions, and was significantly less flexible than subdomain SL2′ at low ionic strength. The PDDFs exhibited the main distance population at 20 Å detected for subdomains SL1 and SL2′ and, as observed with SL2′, Dmax increased 1.7 times at higher ionic strength relative to the low ionic strength value (244 versus 148 Å) (Table 1; Fig. 2I). Molecular weight calculations based on the scattering profiles also indicated the presence of monomers at low salt concentration and dimers at higher ionic strength (Supplemental Table S1).
All-atom models were generated using base-pair restraints supported by NMR observations of the same sequence under similar solution conditions (Cantero-Camacho and Gallego 2015), and were selected based on fitting the theoretical scattering profiles to the experimental curves. The best energy-minimized atomic models fitted well into the SAXS three-dimensional envelopes (Fig. 5). At low ionic strength, the SAXS shape was elongated, and the atomic models giving rise to the best fits with the experimental SAXS profile corresponded to a monomeric structure comprising two coaxially stacked SL1′ and SL2′ stems (Fig. 5C). Atomic models containing three stems (SL1, SL2, and SL3; Fig. 1B) were not supported by the NMR data and gave rise to poorer fitting with the experimental curve (χ2 = 1.46, relative to 0.64 for two-stem models) as well as less overlap with the SAXS envelope and significantly poorer interdomain base stacking (Supplemental Fig. S5). Likewise, the fit worsened significantly when using 3′X dimer atomic models, and the fitting of a monomer/dimer mixture indicated 93% monomer content (Supplemental Table S2), with no observable fitting improvement relative to the monomer model (Fig. 5E).
FIGURE 5.

Solution structure of full-length domain 3′X of HCV RNA. (A,B) Secondary structures of domain 3′X supported by NMR experiments carried out under conditions of low (A) and higher (B) ionic strength. This sequence was found to form monomers or symmetric dimers under these respective solution conditions (Cantero-Camacho and Gallego 2015). (C,D) Average ab initio envelopes of domain 3′X obtained from the SAXS profiles at low (C) and higher (D) salt concentration, superposed with the best energy-minimized respective atomic models, selected by fitting the computed SAXS profiles to the experimental curves. The NSDs were 0.573 and 0.783, respectively. (E,F) Experimental SAXS profiles of domain 3′X obtained at low (E) and higher (F) salt concentration (black dots), overlaid with the theoretical curves calculated from the best energy-minimized monomer (red lines) and dimer (blue lines) models, and from a mixture of monomer and dimer models (green lines). In F, the relatively higher χ2 value is due to a smaller experimental error of the measured curve (see χ-function in Schneidman-Duhovny et al. 2010).
As detected for subdomain SL2′, at higher ionic strength the 3′X envelope almost doubled in size (see Fig. 5D). In these conditions, NMR together with gel electrophoresis and size-exclusion chromatography experiments detected the formation of extended dimers by the full-length sequence comprising a symmetric SL2′ duplex flanked on both sides by SL1′ stem–loops (Fig. 5B; Supplemental Fig. S2B; Cantero-Camacho and Gallego 2015). The theoretical scattering curves of atomic models generated using these secondary structure restrictions had a very good fit with the experimental profile (Fig. 5F), confirming again the consistency of the results obtained with both methodologies. As observed for subdomain SL2′, the fitting of the scattering curve obtained in these conditions with 3′X monomer models was significantly poorer, and no improvement was detected with a mixture of dimer and monomer models (Fig. 5F; Supplemental Table S2).
To further support the results obtained with the scattering data of the full-length sequence, we reconstructed the structure of the entire domain with the envelopes of the individual subdomains. The low-salt average envelopes of subdomains SL1 and SL2′ superposed quite well on the envelope of the domain monomer (Supplemental Fig. S6A). Likewise, the envelope of the full-length 3′X dimer could be reconstructed using the shapes of the SL1 and SL2′ sequences obtained at higher ionic strength, with one SL2′ dimer located in the middle and two SL1 subdomains placed on each side (Supplemental Fig. S6B). Thus the ab initio envelopes provided direct guidance for obtaining the three-dimensional structure adopted by the full-length sequence at each ionic strength condition. Together with the fitting of the atomic models to the experimental SAXS data, they indicated that the domain monomers contained two SL1′ and SL2′ stems stabilized by coaxial stacking, and the 3′X dimers comprised an SL2′ antiparallel dimer flanked by intramolecular SL1′ hairpins.
DISCUSSION
In this report, we have studied the stoichiometry, structure and flexibility of the full-length 3′X-tail of HCV and two smaller subdomain sequences as a function of ionic strength, and provide a view of the three-dimensional structure adopted by the domain, which was previously unavailable. The three-dimensional envelopes and atomic models generated with the SAXS data have allowed us to extract important conclusions regarding the structure of the monomeric and dimeric forms of the domain, as well as their possible impact on the virus life cycle.
Under low ionic strength conditions, the experimental scattering data were consistent with the adoption by the full-length 3′X sequence of an extended monomeric structure containing two stems. This was revealed by SAXS-based molecular weight calculations (Supplemental Table S1) and by the Dmax value detected in the PDDF curve (148 Å; Table 1; Fig. 2I). The three-dimensional envelope calculated from the scattering data was elongated, and the atomic models giving rise to the best fit with the experimental scattering curve were monomeric and comprised two coaxially stacked SL1′ and SL2′ stems (Fig. 5C,E). As these models were generated using as restraints the base pairs detected by NMR spectroscopy for the same sequence under similar solution conditions (Fig. 5A; Cantero-Camacho and Gallego 2015), the agreement between both methodologies adds further support to this conclusion.
Interestingly, the profiles of the Porod–Debye and Kratky plots revealed that at low ionic strength the structure of the isolated SL2′ subdomain monomers was more flexible than that of subdomain SL1 or the full-length domain monomers (Fig. 2; Supplemental Fig. S3), and significantly better fits with the experimental scattering curve were obtained after exploring the flexibility of monomeric SL2′ atomic models (Fig. 4E; Supplemental Fig. S4). These results indicate that, in monomeric form, the structure of the isolated SL2′ sequence was flexible and became stabilized in the context of the full-length domain, likely because of the observed coaxial stacking between the SL2′ and SL1′ stems.
In the structure of the 3′X monomers detected by SAXS and NMR, subdomain SL1′ is shortened relative to the extended SL1 stem present in the three-stem domain conformation (Fig. 1B), and contains three unpaired AGU residues at the 3′ end of the base-paired stem, which correspond to the terminal nt of the HCV genome (Fig. 5). This conformation is compatible with replication, since the presence of at least 3 nt after a stable secondary structure has been shown to promote primer-independent initiation of RNA synthesis by the viral NS5B polymerase (Kao et al. 2000). De novo initiation at the 3′ end of the viral RNA strand is thought to be the physiological mode of launching RNA synthesis in infected cells (Lohmann 2013) and requires a purine at the 5′ end of the nascent RNA chain (Zhong et al. 2000; Cai et al. 2004). Note that the terminal nt of the viral genomic RNA would be base-paired in the alternative three-stem domain fold (Fig. 1B), disfavoring replication initiation. At the 3′ end of the “negative” strand, several terminal unpaired nt follow a shorter stem–loop in most HCV genotypes, and the first nt of the nascent positive chain is usually a G (Cai et al. 2004). In this regard, the NS5B polymerase has been shown to replicate more efficiently from the 3′ end of the negative strand (Reigadas et al. 2001), which serves to initiate the synthesis of multiple copies of positive sense RNA to be packaged into new virions.
One important question remains to be answered regarding the structure of domain 3′X in the monomeric state. Genetic evidence indicates that the 7-nt k sequence of the domain pairs with a complementary k′ sequence located in the apical loop of stem–loop 5BSL3.2 through a kissing-loop interaction (Friebe et al. 2005; You and Rice 2008; Murayama et al. 2010; Palau et al. 2013; Shetty et al. 2013). This distal contact is functionally relevant, as it has been shown to influence the replication and translation processes (Friebe et al. 2005; You and Rice 2008; Murayama et al. 2010; Tuplin et al. 2012, 2015). In the structure detected by SAXS and NMR, the k nt are base-paired in the upper stem of subdomain SL2′ (Fig. 5). The formation of the distal contact would thus necessarily entail a conformational change in the domain to allow sequence k to pair with k′. So far we have not obtained evidence supporting the adoption by the isolated domain of a conformation that would permit the k–k′ contact. In particular, the alternative structure with three separate SL1, SL2, and SL3 stems exposing the k sequence in the apical loop of SL2 (Fig. 1B) gave rise to a poorer fit with the experimental scattering data (Supplemental Fig. S5) and was not detected by NMR spectroscopy (Cantero-Camacho and Gallego 2015). It is thus likely that the 3′X conformation compatible with the distal contact is not significantly populated by the isolated domain and requires the presence of the 5BSL3.2 loop (or alternatively a protein cofactor) to become stabilized. Further work is currently in progress in our laboratory to elucidate the structural details of the distal interaction between 3′X and 5BSL3.2.
The experimentally observed two-stem conformation of domain 3′X promotes viral RNA dimerization. This structure hides the k sequence, but exposes the central palindromic C29UAG32 tract of the DLS in the apical tetraloop of the SL2′ hairpin (Fig. 5). Dimerization of 3′X RNA has been shown to involve a kissing complex intermediate that likely requires two palindromic SL2′ tetraloops (Shetty et al. 2010). In vitro experiments have shown that this intermediate is subsequently converted into a more stable extended duplex conformation in the presence of the core protein of the virus (Cristofari et al. 2004; Ivanyi-Nagy et al. 2006; Shetty et al. 2010). A similar dimerization mechanism has been proposed for HIV-1 and other retroviruses (Greatorex 2004; Moore and Hu 2009; Johnson and Telesnitsky 2010).
In this regard, an important result of this report is the determination of the three-dimensional shape of the extended dimers formed by the 3′X domain. Under higher ionic strength conditions, the Dmax value detected in the PDDF plot of the full-length sequence was 244 Å, 1.7 times greater than the value obtained at low salt (Table 1; Fig. 2I), the molecular weight calculated from the scattering profiles approximately doubled (Supplemental Table S1), and the scattering envelope was correspondingly longer (Fig. 5D). In these conditions, NMR experiments detected the formation by the 3′X sequence of extended symmetric dimers (Fig. 5B; Cantero-Camacho and Gallego 2015), and all-atom models fulfilling these secondary structure restraints gave rise to a very good fit with the experimental SAXS profile and matched the three-dimensional envelope (Fig. 5D,F). The 3′X dimers are structurally related to the monomers, and comprise two antiparallel-paired SL2′ strands containing a palindromic DLS duplex in the middle. Both strands fold into intramolecular SL1′ hairpins that stack coaxially on each side of the SL2′ duplex. The three-dimensional structure of the extended 3′X dimer is elongated, with the two SL1′ stems forming a continuous stack with the 110-nt antiparallel SL2′ duplex occupying the core of the dimer (Fig. 5B,D).
The isolated SL2′ subdomain sequence formed similar extended symmetric dimers at higher ionic strength (Fig. 4B,D). In this case, the flexibility observed in the isolated SL2′ monomers decreased significantly upon dimer formation (Fig. 2E; Supplemental Fig. S3B), likely due to the stabilizing effect of the continuous 16-bp DLS self-complementary duplex in the middle of the dimer (Fig. 4B).
The presence of a DLS in the 3′UTR of HCV, absolutely conserved in all viral isolates, raises interesting questions. The palindromic DLS of HCV (16 nt) (Fig. 1B; Supplemental Fig. S1) is longer than most DLS palindromes identified in retroviruses; for example, the DLS palindrome of HIV-1 (termed dimer initiation sequence, or DIS) is 6-nt long. The 110-nt SL2′ duplex forming the core of the extended HCV dimer (Fig. 5) is likewise longer than most extended duplexes predicted to be formed by retroviruses (Greatorex 2004) (70 nt in the case of HIV-1). However, while retroviruses pack two RNA genomes (Greatorex 2004; Moore and Hu 2009; Johnson and Telesnitsky 2010), HCV virions are widely thought to contain a single copy of genomic RNA, although to our knowledge there is no direct evidence of this fact. The core protein of HCV has been shown to promote the formation of extended 3′X dimers in vitro (Cristofari et al. 2004; Ivanyi-Nagy et al. 2006; Shetty et al. 2010) and is thought to drive packaging, as it coats the RNA forming with it the nucleocapsid of the virus particles (Kunkel et al. 2001; Ivanyi-Nagy et al. 2006). This resembles the roles of the nucleocapsid protein in retroviruses, where RNA dimerization and packaging are tightly coupled processes (Moore and Hu 2009; Johnson and Telesnitsky 2010). In these viruses, the packaging signal and the DLS map to the same 100–300 nt region (Johnson and Telesnitsky 2010). In HCV the mechanism of RNA encapsidation is not as well characterized. The 3′UTR has been recently proposed to act as a cis-acting element for RNA packaging (Shi et al. 2016), and this would be consistent with the presence of a functional DLS in domain 3′X. However, it is also possible that other structures across the virus genome contribute to RNA-core binding and act cooperatively to drive encapsidation (Stewart et al. 2016).
Another intriguing fact is that in HCV the DLS is located at the 3′UTR, while in retroviruses the dimerization and packaging determinants are found at the 5′ end of their genomes. In these later viruses, however, RNA transcription is catalyzed in the nucleus by cellular RNA polymerase II, and is a tightly regulated step coupled to capping, splicing, and export processes that influence the composition and the fate of the retroviral RNA transcripts (see, e.g., Pocock et al. 2016). In contrast, the RNA of HCV is synthesized in the cytoplasm by the viral polymerase, and this process can generate subgenomic molecules (Shimizu et al. 2006). Thus, the presence of the DLS at the 3′UTR might have evolved as a mechanism to ensure that only full-length RNA molecules are packaged into virions. Alternatively, it is also possible that the presence of the DLS at the 3′UTR contributes to control the replication and translation activities of the virus (Ivanyi-Nagy et al. 2006; Shetty et al. 2010; Masante et al. 2015): Since the cytoplasmic magnesium concentration ranges from 1 to 5 mM (approximately corresponding to the higher salt condition in this work), the main factors modulating the 3′X monomer/dimer equilibrium in a cellular environment would likely be local RNA concentration, which would drive the formation of the SL2′ kissing intermediate, and the presence of the core protein, which would stabilize the extended dimer form. Therefore, once the virus-encoded RNA and proteins reach sufficiently high concentrations in the cytoplasm, formation of mature 3′X dimers with an extended SL2′ duplex might diminish negative RNA synthesis and translation by obstructing polymerase progression and by sequestering the k sequence and altering the network of functional distal interactions across the viral genome (Fig. 1A). This would favor core protein binding and RNA packaging. It remains to be seen if HCV DLS dimerization occurs in vivo and fulfills these still hypothetical functions.
MATERIALS AND METHODS
Sequences
The sequences analyzed in this study correspond to HCV genotype 1b and were obtained from vector pFK-I389FLUCNS3-3′ET (Lohmann et al. 1999), kindly provided by Dr. R. Bartenschlager (University of Heidelberg, Germany).
Preparation of RNA samples
The SL1 (46 nt) and SL2′ (55 nt) subdomain RNA sequences (Supplemental Fig. S1) were prepared by T7-polymerase in vitro transcription using synthetic oligonucleotide DNA templates. Construct 3′X (the full-length 98-nt domain) was transcribed from a pUC19 plasmid containing a ScaI restriction site for linearization and run-off transcription. All constructs were purified on denaturing gels containing 20% acrylamide and 8 M urea. After electroelution from the gel, the RNAs were ethanol-precipitated two times and desalted with Sephadex G-25 cartridges. Prior to SAXS experiments, all samples were transferred by diafiltration into aqueous solutions containing 10 mM Tris–HCl (pH 7.0) and 0.1 mM EDTA with no added salts, or additionally containing either 2 mM MgCl2 (subdomains SL1 and SL2′) or 2 mM MgCl2 with 50 mM NaCl (full-length domain 3′X). Under higher ionic strength conditions, the full-length 3′X dimers were isolated by size-exclusion chromatography to obtain homogeneous samples (Supplemental Fig. S2B) and concentrated by diafiltration. Each SAXS sample was prepared at three different RNA concentrations, ranging from 0.375 to 2 mg mL−1 (∼6 to 134 μM). Before data acquisition, the samples were heated at 95°C for 5 min and immediately placed on ice (for low salt conditions) or cooled down slowly (higher salt conditions).
SAXS data acquisition
X-ray scattering measurements were carried out at room temperature using an in-house instrument of the SAXS Core Facility at the Frederick Laboratory of Cancer Research, and beamline 12D-ID-B of the Advanced Photon Source at the Argonne National Laboratory. The samples were initially screened and examined at the SAXS Core Facility using a photon energy of 8 keV and an exposure time of 1 h. Selected samples were measured at the 12-ID-B beamline with a photon energy of 14 keV and an off-center Pilatus 2M detector. Simultaneous WAXS data were recorded, and the total q range examined was ∼0.005 < q < 2.8 Å−1. To evaluate the magnitude of possible interparticle interactions, each RNA was measured at three different concentrations. A total of 30 sequential data frames were recorded for each RNA sample and condition using a flow cell and an exposure time of 1–1.5 sec to minimize radiation damage and obtain good signal-to-noise ratios. The 2D scattering patterns were corrected and reduced to 1D scattering profiles using on-site software. No radiation damage was observed as evidenced by the absence of systematic signal changes in sequential SAXS profiles; this was subsequently confirmed by gel electrophoresis after the X-ray measurements. The scattering profiles of RNAs were calculated by subtracting the background buffer contribution from the sample solution profile. The WAXS data were used to guide accurate background subtraction for the SAXS data using NCI IGOR Pro macros.
SAXS data analysis
For each RNA sequence and ionic strength condition, the radius of gyration (Rg) was obtained across the three different RNA concentrations with PRIMUS (Konarev et al. 2003) by using the Guinier approximation at the low scattering angle region of the profiles. We checked the aggregation state of the samples by evaluating the linearity of the Guinier plots. The concentration effects were eliminated by extrapolating data to infinite dilution using PRIMUS (Konarev et al. 2003) or an in-house Matlab script. To increase the fitting range in the lower q region, the dispersion data were extrapolated to lower q values based on Guinier linear fitting. The program GNOM (Svergun 1992) was then applied using a maximum q value of 0.3 Å−1 to calculate the pair distance distribution function (PDDF) and the maximum intramolecular distance (Dmax). Dmax was monitored in steps of 2 Å until a good fit to the experimental data was obtained and the PDDF curve fell smoothly to zero. The molecular weight of the RNA molecules was estimated from the scattering data using three different methods (Fischer et al. 2010; Petoukhov et al. 2012; Rambo and Tainer 2013).
Ab initio three-dimensional modeling of SAXS data
Twenty low-resolution envelope models were calculated for each sequence and condition from the GNOM output using DAMMIM (Svergun 1999) in “slow” mode. The resulting ab initio shape models were superimposed with SUPCOMB (Kozin and Svergun 2001), averaged with DAMAVER (Volkov and Svergun 2003) based on normalized spatial discrepancy (NSD) criteria, and filtered with DAMFILT (Volkov and Svergun 2003). For each RNA sequence and condition, the resulting DAMFILT envelope was compared with the most representative envelope (NSD = 0) and with a refined envelope generated with DAMSTART (Volkov and Svergun 2003). The shapes were approximately similar for all systems (data not shown).
Secondary structure restraints
The secondary structure adopted by each RNA sequence in each ionic strength condition was previously determined by NMR spectroscopy experiments using the same sequences under similar solution conditions (Cantero-Camacho and Gallego 2015). Specifically, 1H-15N HSQC and HNN-COSY as well as homonuclear NOESY and TOCSY spectra allowed detection and assignment of the base pairs formed by each system.
Building of atomic models
The MC-Sym web server (Parisien and Major 2008) was used to generate either 1000 or 10,000 all-atom models of each RNA molecule with base-pair restrictions based on previous NMR analyses (Cantero-Camacho and Gallego 2015). Specifically, we used as restraints the base-pairing interactions indicated in Figures 3A, 4A,B, and 5A,B. Theoretical scattering profiles were then generated for these three-dimensional structures and compared with the experimental SAXS data at q < 0.5 Å−1 using the FoXS server (Schneidman-Duhovny et al. 2010). We selected the 20 best models (with the lowest χ2 fit values) of each RNA sequence. These models were subsequently refined by progressively minimizing their potential energy with the ff10 force field of AMBER 8.0 (Case et al. 2005) and a generalized Born model for solvent simulation. The best refined model was evaluated with MolProbity (Davis et al. 2007) and overlaid with the average ab initio envelope with SUPCOMB (Kozin and Svergun 2001). Flexible refinement of the best SL2′ monomer model based on SAXS data was carried out with SREFLEX (Panjkovich and Svergun 2016). The resulting model was energy-minimized and evaluated as described before. The monomer/dimer content of the RNA samples was calculated by comparing the experimental SAXS profiles with the theoretical scattering curves of monomer, dimer, or mixture atomic models, generated using FoXS (Schneidman-Duhovny et al. 2010) for monomer or dimer models, and by combining MultiFoXS (Schneidman-Duhovny et al. 2016) and MES (Pelikan et al. 2009) for mixture models.
DATA DEPOSITION
Experimental SAXS data and three-dimensional models of full-length domain 3′X monomers and dimers, subdomain SL2′ monomers and dimers, and subdomain SL1 monomers have been deposited in the Small Angle Scattering Biological Data Bank (SASBDB) with accession codes SASDBM7, SASDBN7, SASDBP7, SASDBQ7, and SASDBR7.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Ministerio de Economía y Competitividad of Spain (grants BFU-2012-30770 and BFU2015-65103-R to J.G.), Universidad Católica de Valencia of Spain (grant PRUCV/2015/629 to J.G. and predoctoral contract 2012-030-001 assigned to A.C.-C.). For SAXS measurements, we gratefully acknowledge use of the SAXS Core facility at the Laboratory of Cancer Research, National Cancer Institute (NCI). The shared beamline 12-ID-B resource was allocated under agreement PUP-24152 between the NCI and the Argonne National Laboratory (ANL). We thank Dr. Xiaobing Zuo (ANL) for his expert support. Use of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility, was operated by ANL under contract DE-AC02-06CH11357.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.060632.117.
REFERENCES
- Ali M, Lipfert J, Seifert S, Herschlag D, Doniach S. 2010. The ligand-free state of the TPP riboswitch: a partially folded RNA structure. J Mol Biol 396: 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R, ed. 2013. Hepatitis C virus: from molecular virology to antiviral therapy. Springer-Verlag, Berlin. [Google Scholar]
- Blight KJ, Rice CM. 1997. Secondary structure determination of the conserved 98-base sequence at the 3′ terminus of hepatitis C virus genome RNA. J Virol 71: 7345–7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JE, Butcher SE. 2012. Nucleic acid structure characterization by small angle X-ray scattering (SAXS). Curr Protoc Nucleic Acid Chem 51: 7.18.1–7.18.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Liang TJ, Luo G. 2004. Effects of mutations of the initiation nucleotides on hepatitis C virus RNA replication in the cell. J Virol 78: 3633–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantero-Camacho Á, Gallego J. 2015. The conserved 3′X terminal domain of hepatitis C virus genomic RNA forms a two-stem structure that promotes viral RNA dimerization. Nucleic Acids Res 43: 8529–8539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Cheatham TE III, Darden T, Gohlke H, Luo R, Merz KM Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. 2005. The Amber biomolecular simulation programs. J Comput Chem 26: 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofari G, Ivanyi-Nagy R, Gabus C, Boulant S, Lavergne JP, Penin F, Darlix JL. 2004. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res 32: 2623–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB III, Snoeyink J, Richardson JS, et al. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35: W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diviney S, Tuplin A, Struthers M, Armstrong V, Elliott RM, Simmonds P, Evans DJ. 2008. A hepatitis C virus cis-acting replication element forms a long-range RNA-RNA interaction with upstream RNA sequences in NS5B. J Virol 82: 9008–9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutkiewicz M, Ciesiolka J. 2005. Structural characterization of the highly conserved 98-base sequence at the 3′ end of HCV RNA genome and the complementary sequence located at the 5′ end of the replicative viral strand. Nucleic Acids Res 33: 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Stagno JR, Bhandari YR, Zuo X, Wang YX. 2015. Small-angle X-ray scattering: a bridge between RNA secondary structures and three-dimensional topological structures. Curr Opin Struct Biol 30: 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer H, de Oliveira Neto M, Napolitano HB, Polikarpov I, Craievich AF. 2010. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J Appl Crystallogr 43: 101–109. [Google Scholar]
- Friebe P, Bartenschlager R. 2002. Genetic analysis of sequences in the 3′ nontranslated region of hepatitis C virus that are important for RNA replication. J Virol 76: 5326–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe P, Boudet J, Simorre JP, Bartenschlager R. 2005. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J Virol 79: 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greatorex J. 2004. The retroviral RNA dimer linkage: different structures may reflect different roles. Retrovirology 1: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Lai MM. 1997. Determination of the secondary structure of and cellular protein binding to the 3′-untranslated region of the hepatitis C virus RNA genome. J Virol 71: 8698–8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanyi-Nagy R, Kanevsky I, Gabus C, Lavergne JP, Ficheux D, Penin F, Fossé P, Darlix JL. 2006. Analysis of hepatitis C virus RNA dimerization and core-RNA interactions. Nucleic Acids Res 34: 2618–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SF, Telesnitsky A. 2010. Retroviral RNA dimerization and packaging: the what, how, when, where, and why. PLoS Pathog 6: e1001007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao CC, Yang X, Kline A, Wang QM, Barket D, Heinz BA. 2000. Template requirements for RNA synthesis by a recombinant hepatitis C virus RNA-dependent RNA polymerase. J Virol 74: 11121–11128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolykhalov AA, Feinstone SM, Rice CM. 1996. Identification of a highly conserved sequence element at the 3′ terminus of hepatitis C virus genome RNA. J Virol 70: 3363–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolykhalov AA, Mihalik K, Feinstone SM, Rice CM. 2000. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J Virol 74: 2046–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36: 1277–1282. [Google Scholar]
- Kozin MB, Svergun DI. 2001. Automated matching of high- and low-resolution structural models. J Appl Crystallogr 34: 33–41. [Google Scholar]
- Kunkel M, Lorinczi M, Rijnbrand R, Lemon SM, Watowich SJ. 2001. Self-assembly of nucleocapsid-like particles from recombinant hepatitis C virus core protein. J Virol 75: 2119–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Ouellet J, Norman DG, Doniach S, Lilley DM. 2008. The complete VS ribozyme in solution studied by small-angle X-ray scattering. Structure 16: 1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann V. 2013. Hepatitis C virus RNA replication. Curr Top Microbiol Immunol 369: 167–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285: 110–113. [DOI] [PubMed] [Google Scholar]
- Lukavsky PJ. 2009. Structure and function of HCV IRES domains. Virus Res 139: 166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masante C, Jaubert C, Palau W, Plissonneau J, Besnard L, Ventura M, Di Primo C. 2015. Mutations of the SL2 dimerization sequence of the hepatitis C genome abrogate viral replication. Cell Mol Life Sci 72: 3375–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MD, Hu WS. 2009. HIV-1 RNA dimerization: It takes two to tango. AIDS Rev 11: 91–102. [PMC free article] [PubMed] [Google Scholar]
- Murayama A, Weng L, Date T, Akazawa D, Tian X, Suzuki T, Kato T, Tanaka Y, Mizokami M, Wakita T, et al. 2010. RNA polymerase activity and specific RNA structure are required for efficient HCV replication in cultured cells. PLoS Pathog 6: e1000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palau W, Masante C, Ventura M, Di Primo C. 2013. Direct evidence for RNA-RNA interactions at the 3′ end of the Hepatitis C virus genome using surface plasmon resonance. RNA 19: 982–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panjkovich A, Svergun DI. 2016. Deciphering conformational transitions of proteins by small angle X-ray scattering and normal mode analysis. Phys Chem Chem Phys 18: 5707–5719. [DOI] [PubMed] [Google Scholar]
- Parisien M, Major F. 2008. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 452: 51–55. [DOI] [PubMed] [Google Scholar]
- Pelikan M, Hura GL, Hammel M. 2009. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen Physiol Biophys 28: 174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI. 2012. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr 45: 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñeiro D, Martinez-Salas E. 2012. RNA structural elements of hepatitis C virus controlling viral RNA translation and the implications for viral pathogenesis. Viruses 4: 2233–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock GM, Becker JT, Swanson CM, Ahlquist P, Sherer NM. 2016. HIV-1 and M-PMV RNA nuclear export elements program viral genomes for distinct cytoplasmic trafficking behaviors. PLoS Pathog 12: e1005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. 2011. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95: 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. 2013. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496: 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reigadas S, Ventura M, Sarih-Cottin L, Castroviejo M, Litvak S, Astier-Gin T. 2001. HCV RNA-dependent RNA polymerase replicates in vitro the 3′ terminal region of the minus-strand viral RNA more efficiently than the 3′ terminal region of the plus RNA. Eur J Biochem 268: 5857–5867. [DOI] [PubMed] [Google Scholar]
- Romero-López C, Berzal-Herranz A. 2009. A long-range RNA-RNA interaction between the 5′ and 3′ ends of the HCV genome. RNA 15: 1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-López C, Barroso-Deljesus A, García-Sacristán A, Briones C, Berzal-Herranz A. 2014. End-to-end crosstalk within the hepatitis C virus genome mediates the conformational switch of the 3′X-tail region. Nucleic Acids Res 42: 567–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Sali A. 2010. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res 38: W540–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A. 2016. FoXS, FoXSDock and MultiFoXS: single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res 44: W424–W429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty S, Kim S, Shimakami T, Lemon SM, Mihailescu MR. 2010. Hepatitis C virus genomic RNA dimerization is mediated via a kissing complex intermediate. RNA 16: 913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty S, Stefanovic S, Mihailescu MR. 2013. Hepatitis C virus RNA: molecular switches mediated by long-range RNA-RNA interactions? Nucleic Acids Res 41: 2526–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi ST, Lai MMC. 2006. HCV 5′ and 3′UTR: when translation meets replication. In Hepatitis C viruses: genomes and molecular biology (ed. Tan SL). Horizon Bioscience, Norfolk, UK. [Google Scholar]
- Shi G, Ando T, Suzuki R, Matsuda M, Nakashima K, Ito M, Omatsu T, Oba M, Ochiai H, Kato T, et al. 2016. Involvement of the 3′ untranslated region in encapsidation of the hepatitis C virus. PLoS Pathog 12: e1005441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu YK, Hijikata M, Oshima M, Shimizu K, Yoshikura H. 2006. Detection of a 5′ end subgenome of hepatitis C virus terminating at nucleotide 384 in patients′ plasma and liver tissues. J Viral Hepat 13: 746–755. [DOI] [PubMed] [Google Scholar]
- Stewart H, Bingham RJ, White SJ, Dykeman EC, Zothner C, Tuplin AK, Stockley PG, Twarock R, Harris M. 2016. Identification of novel RNA secondary structures within the hepatitis C virus genome reveals a cooperative involvement in genome packaging. Sci Rep 6: 22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 25: 495–503. [Google Scholar]
- Svergun DI. 1999. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J 76: 2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Kato N, Cho MJ, Shimotohno K. 1995. A novel sequence found at the 3′ terminus of hepatitis C virus genome. Biochem Biophys Res Commun 215: 744–749. [DOI] [PubMed] [Google Scholar]
- Tuplin A, Struthers M, Simmonds P, Evans DJ. 2012. A twist in the tail: SHAPE mapping of long-range interactions and structural rearrangements of RNA elements involved in HCV replication. Nucleic Acids Res 40: 6908–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuplin A, Struthers M, Cook J, Bentley K, Evans DJ. 2015. Inhibition of HCV translation by disrupting the structure and interactions of the viral CRE and 3′ X-tail. Nucleic Acids Res 43: 2914–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. 2003. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr 36: 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada N, Tanihara K, Takada A, Yorihuzi T, Tsutsumi M, Shimomura H, Tsuji T, Date T. 1996. Genetic organization and diversity of the 3′ noncoding region of the hepatitis C virus genome. Virology 223: 255–261. [DOI] [PubMed] [Google Scholar]
- Yi M, Lemon SM. 2003a. 3′ nontranslated RNA signals required for replication of hepatitis C virus RNA. J Virol 77: 3557–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Lemon SM. 2003b. Structure-function analysis of the 3′ stem–loop of hepatitis C virus genomic RNA and its role in viral RNA replication. RNA 9: 331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You S, Rice CM. 2008. 3′ RNA elements in hepatitis C virus replication: kissing partners and long poly(U). J Virol 82: 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Uss AS, Ferrari E, Lau JY, Hong Z. 2000. De novo initiation of RNA synthesis by hepatitis C virus nonstructural protein 5B polymerase. J Virol 74: 2017–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.