

Abstract
Objective
The aim of this study was to characterize the demographic, behavioural, clinical and immunogenetic determinants of HIV-1 superinfection in a high-risk cohort of MSM.
Design
A retrospective cohort study of prospectively followed MSM.
Methods
Ninety-eight MSM with acute or early HIV-1 monoinfection were followed for a median of 15.6 months. Demographic and human leukocyte antigen (HLA) genotype data were collected at enrolment. Sexual behaviour, clinical and the infection status (monoinfection or superinfection) data were recorded at each visit (at enrolment and thereafter at a median of 4.2-month intervals). HIV-1 superinfection risk was determined by Cox regression and Kaplan–Meier survival analysis.
Results
Ten individuals (10.2%) had superinfection during follow-up. Cox regression did not show significantly increased superinfection risk for individuals with an increased amount of condomless anal intercourse, lower CD4+ T-cell count or higher viral load, but higher number of sexual contacts demonstrated a trend towards significance [hazard ratio, 4.74; 95% confidence interval (95% CI), 0.87–25.97; P = 0.073]. HLA-A*29 (hazard ratio, 4.10; 95% CI, 0.88–14.76; P = 0.069), HLA-B*35 (hazard ratio, 4.64; 95% CI, 1.33–18.17; P = 0.017), HLA-C*04 (hazard ratio, 5.30; 95% CI, 1.51–20.77; P = 0.010), HLA-C*16 (hazard ratio, 4.05; 95% CI, 0.87–14.62; P = 0.071), HLA-DRB1*07 (hazard ratio, 3.29; 95% CI, 0.94–12.90; P = 0.062) and HLA-DRB1*08 (hazard ratio, 15.37; 95% CI, 2.11–79.80; P = 0.011) were associated with an increased risk of superinfection at α = 0.10, whereas HLA-DRB1*11 was associated with decreased superinfection risk (hazard ratio, 0.13; 95% CI, 0.00–1.03; P = 0.054).
Conclusion
HLA genes may, in part, elucidate the genetic basis of differential superinfection risk, and provide important information for the development of efficient prevention and treatment strategies of HIV-1 superinfection.
Keywords: CD4+ cell count, HIV-1 superinfection, human leukocyte antigen, MSM, sexual behaviour, viral load
Introduction
HIV-1 superinfection is a form of dual infection that occurs when a person with established HIV infection becomes reinfected. Dual infection that involves simultaneous (or nearly so) infection with a second strain of HIV-1 before an immune response to the first strain has been mounted is classified as coinfection, while sequential infection after an immune response to the first strain has developed is classified as superinfection [1]. Ultra-deep sequencing techniques have successfully been used to determine whether the individual is infected by one or more HIV-1 strains [2–5]. The risk of superinfection has been demonstrated to be greatest in recently infected individuals who have not yet developed a mature HIV-specific cell-mediated and humoral immune response [1,6]. In the setting of primary HIV-1 infection, evolving and incomplete immunity, due to lack of efficiently neutralizing antibodies and primed CD8+ T lymphocytes that are incapable of killing all infected cells, may generate a state of relative permissiveness to HIV superinfection. HIV-1 superinfection has also been shown to occur at a similar rate to that observed in primary infection in a high-risk group of female sex workers in Uganda [7], whereas the rate of superinfection in high-risk female bar workers in Kenya was found to be significantly lower than the rate observed in primary infection [8].
HIV-1 superinfection has been associated with lower CD4+ T-cell counts, higher viral loads and a shorter time to adverse clinical events than monoinfected persons [9]. In addition, viral recombination in the cells of dually infected persons may occur [10,11] resulting in recombinant strains that are resistant to currently available antiretroviral therapies [12] and may be associated with more rapid disease progression [13–15]. A recent study showed that viral load increase was faster in the superinfection group than in the monoinfected group of Kenyan female sex workers infected with viruses of subtypes A, C and D, and a borderline association with accelerated CD4+ cell count decline was observed, but time to clinical progression events was not different between monoinfected and superinfected women [16].
Although a number of studies have aimed to characterize determinants of superinfection, no definitive risk factors have been reported. The main drawback has been the lack of sensitive methods to detect minor virus populations of the second HIV-1 virus, commonly resulting in under-diagnoses of superinfection. It remains unclear whether superinfection is the result of ongoing high-risk behaviour, or the consequence of suboptimal immunologic responses, particularly during early HIV-1 infection. Our previous case–control study of 18 monoinfected and seven superinfected individuals provided some evidence that immunogenetic factors at the human leukocyte antigen (HLA) loci may play a role in the risk of HIV-1 superinfection. Higher frequencies of HLA-A*29, HLA-B*35 and HLA-C*16, as well as lower frequencies of HLA-DRB1*11 were observed in the superinfected group than in the monoinfected group, although observed differences were not statistically significant but showed a trend towards an association with superinfection [4]. These data also showed that the superinfected group had a faster viral load increase over time than the monoinfected group, but there was no difference in the CD4+ cell count decline between the monoinfected and superinfected groups. To gain a better understanding about these correlates, we prospectively followed a high-risk cohort of MSM who were infected with HIV-1, subtype B, and characterized the demographic, behavioural, clinical and immunogenetic determinants of HIV-1 superinfection.
Materials and methods
Study participants
All study participants were those of the San Diego Primary HIV Infection Program, and were eligible if they had at least one longitudinal follow-up visit, were at least 16 years, were newly diagnosed with recent HIV-1 and were MSM. The study cohort consisted of 98 MSM with acute or early HIV-1, subtype B infection residing in San Diego County, California, USA, who met the inclusion criteria. The study was approved by the UCSD Human Research Protection Program, and written informed consent was obtained from all participants. Guidelines from the US Department of Health and Human Services were followed throughout the project. Demographic data (age and self-reported race/ethnicity) were collected and recorded at enrolment. Behavioural data included the number of sexual contacts and frequency of condomless anal intercourse (receptive and insertive) and were collected at enrolment and at each visit during the follow-up period. Peripheral blood mononuclear cell (PBMC) DNA samples were collected at enrolment and at each follow-up visit. The estimated date of infection (EDI) was calculated using an established protocol [17,18]. Participants were enrolled between June 1996 and December 2005. As the superinfection risk is affected by antiretroviral therapy (ART), participants were censored after the start of ART. Ten individuals out of 98 became superinfected with HIV-1, subtype B, during the follow-up period. Demographics, HLA characteristics, behavioural risk factors and clinical characteristics were analysed individually (univariate), and statistical analyses were performed with the SAS 9.3 software package (http://support.sas.com/software/93/).
Clinical and genetic data
Peripheral blood samples were collected at enrolment and thereafter at a median of 4.2-month intervals [interquartile range (IQR): 1.8–12.2 months]. Viral loads (Amplicor; Roche Molecular Systems, Inc., Pleasanton, California, USA) and CD4+ T-cell counts (flow cytometry; VA San Diego) were measured at enrolment and at each visit during the follow-up period using standard protocols. The dual infection status of study participants was determined using ultra-deep sequencing and single genome sequencing [4,5]. Briefly, partial coding regions within HIV-1 gag, pol and env were isolated, amplified and sequenced from longitudinal blood plasma samples, and resulting sequence datasets underwent bioinformatic and phylogenetic analyses [4,19]. Dual infection was identified within a sample and within an individual when viral sequences demonstrated divergent clustering on a background of epidemiologically unrelated HIV-1 sequences. HIV-1 superinfection was confirmed when an individual’s viral sequences from later timepoints demonstrated divergent clustering from enrolment viral sequences. Participants with evidence of dual infection at enrolment (i.e. coinfection) were excluded from the study. HLA genotyping of HLA-A, HLA-B, HLA-C and HLA-DRB1 alleles was performed from genomic DNA in all individuals using a combination of PCR-SSOP method as described previously [20]. Due to the limited number of individuals and power of the study, two-digit codes of HLA lineage groups were used in the analyses.
Cox proportional hazards regression and Kaplan–Meier survival analysis
Cox proportional hazards regression was used to test the association between the development of superinfection and demographics (age, race/ethnicity), immunogenetic (HLA) characteristics, behavioural risk factors (number of sexual contacts and condomless anal intercourse) and clinical characteristics (CD4+ T-cell count and viral load). Clinical, behavioural or demographics that were found to be significant (P < 0.10) in univariate analyses were entered into a multivariate analysis using a Cox proportional hazards model to identify risk factors associated with the risk of superinfection. As the sexual-behaviour characteristics, viral load and CD4+ T-cell count were captured longitudinally, they were included in the model as time-dependent covariates, and the hazard of superinfection at time t depended on thevalues of these covariates at time t or at the closest preceding values. Kaplan–Meier survival analysis was used to test the difference between individuals who carried a given HLA allele and noncarriers. Timepoints after participants were lost to follow-up (n = 27/27.6%), and after the start of ART (n = 61/62.2%) or detection of superinfection (n = 10/10.2%), whichever occurred first, were censored, and the midpoint between the last monoinfection result and first superinfection result was used as the estimated date of superinfection. P values were determined by the partial-likelihood ratio test (Cox proportional hazards regression) and the log-rank test (Kaplan–Meier survival analysis). HLA alleles, carried by more than one individual, with hazard ratio more than 1.0 and P value less than 0.10, were named as high-risk alleles. HLA alleles with hazard ratio less than 1.0 and P value less than 0.10 were considered low-risk alleles. P values were not adjusted for multiple comparisons, as obtained results represent preliminary, exploratory analysis.
Homozygosity and linkage disequilibrium analyses
The effect of homozygosity of each HLA locus on the superinfection risk was assessed by the Cox proportional hazards regression, and the P values were determined by the partial-likelihood ratio test. Linkage disequilibrium analyses were determined in all 98 study participants using the Allele procedure in SAS.
Results
Characteristics of study participants
We prospectively identified recently HIV-1 infected persons and analysed a subset of MSM with documented monoinfection at presentation. A total of 98 recently infected high-risk MSM were followed for a median of 15.6 months (range: 0.7–73.9 months; Table 1) in order to identify factors associated with increased superinfection risk. The median time from EDI to presentation (i.e. baseline) was 70 days (range: 10–170 days). Seventy-three individuals (74.5%) were non-Hispanic whites, 19 (19.4%) were Hispanic and six (6.1%) were other race/ ethnicity and/or multiracial. These demographic data are consistent with those of our entire cohort [17,21] and reflect the demographics of HIV in San Diego County (http://www.sandiegocounty.gov/content/dam/sdc/hhsa/programs/phs/documents/HIV_Epi_Report_2015_FINAL.PDF). Approximately one-third of individuals belonged to the each age group of 19–28, 29–34 and 35–58 years (Table 1). Determination of superinfection was performed as previously described [4,5] using next-generation sequencing (NGS) and single genome sequencing of three HIV-1 genes (gag, pol, env) [2,3]. Participants were followed while antiretroviral naive and were censored if they started ARTor were lost to follow-up. During the follow-up time, 10 individuals became superinfected with HIV-1, subtype B, at a median time from the EDI of 10.4 months (range: 3.5–21.1 months).
Table 1.
Characteristics and Cox regression of study participants.
| Variable | Individuals at enrolment | HR (95% CI) | P |
|---|---|---|---|
| From EDI to enrolment (days) | |||
| Median (range) | 70.0 (10–170) | ND | ND |
| Follow-up time (months) | |||
| Median (range) | 15.6 (0.7–73.9) | ND | ND |
| Race/ethnicity, no. (%) | |||
| White/non-Hispanic | 73 (74.5) | Reference | Reference |
| Hispanic | 19 (19.4) | 0.98 (0.20–4.74) | 0.984 |
| Other | 6 (6.1) | 1.69 (0.21–13.70) | 0.626 |
| Age groups, no. (%) | |||
| 19–28 | 34 (34.7) | 2.10 (0.38–11.48) | 0.393 |
| 29–34 | 32 (32.7) | 2.66 (0.49–14.64) | 0.260 |
| 35–58 | 32 (32.7) | Reference | Reference |
| Number of sexual contacts, no. (%)a | |||
| 0–1 | 29 (30.9) | Reference | Reference |
| 2–3 | 29 (30.9) | 1.97 (0.27–14.27) | 0.504 |
| >3 | 36 (38.3) | 4.74 (0.87–25.97) | 0.073 |
| Condomless anal intercourse, no. (%)b | 77 (81.1) | 1.71 (0.38–7.59) | 0.482 |
| CD4+ cell count (cells/μl) | |||
| Median (IQR) | 541.5 (460–719) | 0.94 (0.80–1.10)c | 0.438 |
| Viral load (log10 copies per ml) | |||
| Median (IQR) | 4.7 (3.8–5.4) | 1.14 (0.59–2.20) | 0.689 |
CI, confidence interval; EDI, estimated date of primary infection; HR, hazard ratio; IQR, interquartile range; ND, not determined. Data available for
94 and
95 individuals.
Square root scale.
Demographic, behavioural and clinical variables
Table 1 displays the univariate hazard ratios for superinfection. Neither age nor race/ethnicity was significantly associated with superinfection acquisition. Specifically, the youngest MSM of ages 19–28 were no more likely than the oldest men (35 to 58) to acquire superinfection (P = 0.393), nor were MSM of ages 29 to 34 (P = 0.260). In addition, we did not have evidence to conclude that Hispanic participants were more at risk to acquire superinfection than non-Hispanic whites (P = 0.984) nor were participants of other race/ethnicities (P = 0.626).
HIV-1 superinfection has been shown to be associated with an accelerated viral load increase in untreated persons but no statistically significant effect of superinfection on time to clinical events (CD4+ T-cell count <200 cells/μl, ART initiation or death) was detected [16]. In addition, high-risk sexual behaviour has been shown to be associated with an increased superinfection risk [7]. To assess whether high-risk sexual behaviour, lower CD4+ cell count or higher viral load are associated with an increased superinfection risk in our MSM cohort, we analysed these risk factors as time-dependent variables using Cox proportional hazards regression. Surprisingly, none of these expected risk factors demonstrated significantly increased risk for HIV-1 superinfection; however, higher number of sexual contacts (more than three contacts per month) was associated with an increased superinfection risk at α = 0.10 [hazard ratio, 4.74; 95% confidence interval (95% CI), 0.87–25.97; P = 0.073; Table 1].
Genetic studies of human leukocyte antigen loci
HLA Class I molecules play a central role in the immune response to HIV-1 by presenting viral antigens to T cells [22–24], but Class II alleles, particularly at the HLA-DRB1 locus, have also been reported to influence HIV-1 control [25–27]. Prior studies have shown that specific HLA alleles are associated with accelerated disease progression, while other HLA alleles have a protective effect on HIV-1 disease [25–32]. To investigate whether certain HLA alleles are associated with an increased superinfection risk, we determined hazard ratios for each allele with Cox proportional hazards regression using genotypes of all 98 individuals. Increased superinfection risk at α = 0.10 was observed in individuals with HLA-A*29 (hazard ratio, 4.10; 95% CI, 0.88–14.76; P = 0.069), HLA-B*35 (hazard ratio, 4.64; 95% CI, 1.33–18.17; P = 0.017), HLA-C*04 (hazard ratio, 5.30; 95% CI, 1.51–20.77; P = 0.010), HLA-C*16 (hazard ratio, 4.05; 95% CI, 0.87–14.62; P = 0.071), HLA-DRB1 07 (hazard ratio, 3.29; 95% CI, 0.94–12.90; P = 0.062) and HLA-DRB1*08 (hazard ratio, 15.37; 95% CI, 2.11–79.80; P = 0.011), whereas decreased HIV-1 superinfection risk was observed in individuals with HLA-DRB1*11 (hazard ratio, 0.13; 95% CI, 0.00–1.03; P = 0.054) (Table 2 and Supplemental Table 1, http://links.lww.com/QAD/B56).
Table 2.
Low-risk and high-risk HLA alleles for HIV-1 superinfection.
| HLA allele | n (%) | HR (95% CI) | P | Allele frequency of study participantsa |
|---|---|---|---|---|
| Low-risk allele | ||||
| HLA-DRB1*11 | 24 (24.5) | 0.13 (0.00–1.03) | 0.054 | 0.128 |
| High-risk allele | ||||
| HLA-A*29 | 10 (10.2) | 4.10 (0.88–14.76) | 0.069 | 0.051 |
| HLA-B*35 | 24 (24.5) | 4.64 (1.33–18.17) | 0.017 | 0.128 |
| HLA-C*04 | 26 (26.5) | 5.30 (1.51–20.77) | 0.010 | 0.153 |
| HLA-C*16 | 12 (12.2) | 4.05 (0.87–14.62) | 0.071 | 0.061 |
| HLA-DRB1*07 | 34 (34.7) | 3.29 (0.94–12.90) | 0.062 | 0.179 |
| HLA-DRB1*08 | 4 (4.1) | 15.37 (2.11–79.80) | 0.011 | 0.020 |
CI, confidence interval; HR, hazard ratio; n, number of individuals with a given allele.
Allele frequency in all 98 participating individuals.
The number of sexual contacts and HLA alleles that were found to be significant (P < 0.10) in the univariate analyses were entered into a multivariate analysis using a Cox proportional hazards model. In multivariate models, HLA-B*35 (P = 0.020), HLA-C*04 (P = 0.033) and HLA-DRB1*08 (P = 0.027) remained significant, regardless of the number of sexual contacts, but the increased risk of superinfection associated with HLA-A*29, HLA-C*16 and HLA-DRB1*07, and the protective effect of HLA-DRB1*11 detected in univariate model did not persist in a multivariate model in combination with the number of sexual contacts.
Kaplan–Meier survival analysis was performed to confirm the results of Cox proportional hazards regression analysis. All results of Kaplan–Meier survival analysis of low-risk and high-risk alleles were consistent with the results obtained from the Cox regression analysis (Fig. 1 and Table 2).
Fig. 1. Kaplan–Meier survival analysis for low-risk and high-risk HLA alleles to explore the risk of HIV-1 superinfection.
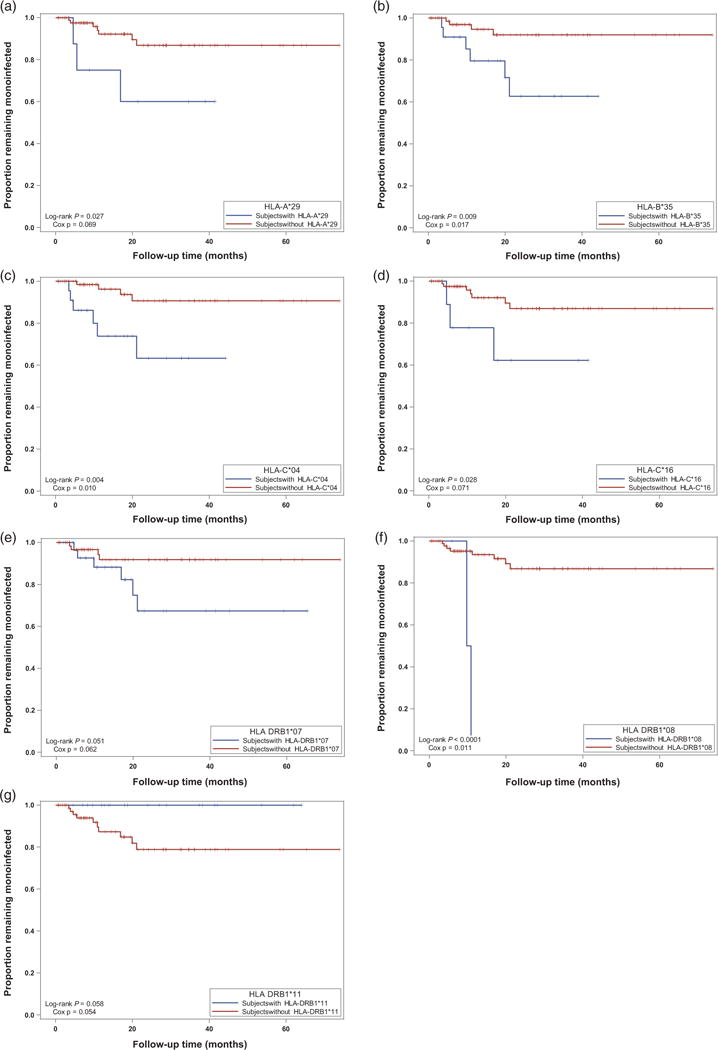
Blue = individuals with the low-risk or high-risk allele; red = individuals without the low-risk or high-risk allele. (a) HLA-A*29; (b) HLA-B*35; (c) HLA-C*04; (d) HLA-C*16; (e) HLA-DRB1*07; (f) HLA-DRB1*08; (g) HLA-DRB1*11. P values were calculated using the log-rank test (Kaplan–Meier) and the partial-likelihood ratio test (Cox).
Homozygosity at HLA loci has been reported to diminish the number of viral epitopes that could serve as a target for cytotoxic T lymphocytes and is associated with accelerated disease progression in individuals with primary infection [28,31–33]. To investigate whether HLA homozygosity increases the risk of HIV-1 superinfection, we performed Cox proportional hazards regression analysis for HLA-A, HLA-B, HLA-C and HLA-DRB1 loci. However, homozygosity at any of these HLA loci did not demonstrate an increased risk for superinfection (Supplemental Table 2, http://links.lww.com/QAD/B56).
Lastly, we assessed how high-risk HLA alleles were distributed in individuals who continued to practice condomless anal intercourse after primary infection and remained monoinfected for at least 2 years (n = 21), and compared their allele distribution to that of superinfected individuals. The 2-year follow-up duration for monoinfected individuals was selected due to previous studies reporting that most superinfections occur within 2 years after primary infection [5,8]. Monoinfected individuals had fewer high-risk HLA-alleles (median: 0.0; IQR: 0–1.5) than the individuals in the superinfected group (median: 3.0; IQR: 2–3) (Table 3 and Supplemental Table 3, http://links.lww.com/QAD/B56).
Table 3.
Distribution of low- and high-risk HLA alleles for HIV-1 superinfection in monoinfected subjects with continued sexual risk behavior and superinfected subjects.
| Contacts per Month Median (Range) | Infection Status | FU Months | HLA-A Allele 1 | HLA-A Allele 2 | HLA-B Allele 1 | HLA-B Allele 2 | HLA-C Allele 1 | HLA-C Allele 2 | HLA-DRB1 Allele 1 | HLA-DRB1 Allele 2 |
|---|---|---|---|---|---|---|---|---|---|---|
| 0.0 (0 – 2) | MI | 45.1 | *03 | *03 | *07 | *07 | *07 | *07 | *07 | *15 |
| 0.5 (0 – 1) | MI | 32.4 | *02 | *23 | *44 | *50 | *05 | *06 | *04 | *04 |
| 1.0 (0– 30) | MI | 24.0 | *02 | *02 | *44 | *51 | *05 | *14 | *04 | *11 |
| 1.0 (1 – 4) | MI | 27.9 | *02 | *03 | *39 | *57 | *06 | *07 | *01 | *07 |
| 1.0 (0 – 2) | MI | 32.8 | *30 | *30 | *35 | *57 | *04 | *18 | *13 | *15 |
| 1.0 (1 – 2) | MI | 34.8 | *02 | *02 | *40 | *46 | *01 | *02 | *09 | *13 |
| 1.0 (1 – 1) | MI | 36.1 | *02 | *02 | *13 | *14 | *07 | *08 | *13 | *15 |
| 1.0 (0 – 1) | MI | 37.4 | *11 | *68 | *07 | *51 | *01 | *07 | *11 | *15 |
| 1.0 (0 – 10) | MI | 38.3 | *02 | *24 | *07 | *18 | *07 | *07 | *01 | *11 |
| 1.0 (1 – 2) | MI | 40.3 | *02 | *11 | *15 | *27 | *02 | *03 | *04 | *04 |
| 1.0 (0 – 15) | MI | 42.1 | *32 | *68 | *40 | *44 | *03 | *07 | *11 | *15 |
| 1.0 (0 – 5) | MI | 59.2 | *01 | *01 | *15 | *49 | *07 | *12 | *07 | *13 |
| 1.5 (1 – 2) | MI | 34.7 | *02 | *29 | *35 | *49 | *04 | *07 | *04 | *04 |
| 1.5 (0 – 20) | MI | 65.4 | *01 | *03 | *14 | *57 | *06 | *08 | *07 | *13 |
| 2.0 (0 – 4) | MI | 28.1 | *30 | *33 | *14 | *53 | *08 | *15 | *03 | *13 |
| 2.0 (1 – 5) | MI | 28.9 | *02 | *68 | *35 | *44 | *04 | *05 | *04 | *11 |
| 2.0 (1 – 4) | MI | 41.5 | *11 | *29 | *27 | *44 | *01 | *16 | *01 | *07 |
| 2.0 (0 – 5) | MI | 42.1 | *01 | *68 | *08 | *44 | *05 | *07 | *03 | *16 |
| 3.0 (2 – 4) | MI | 32.7 | *30 | *30 | *18 | *35 | *04 | *05 | *03 | *13 |
| 3.0 (1 – 5) | MI | 53.7 | *02 | *68 | *27 | *44 | *01 | *05 | *04 | *11 |
| 5.0 (0 – 50) | MI | 41.5 | *25 | *68 | *18 | *58 | *06 | *12 | *13 | *13 |
|
| ||||||||||
| 0.0 (0 – 1) | SI | 11.2 | *26 | *32 | *07 | *38 | *07 | *12 | *09 | *15 |
| 1.0(1 – 3) | SI | 19.9 | *01 | *68 | *35 | *57 | *07 | *07 | *04 | *07 |
| 1.0 (1 – 4) | SI | 21.1 | *24 | *31 | *35 | *51 | *04 | *15 | *04 | *07 |
| 2.01 | SI | 3.5 | *24 | *66 | *35 | *41 | *04 | *17 | *13 | *15 |
| 2.0(1 – 3) | SI | 16.9 | *02 | *29 | *44 | *44 | *05 | *16 | *01 | *07 |
| 4.01 | SI | 4.6 | *23 | *29 | *44 | *44 | *04 | *16 | *04 | *07 |
| 5.01 | SI | 3.8 | *01 | *03 | *08 | *35 | *04 | *07 | *01 | *03 |
| 6.5 (3 – 10) | SI | 10.9 | *02 | *68 | *35 | *40 | *03 | *04 | *08 | *16 |
| 8.01 | SI | 5.5 | *03 | *29 | *44 | *57 | *16 | *18 | *01 | *07 |
| NA | SI | 9.8 | *03 | *32 | *35 | *47 | *04 | *07 | *07 | *08 |
FU, follow-up; MI, monoinfection; NA, not available; SI, superinfection.
Only one data point available; blue, low-risk HLA allele; red, high-risk HLA allele. Contact numbers are median (range) values during the follow-up period.
Discussion
Due to the potential impact of superinfection on disease progression, we endeavoured to characterize the determinants of HIV-1 superinfection. During the follow-up time, 10 individuals became superinfected with HIV-1 within 2 years after primary infection, which is in agreement with previous studies demonstrating that most superinfections occur within the first 2 years of incident infection [5,8]. One of these superinfected individuals (the first superinfected individual in Table 3) has previously been shown to display evidence for a recombination event between original and superinfected viral populations [34]. The univariate results of our study suggest that a higher number of sexual contacts (more than three contacts per month) and specific HLA alleles may play an important role in the risk of HIV-1 superinfection, whereas race/ethnicity, age, higher frequency of condomless anal intercourse, lower CD4+ T-cell count and higher viral load are not significantly associated with increased superinfection risk in this high-risk cohort of MSM. Subanalyses using multivariate models for the identified risk factors that were found to be significant (P < 0.10) confirmed that the increased risk of superinfection is significantly associated with HLA-B*35, HLA-C*04 and HLA-DRB1*08, regardless of the number of sexual contacts. In contrast, the protective effect of HLA-DRB1*11 found in our univariate analysis was not confirmed in multivariate models. Further analyses in a larger cohort would be necessary to fully define the significance of HLA alleles associated with superinfection.
Prior studies have shown that specific HLA-alleles are associated with accelerated disease progression, while other HLA alleles have a protective effect on HIV-1 disease in monoinfected individuals [25–32]. Of the high-risk alleles, HLA-B 35 has most consistently been associated with accelerated disease progression following incident infection [28,29,35,36], but its association with superinfection has not been previously demonstrated. The HLA allele that was associated with decreased superinfection risk in the univariate analysis, HLA-DRB1*11, has been shown to be associated with resistance to chronic hepatitis C virus (HCV) infection and protection from HCV infection [37,38], suggesting that HLA-DRB1*11 may play a role in the resistance to more than one single-stranded RNA virus. Interestingly, HLA-B*27 and HLA-B*57, two alleles that have been shown to be associated with slower disease progression [30,36,39], did not demonstrate a protective effect on superinfection. Therefore, it is possible that superinfection risk may involve pathological mechanisms that are specific to the second HIV-1 infection. Expression studies have shown that higher HLA-C expression protects against HIV infection [40,41] and controls viral load in monoinfected individuals [42]. Numerous pathogenic infections have also been shown to downregulate HLA expression to evade T-cell recognition [43–45]. In addition, in-vitro studies have shown that HIV infection downregulates HLA-A and HLA-B expression [46]. These observations suggest that primary HIV-1 infection may downregulate expression of specific HLA alleles at transcriptional, translational or posttranslational levels resulting in an increased risk for superinfection (individuals with high-risk alleles), whereas upregulation of other alleles may decrease the superinfection risk (individuals with low-risk alleles). Lower HLA expression may result in less efficient presentation of HIVepitopes to cytotoxic T lymphocytes (CTLs) and reduced initiation of T-cell response. The mechanisms by which HIV infection affects HLA expression are currently unknown, but the HLA-C expression studies suggest that noncoding micro RNAs (miRNAs) may play an important role in the regulation of cell surface HLA-C expression [47]. It is possible that similar mechanisms exist between other HLA molecules and miRNAs, and this might explain why some individuals are more prone to HIV-1 superinfection than others. Further studies are required to elucidate if HLA expression levels are critical in superinfection risk, and if miRNAs are responsible for differential expression of HLA molecules after primary infection.
The observation that individuals who remained monoinfection for over 2 years carried fewer high-risk HLA alleles when compared with those who became superinfection further suggests that individuals with specific high-risk HLA alleles are at an increased risk for HIV-1 SI. Interestingly, two monoinfected individuals who continued high-risk sexual behaviour and carried three high-risk alleles remained monoinfected for over 2 years (34.7 and 41.5 months; Table 3), suggesting that they may be carriers of certain protective HLA alleles for HIV-1 superinfection, whose protective effects were not detected in our analyses possibly due to an insufficient cohort size. Alternatively, these two individuals may have engaged in sexual risk behaviours in lower sexual risk networks (i.e. fewer HIV-infected persons, lower network-level viral load or fewer associated sexually transmitted diseases). In addition, it is possible that genetic properties of the donor virus play a role in the superinfection risk and individuals with protective HLA haplotypes are more resistant to the infecting virus, whereas other HLA allele combinations can result in a higher risk of reinfection.
Strong linkage disequilibrium between HLA loci makes the identification of specific risk alleles challenging. A highly significant linkage disequilibrium was observed between all Class I HLA loci (P < 0.0001) and between HLA-B and HLA-DRB1 (P = 0.024) (Supplemental Table 4, http://links.lww.com/QAD/B56). For example, five out of six (83.3%) superinfected individuals and all four monoinfected individuals with continued high-risk sexual behaviour, who carried the HLA-C*04 allele were also the carriers of the well characterized HLA-B*35 allele, and all three superinfected individuals and one monoinfected individual with continued high-risk sexual behaviour who carried the HLA-A*29 high-risk allele carried also the HLA-C*16 and HLA-DRB1*07 high-risk alleles, but not HLA-B*35 (Table 3). Therefore, it is possible that some of the observed high-risk alleles for HIV-1 superinfection may be due to a strong linkage disequilibrium between HLA loci. This kind of phenomenon has been reported between several HLA alleles, including HLA-B*35 and HLA-C*04, where the association between HLA-C*04 and rapid disease progression in individuals with primary HIV-1 infection was shown to be due to a strong linkage disequilibrium with HLA-B*35 [28,30,35]. Due to the complex linkage disequilibrium patterns, particular care must be taken when deciphering the results of association studies of the HLA loci in order to avoid confounding results. However, despite this limitation, association analyses represent an essential step when elucidating immunogenetic determinants of HIV-1 superinfection for further studies.
Another limitation of this study is that HLA lineage groups (two-digit allele groups) were used in statistical analyses instead of individual alleles, due to limited number of participants in the study resulting in limited statistical power. Therefore, we were not able to dissect what individual alleles are associated with increased and decreased HIV-1 superinfection risk in our MSM cohort. In addition, the number of individuals who became superinfected during the follow-up period was limited (n = 10/10.2%). Due to these limitations, it would be desirable to confirm these results in a larger study cohort with a larger person-time of follow-up and a higher number of events to be able to perform more detailed genetic analyses of HLA loci and to dissect the effect of each individual HLA risk-allele on HIV-1 superinfection. In conclusion, the results of our study suggest that a high number of sexual contacts and specific HLA alleles may play a significant role in the risk of HIV-1 superinfection, whereas race/ethnicity, age, lower CD4+ cell count and higher viral load are not significantly associated with an increased superinfection risk in this high-risk cohort of MSM. These results can be useful when determining the risk of potential superinfection susceptibility in patients with repeated exposure to HIV-1 after primary infection. In addition, a better understanding of superinfection will also provide important information for vaccine development of HIV-1, as HIV-1 superinfection can be considered a vaccine failure because the primary infection is not able to prevent the reinfection during the early stages of HIV-1 disease.
Supplementary Material
Acknowledgments
The authors give special thanks to all participating participants who made this study possible.
This study was funded by NIH grants AI043638, AI074621, AI106039, MH100974, AI036214 and the California HIV Research Program grant RN07-SD-702.
Footnotes
Conflicts of interest
D.D.R. has served as a consultant for Chimerix, Gilead Sciences and Monogram Biosciences. D.M.S. has served as a consultant for Gen-Probe and Testing Talent Services, and has received research funding from ViiV Pharmaceuticals. S.J.L. has received research grant funding from Gilead Sciences.
J.V. contributed to the study design and data interpretation, performed data analysis and wrote the manuscript. A.C. contributed to the data analysis, data interpretation and the writing of the manuscript. G.A.W. generated the time-dependent database, and contributed to the data analysis and writing of the manuscript. C.M.A. designed statistical analyses, performed data analysis and contributed to data interpretation and the writing of the manuscript. D.D.R. and D.M.S. contributed to the study design, data analysis and the writing of the manuscript. S.J.L. designed and conceived the study, directed the data analysis, contributed to data interpretation and manuscript writing, and was responsible for the overall study.
References
- 1.Smith DM, Richman DD, Little SJ. HIV superinfection. J Infect Dis. 2005;192:438–444. doi: 10.1086/431682. [DOI] [PubMed] [Google Scholar]
- 2.Piantadosi A, Chohan B, Chohan V, McClelland RS, Overbaugh J. Chronic HIV-1 infection frequently fails to protect against superinfection. PLoS Pathog. 2007;3:e177. doi: 10.1371/journal.ppat.0030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piantadosi A, Ngayo MO, Chohan B, Overbaugh J. Examination of a second region of the HIV type 1 genome reveals additional cases of superinfection. AIDS Res Hum Retroviruses. 2008;24:1221. doi: 10.1089/aid.2008.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pacold ME, Pond SL, Wagner GA, Delport W, Bourque DL, Richman DD, et al. Clinical, virologic, and immunologic correlates of HIV-1 intraclade B dual infection among men who have sex with men. AIDS. 2012;26:157–165. doi: 10.1097/QAD.0b013e32834dcd26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagner GA, Pacold ME, Kosakovsky Pond SL, Caballero G, Chaillon A, Rudolph AE, et al. Incidence and prevalence of intrasubtype HIV-1 dual infection in at-risk men in the United States. J Infect Dis. 2014;209:1032–1038. doi: 10.1093/infdis/jit633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chohan BH, Piantadosi A, Overbaugh J. HIV-1 superinfection and its implications for vaccine design. Curr HIV Res. 2010;8:596–601. doi: 10.2174/157016210794088218. [DOI] [PubMed] [Google Scholar]
- 7.Redd AD, Ssemwanga D, Vandepitte J, Wendel SK, Ndembi N, Bukenya J, et al. Rates of HIV-1 superinfection and primary HIV-1 infection are similar in female sex workers in Uganda. AIDS. 2014;28:2147–2152. doi: 10.1097/QAD.0000000000000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ronen K, McCoy CO, Matsen FA, Boyd DF, Emery S, Odem-Davis K, et al. HIV-1 superinfection occurs less frequently than initial infection in a cohort of high-risk Kenyan women. PLoS Pathog. 2013;9:e1003593. doi: 10.1371/journal.ppat.1003593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ssemwanga D, Lyagoba F, Ndembi N, Mayanja BN, Larke N, Wang S, et al. Multiple HIV-1 infections with evidence of recombination in heterosexual partnerships in a low risk Rural Clinical Cohort in Uganda. Virology. 2011;411:113–131. doi: 10.1016/j.virol.2010.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hue S, Hassan AS, Nabwera H, Sanders EJ, Pillay D, Berkley JA, et al. HIV type 1 in a rural coastal town in Kenya shows multiple introductions with many subtypes and much recombination. AIDS Res Hum Retroviruses. 2012;28:220–224. doi: 10.1089/aid.2011.0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koning FA, Badhan A, Shaw S, Fisher M, Mbisa JL, Cane PA. Dynamics of HIV type 1 recombination following superinfection. AIDS Res Hum Retroviruses. 2013;29:963–970. doi: 10.1089/aid.2013.0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith DM, Wong JK, Hightower GK, Ignacio CC, Koelsch KK, Petropoulos CJ, et al. HIV drug resistance acquired through superinfection. AIDS. 2005;19:1251–1256. doi: 10.1097/01.aids.0000180095.12276.ac. [DOI] [PubMed] [Google Scholar]
- 13.Jost S, Bernard MC, Kaiser L, Yerly S, Hirschel B, Samri A, et al. A patient with HIV-1 superinfection. N Engl J Med. 2002;347:731–736. doi: 10.1056/NEJMoa020263. [DOI] [PubMed] [Google Scholar]
- 14.Gottlieb GS, Nickle DC, Jensen MA, Wong KG, Grobler J, Li F, et al. Dual HIV-1 infection associated with rapid disease progression. Lancet. 2004;363:619–622. doi: 10.1016/S0140-6736(04)15596-7. [DOI] [PubMed] [Google Scholar]
- 15.Cornelissen M, Pasternak AO, Grijsen ML, Zorgdrager F, Bakker M, Blom P, et al. HIV-1 dual infection is associated with faster CD4+ T-cell decline in a cohort of men with primary HIV infection. Clin Infect Dis. 2012;54:539–547. doi: 10.1093/cid/cir849. [DOI] [PubMed] [Google Scholar]
- 16.Ronen K, Richardson BA, Graham SM, Jaoko W, Mandaliya K, McClelland RS, et al. HIV-1 superinfection is associated with an accelerated viral load increase but has a limited impact on disease progression. AIDS. 2014;28:2281–2286. doi: 10.1097/QAD.0000000000000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le T, Wright EJ, Smith DM, He W, Catano G, Okulicz JF, et al. Enhanced CD4+ T-cell recovery with earlier HIV-1 antiretroviral therapy. N Engl J Med. 2013;368:218–230. doi: 10.1056/NEJMoa1110187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith DM, Strain MC, Frost SD, Pillai SK, Wong JK, Wrin T, et al. Lack of neutralizing antibody response to HIV-1 predisposes to superinfection. Virology. 2006;355:1–5. doi: 10.1016/j.virol.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 19.Wagner GA, Pacold ME, Vigil E, Caballero G, Morris SR, Kosakovsky Pond SL, et al. Using ultradeep pyrosequencing to study HIV-1 coreceptor usage in primary and dual infection. J Infect Dis. 2013;208:271–274. doi: 10.1093/infdis/jit168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahuja SK, Kulkarni H, Catano G, Agan BK, Camargo JF, He W, et al. CCL3L1-CCR5 genotype influences durability of immune recovery during antiretroviral therapy of HIV-1-infected individuals. Nat Med. 2008;14:413–420. [Google Scholar]
- 21.Morris SR, Little SJ, Cunningham T, Garfein RS, Richman DD, Smith DM. Evaluation of an HIV nucleic acid testing program with automated Internet and voicemail systems to deliver results. Ann Intern Med. 2010;152:778–785. doi: 10.1059/0003-4819-152-12-201006150-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomiyama H, Yamada N, Komatsu H, Hirayama K, Takiguchi M. A single CTL clone can recognize a naturally processed HIV-1 epitope presented by two different HLA class I molecules. Eur J Immunol. 2000;30:2521–2530. doi: 10.1002/1521-4141(200009)30:9<2521::AID-IMMU2521>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 23.Ueno T, Tomiyama H, Takiguchi M. Single T cell receptor-mediated recognition of an identical HIV-derived peptide presented by multiple HLA class I molecules. J Immunol. 2002;169:4961–4969. doi: 10.4049/jimmunol.169.9.4961. [DOI] [PubMed] [Google Scholar]
- 24.Hickman HD, Luis AD, Bardet W, Buchli R, Battson CL, Shearer MH, et al. Cutting edge: class I presentation of host peptides following HIV infection. J Immunol. 2003;171:22–26. doi: 10.4049/jimmunol.171.1.22. [DOI] [PubMed] [Google Scholar]
- 25.Malhotra U, Holte S, Dutta S, Berrey MM, Delpit E, Koelle DM, et al. Role for HLA class II molecules in HIV-1 suppression and cellular immunity following antiretroviral treatment. J Clin Invest. 2001;107:505–517. doi: 10.1172/JCI11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Julg B, Moodley ES, Qi Y, Ramduth D, Reddy S, Mncube Z, et al. Possession of HLA class II DRB1*1303 associates with reduced viral loads in chronic HIV-1 clade C and B infection. J Infect Dis. 2011;203:803–809. doi: 10.1093/infdis/jiq122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ranasinghe S, Cutler S, Davis I, Lu R, Soghoian DZ, Qi Y, et al. Association of HLA-DRB1-restricted CD4(+) T cell responses with HIV immune control. Nat Med. 2013;19:930–933. doi: 10.1038/nm.3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carrington M, Nelson GW, Martin MP, Kissner T, Vlahov D, Goedert JJ, et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283:1748–1752. doi: 10.1126/science.283.5408.1748. [DOI] [PubMed] [Google Scholar]
- 29.Lazaryan A, Song W, Lobashevsky E, Tang J, Shrestha S, Zhang K, et al. The influence of human leukocyte antigen class I alleles and their population frequencies on human immunodeficiency virus type 1 control among African Americans. Hum Immunol. 2011;72:312–318. doi: 10.1016/j.humimm.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaslow RA, Carrington M, Apple R, Park L, Munoz A, Saah AJ, et al. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat Med. 1996;2:405–411. doi: 10.1038/nm0496-405. [DOI] [PubMed] [Google Scholar]
- 31.Hendel H, Caillat-Zucman S, Lebuanec H, Carrington M, O’Brien S, Andrieu JM, et al. New class I and II HLA alleles strongly associated with opposite patterns of progression to AIDS. J Immunol. 1999;162:6942–6946. [PubMed] [Google Scholar]
- 32.Keet IP, Tang J, Klein MR, LeBlanc S, Enger C, Rivers C, et al. Consistent associations of HLA class I and II and transporter gene products with progression of human immunodeficiency virus type 1 infection in homosexual men. J Infect Dis. 1999;180:299–309. doi: 10.1086/314862. [DOI] [PubMed] [Google Scholar]
- 33.Tang J, Costello C, Keet IP, Rivers C, Leblanc S, Karita E, et al. HLA class I homozygosity accelerates disease progression in human immunodeficiency virus type 1 infection. AIDS Res Hum Retroviruses. 1999;15:317–324. doi: 10.1089/088922299311277. [DOI] [PubMed] [Google Scholar]
- 34.Chaillon A, Wagner GA, Hepler NL, Little SJ, Kosakovsky Pond SL, Caballero G, et al. Dynamics of viral evolution and neutralizing antibody response after HIV-1 superinfection. J Virol. 2013;87:12737–12744. doi: 10.1128/JVI.02260-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao X, Nelson GW, Karacki P, Martin MP, Phair J, Kaslow R, et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N Engl J Med. 2001;344:1668–1675. doi: 10.1056/NEJM200105313442203. [DOI] [PubMed] [Google Scholar]
- 36.International HIVCS. Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yenigun A, Durupinar B. Decreased frequency of the HLA-DRB1*11 allele in patients with chronic hepatitis C virus infection. J Virol. 2002;76:1787–1789. doi: 10.1128/JVI.76.4.1787-1789.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thursz M, Yallop R, Goldin R, Trepo C, Thomas HC. Influence of MHC class II genotype on outcome of infection with hepatitis C virus. The HENCORE group. Hepatitis C European Network for Cooperative Research. Lancet. 1999;354:2119–2124. doi: 10.1016/s0140-6736(99)91443-5. [DOI] [PubMed] [Google Scholar]
- 39.Emu B, Sinclair E, Hatano H, Ferre A, Shacklett B, Martin JN, et al. HLA class I-restricted T-cell responses may contribute to the control of human immunodeficiency virus infection, but such responses are not always necessary for long-term virus control. J Virol. 2008;82:5398–5407. doi: 10.1128/JVI.02176-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas R, Apps R, Qi Y, Gao X, Male V, O’HUigin C, et al. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat Genet. 2009;41:1290–1294. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Specht A, Telenti A, Martinez R, Fellay J, Bailes E, Evans DT, et al. Counteraction of HLA-C-mediated immune control of HIV-1 by Nef. J Virol. 2010;84:7300–7311. doi: 10.1128/JVI.00619-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. 1996;2:338–342. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- 44.Bennett EM, Bennink JR, Yewdell JW, Brodsky FM. Cutting edge: adenovirus E19 has two mechanisms for affecting class I MHC expression. J Immunol. 1999;162:5049–5052. [PubMed] [Google Scholar]
- 45.Ishido S, Wang C, Lee BS, Cohen GB, Jung JU. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J Virol. 2000;74:5300–5309. doi: 10.1128/jvi.74.11.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Apps R, Meng Z, Del Prete GQ, Lifson JD, Zhou M, Carrington M. Relative expression levels of the HLA class-I proteins in normal and HIV-infected cells. J Immunol. 2015;194:3594–3600. doi: 10.4049/jimmunol.1403234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kulkarni S, Savan R, Qi Y, Gao X, Yuki Y, Bass SE, et al. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011;472:495–498. doi: 10.1038/nature09914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.