

Abstract
Tumor initiating cells (TICs) represent cancer stem-like cell (CSC) subpopulations within tumors that are thought to give rise to recurrent cancer after therapy. Identifying key regulators of TIC/CSC maintenance is essential for the development of therapeutics designed to limit recurrence. The steroid receptor coactivator 3 (SRC-3) is overexpressed in a wide range of cancers, driving tumor initiation, cell proliferation, and metastasis. Here we report that SRC-3 supports the TIC/CSC state and induces an epithelial to mesenchymal transition (EMT) by driving expression of the master EMT regulators and stem cell markers. We also show that inhibition of SRC-3 and SRC-1 with SI-2, a second-generation SRC-3/SRC-1 small molecule inhibitor, targets the CSC/TIC population both in vitro and in vivo. Collectively, these results identify SRC coactivators as regulators of stem-like capacity in cancer cells and that these coactivators can serve as potential therapeutic targets to prevent the recurrence of cancer.
Keywords: Tumor-initiating cells, steroid receptor coactivator, cancer stem-like cells, epithelial to mesenchymal transition, breast cancer
Introduction
Tumor-initiating cells (TICs) or cancer stem-like cells (CSCs) frequently exist as a subpopulation of cells in a tumor that is resistant to existing treatments. Their survival and outgrowth after therapy is thought to be responsible for recurrent disease that is ultimately responsible for the vast majority of cancer deaths (1,2). CSCs are characterized by the ability of cells to self-renew and to drive tumor initiation and metastasis (3). Among solid tumors, they were first discovered in breast cancer (3) and have since been identified in other solid tumors (4). In vitro, CSCs are characterized by their cell surface marker profiles (3), their ability to form colonies in suspension culture (5) and their increased resistance to chemotherapeutic agents (2,6). The induction of an epithelial to mesenchymal transition (EMT) in neoplastic epithelial cells also is thought to enrich for these cancer stem-like cells (7). During EMT, cancer cells lose their cell-cell adhesions, cell polarity and acquire a more motile mesenchymal phenotype (8), thus enhancing migratory and invasive properties that are important for a cancer cell’s metastatic potential. EMT is characterized by the repression of epithelial markers such as E-cadherin and enhanced expression of mesenchymal markers such as snail, slug and vimentin (8). Importantly, EMT has been shown to be linked to the development of chemo-resistance in cancers such as, breast (9) and pancreatic neoplasias (10). Hence, identifying key regulators of CSC maintenance and chemo-resistance is important for the development of new therapeutic strategies that can both block EMT and eliminate resistant CSC/TIC populations of tumor cells.
The steroid receptor coactivator (SRC) family consists of three members: SRC-1 (NCOA1) (11), SRC-2 (NCOA2/TIF2/GRIP1) (12) and SRC-3 (NCOA3/AIB1/RAC3/ACTR/pCIP) (13) and function as key ‘platform’ coactivators that assemble multi-protein complexes to drive nuclear receptor (NR) mediated transcription. All three SRCs have established roles in regulating nuclear receptor-mediated gene transcription as well as transcriptional programs driven by other transcription factors (14). As a result of their broad utilization by a diverse array of transcription factors, the SRCs regulate multiple physiological and pathophysiological processes. All three SRCs are frequently overexpressed or amplified in different cancers. For instance, SRC-1 is overexpressed in breast cancer and promotes lung metastasis through increased cancer cell migration and invasion (14). SRC-2 is amplified in ~8% of primary prostate tumors and overexpressed in ~ 37% metastatic prostate tumors in patients (15), stimulating cancer cell growth through both androgen receptor (AR) dependent and independent metabolic mechanisms. Of the three SRCs, SRC-3 is the most prominently implicated in cancer, being overexpressed in breast (16), prostate (17), pancreatic cancer (18) and many other cancers (19) and can promote tumor initiation, progression and metastasis. Additionally, overexpression of SRC-3 has been linked to endocrine resistance in breast cancers (14).
As coactivators, the SRCs do not directly bind DNA but instead interact with activated transcription factors and form a protein-protein interaction scaffold upon which other co-coactivators such as CBP, p300, CARM1 can be assembled into a larger multi-protein coactivator complex to activate a vast array of genes important for diverse physiological processes (20). The SRCs previously have been considered to be difficult drug targets because of their large size and flexible structure and their reliance on protein-protein interactions (21). However, our lab has recently executed a high throughput screening campaign that has led to the identification of several small molecule inhibitors (SMIs) (21,22) and stimulators (23) for the SRCs. Importantly, we have shown that several of these SRC SMIs can inhibit cancer cell proliferation in vitro and in mouse xenograft models (21,22). While other groups have sought to develop coactivator binding inhibitors (CB1s) (24) designed to block the receptor-SRC protein-protein interface, SMIs designed to specifically target SRCs represents a distinct niche of novel class of anti-cancer agents.
While SRCs have been broadly implicated in tumor initiation and progression, not much is known about their function in regulating the tumor initiating capacity or ‘stem-like states’ of cancer cells responsible for resistance to first-line therapy and cancer recurrence. In a recent study SRC-3 was found to function as a coactivator for the estrogen related receptor-β (ESRRB) and was reported to maintain genes directing embryonic stem cell self-renewal and pluripotency (25). Additionally, SRC-3 expression is negatively correlated with the epithelial marker E-cadherin in pancreatic adenocarcinomas (26) and in the MMTV-PyMt mouse model of breast cancer (27). Thus, we hypothesized that SRC-3 may promote the stem-like state of CSCs and support the induction of EMT.
Here, we show that SRC-3 drives the formation of CSCs and supports tumor outgrowth. In concert with this, SRC-3 induces EMT by stimulating the expression of transcription factors, such as snail and slug, which are key factors that support the mesenchymal state. Importantly, we also demonstrate that by inhibiting SRC-3 activity with a second-generation SRC SMI we can block TICs which are prominent in the emergence of drug resistant, recurrent tumors that arise after treatment with first-line therapies.
Materials and Methods
Cell lines
The lung cancer cell lines A549 (adenocarcinoma), H1299 (non-small cell lung cancer) and H358 (non-small cell lung cancer); the breast cancer cell lines MCF-7 (estrogen receptor positive, luminal), MDA-MB-231 (triple negative, basal), SKBR3 (Her2 positive) and MDA-MB-468 (triple negative, basal) and 293T cells (human embryonic fibroblasts) were all purchased from ATCC and grown at the Tissue culture core at Baylor College of Medicine (BCM) where they are tested for mycoplasma every three to four months using the Mycoalert mycoplasma detection kit (Lonza). MCF-7 (received in 1996, used between passage 60 to 85), MDA-MB-231 (received in 1994, used between passage 32 to 90), SKBR3 (received in 2005, used between passage 39 to 56) and 293T (received in 2004, used between passage 20 to 50) cell lines were grown in DMEM (Cellgro), H1299 (received from ATCC in 2012 and used between passage 1 to 5 since thaw) and H358 (received from ATCC in 2016 and used between passage 1 to 5 since thaw) were grown in RPMI1640 (Cellgro) and A549 (received in 1993, passage 86 to 110) was grown in Kaighin’s medium supplemented with 10 % fetal calf serum (FCS) (Cellgro) and penicillin and streptomycin (Gibco). The MDA-MB-468 (received from ATCC in 2005 and used between passage 1 to 8 since thaw) cell line was grown in Leibovitz’s L-15 media supplemented with 10 % FCS, penicillin and streptomycin. Additionally, the MDA-MB-468 cells were grown in the absence of carbon dioxide. All cell identities were verified using the short tandem repeat (STR) analysis done by the tissue culture core at BCM. Stable MCF-7 cell lines expressing either SRC-3 shRNA (shSRC-3) or a non-targeting shRNA (NT shRNA) were generated by infection with lentivirus particles. Briefly, 293T cells were transfected with either pLKO.1 NT shRNA (SHC-016, Sigma) or pLKO.1 shSRC-3 (NM_006534.2-4717s21c1, CCGGTTCCACCTCCTAGGATATAACTCGAGTTATATCCCTAGGAGGTGGAATTTTTG, Sigma) plasmids together with pMD2.G and psPAX2 second generation packaging vectors using lipofectamine 2000 (Life Technologies). Forty-eight hours after transfection, supernatants were collected and filtered. MCF-7 cells were then transduced with the respective filtered supernatants in the presence of 4 μg/ml polybrene (Santa Cruz Biotechnology) and selected with 1 μg/ml puromycin (Gibco). For reporter assays, stable MCF-7 cells expressing either NT shRNA or shSRC-3 were grown in phenol red free DMEM (Cellgro) with 5% charcoal stripped FCS (Cellgro).
Reagents
Antibodies used include β-actin (Cell Signaling #4970), FLAG (Sigma #F1804), e-cadherin, snail, slug, vimentin, n-cadherin (Cell Signaling, EMT Ab sampler kit #9782S), SRC-1 (Santa Cruz Biotechnology #sc-32789), SRC-2 (BD Biosciences #610985) and SRC-3 (in-house monoclonal antibody created by the BCM Monoclonal Antibody Core). HRP conjugated donkey anti-rabbit and sheep anti-mouse secondary antibodies were from Pierce. pCMV-Flag-SRC-3 has been previously described (28). SBE4–luc, Flag-Smad3 full length, Flag-Smad3-NL, and Flag-Smad3-C plasmids were obtained from Addgene. The siGENOME SMARTpool siRNAs targeting SRC-1 (M-005196-03-0005), SRC-2 (M-020159-01-0005) or SRC-3 (M-003759-02-0010) and a non-targeting siRNA (pool #2, D-001206-14-05 or siRNA #3, D-001210-03-05) were purchased from Dharmacon. SI-2 was synthesized in our laboratory at BCM.
In vivo limiting dilution assay
MDA-MB-231 cells were treated with 250 nM SI-2 or DMSO for three days following which the drug was washed off and the cells were allowed to recover for an additional three days. After recovery the cells were washed, trypsinized, stained with trypan blue and counted. Viable cells were injected at limiting dilutions of 250,000, 25,000 or 2,500 cells into the second pair of mammary fat pads of 5–6 week old female athymic nude mice obtained from Envigo International. 5 mice per group were injected in two sites. All animal experiments were in compliance with the IACUC guidelines. Tumor initiation and growth was monitored for 8 weeks. The tumor initiation frequency was calculated using the ELDA software (29).
Flow cytometry
An aldeflour kit (Stem cell technologies) was used as per the manufacturer’s protocol to assay for ALDH activity. Briefly, cells were trypsinized following which 500,000 – 1 million cells were resuspended in 1 ml aldeflour assay buffer. Aldeflour reagent was added to this cell suspension. Half of the cell suspension was then transferred to a fresh tube containing DEAB. The cells were incubated at 37°C for 45 mins, washed and then subjected to flow cytometry using the BD LSRII flow cytometer (BD Biosciences). For CD44/CD24 detection, cells were washed, trypsinized and counted. 500,000 cells were resuspended in D-PBS with 2% FCS (Cellgro) with APC-CD44 and CD24-PE (BD Pharmingen #559952 and #559942) conjugated antibodies at dilutions of 1:20 and incubated at 37°C for 30 minutes. Cells were then washed three times with cold D-PBS with 2% FCS, resuspended and stained with Sytox Blue (Life Technologies). Flow cytometry was carried out using the BD LSRII flow cytometer (BD Biosciences).
Tumorsphere assay
Cells were plated in 6-well or 96-well low attachment plates (Corning) in DME/F12 with 1% methylcellulose (Sigma), bFGF (Peprotech), hEGF (R & D Systems), B27 supplement (Life Technologies) and Penicillin/streptomycin (Gibco). Fresh media was added every three days and the number of tumorspheres were counted and photographed after 8–10 days. A Zeiss light microscope was used to count and quantify the tumorspheres.
Reporter Assay
Cell lysates were isolated using the Reporter lysis Buffer assay system from Promega as per the manufacturer’s protocol. Briefly, cells were rinsed twice with DPBS and lysed with 1X reporter lysis buffer. Following a freeze-thaw cycle, lysed cells were collected and spun down at 14,000 rpm for 10 mins. Supernatants were subjected to protein quantification using the Bio-Rad protein assay dye (Bio-Rad) and to a luciferase assay using a luciferase substrate (Promega).
Immunoblotting
Total protein was isolated from cells using NETN (20 mM Tris, pH 7.5, 1 mM EDTA, 150 mM NaCl, 0.5% Igepal-CA630) lysis buffer with 10% glycerol, protease and phosphatase inhibitors (Roche). Cell lysates were incubated at 4°C for 30 minutes followed by a brief sonication. Lysates were cleared by centrifugation at 12,000 rpm for 10 mins. Total protein was quantified using the Bio-Rad protein assay dye (Bio-Rad). Proteins were separated using 4–15% Mini-PROTEAN® TGX™ Precast gels (Bio-Rad) and transferred onto PVDF membranes (Bio-Rad). Ponceau S (Sigma) staining was carried out to visualize transfer efficiency. Membranes were incubated with primary antibodies at 4°C overnight followed by incubation with either anti-mouse or anti-rabbit HRP conjugated secondary antibodies. Enhanced ECL (Pierce) was used to visualize blots either using X-ray film (Denville) or a Bio-Rad Chemi-doc imager.
GST Pull-Down Assays
Expression and purification of GST fusion SRC-3 fragment proteins were performed as previously described (30). Flag-Smad3 expressed cell lysates were precleared with glutathione-agarose beads and then incubated with GST fusion protein-conjugated glutathione-agarose beads overnight at 4°C. The beads were then washed five times with STE buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, and 0.5% NP-40) containing 1 mM PMSF, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 1 mM sodium orthovanadate. The samples were boiled in 2×SDS loading buffer prior to SDS-PAGE, followed by immunoblot detection with anti-Flag antibody.
Co-Immunoprecipitation Assay
Cells were washed twice with ice-cold PBS and lysed in 1 ml of RIPA lysis buffer. After centrifugation at 13,000 rpm for 10 min at 4°C, the supernatants were precleared with protein A-G (2:1) beads for 30 min, centrifuged to remove the beads, and then incubated with appropriate antibodies and protein A-G beads overnight at 4°C with constant rotation. The beads were washed three times with RIPA buffer, boiled in 2×SDS loading buffer, analyzed by SDS-PAGE and blotted with appropriate antibodies.
Cell Viability
MCF-7 or MDA-MB-231 cells were seeded in 96-well plates and allowed to reach 60 % to 70 % confluency. After indicated compound treatments for 72 hours, relative numbers of viable cells were measured by MTS assay using the Cell Titer 96 Aqueous One Solution Cell Proliferation Assay (Promega).
RNA isolation and qPCR
Total RNA was isolated using Trizol (Invitrogen) and purified using the Direct-zol RNA miniprep kit (Genesee Scientific). The superscript first strand synthesis kit (Invitrogen) was used to make cDNA. Quantitative real time (qPCR) was performed using the Applied Biosystems Step One Plus fast real-time PCR system (Applied Biosystems) and Taqman (Invitrogen) or SYBR green (Applied Biosystems) reagents. Relative amounts of transcripts were quantified by the comparative threshold cycle method (ΔΔCt) with GAPDH or β-actin as the endogenous reference control. The primers for gene expression analysis were designed using the Roche Universal Probe library and are listed in supplemental table 1.
Transfections
Lipofectamine 3000 (Life Technologies) was used to transfect H1299, H358, MCF7, MDA-MB-468 and MDA-MB-231 cells as per the manufacturer’s protocol. Lipofectamine 2000 (Life Technologies) was used to transfect 293T and A549 cells as per the manufacturer’s protocol. Optimem reduced serum was used to make DNA-liposome complexes. Cells were incubated with complexes for 48 hours prior to further analysis.
Immunofluorescence
Tumorspheres were collected on a 30 μm filter and washed 2 times with D-PBS. Tumorspheres were then fixed in formalin and permeabilized with 0.5 % Triton X-100 (Sigma) in D-PBS for either 5 mins or overnight. Tumorspheres were then washed, blocked for 1 hour in 1 % BSA with 0.1 % Tweem-20 (Fisher Scientific) in D-PBS and incubated with primary antibodies to SRC-3, Snail or E-cadherin overnight in blocking solution. Tumorspheres were then washed with D-PBS and stained with secondary antibodies; Alexa-flour conjugated 488 or 594 (Abcam) for 1 hour in blocking solution. Following washes with D-PBS, DNA was stained with DAPI and stained tumorspheres were mounted on cavity slides with mounting media. Tumorspheres were imaged using the Nikon A1-R confocal microscope.
ChIP-qPCR
ChIP assay was performed as previously described (31). The soluble chromatin was prepared from a total of 2×107 cells. The pre-cleared chromatin solution was subjected to immunoprecipitation with anti-Flag antibody or anti-mouse IgG (Millipore #12-371). Following multiple washes, the antibody-protein-DNA complex was eluted from the beads; the DNA was purified and subjected to real-time PCR with primers listed in supplemental table 2.
Statistics
Significance testing was done using the Student’s t-test for means between two groups or one way ANOVA in conjunction with Tukey’s multiple comparison test for means between more than two groups. Error bars represent standard error of means.
Results
SRCs promote stem-like characteristics in cancer cells
To assess the role of the SRCs in CSCs, we first examined the expression of SRC -1, -2 and -3 in cells growing as tumorspheres under ultra-low attachment conditions compared to cells grown under standard monolayer culture conditions. Tumorspheres exhibited increased expression of the stem cell markers Oct4, Sox2 and Nanog (Fig. 1A). Concomitantly, expression of all three SRCs was increased in tumorspheres compared to cells grown in monolayer culture. To ascertain if the observed increases of any of the SRCs might underlie CSC tumorsphere formation, we knocked down SRC-1, -2 or -3 in lung and breast cancer cells and determined the tumorsphere forming capacity of these cells. Decreased expression of SRC-1 and -3 but not -2, significantly reduced the number of tumorspheres formed on the low attachment substrate (Fig. 1B–D). This suggested that despite the increased expression of all three SRCs in CSC tumorspheres, SRC-1 and -3 may be most responsible for CSC tumorsphere formation. Since CSCs are characterized by the expression of aldehyde dehydrogenase (ALDH) (32), we also measured the percentage of ALDH+ cells after knockdown of SRC-1, -2 or -3. Interestingly, we found that knockdown of only SRC-3 but not SRC-1 or -2 significantly reduced the proportion of ALDH+ breast CSCs (Fig. 2A–C). Collectively, our data suggests that SRC-3 may primarily be important for the maintenance of stem-like characteristics in cancer cells. These data also suggest that SRC-1 is important for the proliferation of cancer stem-like cells.
Figure 1. SRCs are important for tumorsphere formation.
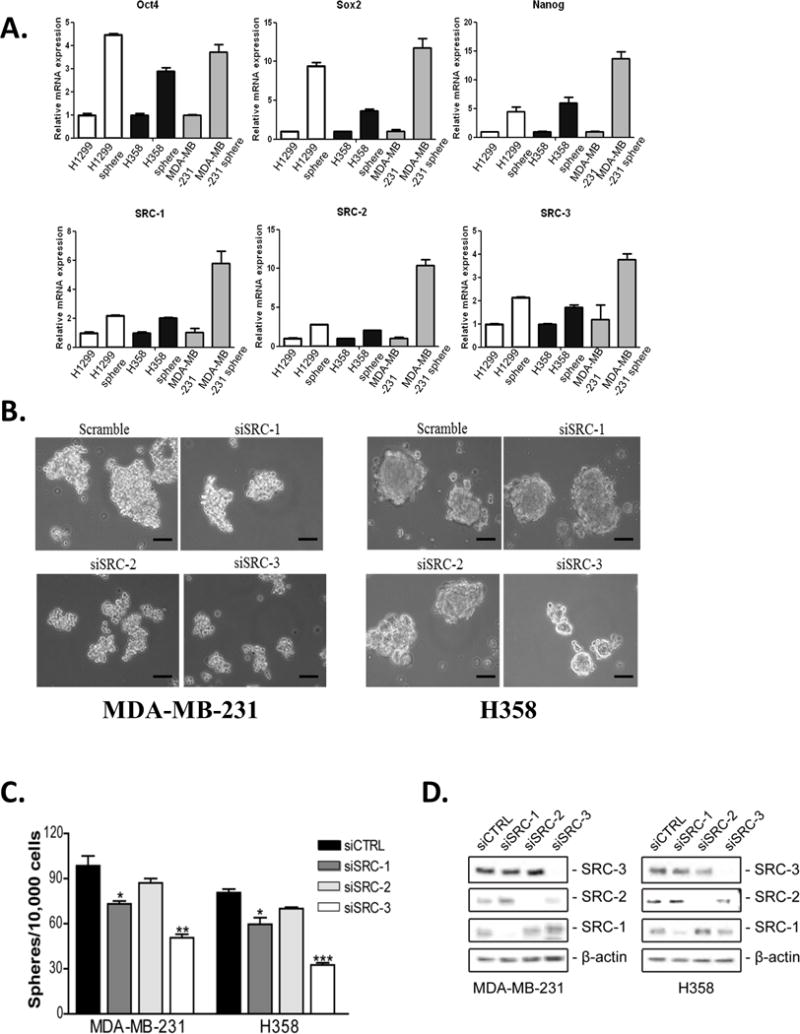
Expression of SRC-1, -2 and -3 is increased in tumorspheres. (A) Total RNA was isolated from H1299, H358 and MDA-MB-231 cells grown either in monolayer culture or in serum free media on ultra-low attachment plates and converted to cDNA. QPCR was performed using primers for GAPDH, Oct4, Sox2, Nanog, SRC-1, -2 and -3. Gene expression was quantified using GAPDH as an internal control. Values shown are the mean ± S.E.M. (n=3). (B-D) Reduced expression of SRC-1 and -3 but not SRC-2 reduces tumorsphere formation. MDA-MB-231 and H358 cells were transfected with either control siRNA or siRNA targeting SRC-1, SRC-2 or SRC-3. (B) Bright field images of siRNA transfected cells plated in serum free media on ultra-low attachment plates after 8 days. Scale bar = 50 μm. (C) Tumorspheres greater than 50 μm were counted and the numbers of spheres formed were graphed. Values shown are the mean ± S.E.M. (n=3). One-way ANOVA followed by Tukey’s multiple comparison tests were used for significance testing. * = p < 0.05, ** = p < 0.01, *** = p < 0.001. (D) 48 hours post transfection total protein was isolated from the cells and subjected to immunoblot analysis with antibodies to SRC-1, SRC-2, SRC-3 and β-actin.
Figure 2. SRC-3 regulates the proportion of CSCs.
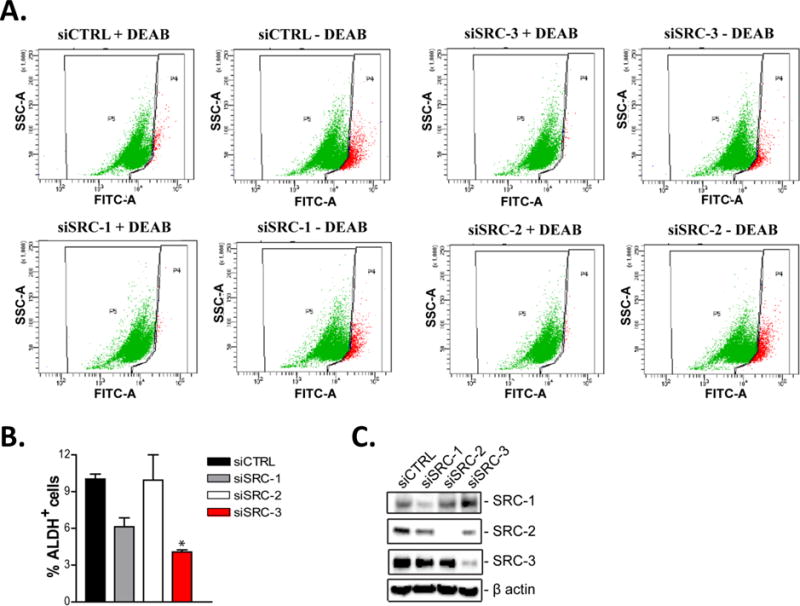
Decreased expression of SRC-3 reduces the % ALDH+ cell population. SKBR3 cells were transiently transfected with either control (siCTRL), SRC-1 (siSRC-1), SRC-2 (siSRC-2) or SRC-3 (siSRC-3) targeting siRNAs for 48 hours. (A-B) Cells were then assayed for ALDH activity using flow cytometry. Representative flow cytometry dot plots are shown. The graphed data represents the mean ± S.E.M. (n=3). One-way ANOVA and Tukey’s multiple comparison tests were used for significance testing. * = p < 0.05. (C) 48 hours post transfection total protein was isolated from the cells and subjected to immunoblot analysis with antibodies to SRC-1, SRC-2, SRC-3 and β-actin.
SRC-3 regulates an EMT network in cancer cells
EMT is associated with the cancer cell stem-like state and can contribute to tumor progression, metastasis and therapy resistance (33). This raises the intriguing possibility that SRC-3 may induce EMT to drive stem-like characteristics in cancer cells. We found that increased expression of SRC-3 in the A549 epithelial cancer cell line induced a mesenchymal phenotype as evidenced by the loss of cell contacts and the acquisition of spindle cell morphology (Fig. 3A). Additionally, genes involved in the EMT network were altered: the protein expression of mesenchymal markers snail, slug, vimentin and N-cadherin were increased, while protein expression of the epithelial marker E-cadherin was reduced (Fig. 3B). QPCR analysis confirmed elevated expression of snail and slug and decreased expression of E-cadherin upon SRC-3 overexpression (Fig. 3C) in lung cancer cells. SRC-3 is frequently amplified and overexpressed in breast cancer and previous studies have identified a role for SRC-3 in regulating EMT in breast cancer (27). Hence, to confirm the role of SRC-3 in inducing EMT in breast cancer, we examined the effect of loss of SRC-3 on expression of EMT markers. We found that decreased expression of SRC-3 in MCF-7 cells upregulated protein expression of e-cadherin and reduced protein expression of snail (Fig. 3D). QPCR analysis confirmed the upregulation of e-cadherin and decrease in snail and slug RNA expression on stable knockdown of SRC-3 expression (Fig. 3E).
Figure 3. SRC-3 induces and regulates an epithelial to mesenchymal transition.
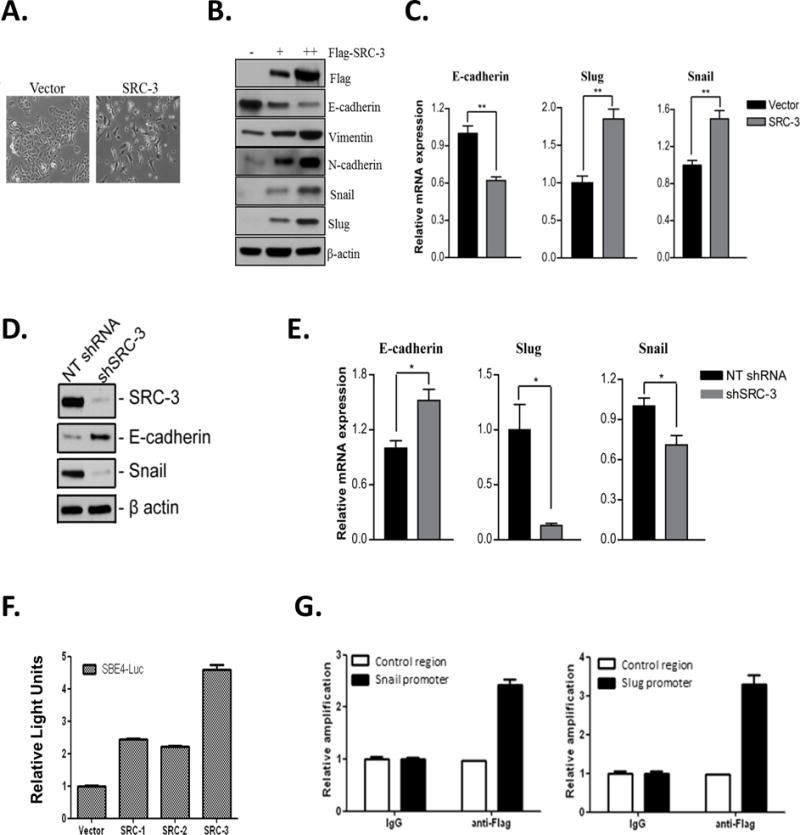
Increased SRC-3 expression increases mesenchymal marker expression. A549 cells were transiently transfected with an expression vector for Flag-SRC-3. (A) Bright field image demonstrating a change from epithelial to mesenchymal morphology 48 hours after transient transfection of Flag-SRC-3 or vector control. (B) Immunoblotting of total protein isolated from cells overexpressing Flag-SRC-3, with + and ++ indicating low and high levels of SRC-3, respectively. Cell lysates were immunoblotted with antibodies for Flag, e-cadherin, vimentin, n-cadherin, snail, slug and β-actin. (C) RNA was isolated from cells and converted to cDNA. QPCR was carried out using primers for e-cadherin, snail, slug and GAPDH. Gene expression was quantified using GAPDH as an internal control. Values shown are the mean ± S.E.M. (n=3). Student’s t-test was used for significance testing. ** = p < 0.01. (D) Decreased expression of SRC-3 increases epithelial marker expression. Total protein was isolated from stable MCF-7 cells that express either non-targeting shRNA or SRC-3 targeting shRNA. Immunoblotting was carried out with antibodies to SRC-3, e-cadherin, snail and β-actin. (E) Total RNA was isolated from stably transfected MCF-7 cells and converted to cDNA. QPCR analysis was carried out using primers for e-cadherin, snail, slug and GAPDH. Gene expression was quantified using GAPDH as an internal loading control. Values shown represent the mean ± S.E.M. (n=3). Student’s t-test was used for significance testing. * = p < 0.05. (F) SRC-3 overexpression stimulates Smad-dependent reporter activity. H1299 cells were co-transfected with a Smad binding element (SBE4-luc) luciferase promoter-reporter construct and either SRC-1, -2 or -3 expression vectors. 48 hours post transfection luciferase activity was measured and normalized to total protein. Values shown are the means ± S.E.M. (n=3). (G) SRC-3 is recruited to the Smad binding element in the promoters of Snail and Slug. H1299 cells were transiently transfected with Flag-SRC-3. Chromatin immunoprecipitation was performed with antibodies against Flag and IgG. DNA was isolated and qPCR was performed using primers to the Smad binding regions in the promoters of snail and slug. Primers to GAPDH were used as a control. Relative occupancy was calculated after normalizing to input and setting the control region to 1. Values shown are means ± S.E.M. (n=3).
The TGFβ signaling pathway is important for stimulating snail and slug expression during EMT and is dependent on Smad transcription factors (34). Thus, we examined the effect of SRC-3 expression on the activity of a luciferase reporter containing four Smad binding elements, SBE4-luc (35). Compared to SRC-1 and -2, overexpression of SRC-3 had a greater effect in stimulating the activity of the SBE4-luc reporter (Fig. 3F). Furthermore, decreased expression of SRC-3 repressed TGFβ mediated induction of the SBE4-luc reporter in lung and breast cancer cells (Supplementary Fig. 1A). The regulation of snail and slug by SRC-3 appeared to be due to the direct recruitment of SRC-3 to the Smad3 response element in their promoters (Fig. 3G), suggesting that SRC-3 functions as a common coactivator for both of these EMT-related transcription factors. Co-immunoprecipitation experiments confirmed an interaction between SRC-3 and Smad3 (Supplementary Fig. 1B). We performed GST pulldowns to identify the region in SRC-3 that is required for its interaction with Smad3 (Supplementary Fig. 1C). GST-tagged SRC-3 fragments for the well characterized bHLH-PAS, RID or CID domains were incubated with lysates from 293T cells overexpressing Flag-Smad3 in a GST pulldown assay. Our results demonstrate that the RID domain of SRC-3 is required for its interaction with Smad3 (Supplementary Fig. 1D). For Smad3, the N-terminal and linker region (NL), C-terminal region (C) or full length (FL) protein were tested for interaction with SRC-3. Co-immunoprecipitation of the different flag-tagged Smad3 constructs with full length SRC-3 in 293T cells revealed that the C- terminal of Smad3 is required for its interaction with SRC-3 (Supplementary Fig. 1E). Together, our data suggest that SRC-3 can regulate EMT in cancer cells likely through the coactivation of Smad3.
SI-2 inhibits SRC-3 and targets the breast CSC population
Recent studies have shown that altering coactivator function is a new entrée into the treatment of cancer. For instance, a newly identified second-generation SRC inhibitor developed in our laboratory, SI-2, can inhibit SRC-3 activity, reduce breast cancer cell migration and block tumor growth (21). However, it is crucial to examine the potential for cancer therapeutics to kill resistant CSC/TIC tumor cells that are responsible for disease recurrence and resistance to first-line therapies. Since our data implicated a role for SRC-3 in the induction of EMT and promotion of the CSC/TIC state, we hypothesized that inhibition of SRC-3 with a SMI may target and incapacitate CSC/TICs, blocking tumor recurrence. Since SRC-3 is amplified or overexpressed in breast cancer (13,16) and we have shown that SI-2 can effectively inhibit breast tumor growth (21), we sought to examine the specific effects of SRC-3 inhibition on breast TIC/CSC potential. For this, hormone receptor positive MCF-7 or triple negative MDA-MB-231 cells were treated with SI-2 for twenty four hours, and then tested in in vitro tumorsphere assays on an ultra-low attachment substrate. Both these cell lines show decreased SRC-3 protein expression (Supplementary Fig. 2A) upon SI-2 treatment and their proliferation was inhibited by SI-2 (Supplementary Fig. 2B). Importantly, inhibiting SRC-3 with SI-2 led to an inhibition of breast CSC tumorsphere formation in MCF-7 (Fig. 4A) and MDA-MB-231 (Fig. 4B) cells. Additionally, protein expression of SRC-3 and snail (Fig. 4C) were reduced while expression of E-cadherin (Fig. 4D) was increased in SI-2 treated tumorspheres compared to control treated tumorspheres.
Figure 4. SI-2 reduces tumorsphere formation.
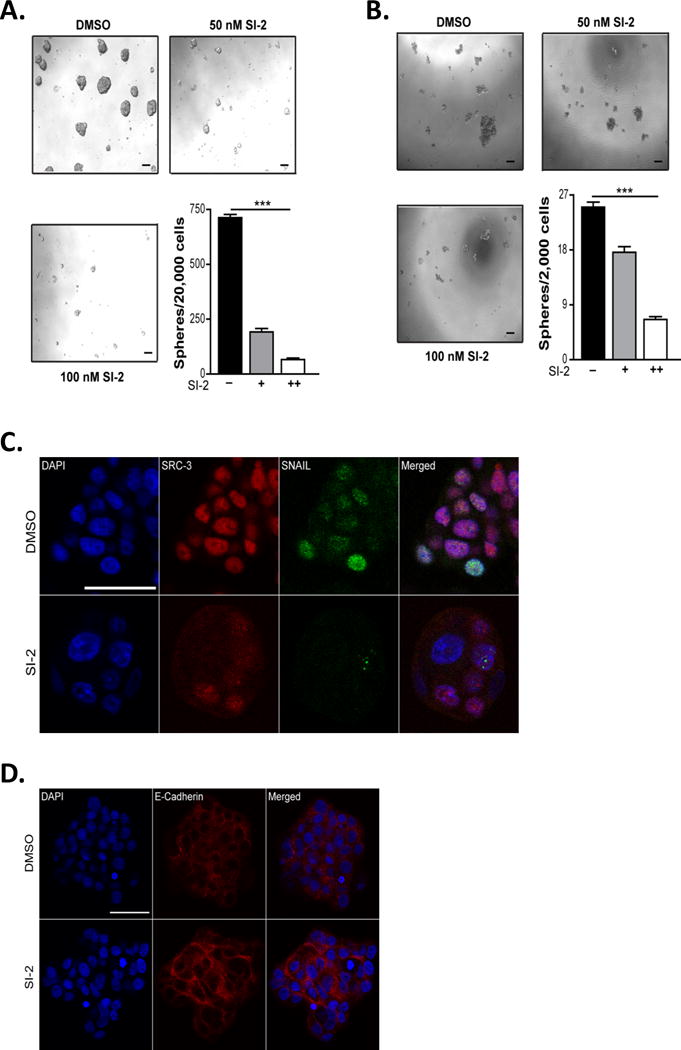
(A-B) Inhibition of SRC-3 with SI-2 decreases tumorsphere formation. MCF-7 or MDA-MB-231 cells were treated with SI-2 or DMSO and 24 hours later cells were trypsinized and counted. Viable cells were plated in serum free media on low attachment plates for 8-10 days. Scale bar = 50 μm. The number of tumorspheres that were greater than 50 μm were counted and graphed. Values represent means ± S.E.M. (n=3). One-way ANOVA was used for significance testing. *** = p < 0.005. (C-D) Treatment with SI-2 regulates expression of EMT associated proteins. DMSO or SI-2 treated tumorspheres from MCF7 cells were isolated, fixed and stained with antibodies to SRC-3, Snail or e-cadherin. DAPI was used to stain the DNA. A Nikon A1-R confocal microscope was used to image the tumorspheres. Representative images are shown (n=5). Scale bar = 100 μm.
In addition to their tumorsphere forming capacity, breast CSCs are characterized by their expression of ALDH, the presence of the cell surface marker CD44, and absence or low expression of the cell surface marker CD24 (3). Previous studies characterized the expression of stem cell markers in a panel of breast cancer cell lines and found high ALDH expression in Her-2 positive SKBR3 and triple negative MDA-MB-468 breast cancer cells and high expression of CD44 in triple negative MDA-MB-231 breast cancer cells (36). To determine whether inhibition of SRC-3 with SI-2 reduces the expression of these markers within the CSC population, triple negative MDA-MB-231 and MDA-MB-468, and Her-2 positive SKBR3 cells were treated with SI-2 or DMSO and examined for CSC marker expression. Indeed, we found that treatment of breast cancer cells with SI-2 reduced the CD44+/CD24-/lo (Fig. 5A) MDA-MB-231 cell populations and the ALDH+ cell populations in SKBR3 (Fig. 5B & C) and MDA-MB-468 (Fig. 5B & C) cell lines respectively. These data confirm that inhibiting SRC-3 activity with SI-2 can selectively interfere with the TIC/CSC state in the three main sub-types of breast cancer cells in addition to its already known ability to block breast cancer cell proliferation (21).
Figure 5. SI-2 reduces the proportion of breast CSCs.

(A) Treatment with SI-2 reduces the percentage of CD44+/CD24− population. MDA-MB-231 cells were treated with DMSO or SI-2 for 48 hours following which the cells were allowed to recover without drug for an additional 48 hours. Cells were then stained with APC-CD44 or PE-CD24 antibodies and Sytox blue and analyzed by flow cytometry. Representative flow cytometry dot plots are shown. The graphed data represents the mean ± S.E.M. (n=3). Student’s t-test was used for significance testing. ** = p < 0.01. (B-C) Treatment with SI-2 reduces the ALDH+ cell population. SKBR3 cells were treated with DMSO or SI-2 for 48 hours and allowed to recover without drug for an additional 48 hours. MDA-MB-468 cells were treated with DMSO or SI-2 for 48 hours. Both cell lines were then assayed for ALDH activity using flow cytometry. Representative flow cytometry dot plots are shown. The graphed data represents the mean ± S.E.M. (n=3). Student’s t-test was used for significance testing. *** = p < 0.005. (D) Treatment with SI-2 in vitro the tumor initiating frequency in vivo. MDA-MB-231 cells were treated with DMSO or SI-2 for 3 days and allowed to recover in drug free media for an additional 3 days. Total protein was isolated from the cells and immunoblotted for SRC-3 and β-actin. The cells were then injected into the second pair of mammary fat pads of SCID mice in limiting dilutions of 250,000, 25,000 or 2,500 cells (n=10/group). Tumor initiation and growth were monitored for 8 weeks. The tumor initiation frequency was calculated using the ELDA software.
Treatment with SI-2 reduces the tumor initiating capacity of breast CSCs
The limiting dilution assay is used to determine tumor initiating capacity of cancer cells in an in vivo setting (37). We previously reported that SI-2 can reduce breast tumor growth in a xenograft model system (21). To determine whether SI-2 treatment can specifically target the TIC/CSC cell population, MDA-MB-231 cells were treated with SI-2 or DMSO in vitro for three days and after a three day recovery, equal numbers of viable cells were injected in limiting dilutions into the mammary fat pads of mice. Decreased expression of SRC-3 in SI-2 treated cells was confirmed by immunoblotting (Fig. 5D). Importantly, SI-2 treated cells had a 15-fold reduction in the tumor initiation frequency compared to control treated cells (Fig. 5D), thus confirming that inhibiting SRC-3 using SI-2 can reduce the tumor initiating capacity of CSCs. In summary, inhibiting SRC-3 activity with SI-2 demonstrates a proof-of-concept that a SRC SMI could be useful to combat cancer recurrence arising from therapy-resistant TIC/CSC cells that survive first-round therapeutic intervention.
Discussion
Metastatic breast cancers arising due to resistance to chemotherapy represent the vast majority of breast cancer mortality cases. In response to many conventional chemotherapies, tumor cells frequently activate stress-related pathways to escape treatment such as EMT, thereby inducing genes that double as drivers of self-renewal leading to the acquisition of stem-like characteristics. This adoption of a stem-like state is linked to a temporary reduced rate of proliferation, morphological changes associated with increased motility and invasion that facilitate cancer cell survival during the brief, but stressful period of cell extravasation and seeding in distant metastatic sites (38). Since SRC-3 has been shown to maintain self-renewal in embryonic stem cells (25), we hypothesized that it may play a role in the induction and maintenance of stem-like characteristics in cancer cells as well. We found that expression of all three SRCs are elevated in tumorspheres when cells are grown on an ultra-low attachment substrate (Fig. 1). Si-RNA mediated knockdown of SRC-1 and -3 but not SRC-2 reduced tumorsphere number, pointing to a more dominant role for SRC-1 and -3 in promoting the TIC/CSC state (Fig. 1). However, in further analysis of the role of the SRCs in CSCs, we found that only SRC-3 is required to maintain an ALDH+ breast CSC population (Fig. 2), demonstrating that similar to its role in embryonic stem cell maintenance, SRC-3 plays a dominant role in maintaining CSCs. While not much is known about the expression and activity of SRC-3 in CSCs from breast and lung cancer patient samples, high expression of SRC-3 is inversely correlated with overall and post-progression survival in patients with triple negative breast cancer (21). Additionally, in ER+ breast cancer patients that receive tamoxifen therapy, high SRC-3 expression leads to worse disease free survival indicative of tamoxifen resistance (39). Furthermore, patients with high SRC-3 expression in non-small cell lung cancer have poor overall and progression free survival (40).
Previous studies have demonstrated that EMT in neoplastic cells can enrich for CSCs (7,41). Consistent with this, we found that in conjunction with its role in CSC maintenance, SRC-3 also induces EMT; evidence exists to suggest that it supports additional EMT-related pathways as well (27). Here, we identify the TGFβ-Smad3 signaling axis as a key signaling network that SRC-3 drives to induce EMT (Fig. 3). Downstream of TGFβ, SRC-3 interacts with Smad3 and regulates expression of the EMT transcription factors, snail and slug, that are required to induce the mesenchymal state. While SRC-1 has been shown to promote EMT through the upregulation of Twist expression (42), we found SRC-3 to be a stronger driver of CSC potential in the breast cancer cell lines we tested.
CSCs are frequently resistant to conventional chemotherapy and radiation treatment (2,43). For instance, it has been shown that treatment with the chemotherapeutic paclitaxel can lead to an undesirable induction and expansion of breast CSCs (44). Consistent with this, recent clinical studies have found that breast tumors show enrichment for CD44+/CD24− cells in response to conventional chemotherapy (1). In an effort to identify drugs that target the TIC tumor cell population, salinomycin was successful in targeting breast CSCs (45). However, the mechanism of action for salinomycin in killing breast CSCs is not well understood; additionally, salinomycin interferes with potassium channels and can be toxic to normal neural and hematopoietic cells (46). Thus, there is a need to identify drugs that can target the abolition of resistant TIC populations. With SRC-3’s ability to regulate both EMT and the CSC state we posited that by targeting SRC-3 we can specifically target this resistant tumor cell population. Indeed, we found that a second generation SMI, SI-2, which inhibits cellular SRC activity with low nanomolar potency (21), is effective in reducing the breast CSC population in a panel of cell lines representing different types of breast cancers. Treatment with SI-2 targeted CSCs as demonstrated by a reduction in tumorsphere formation and the proportion of ALDH+ and CD44+/CD24− breast CSCs (Fig. 4–5); inhibiting SRC-3 with SI-2 pushed the CD44+/CD24− cells from an undifferentiated state into the more differentiated CD44+/CD24+ state (47). Importantly, treatment of breast cancer cells with SI-2 in vitro was able to reduce the tumor initiating capacity of breast CSCs in vivo (Fig. 5). This was a sustained, durable effect since a single treatment with SI-2 in vitro could prevent tumor initiation in vivo for up to 8 weeks.
The fact that CSCs are frequently resistant and can indeed even be enriched for by conventional chemotherapeutic interventions provides a strong impetus to incorporate TIC/CSC targeted therapeutics into existing first-line clinical treatment to pro-actively eliminate this population of tumor cells. An effective therapeutic strategy could be envisioned by using a drug combination that targets both the CSC and non-CSC tumor cells. Because SRC-3 drives breast cancer in numerous ways such as by promoting tumor initiation, cancer cell proliferation, metastasis, drug resistance (48,49) and CSC maintenance, a SRC-3 SMI such as SI-2 might be able to both inhibit bulk tumor cell growth and also the formation of tumor TICs/CSCs. CSCs can be induced and maintained by other oncogenes such as RhoC (50) and it is possible that treatment with SI-2 may give rise to resistant CSCs that are driven by other factors. Hence, it will be better to explore the integration of SRC-3 SMIs as drug combination paradigms into existing standards of care for breast cancer that utilize conventional chemotherapeutic agents or in combination with other potential CSCs targeting agents. In this way, the pro-active targeting of TICs/CSCs that would otherwise be induced by drugs such as paclitaxel can be used to inhibit the eventual development of recurrent, metastatic disease responsible for the vast majority of breast cancer deaths.
Supplementary Material
Acknowledgments
Grant Support
This work was supported by funding from the Susan G. Komen Foundation (PG12221410), the Prostate Cancer Foundation, the Department of Defense Breast Cancer Research Program (BC120894), the Cancer Prevention and Research Institute of Texas (RP100348 and RP101251), and from the National Institutes of Health (DK059820) to B.W. O’Malley; and from the National Institutes of Health (HD076596) to D.M. Lonard; and from the Cancer Prevention and Research Institute of Texas (RP120732), the National Institutes of Health (CA112403 and CA193455) to J. Xu and (R01-GM115622) and (R01-CA207701) to J. Wang. This project was supported by the Integrated Microscopy Core at Baylor College of Medicine with funding from the NIH (DK56338, and CA125123), CPRIT (RP150578) the Dan L. Duncan Comprehensive Cancer Center, and the John S. Dunn Gulf Coast Consortium for Chemical Genomics. This project was also supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574) and the expert assistance of Joel M. Sederstrom.
Footnotes
Conflict of Interest
L.W., J.W., D.M.L., and B.W.O. are co-founders and hold stock in Coregon, Inc., which is developing steroid receptor coactivator regulators for clinical use.
References
- 1.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu M-F, et al. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. JNCI J Natl Cancer Inst. 2008;100:672–9. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 2.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26:2839–45. doi: 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–68. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 5.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–70. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 7.Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalluri R, Weinberg RA, Kalluri R, Neilson EG, Hay ED, Lipschutz JH, et al. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–6. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–30. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oñate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354–7. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 12.Voegel JJ, Heine MJ, Zechel C, Chambon P, Gronemeyer H. TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J. 1996;15:3667–75. [PMC free article] [PubMed] [Google Scholar]
- 13.Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–8. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 14.Xu J, Wu R-C, O’Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer. 2009;9:615–30. doi: 10.1038/nrc2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bautista S, Vallès H, Walker RL, Anzick S, Zeillinger R, Meltzer P, et al. In breast cancer, amplification of the steroid receptor coactivator gene AIB1 is correlated with estrogen and progesterone receptor positivity. Clin Cancer Res. 1998;4:2925–9. [PubMed] [Google Scholar]
- 17.Zhou H-J, Yan J, Luo W, Ayala G, Lin S-H, Erdem H, et al. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res. 2005;65:7976–83. doi: 10.1158/0008-5472.CAN-04-4076. [DOI] [PubMed] [Google Scholar]
- 18.Ghadimi BM, Schröck E, Walker RL, Wangsa D, Jauho A, Meltzer PS, et al. Specific chromosomal aberrations and amplification of the AIB1 nuclear receptor coactivator gene in pancreatic carcinomas. Am J Pathol. 1999;154:525–36. doi: 10.1016/S0002-9440(10)65298-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lonard DM, O’Malley BW. Molecular Pathways: Targeting Steroid Receptor Coactivators in Cancer. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu R-C, Smith CL, O’Malley BW. Transcriptional Regulation by Steroid Receptor Coactivator Phosphorylation. Endocr Rev. 2005;26:393–9. doi: 10.1210/er.2004-0018. [DOI] [PubMed] [Google Scholar]
- 21.Song X, Chen J, Zhao M, Zhang C, Yu Y, Lonard DM, et al. Development of potent small-molecule inhibitors to drug the undruggable steroid receptor coactivator-3. Proc Natl Acad Sci U S A. 2016;113:4970–5. doi: 10.1073/pnas.1604274113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Lonard DM, Yu Y, Chow D-C, Palzkill TG, Wang J, et al. Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res. 2014;74:1506–17. doi: 10.1158/0008-5472.CAN-13-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Yu Y, Chow D-C, Yan F, Hsu C-C, Stossi F, et al. Characterization of a Steroid Receptor Coactivator Small Molecule Stimulator that Overstimulates Cancer Cells and Leads to Cell Stress and Death. Cancer Cell. 2015;28:240–52. doi: 10.1016/j.ccell.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shao D, Berrodin TJ, Manas E, Hauze D, Powers R, Bapat A, et al. Identification of novel estrogen receptor alpha antagonists. J Steroid Biochem Mol Biol. 2004;88:351–60. doi: 10.1016/j.jsbmb.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 25.Percharde M, Lavial F, Ng J-H, Kumar V, Tomaz RA, Martin N, et al. Ncoa3 functions as an essential Esrrb coactivator to sustain embryonic stem cell self-renewal and reprogramming. Genes Dev. 2012;26:2286–98. doi: 10.1101/gad.195545.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo S, Xu J, Xue R, Liu Y, Yu H. Overexpression of AIB1 correlates inversely with E-cadherin expression in pancreatic adenocarcinoma and may promote lymph node metastasis. Int J Clin Oncol. 2014;19:319–24. doi: 10.1007/s10147-013-0549-2. [DOI] [PubMed] [Google Scholar]
- 27.Qin L, Liao L, Redmond A, Young L, Yuan Y, Chen H, et al. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol Cell Biol. 2008;28:5937–50. doi: 10.1128/MCB.00579-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu R-C, Qin J, Yi P, Wong J, Tsai SY, Tsai M-J, et al. Selective Phosphorylations of the SRC-3/AIB1 Coactivator Integrate Genomic Reponses to Multiple Cellular Signaling Pathways. Mol Cell. 2004;15:937–49. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 29.Hu Y, Smyth GK. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–8. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 30.Yi P, Wang Z, Feng Q, Pintilie GD, Foulds CE, Lanz RB, et al. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol Cell. 2015;57:1047–58. doi: 10.1016/j.molcel.2015.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brandl M, Seidler B, Haller F, Adamski J, Schmid RM, Saur D, et al. IKK(α) controls canonical TGF(ß)-SMAD signaling to regulate genes expressing SNAIL and SLUG during EMT in panc1 cells. J Cell Sci. 2010;123:4231–9. doi: 10.1242/jcs.071100. [DOI] [PubMed] [Google Scholar]
- 32.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–67. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith B, Bhowmick N. Role of EMT in Metastasis and Therapy Resistance. J Clin Med. 2016;5:17. doi: 10.3390/jcm5020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nawshad A, Lagamba D, Polad A, Hay ED. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs. 2005;179:11–23. doi: 10.1159/000084505. [DOI] [PubMed] [Google Scholar]
- 35.Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, et al. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–7. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 36.Hwang-Verslues WW, Kuo W-H, Chang P-H, Pan C-C, Wang H-H, Tsai S-T, et al. Multiple Lineages of Human Breast Cancer Stem/Progenitor Cells Identified by Profiling with Stem Cell Markers. PLoS One. 2009;4:e8377. doi: 10.1371/journal.pone.0008377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolev VN, Wright QG, Vidal CM, Ring JE, Shapiro IM, Ricono J, et al. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer Res. 2015;75:446–55. doi: 10.1158/0008-5472.CAN-14-1223. [DOI] [PubMed] [Google Scholar]
- 38.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–64. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 39.Cai D, Shames DS, Raso MG, Xie Y, Kim YH, Pollack JR, et al. Steroid receptor coactivator-3 expression in lung cancer and its role in the regulation of cancer cell survival and proliferation. Cancer Res. 2010;70:6477–85. doi: 10.1158/0008-5472.CAN-10-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SAW, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95:353–61. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 41.Morel A-P, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS One. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin L, Liu Z, Chen H, Xu J. The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res. 2009;69:3819–27. doi: 10.1158/0008-5472.CAN-08-4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diehn M, Clarke MF. Cancer stem cells and radiotherapy: new insights into tumor radioresistance. J Natl Cancer Inst. 2006;98:1755–7. doi: 10.1093/jnci/djj505. [DOI] [PubMed] [Google Scholar]
- 44.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10:R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–59. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou S, Wang F, Wong ET, Fonkem E, Hsieh T-C, Wu JM, et al. Salinomycin: a novel anti-cancer agent with known anti-coccidial activities. Curr Med Chem. 2013;20:4095–101. doi: 10.2174/15672050113109990199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ricardo S, Vieira AF, Gerhard R, Leitão D, Pinto R, Cameselle-Teijeiro JF, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol. 2011;64:937–46. doi: 10.1136/jcp.2011.090456. [DOI] [PubMed] [Google Scholar]
- 48.Xu J, Wu R-C, O’Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer. 2009;9:615–30. doi: 10.1038/nrc2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.García-Becerra R, Santos N, Díaz L, Camacho J. Mechanisms of resistance to endocrine therapy in breast cancer: focus on signaling pathways, miRNAs and genetically based resistance. Int J Mol Sci. 2012;14:108–45. doi: 10.3390/ijms14010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosenthal DT, Zhang J, Bao L, Zhu L, Wu Z, Toy K, et al. RhoC Impacts the Metastatic Potential and Abundance of Breast Cancer Stem Cells. PLoS One. 2012;7:e40979. doi: 10.1371/journal.pone.0040979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.