

Abstract
Increased concentrations of kynurenic acid (KYNA) in the prefrontal cortex (PFC) are thought to contribute to the development of cognitive deficits observed in schizophrenia. Although this view is consistent with preclinical studies showing a negative impact of prefrontal KYNA elevation on executive function, the mechanism underlying such a disruption remains unclear. Here, we measured changes in local field potential (LFP) responses to ventral hippocampal stimulation in vivo and conducted whole-cell patch-clamp recordings in brain slices to reveal how nanomolar concentrations of KYNA alter synaptic transmission in the PFC of male adult rats. Our data show that prefrontal infusions of KYNA attenuated the inhibitory component of PFC LFP responses, a disruption that resulted from local blockade of α7-nicotinic acetylcholine receptors (α7nAChR). At the cellular level, we found that the inhibitory action exerted by KYNA in the PFC occurred primarily at local GABAergic synapses through an α7nAChR-dependent presynaptic mechanism. As a result, the excitatory–inhibitory ratio of synaptic transmission becomes imbalanced in a manner that correlates highly with the level of GABAergic suppression by KYNA. Finally, prefrontal infusion of a GABAAR positive allosteric modulator was sufficient to overcome the disrupting effect of KYNA and normalized the pattern of LFP inhibition in the PFC. Thus, the preferential inhibitory effect of KYNA on prefrontal GABAergic transmission could contribute to the onset of cognitive deficits observed in schizophrenia because proper GABAergic control of PFC output is one key mechanism for supporting such cortical functions.
SIGNIFICANCE STATEMENT Brain kynurenic acid (KYNA) is an astrocyte-derived metabolite and its abnormal elevation in the prefrontal cortex (PFC) is thought to impair cognitive functions in individuals with schizophrenia. However, the mechanism underlying the disrupting effect of KYNA remains unclear. Here we found that KYNA biases the excitatory–inhibitory balance of prefrontal synaptic activity toward a state of disinhibition. Such disruption emerges as a result of a preferential suppression of local GABAergic transmission by KYNA via presynaptic inhibition of α7-nicotinic acetylcholine receptor signaling. Therefore, the degree of GABAergic dysregulation in the PFC could be a clinically relevant contributing factor for the onset of cognitive deficits resulting from abnormal increases of cortical KYNA.
Keywords: GABA, kynurenic acid, local field potentials, nicotinic receptor, prefrontal cortex, ventral hippocampus
Introduction
The functional maturation of GABAergic circuits in the prefrontal cortex (PFC) is one key mechanism for enabling proper processing of ventral hippocampal afferent information (Thomases et al., 2013; Caballero et al., 2014) and for supporting PFC-dependent cognitive functions (Floresco, 2013; Tse et al., 2015). It is thought that a disruption of such GABAergic control in the PFC underlies the onset of cognitive deficits seen in a variety of psychiatric disorders including schizophrenia (Tseng et al., 2009; Tse et al., 2015; Caballero et al., 2016). Further understanding of the different synaptic mechanisms that enable proper regulation of GABAergic function in the PFC is therefore a critical step toward gaining insights on how to restore normal cognition in schizophrenia and related psychiatric syndromes (Caballero and Tseng, 2016).
Of special interest in this context is the abnormal elevation of the tryptophan metabolite kynurenic acid (KYNA) in the PFC of individuals with schizophrenia, a biochemical dysregulation that is not secondary to antipsychotic medication (Schwarcz et al., 2001; Ceresoli-Borroni et al., 2006; Sathyasaikumar et al., 2011; Larsson et al., 2015). Brain KYNA is an astrocyte-derived metabolite that is present in the mammalian CNS at nanomolar concentrations (Moroni et al., 1988; Turski et al., 1988) and negatively modulates α7-nicotinic acetylcholine receptor (α7nAChR) function under physiological conditions (Albuquerque and Schwarcz, 2013). At higher concentrations, KYNA can also function as a competitive inhibitor of the glycine coagonist (glycine-B) site of the NMDA receptor (NMDAR) (Kessler et al., 1989; Parsons et al., 1997). Therefore, the increased PFC levels of KYNA observed in schizophrenia may be clinically relevant and mechanistically linked to the onset of cognitive impairments as a result of deficits in α7nAChR and/or NMDAR function in cortical circuits (Robbins and Murphy, 2006; Timofeeva and Levin, 2011). Accordingly, studies from animal models converge to indicate that increased cortical levels of KYNA can lead to deficits in executive function (Chess et al., 2007; Akagbosu et al., 2012; Alexander et al., 2012, 2013; Pocivavsek et al., 2012; Pershing et al., 2015; Pershing et al., 2016). However, the precise mechanism underlying the disrupting action of KYNA in cortical circuits remains elusive despite the fact that fluctuations in endogenous KYNA levels in the PFC are known to modulate bidirectionally the extracellular concentrations of glutamate (Konradsson-Geuken et al., 2010; Wu et al., 2010), GABA (Beggiato et al., 2014), dopamine (Pocivavsek et al., 2016; Valentini et al., 2016), and acetylcholine (Zmarowski et al., 2009).
The goal of the present study is to gain insights into how KYNA disrupts cortical circuits by determining the functional impact of elevations of nanomolar concentrations of KYNA in the PFC. We used local field potential (LFP) recordings to assess disruptions in the pattern of PFC response to ventral hippocampal train stimulation (10, 20, and 40 Hz) following PFC infusions of KYNA in vivo. This protocol of train stimulation was chosen because of its sensitivity in revealing changes in the relative contribution of excitatory and inhibitory transmission in the PFC (Cass et al., 2013; Thomases et al., 2013). At the cellular level, whole-cell patch-clamp recordings in brain slices were then used to identify the mechanisms by which nanomolar concentrations of KYNA regulate synaptic transmission onto pyramidal output neurons in the PFC.
Materials and Methods
Procedures.
All experimental procedures were approved by the Rosalind Franklin University Institutional Animal Care and Use Committee in accordance with National Institutes of Health guidelines. All recordings were conducted from adult (>80-d-old) male Sprague Dawley rats (Harlan Laboratories). Rats were allowed to habituate for at least 7 d upon arrival, group housed (3/cage), and maintained under constant temperature (21–23°C) in a 12/12 h light/dark cycle with food and water available ad libitum. All chemicals were purchased from Sigma-Aldrich except for Indiplon, which was obtained from Tocris Bioscience.
In vivo recordings of LFPs in the medial PFC.
All LFP recordings were conducted in the medial PFC (prelimbic and infralimbic regions) using a concentric bipolar electrode (SNE-100x 50 mm; Rhodes Medical Instruments) attached to a 28-gauge cannula to enable local administration of artificial CSF (aCSF)-containing agonists and antagonists as described previously (Thomases et al., 2013). Here, single infusions of 0.8 μl of aCSF alone (control) or in combination with one of the following neuromodulators were delivered into the PFC at 0.1 μl/min: (1) KYNA (100–300 nm), (2) methyllycaconitine (MLA; 300 nm), (3) 7-chlorokynurenic acid (7Cl-KYNA; 300 nm), (4) Indiplon (10 μm), (5) KYNA + Indiplon, or (6) MLA + Indiplon. The chemical composition of the aCSF solution was as follows (in mm): 122.5 NaCl, 3.5 KCl, 25 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, 1 MgCl2, 20 glucose, and 1 ascorbic acid, pH 7.40, 295–305 mOsm. For stimulation, another concentric bipolar electrode (NE-100x 50 mm; Rhodes Medical Instruments) was lowered into the ventral hippocampus (Fig. 1) and trains of electrical pulses (300 μs square pulses at 0.75 mA) were delivered every 15 s through a computer-controlled pulse generator (Master-8; A.M.P.I.). Changes in the pattern of LFP at 10, 20, and 40 Hz were assessed by the amplitude of the evoked LFP as described previously (Thomases et al., 2013). All example traces of LFP shown are averages of six evoked responses.
Figure 1.

Ventral hippocampal-evoked LFP responses in the adult PFC. a, coronal sections of the ventral hippocampus and PFC showing the anatomical location (asterisks) of the stimulating and recording electrodes, respectively. b, Characteristic response pattern of LFP in the adult PFC (n = 6) elicited by ventral hippocampal train stimulation at 10, 20, and 40 Hz (calibration: 10 mV/100 ms at 10 Hz; 10 mV/50 ms at 20 Hz; 10 mV/25 ms at 40 Hz).
Whole-cell patch-clamp recordings of IPSCs in the medial PFC.
All experimental procedures (brain slicing and patch-clamp recordings) were conducted as described previously (Cass et al., 2014; Flores-Barrera et al., 2014). Briefly, all recordings were conducted from layer V pyramidal neurons of the medial PFC (infralimbic and prelimbic regions; 350-μm-thick coronal slices) at 33–35°C using a cesium-based internal solution containing 0.1% Neurobiotin (Vector Laboratories) and the following (in mm): 140 CsCl, 10 HEPES, 2 MgCl2, 5 NaATP, 0.6 NaGTP, and 3 QX-314, pH 7.23–7.28, 280–282 mOsm. The recording aCSF contained 10 μm CNQX and 50 μm APV and the following (in mm): 122.5 NaCl, 3.5 KCl, 25 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, 1 MgCl2, 20 glucose, and 1 ascorbic acid, pH 7.40–7.43, 295–305 mOsm. Both GABA-A receptor-mediated spontaneous and evoked IPSCs were obtained in voltage-clamp mode (holding potential: −70 mV). Only neurons exhibiting stable baseline activity (i.e., 10 min) were included for analyses. Typically, the mean baseline IPSC frequency obtained from at least two noncontiguous epochs of 60 s each was compared with equivalent measures taken 10 min after bath application of 300 nm KYNA, MLA, or 7Cl-KYNA. The effects of KYNA, MLA, and 7Cl-KYNA were also assessed by measuring changes in the amplitude of electrically evoked IPSCs. Data from the evoked response were collected from another cohort of neurons by means of local stimulation elicited every 10 s using a Teflon-coated bipolar electrode placed ∼200 μm from the cell body along the apical dendritic axis. The stimulation intensity (300 μs square pulses at 50–100 μA range) was chosen from the minimum current required to elicit an IPSC response with <20% variability in amplitude. For the frequency analysis, relative changes from baseline IPSC amplitude were determined after 10 min bath application of KYNA or MLA.
Whole-cell patch-clamp recordings of excitatory and inhibitory synaptic currents within a single pyramidal neuron in the medial PFC.
All recordings were conducted from layer V pyramidal neurons using a low-chloride-based internal solution and an external solution free of glutamate and GABA blockers to enable concurrent acquisition of excitatory and inhibitory synaptic currents at the single-cell level. The low-chloride-based internal solution contained 0.1% Neurobiotin (Vector Laboratories) and the following (in mm): 10 CsCl, 130 Gluconic acid, 10 HEPES, 2 MgCl2, 5 NaATP, 0.6 NaGTP, and 3 QX-314, pH 7.23–7.28, 280–282 mOsm. As a result, both spontaneous glutamatergic and GABAergic events could be assessed readily by recording the frequency of postsynaptic currents (PSCs) at the −60 mV (PSC−60 mV) and +15 mV (PSC+15 mV) holding potentials, respectively. As described above, only neurons with at least 10 min of stable baseline activity were included for analyses. The frequency of PSC−60 mV and PSC+15 mV events from at least two noncontiguous baseline epochs of 60 s each was compared with equivalent measures taken 10 min after bath application of KYNA, KYNA + MLA, or KYNA + 7Cl-KYNA.
The impact of KYNA was also assessed on locally evoked PSC−60 mV and PSC+15 mV at the single-cell level following the same experimental design used to record glutamatergic and GABAergic events. This dataset was obtained from another cohort of layer V pyramidal neurons by means of local stimulation elicited every 10 s (as described above) and only neurons with at least 15 min of stable baseline recordings were included for analyses. Relative changes of evoked PSC−60 mV and PSC+15 mV amplitude were determined after 10 min bath application of KYNA or KYNA + Indiplon.
Experimental design and statistical analysis.
Data were summarized as mean ± SEM and differences among experimental conditions (within- and between-subjects design) were considered statistically significant at p < 0.05. Paired t test was used for two-group within-subject comparison involving a single continuous variable (e.g., predrug vs postdrug application), whereas one- and two-way ANOVAs were applied to assess between-subject comparisons along three or more dependent variables (e.g., vehicle vs drug A vs drug B), followed by the appropriate post hoc tests (StatSoft).
Results
We first determined how single PFC infusion of KYNA (50–300 nm) affects LFP responses to ventral hippocampal train stimulation at 10, 20, and 40 Hz (Fig. 1). These frequencies of stimulation were chosen to reveal changes in the balance of excitation and inhibition in the PFC in vivo (Thomases et al., 2013). Relative to aCSF, PFC infusions with 50, 100, and 300 nm KYNA failed to alter the pattern of LFP facilitation observed at 10 Hz (Fig. 2a). This was not the case at 20 Hz. Although the normal pattern of transient inhibition (i.e., second pulse) remained unchanged after PFC infusion of 50 nm KYNA, a marked LFP facilitation emerged when 300 nm KYNA was administered (Fig. 2b). Notably, the 100 nm KYNA concentration was sufficient to block the transient inhibition without causing any LFP facilitation. A similar dose-dependent effect was observed at 40 Hz after PFC infusion of KYNA (Fig. 2c). Typically, ventral hippocampal stimulation at 40 Hz suppresses the amplitude of LFP in the PFC (Thomases et al., 2013). Although the overall pattern of PFC response at 40 Hz remained largely unaffected by KYNA, the magnitude of LFP suppression began to appear slightly diminished at 100 nm. Such attenuation of LFP inhibition became markedly significant after PFC infusion of 300 nm KYNA (Fig. 2c). Together, these results indicate that increasing prefrontal KYNA levels within nanomolar concentrations is sufficient to disrupt PFC processing of afferent drive in a frequency-dependent manner, as revealed by the reduced inhibitory control of ventral hippocampal-evoked LFP responses.
Figure 2.

Disruption of prefrontal LFP response by local infusion of KYNA. a, PFC infusions of aCSF (n = 6) or KYNA (50, 100, and 300 nm; n = 5–8/group) failed to alter the pattern of LFP facilitation resulting from ventral hippocampal train stimulation at 10 Hz. Note the similar magnitude of LFP facilitation across the different treatment groups (*p < 0.05 vs first pulse, LSD post hoc test after significant ANOVA). Inset traces of LFP show the response pattern elicited by hippocampal stimulation at 10 Hz (calibration: 5 mV/100 ms). b, At 20 Hz, PFC infusions of aCSF or 50 nm KYNA resulted in similar patterns of transient LFP inhibition (*p < 0.05 vs first pulse, LSD post hoc test after significant ANOVA). Such a pattern of LFP inhibition was absent after the infusion of 100 nm KYNA, whereas a facilitation emerged with 300 nm KYNA (*p < 0.05 vs first pulse, +p < 0.05/++p < 0.005 vs aCSF, LSD post hoc test; main effect of treatment F(1,120) = 57.2, p < 0.0001, two-way ANOVA). Bar graph summarizes the mean LFP response calculated from pulses 2–10 (**p < 0.005 vs any other group, Tukey's post hoc test; F(3,20) = 9.6, p < 0.0005, one-way ANOVA). Inset traces illustrate the abnormal LFP facilitation at 20 Hz resulting from PFC infusion of 300 nm KYNA (calibration: 5 mV/50 ms). c, Stimulation at 40 Hz typically suppresses LFP in the PFC. Both the aCSF and 50 nm KYNA groups showed similar patterns of LFP inhibition (++p < 0.005/+p < 0.05 vs aCSF, ***p < 0.0005 vs first pulse, LSD post hoc test after significant ANOVA). However, the magnitude of LFP suppression was markedly attenuated following PFC infusion of 300 nm KYNA (+++p < 0.0005/++p < 0.005/+p < 0.05 vs aCSF, ***p < 0.0005 vs first pulse, LSD post hoc test; main effect of treatment, F(1,120) = 60.7, p < 0.0001, two-way ANOVA). Bar graph summarizes the mean LFP response calculated from pulses 2–10 (**p < 0.005 vs any other group, Tukey's post hoc test; F(3,20) = 7.4, p < 0.002, one-way ANOVA). Inset traces of 40 Hz-induced LFP illustrate the disruption elicited by 300 nm KYNA (calibration: 5 mV/30 ms).
Nanomolar concentrations of KYNA are known to negatively modulate α7nAChRs (Hilmas et al., 2001) and to block the glycine-B site of the NMDAR (Kessler et al., 1989; Hilmas et al., 2001). Therefore, inhibition of these receptors could contribute to the frequency-dependent disruption of LFP inhibition observed following PFC infusions of KYNA. To begin testing these mechanisms, we first assessed the impact of 7Cl-KYNA, which blocks the glycine-B site of the NMDAR without affecting the α7nAChR (Kemp et al., 1988; Hilmas et al., 2001). Results revealed no apparent changes in the pattern of LFP response after PFC infusions of 300 nm 7Cl-KYNA (Fig. 3a–c). The magnitude of LFP facilitation (10 Hz) and suppression (20 and 40 Hz) recorded in the presence of 7Cl-KYNA were indistinguishable from aCSF controls. In contrast, PFC infusion of the α7nAChR antagonist MLA (300 nm) effectively disrupted the pattern of LFP response in a frequency-dependent manner resembling that induced by KYNA (Fig. 3d–f). Similar to the latter, PFC infusion of MLA revealed a pattern of sustained LFP facilitation at 20 Hz (Fig. 3e) and a marked attenuation of LFP suppression at 40 Hz (Fig. 3f). Collectively, these results indicate that the disrupting effects of KYNA in the PFC could result from local blockade of α7nAChR function (Fig. 3g–i).
Figure 3.

PFC infusion of the α7nAChR antagonist MLA mimics the frequency-dependent disruption induced by KYNA. a–c, Summary of the results obtained after PFC infusion of 300 nm 7Cl-KYNA (n = 5). Note that the patterns of LFP response to ventral hippocampal stimulation after 7Cl-KYNA infusions are indistinguishable from the aCSF controls (*p < 0.05/***p < 0.0005 vs first pulse, LSD post hoc test after significant ANOVA). d–f, In contrast, PFC infusion of 300 nm MLA (n = 5) disrupted the LFP response at 20 and 40 Hz without altering the pattern of facilitation at 10 Hz. Two-way ANOVA revealed a main effect of pulse at 10 Hz (F(1,90) = 10.6, p < 0.0001) and main effects of treatment at both 20 Hz (F(1,90) = 54.8, p < 0.0001) and 40 Hz (F(1,90) = 95.6, p < 0.0001). A main effect of pulse (F(9,90) = 106.9, p < 0.0001) and treatment × pulse were also observed at 40 Hz (F(9,90) = 3.3, p < 0.002). LSD post hoc tests: ***p < 0.0005/*p < 0.05 versus first pulse, +++p < 0.0005/++p < 0.005/+p < 0.05 versus aCSF. g–i, Summary of the frequency-dependent LFP changes calculated from pulses 2–10 (**p < 0.005/*p < 0.05 vs aCSF or 7Cl-KYNA, Tukey's post hoc test after significant one-way ANOVA: F(3,20) = 12.4, p < 0.0001 at 20 Hz and F(3,20) = 9.5, p < 0.0005 at 40 Hz). Inset traces illustrate the pattern of LFP response following PFC infusions of 7Cl-KYNA or MLA (calibration: 5 mV/100 ms at 10 Hz; 5 mV/50 ms at 20 Hz; 5 mV/30 ms at 40 Hz).
GABAergic function in the PFC plays a critical role in mediating the distinctive frequency-dependent pattern of LFP inhibition resulting from ventral hippocampal train stimulation (Thomases et al., 2013). Therefore, it is conceivable that a GABAergic component contributes to the disinhibitory action of KYNA in the PFC. To test this, we conducted whole-cell patch-clamp recordings in PFC brain slices obtained from adult rats to assess the impact of KYNA on GABAAR-mediated IPSC onto layer V pyramidal neurons. Relative to baseline activity, bath application of 300 nm KYNA significantly reduced the number of spontaneous IPSC events (Fig. 4a,e). A comparable level of IPSC suppression was observed after bath application of MLA (Fig. 4b,e,f), but not of 7Cl-KYNA (Fig. 4c,e,f). However, the mean amplitude of spontaneous IPSC events remained unchanged after bath application of KYNA (from 24.9 ± 3.1 pA to 25.1 ± 3.4 pA), MLA (from 24.5 ± 3.2 pA to 24.8 ± 3.6 pA), or 7Cl-KYNA (from 24.6 ± 3.8 pA to 25.1 ± 3.5 pA). Notably, the inhibitory effect of KYNA on IPSC frequency was occluded by MLA (Fig. 4d). Similarly, bath application of 300 nm KYNA diminished the amplitude of locally evoked IPSC concurrently with a facilitation of the paired-pulse ratio (Fig. 4g,h). No further suppression of IPSC amplitude by KYNA was observed when recordings were conducted in the presence of MLA (Fig. 4i). Together, these results indicate that the inhibitory action of KYNA on PFC GABAergic transmission occurs via presynaptic α7nAChR antagonism, as revealed by the reduction in IPSC frequency and increased paired-pulse ratio accompanying the attenuation of the evoked IPSC amplitude.
Figure 4.

KYNA-induced attenuation of GABAergic transmission in the PFC is occluded by MLA. a, Bath application of 300 nm KYNA (10 min, n = 7) reduced the frequency of spontaneous IPSCs in layer V pyramidal neurons of the PFC (***p < 0.0005 vs baseline, paired t test). b, A similar degree of inhibition was observed after bath application of 300 nm MLA (10 min, n = 7; ***p < 0.0005 vs baseline, paired t test). c, Conversely, bath application of 300 nm 7Cl-KYNA (10 min, n = 5) failed to change the frequency of IPSCs. d, As expected from b, the addition of MLA into the baseline aCSF reduced the frequency of IPSCs. However, bath application of 300 nm KYNA failed to diminish the IPSC frequency further (n = 6). e, Traces of IPSC recordings illustrating the effects of bath application of KYNA, MLA, and 7Cl-KYNA (calibration: 10 pA/0.5 s). f, Bar graph summarizing the effects of KYNA, MLA, and 7Cl-KYNA on IPSC frequency as a percentage change from baseline (***p < 0.0005 vs 7Cl-KYNA, Tukey's post hoc test; F(2,16) = 23.6, p < 0.0001, one-way ANOVA). g, Recording design used to elicit IPSC in layer V pyramidal neurons by local electrical stimulation. h, Bath application of 300 nm KYNA (n = 6) elicited a significant attenuation of the evoked IPSC amplitude (***p < 0.0005 vs baseline, paired t test) concurrent with an elevation of the IPSC paired-pulse ratio (PPR: IPSC2/IPSC1 at 50 ms intervals; **p < 0.005 vs baseline, paired t test). Inset: Traces of evoked IPSC illustrating the inhibitory action of KYNA (calibration: 20 pA/25 ms). i, Note that the inhibitory effect of KYNA was not longer apparent when recordings were conducted in the presence of MLA (n = 7).
In addition to attenuating prefrontal GABAergic function, nanomolar concentrations of KYNA could also diminish the level of glutamatergic drive onto pyramidal output neurons. The balance of excitatory–inhibitory (E–I) synaptic activity within a single pyramidal neuron is expected to remain unaltered if KYNA's inhibition of GABAergic and glutamatergic transmission is comparable. To address this, recordings from layer V pyramidal neurons in the PFC were collected using a protocol that enables the acquisition of GABAergic and glutamatergic synaptic activity within a single cell (Fig. 5a,b; see Materials and Methods for details). Bath application of 300 nm KYNA also reduced the frequency of glutamatergic synaptic activity, as determined by the number of spontaneous postsynaptic current events recorded at the −60 mV (PSC−60 mV) holding potential (Fig. 5c). However, KYNA induced a much greater suppression of GABAergic synaptic activity (i.e., picrotoxin-sensitive PSC+15 mV) within the same cell (Fig. 5d) such that a major shift toward a higher E–I ratio emerged (Fig. 5e). Further analyses of the data revealed that the magnitude of E–I imbalance elicited by KYNA correlates highly with the level of IPSC (PSC+15 mV) frequency suppression (Fig. 5f). A similar inhibitory effect was found with MLA, but not with 7Cl-KYNA (Fig. 5g). Notably, bath application of KYNA failed to further reduce the diminished PSC+15 mV frequency elicited by MLA (Fig. 5g). Such an occlusion was not observed when recordings were conducted in the presence of 7Cl-KYNA (Fig. 5g). Together, these results indicate that the E–I imbalance induced by KYNA in the PFC results from a preferential suppression of local GABAergic transmission via presynaptic α7nAChR antagonism (Fig. 5h).
Figure 5.

Preferential inhibition of PFC GABAergic over glutamatergic transmission by KYNA. a, Concurrent acquisition of inhibitory (PSC+15 mV) and excitatory (PSC−60 mV) postsynaptic currents at layer V pyramidal neurons in the PFC using low-chloride-based internal solution. Note that bath application of the GABAAR antagonist picrotoxin (50 μm, 10 min) suppressed PSC+15 mV events (n = 6; p < 0.0001 vs baseline, paired t test) without altering the frequency of synaptic activity recorded at the −60 mV holding potential (PSC−60 mV). b, Example traces of PSC+15 mV and PSC−60 mV recorded from the same pyramidal neuron illustrating the effect of picrotoxin shown in a (calibration: 20 pA/1 s). c, KYNA (300 nm, 10 min) exerted a small but significant attenuation of PSC−60 mV frequency (n = 5; *p < 0.05 vs baseline, paired t test). d, However, the extent of KYNA-induced synaptic inhibition at PSC+15 mV was significantly larger (n = 5; **p < 0.005 vs baseline, paired t test). e, Accordingly, KYNA increases the E–I ratio of synaptic activity in layer V pyramidal neurons (n = 5; **p < 0.005 vs baseline, paired t test). f, Further analysis revealed a significant correlation between the E–I ratio and PSC+15 mV (i.e., IPSC) frequency obtained after bath application of KYNA. g, Consistent with data shown in Figure 4d, the inclusion of MLA (300 nm, n = 6) into the baseline aCSF reduced the frequency of PSC+15 mV and occluded the inhibitory effect of KYNA. Conversely, 7Cl-KYNA (300 nm, n = 6) failed to alter the frequency of both PSC−60 mV and PSC+15 mV events. Subsequent bath application of KYNA significantly diminished the number of PSC+15 mV (**p < 0.005 vs baseline, paired t test) without disrupting the PSC−60 mV frequency. h, Summary of the E–I ratio data obtained following bath application of KYNA, MLA, and 7Cl-KYNA.
If the frequency-dependent disruption elicited by KYNA is due to a presynaptic attenuation of GABAergic function, it is anticipated that a postsynaptic facilitation of GABAAR transmission would restore the normal pattern of LFP inhibition in the PFC. To test this hypothesis, we first investigated whether the inclusion of the GABAAα1-positive allosteric modulator Indiplon (10 μm) (Thomases et al., 2013) into the perfusion solution prevents the disrupting effect of KYNA on locally evoked E–I synaptic events using the same protocol as in Figure 5, which enables the acquisition of GABAergic and glutamatergic transmission (Fig. 6a). Data show that KYNA reduced the amplitude of locally evoked PSC+15 mV without altering the PSC−60 mV response (Fig. 6b). As a result, the balance of the evoked postsynaptic currents becomes disrupted and a shift toward a higher E–I ratio emerges (Fig. 6c). The inclusion of Indiplon effectively prevented the effects of KYNA at both PSC+15 mV amplitude (Fig. 6d) and E–I ratio of the evoked response (Fig. 6e). Next, Indiplon was coinfused with KYNA into the PFC and changes in hippocampal-evoked LFP were compared with those induced by KYNA alone (Fig. 7). Whereas the pattern of LFP response at 10 Hz remained unaffected (Fig. 7a), the abnormal facilitation of LFP at 20 Hz resulting from PFC infusions of KYNA was no longer apparent with the inclusion of Indiplon (Fig. 7b). Similarly, the addition of Indiplon was sufficient to prevent the attenuated LFP suppression induced by KYNA at 40 Hz (Fig. 7c). Indiplon alone does not alter the pattern of LFC response in the PFC. Together, these results provide a mechanistic link between the level of prefrontal GABAergic function and the frequency-dependent LFP disruption elicited by nanomolar concentrations of KYNA in the PFC.
Figure 6.

Reversal of KYNA-induced E–I imbalance by Indiplon. a, Recording of locally evoked inhibitory (PSC+15 mV) and excitatory (PSC−60 mV) postsynaptic currents at layer V pyramidal neurons using low-chloride-based internal solution. As in Figure 5, bath application of picrotoxin (50 μm, 10 min) suppressed the amplitude of locally evoked PSC+15 mV (n = 5; p < 0.001 vs baseline, paired t test) without altering the PSC−60 mV response (calibration: 100 pA/100 ms). b, KYNA (300 nm, 10 min) failed to reduce the amplitude of locally evoked PSC−60 mV, but diminished the PSC+15 mV amplitude by ∼30% (n = 7; **p < 0.001 vs baseline, paired t test). c, As a result, KYNA increases the E–I ratio of the evoked synaptic response (**p < 0.002 vs baseline, paired t test), as estimated from the normalized PSC amplitude to baseline. d, The addition of Indiplon into the perfusion solution effectively blocked the inhibitory effect of KYNA on locally evoked PSC+15 mV (n = 5). e, Accordingly, the E–I ratio of evoked postsynaptic currents remained unchanged.
Figure 7.
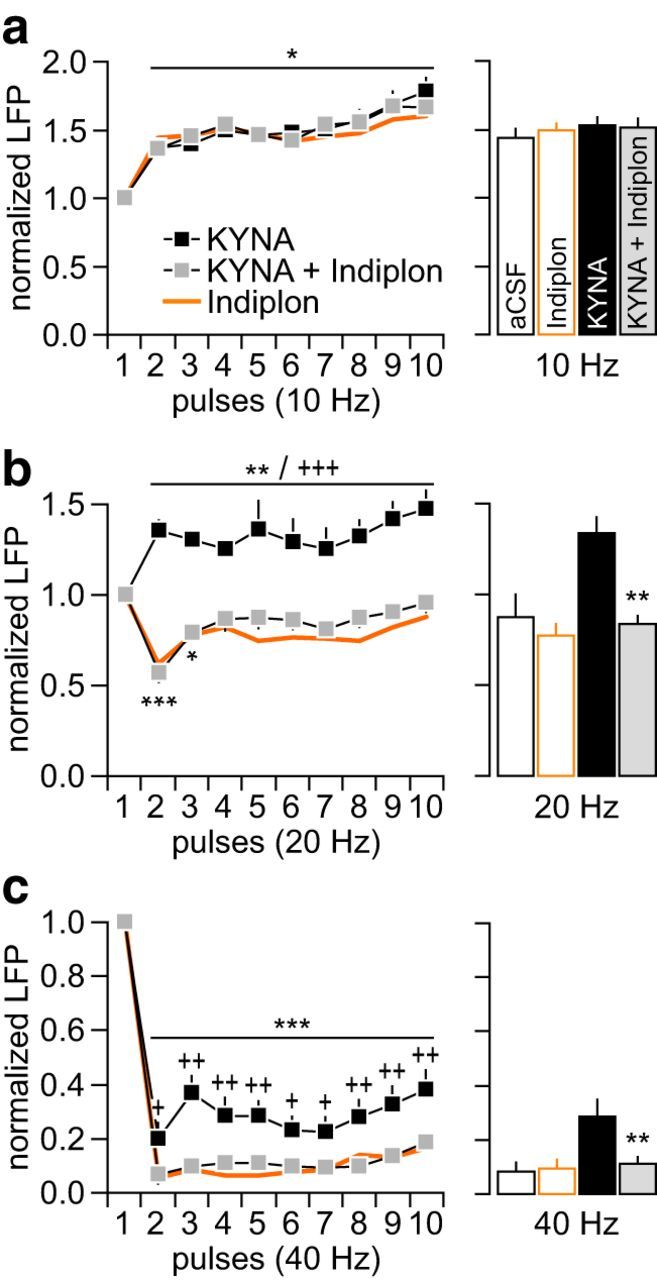
PFC infusion of Indiplon prevents the disrupting effects of KYNA. a, Normal pattern of LFP facilitation at 10 Hz remained unaltered by the addition of Indiplon (10 μm) to the KYNA infusion solution (KYNA + Indiplon: n = 8; *p < 0.05 vs first pulse, LSD post hoc test after significant main effect of pulse, two-way ANOVA). The pattern of LFP facilitation elicited by Indiplon alone (n = 5) is included for comparison. Bar graph summarizes the mean LFP response calculated from pulses 2–10. Data from the aCSF group are included for comparison. b, At 20 Hz, the abnormal LFP facilitation induced by KYNA is no longer apparent with the addition of Indiplon (*p < 0.05/**p < 0.005/***p < 0.0005 vs first pulse, +++p < 0.0005 vs KYNA + Indiplon, LSD post hoc test; main effect of treatment: F(1,140) = 230.3, p < 0.0001, two-way ANOVA). Indiplon alone does not affect the normal pattern of LFP suppression at 20 Hz. Bar graph of the mean LFP response (from pulses 2–10) summarizes the reversal effect of Indiplon (**p < 0.005 vs KYNA, Tukey's post hoc test; F(3,26) = 14.31, p < 0.0001, one-way ANOVA). c, Attenuated LFP suppression at 40 Hz was no longer observed when Indiplon was coadministered with KYNA into the PFC (***p < 0.0005 vs first pulse, +p < 0.05/++p < 0.005/+++p < 0.0005 vs KYNA + Indiplon, LSD post hoc test; main effect of treatment: F(1,140) = 63.1, p < 0.0001, two-way ANOVA). Indiplon alone does not alter the pattern of LFP suppression at 40 Hz. Bar graph of the mean LFP response (from pulses 2 to 10) summarizes the reversal effect of Indiplon (**p < 0.005 vs KYNA, Tukey's post hoc test; F(3,26) = 11.09, p < 0.0001, one-way ANOVA).
Discussion
The present study showed that local administration of nanomolar concentrations of KYNA into the PFC attenuated the inhibitory component of LFP responses (20–40 Hz) without affecting the pattern of LFP facilitation. This is consistent with data obtained from PFC brain slices revealing an inhibitory action of KYNA, which primarily reduces GABAergic transmission through a presynaptic α7nAChR-dependent mechanism. Accordingly, PFC infusion of the GABAAα1 positive allosteric modulator Indiplon effectively prevented the disrupting effect of KYNA and restored the normal pattern of LFP inhibition. Together, these results indicate that local prefrontal GABAergic function is the preferential target of KYNA's inhibitory effect in the PFC.
Brain KYNA is an astrocyte-derived metabolite of the kynurenine pathway of tryptophan degradation and its dysregulation is thought to be implicated in schizophrenia (Plitman et al., 2017). Evidence for such a pathophysiological link arises from studies showing increased levels of KYNA in the brain and CSF (Erhardt et al., 2001; Schwarcz et al., 2001; Linderholm et al., 2012), and abnormal expression and activity of key kynurenine pathway enzymes in the brain (Miller et al., 2004; Sathyasaikumar et al., 2011; Wonodi et al., 2011) of patients with schizophrenia. Certainly, a causal link between relatively modest elevations of cortical KYNA and deficits in executive function has been well documented in preclinical studies (Chess et al., 2007; Akagbosu et al., 2012; Alexander et al., 2012, 2013). At the mechanistic level, KYNA is known to modulate α7nAChR negatively (Hilmas et al., 2001) and to block the glycine coagonist site of the NMDAR (Parsons et al., 1997). Notably, however, the negative impact of KYNA on PFC-related cognitive functions has been suggested to occur mainly through inhibition of α7nAChR signaling (Alexander et al., 2012, 2013). This interpretation is consistent with the results of the present study showing that the disrupting effect of KYNA in the PFC is also α7nAChR dependent, primarily at local GABAergic synapses. Thus, the degree of GABAergic dysregulation in the PFC could be a clinically relevant contributing factor for the onset of cognitive deficits resulting from abnormal increases of cortical KYNA as seen in schizophrenia.
In addition to downregulating GABAergic function and decreasing extracellular GABA levels (Beggiato et al., 2014), nanomolar KYNA also reduces extracellular glutamate levels in the PFC (Konradsson-Geuken et al., 2010; Wu et al., 2010). However, the extent of GABAergic synaptic inhibition by KYNA was much greater than the effect on glutamatergic synapses. As a result, a disinhibitory imbalance of E–I synaptic activity emerges in the PFC. Accordingly, the frequency-dependent LFP disinhibition elicited by PFC infusion of KYNA is indistinguishable from that induced by the GABAAR antagonist picrotoxin (Cass et al., 2013; Thomases et al., 2013). The disruption of LFP by KYNA was no longer apparent after strengthening of prefrontal GABAAα1R function with Indiplon, indicating that local GABAergic synapses are more sensitive to the inhibitory effect of KYNA. Future studies will determine whether distinct functional expression of α7nAChR between excitatory and inhibitory synapses contributes to bias the inhibitory action of KYNA onto GABAergic transmission in the PFC. It is also possible that differences in the localization rather than expression per se of α7nAChR receptors are critical to the observed effects (Frazier et al., 1998).
Proper maturation of prefrontal GABAergic function during adolescence and its control of PFC output are critical for supporting adult cognitive functions such as working memory, decision making, and impulse control (Tse et al., 2015; Caballero et al., 2016). Although massive functional remodeling takes place in the PFC during adolescence (Caballero and Tseng, 2016), it is also the local GABAergic system that renders the PFC labile during this developmental period (Caballero et al., 2016). Therefore, it is possible that persistent elevations of KYNA in the PFC during adolescence may elicit enduring GABAergic dysfunction and disrupt the acquisition of inhibitory control that normally emerges in the adult PFC (Caballero et al., 2016). Elevations of KYNA levels during sensitive periods of perinatal development (Pocivavsek et al., 2012; Alexander et al., 2013; Forrest et al., 2013; Liu et al., 2014; Pisar et al., 2014; Pershing et al., 2015, 2016) or adolescence (Akagbosu et al., 2012; DeAngeli et al., 2014, Pershing et al., 2016) may alter the balance of E–I in the PFC and contribute to the onset of cognitive deficits later in life, as seen in schizophrenia and other major psychiatric disorders.
Collectively, the results of the present study suggest a mechanism by which endogenous KYNA could affect PFC-dependent cognitive functions adversely. We showed for the first time that the inhibitory effect of KYNA in the PFC occurs primarily at GABAergic synapses through a presynaptic blockade of α7nAChR signaling. Although KYNA can also function as a competitive inhibitor of NMDAR function via the glycine-B site (Kessler et al., 1989; Parsons et al., 1997), its contribution to disrupting PFC synaptic activity is not apparent because both the pattern of LFP responses and GABAergic transmission remained unaltered after application of the highly selective glycine-B site antagonist 7-Cl-KYNA (Kemp et al., 1988).
Footnotes
This work was supported by Rosalind Franklin University and the National Institutes of Health (Grant R01-MH086507 to K.Y.T.).
The authors declare no competing financial interests.
References
- Akagbosu CO, Evans GC, Gulick D, Suckow RF, Bucci DJ (2012) Exposure to kynurenic acid during adolescence produces memory deficits in adulthood. Schizophr Bull 38:769–778. 10.1093/schbul/sbq151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, Schwarcz R (2013) Kynurenic acid as an antagonist of alpha7 nicotinic acetylcholine receptors in the brain: facts and challenges. Biochem Pharmacol 85:1027–1032. 10.1016/j.bcp.2012.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander KS, Wu HQ, Schwarcz R, Bruno JP (2012) Acute elevations of brain kynurenic acid impair cognitive flexibility: normalization by the alpha7 positive modulator galantamine. Psychopharmacology (Berl) 220:627–637. 10.1007/s00213-011-2539-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander KS, Pocivavsek A, Wu HQ, Pershing ML, Schwarcz R, Bruno JP (2013) Early developmental elevations of brain kynurenic acid impair cognitive flexibility in adults: reversal with galantamine. Neuroscience 238:19–28. 10.1016/j.neuroscience.2013.01.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggiato S, Tanganelli S, Fuxe K, Antonelli T, Schwarcz R, Ferraro L (2014) Endogenous kynurenic acid regulates extracellular GABA levels in the rat prefrontal cortex. Neuropharmacology 82:11–18. 10.1016/j.neuropharm.2014.02.019 [DOI] [PubMed] [Google Scholar]
- Caballero A, Tseng KY (2016) GABAergic function as a limiting factor for prefrontal maturation during adolescence. Trends Neurosci 39:441–448. 10.1016/j.tins.2016.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero A, Thomases DR, Flores-Barrera E, Cass DK, Tseng KY (2014) Emergence of GABAergic-dependent regulation of input-specific plasticity in the adult rat prefrontal cortex during adolescence. Psychopharmacology (Berl) 231:1789–1796. 10.1007/s00213-013-3216-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero A, Granberg R, Tseng KY (2016) Mechanisms contributing to prefrontal cortex maturation during adolescence. Neurosci Biobehav Rev 70:4–12. 10.1016/j.neubiorev.2016.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass DK, Thomases DR, Caballero A, Tseng KY (2013) Developmental disruption of gamma-aminobutyric acid function in the medial prefrontal cortex by noncontingent cocaine exposure during early adolescence. Biol Psychiatry 74:490–501. 10.1016/j.biopsych.2013.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass DK, Flores-Barrera E, Thomases DR, Vital WF, Caballero A, Tseng KY (2014) CB1 cannabinoid receptor stimulation during adolescence impairs the maturation of GABA function in the adult rat prefrontal cortex. Mol Psychiatry 19:536–543. 10.1038/mp.2014.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceresoli-Borroni G, Rassoulpour A, Wu HQ, Guidetti P, Schwarcz R (2006) Chronic neuroleptic treatment reduces endogenous kynurenic acid levels in rat brain. J Neural Transm (Vienna) 113:1355–1365. 10.1007/s00702-005-0432-z [DOI] [PubMed] [Google Scholar]
- Chess AC, Simoni MK, Alling TE, Bucci DJ (2007) Elevations of endogenous kynurenic acid produce spatial working memory deficits. Schizophr Bull 33:797–804. 10.1093/schbul/sbl033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAngeli NE, Todd TP, Chang SE, Yeh HH, Yeh PW, Bucci DJ (2014) Exposure to kynurenic acid during adolescence increases sign-tracking and impairs long-term potentiation in adulthood. Front Behav Neurosci 8:451. 10.3389/fnbeh.2014.00451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt S, Blennow K, Nordin C, Skogh E, Lindström LH, Engberg G (2001) Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci Lett 313:96–98. 10.1016/S0304-3940(01)02242-X [DOI] [PubMed] [Google Scholar]
- Flores-Barrera E, Thomases DR, Heng LJ, Cass DK, Caballero A, Tseng KY (2014) Late adolescent expression of GluN2B transmission in the prefrontal cortex is input-specific and requires postsynaptic protein kinase A and D1 dopamine receptor signaling. Biol Psychiatry 75:508–516. 10.1016/j.biopsych.2013.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB. (2013) Prefrontal NMDA receptors and cognition: working 2B remembered. Neuron 77:603–605. 10.1016/j.neuron.2013.01.029 [DOI] [PubMed] [Google Scholar]
- Forrest CM, Khalil OS, Pisar M, McNair K, Kornisiuk E, Snitcofsky M, Gonzalez N, Jerusalinsky D, Darlington LG, Stone TW (2013) Changes in synaptic transmission and protein expression in the brains of adult offspring after prenatal inhibition of the kynurenine pathway. Neuroscience 254:241–259. 10.1016/j.neuroscience.2013.09.034 [DOI] [PubMed] [Google Scholar]
- Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV (1998) Acetylcholine activates an alpha-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci 18:1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX (2001) The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci 21:7463–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp JA, Foster AC, Leeson PD, Priestley T, Tridgett R, Iversen LL, Woodruff GN (1988) 7-Chlorokynurenic acid is a selective antagonist at the glycine modulatory site of the N-methyl-D-aspartate receptor complex. Proc Natl Acad Sci U S A 85:6547–6550. 10.1073/pnas.85.17.6547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler M, Terramani T, Lynch G, Baudry M (1989) A glycine site associated with N-methyl-D-aspartic acid receptors: characterization and identification of a new class of antagonists. J Neurochem 52:1319–1328. 10.1111/j.1471-4159.1989.tb01881.x [DOI] [PubMed] [Google Scholar]
- Konradsson-Geuken A, Wu HQ, Gash CR, Alexander KS, Campbell A, Sozeri Y, Pellicciari R, Schwarcz R, Bruno JP (2010) Cortical kynurenic acid bi-directionally modulates prefrontal glutamate levels as assessed by microdialysis and rapid electrochemistry. Neuroscience 169:1848–1859. 10.1016/j.neuroscience.2010.05.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson MK, Schwieler L, Goiny M, Erhardt S, Engberg G (2015) Chronic antipsychotic treatment in the rat: effects on brain interleukin-8 and kynurenic acid. Int J Tryptophan Res 8:49–52. 10.4137/IJTR.S25915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linderholm KR, Skogh E, Olsson SK, Dahl ML, Holtze M, Engberg G, Samuelsson M, Erhardt S (2012) Increased levels of kynurenine and kynurenic acid in the CSF of patients with schizophrenia. Schizophr Bull 38:426–432. 10.1093/schbul/sbq086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XC, Holtze M, Powell SB, Terrando N, Larsson MK, Persson A, Olsson SK, Orhan F, Kegel M, Asp L, Goiny M, Schwieler L, Engberg G, Karlsson H, Erhardt S (2014) Behavioral disturbances in adult mice following neonatal virus infection or kynurenine treatment–role of brain kynurenic acid. Brain Behav Immun 36:80–89. 10.1016/j.bbi.2013.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, Llenos IC, Dulay JR, Barillo MM, Yolken RH, Weis S (2004) Expression of the kynurenine pathway enzyme tryptophan 2,3-dioxygenase is increased in the frontal cortex of individuals with schizophrenia. Neurobiol Dis 15:618–629. 10.1016/j.nbd.2003.12.015 [DOI] [PubMed] [Google Scholar]
- Moroni F, Russi P, Lombardi G, Beni M, Carlà V (1988) Presence of kynurenic acid in the mammalian brain. J Neurochem 51:177–180. 10.1111/j.1471-4159.1988.tb04852.x [DOI] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Quack G, Hartmann S, Lorenz B, Wollenburg C, Baran L, Przegalinski E, Kostowski W, Krzascik P, Chizh B, Headley PM (1997) Novel systemically active antagonists of the glycine site of the N-methyl-D-aspartate receptor: electrophysiological, biochemical and behavioral characterization. J Pharmacol Exp Ther 283:1264–1275. [PubMed] [Google Scholar]
- Pershing ML, Bortz DM, Pocivavsek A, Fredericks PJ, Jørgensen CV, Vunck SA, Leuner B, Schwarcz R, Bruno JP (2015) Elevated levels of kynurenic acid during gestation produce neurochemical, morphological, and cognitive deficits in adulthood: implications for schizophrenia. Neuropharmacology 90:33–41. 10.1016/j.neuropharm.2014.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pershing ML, Phenis D, Valentini V, Pocivavsek A, Lindquist DH, Schwarcz R, Bruno JP (2016) Prenatal kynurenine exposure in rats: age-dependent changes in NMDA receptor expression and conditioned fear responding. Psychopharmacology (Berl) 233:3725–3735. 10.1007/s00213-016-4404-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phenis D, Schumacher JD, Valentini V, Bruno JP (2016) Positive allosteric modulators of the alpha7 nicotinic acetylcholine receptor reinstate cognitive control and potentiate glutamate levels in prefrontal cortex. Soc Neurosci Abstr 42:364.309. [Google Scholar]
- Pisar M, Forrest CM, Khalil OS, McNair K, Vincenten MC, Qasem S, Darlington LG, Stone TW (2014) Modified neocortical and cerebellar protein expression and morphology in adult rats following prenatal inhibition of the kynurenine pathway. Brain Res 1576:1–17. 10.1016/j.brainres.2014.06.016 [DOI] [PubMed] [Google Scholar]
- Plitman E, Iwata Y, Caravaggio F, Nakajima S, Chung JK, Gerretsen P, Kim J, Takeuchi H, Chakravarty MM, Remington G, Graff-Guerrero A (2017) Kynurenic acid in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 43:764–777. 10.1093/schbul/sbw221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocivavsek A, Wu HQ, Elmer GI, Bruno JP, Schwarcz R (2012) Pre- and postnatal exposure to kynurenine causes cognitive deficits in adulthood. Eur J Neurosci 35:1605–1612. 10.1111/j.1460-9568.2012.08064.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocivavsek A, Notarangelo FM, Wu HQ, Bruno JP, Schwarcz R (2016) Astrocytes as pharmacological targets in the treatment of schizophrenia: focus on kynurenic acid. In: Handbook of behavioral neuroscience (Pletnikov MV, Waddington JL, eds.), pp 423–443. New York: Elsevier. [Google Scholar]
- Robbins TW, Murphy ER (2006) Behavioural pharmacology: 40+ years of progress, with a focus on glutamate receptors and cognition. Trends Pharmacol Sci 27:141–148. 10.1016/j.tips.2006.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyasaikumar KV, Stachowski EK, Wonodi I, Roberts RC, Rassoulpour A, McMahon RP, Schwarcz R (2011) Impaired kynurenine pathway metabolism in the prefrontal cortex of individuals with schizophrenia. Schizophr Bull 37:1147–1156. 10.1093/schbul/sbq112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Rassoulpour A, Wu HQ, Medoff D, Tamminga CA, Roberts RC (2001) Increased cortical kynurenate content in schizophrenia. Biol Psychiatry 50:521–530. 10.1016/S0006-3223(01)01078-2 [DOI] [PubMed] [Google Scholar]
- Thomases DR, Cass DK, Tseng KY (2013) Periadolescent exposure to the NMDA receptor antagonist MK-801 impairs the functional maturation of local GABAergic circuits in the adult prefrontal cortex. J Neurosci 33:26–34. 10.1523/JNEUROSCI.4147-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeeva OA, Levin ED (2011) Glutamate and nicotinic receptor interactions in working memory: importance for the cognitive impairment of schizophrenia. Neuroscience 195:21–36. 10.1016/j.neuroscience.2011.08.038 [DOI] [PubMed] [Google Scholar]
- Tse MT, Piantadosi PT, Floresco SB (2015) Prefrontal cortical gamma-aminobutyric acid transmission and cognitive function: drawing links to schizophrenia from preclinical research. Biol Psychiatry 77:929–939. 10.1016/j.biopsych.2014.09.007 [DOI] [PubMed] [Google Scholar]
- Tseng KY, Chambers RA, Lipska BK (2009) The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav Brain Res 204:295–305. 10.1016/j.bbr.2008.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turski WA, Nakamura M, Todd WP, Carpenter BK, Whetsell WO Jr, Schwarcz R (1988) Identification and quantification of kynurenic acid in human brain tissue. Brain Res 454:164–169. 10.1016/0006-8993(88)90815-3 [DOI] [PubMed] [Google Scholar]
- Valentini V, Schumacher JD, Phenis D, Bortz D, Bruno JP (2016) Accumbens-prefrontal interactions in the regulation of multiple transmitter systems: implications for cognitive deficits in schizophrenia. Soc Neurosci Abstr 42:364.323. [Google Scholar]
- Wonodi I, Stine OC, Sathyasaikumar KV, Roberts RC, Mitchell BD, Hong LE, Kajii Y, Thaker GK, Schwarcz R (2011) Downregulated kynurenine 3-monooxygenase gene expression and enzyme activity in schizophrenia and genetic association with schizophrenia endophenotypes. Arch Gen Psychiatry 68:665–674. 10.1001/archgenpsychiatry.2011.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HQ, Pereira EF, Bruno JP, Pellicciari R, Albuquerque EX, Schwarcz R (2010) The astrocyte-derived alpha7 nicotinic receptor antagonist kynurenic acid controls extracellular glutamate levels in the prefrontal cortex. J Mol Neurosci 40:204–210. 10.1007/s12031-009-9235-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmarowski A, Wu HQ, Brooks JM, Potter MC, Pellicciari R, Schwarcz R, Bruno JP (2009) Astrocyte-derived kynurenic acid modulates basal and evoked cortical acetylcholine release. Eur J Neurosci 29:529–538. 10.1111/j.1460-9568.2008.06594.x [DOI] [PubMed] [Google Scholar]