

Abstract
The revolution in CRISPR-mediated genome editing has enabled the mutation and insertion of virtually any DNA sequence, particularly in cell culture where selection can be used to recover relatively rare homologous recombination events. The efficient use of this technology in animal models still presents a number of challenges, including the time to establish mutant lines, mosaic gene editing in founder animals, and low homologous recombination rates. Here we report a method for CRISPR-mediated genome editing in Xenopus oocytes with homology-directed repair (HDR) that provides efficient non-mosaic targeted insertion of small DNA fragments (40-50 nucleotides) in 4.4-25.7% of F0 tadpoles, with germline transmission. For both CRISPR/Cas9-mediated HDR gene editing and indel mutation, the gene-edited F0 embryos are uniformly heterozygous, consistent with a mutation in only the maternal genome. In addition to efficient tagging of proteins in vivo, this HDR methodology will allow researchers to create patient-specific mutations for human disease modeling in Xenopus.
KEY WORDS: CRISPR/Cas9, F0, Xenopustropicalis, Xenopuslaevis, Homology-directed repair (HDR), Non-mosaic
Summary: Genome editing in Xenopus oocytes employing homology-directed repair and DNA ligase inhibition effciently generates non-mosaic mutations with germline transmission.
INTRODUCTION
CRISPR/Cas9 genome modification is a powerful approach for generating both targeted indel mutations by non-homologous end-joining (NHEJ) repair and more precise gene editing by the introduction of specific sequences through homology-directed repair (HDR) (Auer et al., 2014; Bassett et al., 2014; Bhattacharya et al., 2015; Blitz et al., 2013; Chen et al., 2013; Friedland et al., 2013; Guo et al., 2014; Hisano et al., 2015; Kimura et al., 2014; Kotani et al., 2015; Li et al., 2015; Nakayama et al., 2013, 2014; Ran et al., 2013; Shi et al., 2015; Wang et al., 2015; Yang et al., 2014). While NHEJ-mediated indel mutations have proven to be effective in animal models including mouse, zebrafish and Xenopus (Blitz et al., 2013; Kotani et al., 2015; Nakayama et al., 2013; Wang et al., 2015; Xue et al., 2014), HDR-mediated targeted DNA insertion has proven more challenging. Microhomology-mediated end-joining (MMEJ)-dependent integration has been demonstrated in Xenopus, but germline transmission of the integrated sequence was not observed and F0 embryos are primarily mosaic (Nakade et al., 2014). A homology-independent targeted integration (HITI) strategy without clean junctional DNA sequences has also been reported in Xenopus (Shi et al., 2015, 2017). In all these reports the efficiency of knock-in events for small DNA sequences is low (Miyaoka et al., 2014; Yu et al., 2014), typically ∼1-2%, and although drug selection or cell sorting can be used to recover these rare integration events in cell lines, this is generally not possible in vertebrate models. As a result, CRISPR/HDR-mediated targeted insertion in animal models requires lengthy, tedious screening of F0 founders. In addition, these F0 animals are usually mosaic, containing a mixture of wild-type alleles, indel mutations and rare gene-edited insertions in different cells, requiring breeding to F1 or F2 generations to obtain uniform germline transmission and/or homozygous mutant animals. These limitations present a major bottleneck in our ability to realize the full potential of CRISPR/Cas9 gene editing in animal models.
To date, HDR-mediated knock-in has not been reported in Xenopus. Here we have developed a novel method that utilizes CRISPR/Cas9 gene editing in Xenopus oocytes, which have higher HDR activity than embryos (Hagmann et al., 1996). Isolated oocytes injected with CRISPR/Cas9 reagents and recombinant DNA are cultured in vitro for several days in the presence of DNA ligase inhibitor to increase HDR activity. This allows efficient editing of the single maternal genome, followed by the host transfer method (Mir and Heasman, 2008; Olson et al., 2012) whereby oocytes are matured in vitro, transplanted into an ovulating host female and then fertilized in vitro. This method enables both efficient indel mutation and precise targeted insertion of epitope tags into a protein-coding sequence with non-mosaic uniformly expressing F0 founders. This approach also eliminates the need to screen many mosaic F0 embryos and reduces the multi-generational breeding normally needed for uniform germline transmission; in our procedure, the F0 animals are uniformly heterozygous in all cells with germline transmission. This approach, along with the availability of sequenced genomes, significantly expands the toolkit available to Xenopus researchers (Grainger, 2012; Harland and Grainger, 2011; Hellsten et al., 2010; Session et al., 2016) and lays the groundwork for efficient insertion of patient-based point mutations for human disease modeling in Xenopus.
RESULTS AND DISCUSSION
Comparison of CRISPR gene editing between embryos and oocytes
It has been reported that the injection of TALEN mRNAs into X. laevis oocytes shows a higher rate of mutagenesis in the F0 generation compared with the conventional injection of embryos (Miyamoto et al., 2015; Nakajima and Yaoita, 2015; Ratzan et al., 2016). However, the resultant F0 embryos had highly mosaic mutations, possibly because the DNA modification occurred after fertilization owing to the time that it takes to translate the TALEN mRNAs (Nakajima and Yaoita, 2015). We hypothesized that CRISPR-mediated genome editing using the Cas9 protein in oocytes would be more efficient. To test this, we first compared CRISPR-mediated indel mutation in oocytes followed by in vitro maturation and fertilization by the host transfer method (Olson et al., 2012) with standard CRISPR/Cas9 injections of fertilized eggs (Fig. 1A). Oocytes of Xenopus laevis and Xenopus tropicalis were defolliculated, injected with Cas9 protein/sgRNA and incubated for 72 h. Oocytes were then matured in vitro with progesterone, stained with a vital dye and then transplanted into a surrogate female frog, where they were ovulated and fertilized in vitro with wild-type sperm (Mir and Heasman, 2008; Olson et al., 2012).
Fig. 1.

Comparison of CRISPR/Cas9 methods in Xenopus oocytes and embryos. (A) Schematic comparing the oocyte isolation/host transfer method (left) with direct embryo injection of CRISPR/Cas9 (right). The generation time for X. laevis is 12-16 months and for X. tropicalis 4-10 months. (B) Overall efficiency of indel generation in F0. The numbers of viable tadpoles from injected oocytes and the percentage of embryos with single indel mutations are shown for the host transfer and embryo injection methods. There is no mosaicism in embryos derived from host transfer method (number of different indel mutations per embryo=1). (C) Examples of genotyping analysis of indel mutations of X. tropicalis F0 embryos. The target sequence is underlined and the protospacer adjacent motif (PAM) is highlighted in blue characters.
T7EI nuclease genotyping of embryos generated from oocyte injections showed a high frequency of indel mutation (∼90%) in both X. laevis and X. tropicalis with four different sgRNAs: X. tropicalis ctnnb1 (19/23), X. tropicalis smad1 (34/36), X. laevis wnt7b.L (33/41) and X. laevis vangl2.S (23/24) (Fig. 1B). Cloning and sequencing of the targeted region from individual host-transferred embryos revealed that ∼50% of the sequences were wild type and the other 50% were a single species of indel, indicating that the embryos were non-mosaic heterozygotes. We grew two X. tropicalis individuals with predicted loss-of-function mutations for ctnnb1 indels (Δ5, Δ23) and two X. laevis individuals with predicted loss-of-function vangl2 indels (Δ13, +9) to adulthood to check for germline transmission. Each F0 founder exhibited germline transmission, and sequencing confirmed that ∼50% of the F1 tadpoles carried the indel, demonstrating a heterozygous germline mutation in the F0 founder (Fig. 1C; data not shown for vangl2). By contrast, injection of CRISPR/Cas9 reagents into one-cell or two-cell stage embryos resulted in indel mutations in ∼55% for the same sgRNAs: ctnnb1 (45%, n=9), smad1 (80%, n=5), wnt7b.L (45%, n=5) or vangl2.S (50%, n=9). Sequencing of PCR products from three individual ctnnb1 sgRNA-injected X. tropicalis embryos revealed a range of different sequences in each embryo, indicating extensive mosaics with mixtures of indel mutations (Fig. 1B,C), consistent with previous reports (Blitz et al., 2013, 2016; Guo et al., 2014; Nakayama et al., 2013). Previous attempts with precise integration into target chromosomes (Nakade et al., 2014) or HITI (Shi et al., 2015) methods were unable to achieve either germline transmission or clean insertion of exogenous DNA fragments into the target site. The recently reported ʻleapfrogging' approach (Blitz et al., 2016), in which genome-edited primordial germ cells are transferred, is a good method for making compound heterozygous knockouts in the F1 generation, but animals will still have multiple mosaic mutations. Our method could be advantageous in that all F1 embryos would contain the same mutant allele.
Efficient HDR-mediated knock-in of epitope tags in oocytes
Since Xenopus oocytes have a higher potential for homologous recombination than fertilized embryos (Hagmann et al., 1996), we next tested whether the host transfer method could be used for efficient HDR-mediated knock-in. We targeted the C-terminus of X. laevis Ctnnb1 (β-catenin), a key cytoskeletal protein and effector of the canonical Wnt pathway, because previous studies have shown that addition of epitope tags to the C-terminus do not affect the function of the resulting fusion protein (Fig. 2A) (Evans et al., 2010; Miller and Moon, 1997). CRISPR components were injected into X. laevis oocytes followed by host transfer or into embryos. Western blotting showed that 6 out of 70 (∼8.5%) host transfer-derived embryos had FLAG-positive protein coincident with Ctnnb1 (Fig. 2B), indicating that the Ctnnb1.L:2×FLAG fusion protein was made. By contrast, we never detected a Ctnnb1:2×FLAG protein in western blots from embryo injections (n=52) (Fig. 2C). Although it is possible that this difference is due in part to the fact that, owing to toxicity in embryos, we can only inject 80 pg of the single-stranded repair oligonucleotide (RO) in embryos compared with 200 pg in oocytes, the most likely explanation is the known higher HDR efficiency in oocytes. Immunostaining with anti-FLAG and anti-Ctnnb1 antibodies revealed FLAG-positive F0 embryos among the oocyte-injected samples in which all epidermal cells expressed the Ctnnb1:2×FLAG fusion protein (Fig. 2D). By contrast, FLAG expression was only observed in a few cells of embryo-injected samples, demonstrating very inefficient and highly mosaic integration (Fig. 2D).
Fig. 2.

Comparative analysis of HDR in Xenopus oocytes and embryos. (A) The procedure for precisely inserting a 2×FLAG tag into the ctnnb1 gene through an HDR-mediated recombination event. (B) Representative western blot of single X. laevis embryos obtained by oocyte injection/host transfer. In the bottom image the green bands represent β-catenin protein and the red band represents the FLAG tag. (C) Representative western blot with of X. laevis embryos derived by embryo injection. (D) Whole-mount immunostaining of epithelial tissues shows expression of the FLAG tag in the same pattern as β-catenin protein in oocyte-injected samples, as expected for heterozygous embryos, and in just a few cells in embryo injected samples, as expected for mosaic embryos. Scale bars: 250 μm (red); 30 μm (white). (E) Real-time PCR (top) shows the stability (mean±s.d. from three independent experiments) of sgRNA in oocytes and western blot (bottom) demonstrates the decay of 600 pg Cas9 protein injected into oocytes and embryos of X. laevis. (F) Sequencing of 20 (for 2×FLAG) or 24 (for V5) clones from the oocyte-injected embryo shows successful knock-in of 2×FLAG in one allele of ctnnb1 and of the V5 epitope in one allele of vangl2.S. PAM sequences are marked in blue; green indicates mismatched nucleotide from the RO; red indicates epitope. No conclusions were drawn from lane 7 owing to damage to the blot. (G) Treatment of oocytes with the DNA ligase IV inhibitor SCR-7 (5 µM) increases the efficiency of successful HDR-mediated knock-in. Representative western blots are shown for ctnnb1.S:2×HA with or without SCR-7 treatment. (H) The overall efficiency of HDR events in F0 tadpoles. Xt, X. tropicalis; Xl, X. laevis.
We postulated that in the oocyte-injected embryos only the maternal genome was mutated because Cas9 protein and/or the sgRNA was degraded over the 72-h culture period. Indeed, RT-PCR and western blots showed that sgRNA and Cas9 protein, respectively, were degraded after 3 days in culture (Fig. 2E). Thus, there is very little Cas9/sgRNA complex remaining in the oocytes after 3 days of culture, suggesting that little if any cutting of the paternal genome is likely to occur in embryos generated from these oocytes. We conclude that CRISPR /Cas9-mediated genome cleavage and indel mutation is probably more efficient in oocytes than in embryos due to the longer incubation time resulting in non-mosaic mutation of the maternal genome. To verify the genotype of knock-in embryos obtained by the oocyte injection method, genomic DNA was PCR amplified and 20 subclones were sequenced. This confirmed the presence of only two types of sequence: one wild-type ctnnb1.L and the other with an in-frame insertion of the tag indicating that the embryos are heterozygous (Fig. 2F). As a second test of HDR-mediated knock-in we sought to insert a V5 tag into the C-terminus of X. laevis Vangl2.S. Genotyping of the samples from the oocyte injection/host transfer method confirmed that the V5 sequence was successfully integrated into the targeted locus (4.4%, 2/45). Again, we were never successful in detecting a V5 knock-in from embryo injections (n=43). These results demonstrate that the oocyte injection method allows non-mosaic precise integration of epitope tags into endogenous loci. We expect that any model system, such as mouse, cow and pig, where it is feasible to culture oocytes and perform in vitro fertilization might exploit the high HDR activity of the oocyte to facilitate genome editing.
Increased efficiency of HDR-mediated knock-in by DNA ligase inhibitor treatment
Since it has been reported that inhibition of DNA ligase activity enhances the rate of HDR over NHEJ in a cell type-specific and context-dependent manner (Chu et al., 2015; Hagmann et al., 1996; Maruyama et al., 2015; Song et al., 2016), we tested whether this could be used to further improve HDR-mediated knock-in efficiency in oocytes. Consistent with this hypothesis, addition of the DNA ligase inhibitor SCR-7 (5 µM) to oocyte culture medium after injection of CRISPR components into oocytes increased the rate of successful knock-in for both ctnnb1 and vangl2 from an average of 7.4% to 22% (Fig. 2G,H, Fig. S1). Interestingly, SCR-7 treatment of CRISPR-injected embryos did not improve HDR-mediated knock-in but rather it was toxic and resulted in lethality. Both control uninjected embryos and CRISPR/Cas9-injected embryos died when cultured in SCR-7, suggesting that the toxicity did not involve failure to repair CRISPR/Cas9-mediated double-stranded breaks.
Germline transmission in X. tropicalis
We next set out to adapt the oocyte knock-in system to the diploid species X. tropicalis, which has a shorter generation time than X. laevis and is more amenable to classical genetics. While the host transfer method is feasible in X. tropicalis (Olson et al., 2012), the smaller size of oocytes makes manual defolliculation more challenging. We aimed to identify an enzymatic method for defolliculation in X. tropicalis. Although it has been reported that Xenopus oocytes isolated by collagenase are not competent to be fertilized (Heasman et al., 1991), we reasoned that this might in part be due to the crude collagenase preparations that are typically used. We tested various pure collagenase subtypes I to XI and found that collagenase VII treatment enabled isolation of X. tropicalis oocytes that could be fertilized after host transfer. We then performed the host transfer method followed by injection of ctnnb1:2×FLAG RO together with Cas9/sgRNA. Two independent knock-in Tg(ctnnb1:2×FLAG) founder females were sequence-validated and grown for 6 months to sexual maturity. To test germline transmission, we mated the transgenic females with wild-type males and immunostained the resulting embryos with anti-FLAG antibody. Approximately half [43% (n=84) and 53% (n=49) from two founder female/wild-type male matings] of the F1 embryos carried ubiquitously expressed β-catenin-FLAG fusion protein in the cortex of all cells, demonstrating non-mosaic germline transmission of the edited allele (Fig. 3).
Fig. 3.
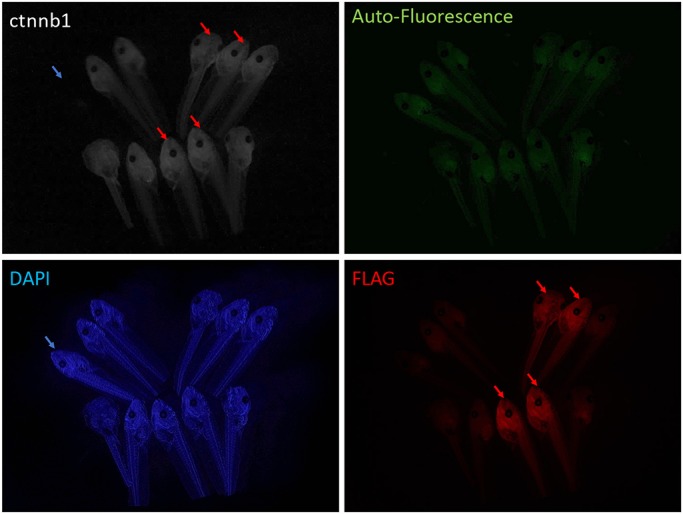
Germline transmission of X. tropicalis Tg(ctnnb1:2×FLAG) knock-in. Representative images showing successful FLAG epitope tagging of β-catenin protein in four embryos (red arrows). The embryo marked with the blue arrow is stained with DAPI only (no primary antibody).
Conclusions
We report a method for efficient non-mosaic CRISPR-mediated indel mutation and precise knock-in of small tags in Xenopus with germline transmission. The next step will be to ascertain whether this method can be used for longer fluorescent tags, such as GFP, as has already been demonstrated in mouse (Miura et al., 2015; Yang et al., 2013) and zebrafish (Li et al., 2015). Looking beyond protein tagging, the method that we have described here should be ideal for the generation of specific point mutations to model human genetic disease or to modify putative transcription factor-binding sites in enhancers.
MATERIALS AND METHODS
CRISPR/Cas9 components
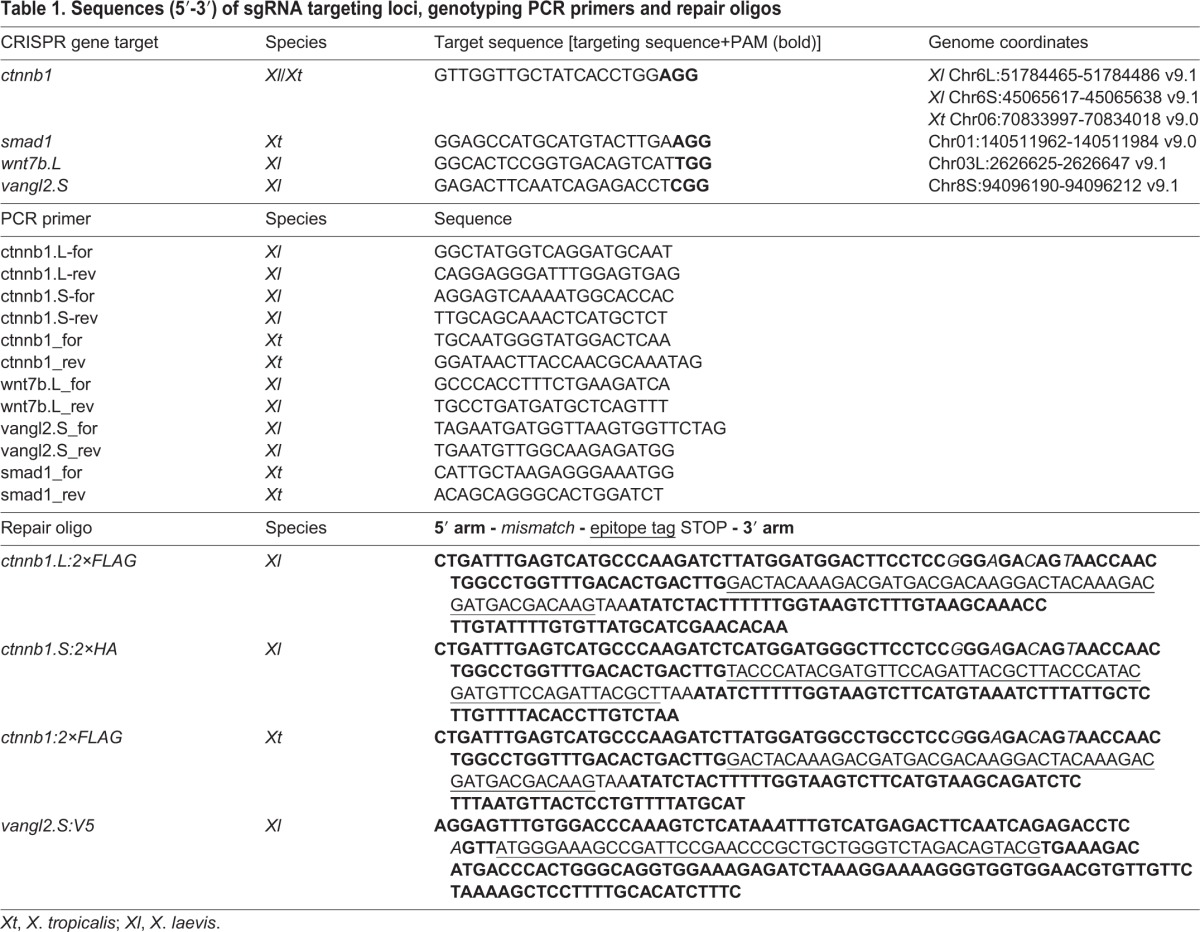
Recombinant SpCas9 protein (PNA Bio) was used in this study as it has less toxicity and creates gene modifications more rapidly and effectively than Cas9 mRNA (Bhattacharya et al., 2015). Four sgRNAs were designed using the E-CRISPR and the CRISPRdirect websites (Heigwer et al., 2014; Naito et al., 2015) for X. laevis and X. tropicalis, respectively, and were synthesized using the MegaShortscript Kit (Ambion). The sequences are summarized in Table 1. Potential off-target sites were screened by allowing two mismatches in sgRNA targeting sequence on the GGGenome website (https://gggenome.dbcls.jp/) and tested by T7E1 endonuclease assay (Mashal et al., 1995). The repair oligo (RO) used is a 200 base single-stranded oligonucleotide (Ultramer, Integrated DNA Technologies) with 40-45 bases of homologous sequence that spans the targeted locus. An epitope tag was added just before the stop codon. Two to four silent mutations in the sgRNA targeting sequence were introduced on the repair construct to prevent the Cas9/sgRNA complex from cutting after successful recombination.
Table 1.
Sequences (5′-3′) of sgRNA targeting loci, genotyping PCR primers and repair oligos
Xenopus oocyte host transfer and embryo manipulations
All animal procedures were performed in accordance with CCHMC institutional approved IACUC protocols. For host transfer experiments, fully grown X. laevis oocytes were isolated manually, or X. tropicalis oocytes were isolated by collagenase VII (Sigma, C2799-15KU, lot SLBG8812V) treatment of dissected ovarian tissue for 2 h with 0.1 KU/ml oocyte culture medium [OCM: 60% L-15 (Sigma; L4386) supplemented with 0.1 mg/ml BSA, 50 UI Pen/Strep, pH 7.8] at 26°C. Oocytes were injected with the CRISPR components: for X. laevis we injected 600 pg Cas9 protein, 300 pg sgRNA and 200 pg RO and cultured the oocytes for 72 h in OCM at 18°C; for X. tropicalis the oocytes were injected with 300 pg Cas9 protein, 200 pg sgRNA and 30 pg RO and cultured at 23°C. Oocytes were then matured by further culture in OCM containing 2 µM progesterone for 12 h for X. laevis or 3 h for X. tropicalis. Matured oocytes were stained with vital dyes, transferred to an ovulating X. laevis host as previously described (Mir and Heasman, 2008) and then fertilized in vitro. For embryo injection, 600 pg Cas9 protein, 80 pg RO and 300 pg sgRNA were injected at the one-cell stage for X. laevis, or 300 pg Cas9 Protein, 10 pg RO and 200 pg sgRNA were injected for X. tropicalis. Different amounts of RO were used for the two species in knock-in experiments to avoid non-specific toxicity of single-stranded DNA oligos (Shuttleworth et al., 1988). Embryos were cultured until tadpole stage NF45 (Nieuwkoop and Faber, 1994) and genotyped. To enhance the frequency of HDR over NHEJ repair, injected oocytes or embryos were cultured in OCM containing the DNA ligase IV inhibitor SCR-7 (5 µM; Selleck Chemicals, S7742; also known as SCR-7 pyrazine) (Chu et al., 2015; Maruyama et al., 2015).
Genotyping, immunostaining and western blot
Genomic DNA from tadpole tails was extracted by the HotSHOT method (Truett et al., 2000), followed by PCR with the primers summarized in Table 1. Tadpole tail tissue was isolated at stage NF45 or later. Tadpoles were anesthetized for 30 s in a solution of 0.025% ethyl 3-aminobenzoate methanesulfonate salt (tricaine; Sigma, MS222) in 0.1× MMR solution (10 mM NaCl, 0.2 mM KCl, 0.1 mM MgSO4, 0.2 mM CaCl2, 0.01 mM EDTA, 0.5 mM HEPES, pH 7.4). During the anesthesia, one-quarter or one-third of the tail was cut on a Petri dish and the tadpoles were closely monitored until complete recovery. The alkaline lysis reagent of the HotSHOT method was used to disrupt the tissue (100 µl for X. tropicalis and 150 µl for X. laevis) by heating at 95°C for 20 min. After the lysis step, an equal volume of the neutralizing reagent of the HotSHOT method was added to stop the reaction. After centrifugation (12,000 g, 5 min) to remove debris, 1-4 µl crude genomic DNA solution was used for PCR amplification. Genomic PCR was performed using the Phusion High-Fidelity PCR Kit (NEB) with HF Buffer, with 40 cycles at an annealing temperature of 64°C (30 s) and extension at 72°C for 40 s. To validate the genotype, PCR products were cloned using the TOPO TA Cloning Kit (Life Technologies) and 20 randomly selected clones were sequenced. Whole-mount immunostaining and western blots were performed using standard protocols as previously described (Wang et al., 2013) with mouse anti-Cas9 (Abcam, ab191468; 1/1000), anti-β-catenin (Santa Cruz, H-102; 1/1000), anti-FLAG M2 (Sigma, F3165; 1/2000), anti-HA 3F10 (Roche, 12158167001; 1/3000), anti-V5 tag 2F11F7 (ThermoFisher, 37-7500; 1/1000), anti-Vangl2 (Santa Cruz, H-55; 1/500) and anti-α-tubulin (DM1A, Neomarker; 1/5000) antibodies. Western blot images were taken with a LICOR Odyssey CLx imaging system and Precision Plus Protein Standards (Bio-Rad, 1610374) were used as protein size markers.
Acknowledgements
We thank Zachary Agricola, Scott Rankin, and Drs Takuya Nakayama, Yueh-Chiang Hu and Robert Grainger for critical reading of the manuscript and providing comments.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: A.M.Z., S.-W.C.; Methodology: E.T., S.-W.C.; Validation: Y.A., S.-W.C.; Formal analysis: Y.A., E.T., S.-W.C.; Investigation: Y.A., E.T.; Resources: A.M.Z., S.-W.C.; Data curation: Y.A.; Writing - original draft: Y.A., S.-W.C.; Writing - review & editing: A.M.Z., S.-W.C.; Supervision: A.M.Z., S.-W.C.
Funding
This work was partially supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (P30 DK078392, K01 DK101618) to S.-W.C. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.152967.supplemental
References
- Auer T. O., Duroure K., De Cian A., Concordet J.-P. and Del Bene F. (2014). Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 24, 142-153. 10.1101/gr.161638.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett A. R., Tibbit C., Ponting C. P. and Liu J.-L. (2014). Mutagenesis and homologous recombination in Drosophila cell lines using CRISPR/Cas9. Biol. Open 3, 42-49. 10.1242/bio.20137120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya D., Marfo C. A., Li D., Lane M. and Khokha M. K. (2015). CRISPR/Cas9: An inexpensive, efficient loss of function tool to screen human disease genes in Xenopus. Dev. Biol. 408, 196-204. 10.1016/j.ydbio.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz I. L., Biesinger J., Xie X. and Cho K. W. Y. (2013). Biallelic genome modification in F(0) Xenopus tropicalis embryos using the CRISPR/Cas system. Genesis 51, 827-834. 10.1002/dvg.22719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz I. L., Fish M. B. and Cho K. W. Y. (2016). Leapfrogging: primordial germ cell transplantation permits recovery of CRISPR/Cas9-induced mutations in essential genes. Development 143, 2868-2875. 10.1242/dev.138057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Gilbert L. A., Cimini B. A., Schnitzbauer J., Zhang W., Li G.-W., Park J., Blackburn E. H., Weissman J. S., Qi L. S. et al. (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479-1491. 10.1016/j.cell.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu V. T., Weber T., Wefers B., Wurst W., Sander S., Rajewsky K. and Kühn R. (2015). Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 33, 543-548. 10.1038/nbt.3198 [DOI] [PubMed] [Google Scholar]
- Evans P. M., Chen X., Zhang W. and Liu C. (2010). KLF4 interacts with beta-catenin/TCF4 and blocks p300/CBP recruitment by beta-catenin. Mol. Cell. Biol. 30, 372-381. 10.1128/MCB.00063-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland A. E., Tzur Y. B., Esvelt K. M., Colaiacovo M. P., Church G. M. and Calarco J. A. (2013). Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 10, 741-743. 10.1038/nmeth.2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger R. M. (2012). Xenopus tropicalis as a model organism for genetics and genomics: past, present, and future. Methods Mol. Biol. 917, 3-15. 10.1007/978-1-61779-992-1_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Zhang T., Hu Z., Zhang Y., Shi Z., Wang Q., Cui Y., Wang F., Zhao H. and Chen Y. (2014). Efficient RNA/Cas9-mediated genome editing in Xenopus tropicalis. Development 141, 707-714. 10.1242/dev.099853 [DOI] [PubMed] [Google Scholar]
- Hagmann M., Adlkofer K., Pfeiffer P., Bruggmann R., Georgiev O., Rungger D. and Schaffner W. (1996). Dramatic changes in the ratio of homologous recombination to nonhomologous DNA-end joining in oocytes and early embryos of Xenopus laevis. Biol. Chem. Hoppe Seyler 377, 239-250. 10.1515/bchm3.1996.377.4.239 [DOI] [PubMed] [Google Scholar]
- Harland R. M. and Grainger R. M. (2011). Xenopus research: metamorphosed by genetics and genomics. Trends Genet. 27, 507-515. 10.1016/j.tig.2011.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman J., Holwill S. and Wylie C. C. (1991). Fertilization of cultured Xenopus oocytes and use in studies of maternally inherited molecules. Methods Cell Biol. 36, 213-230. 10.1016/S0091-679X(08)60279-4 [DOI] [PubMed] [Google Scholar]
- Heigwer F., Kerr G. and Boutros M. (2014). E-CRISP: fast CRISPR target site identification. Nat. Methods 11, 122-123. 10.1038/nmeth.2812 [DOI] [PubMed] [Google Scholar]
- Hellsten U., Harland R. M., Gilchrist M. J., Hendrix D., Jurka J., Kapitonov V., Ovcharenko I., Putnam N. H., Shu S., Taher L. et al. (2010). The genome of the Western clawed frog Xenopus tropicalis. Science 328, 633-636. 10.1126/science.1183670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano Y., Sakuma T., Nakade S., Ohga R., Ota S., Okamoto H., Yamamoto T. and Kawahara A. (2015). Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Sci. Rep. 5, 8841 10.1038/srep08841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y., Hisano Y., Kawahara A. and Higashijima S. (2014). Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Sci. Rep. 4, 6545 10.1038/srep06545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani H., Taimatsu K., Ohga R., Ota S. and Kawahara A. (2015). Efficient multiple genome modifications induced by the crRNAs, tracrRNA and Cas9 protein complex in zebrafish. PLoS ONE 10, e0128319 10.1371/journal.pone.0128319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Zhang B., Bu J. and Du J. (2015). Intron-based genomic editing: a highly efficient method for generating knockin zebrafish. Oncotarget 6, 17891-17894. 10.18632/oncotarget.4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T., Dougan S. K., Truttmann M. C., Bilate A. M., Ingram J. R. and Ploegh H. L. (2015). Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33, 538-542. 10.1038/nbt.3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashal R. D., Koontz J. and Sklar J. (1995). Detection of mutations by cleavage of DNA heteroduplexes with bacteriophage resolvases. Nat. Genet. 9, 177-183. 10.1038/ng0295-177 [DOI] [PubMed] [Google Scholar]
- Miller J. R. and Moon R. T. (1997). Analysis of the signaling activities of localization mutants of beta-catenin during axis specification in Xenopus. J. Cell Biol. 139, 229-243. 10.1083/jcb.139.1.229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir A. and Heasman J. (2008). How the mother can help: studying maternal Wnt signaling by anti-sense-mediated depletion of maternal mRNAs and the host transfer technique. Methods Mol. Biol. 469, 417-429. 10.1007/978-1-60327-469-2_26 [DOI] [PubMed] [Google Scholar]
- Miura H., Gurumurthy C. B., Sato T., Sato M. and Ohtsuka M. (2015). CRISPR/Cas9-based generation of knockdown mice by intronic insertion of artificial microRNA using longer single-stranded DNA. Sci. Rep. 5, 12799 10.1038/srep12799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto K., Suzuki K. T., Suzuki M., Sakane Y., Sakuma T., Herberg S., Simeone A., Simpson D., Jullien J., Yamamoto T. et al. (2015). The expression of TALEN before fertilization provides a rapid knock-out phenotype in xenopus laevis founder embryos. PLoS ONE 10, e0142946 10.1371/journal.pone.0142946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaoka Y., Chan A. H., Judge L. M., Yoo J., Huang M., Nguyen T. D., Lizarraga P. P., So P.-L. and Conklin B. R. (2014). Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat. Methods 11, 291-293. 10.1038/nmeth.2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito Y., Hino K., Bono H. and Ui-Tei K. (2015). CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 31, 1120-1123. 10.1093/bioinformatics/btu743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakade S., Tsubota T., Sakane Y., Kume S., Sakamoto N., Obara M., Daimon T., Sezutsu H., Yamamoto T., Sakuma T. et al. (2014). Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 5, 5560 10.1038/ncomms6560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K. and Yaoita Y. (2015). Highly efficient gene knockout by injection of TALEN mRNAs into oocytes and host transfer in Xenopus laevis. Biol. Open 4, 180-185. 10.1242/bio.201410009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T., Fish M. B., Fisher M., Oomen-Hajagos J., Thomsen G. H. and Grainger R. M. (2013). Simple and efficient CRISPR/Cas9-mediated targeted mutagenesis in Xenopus tropicalis. Genesis 51, 835-843. 10.1002/dvg.22720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T., Blitz I. L., Fish M. B., Odeleye A. O., Manohar S., Cho K. W. and Grainger R. M. (2014). Cas9-based genome editing in Xenopus tropicalis. Methods Enzymol. 546, 355-375. 10.1016/B978-0-12-801185-0.00017-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop P. D. and Faber J. (1994). Normal Table of Xenopus laevis (Daudin): A Systematical and Chronological Survey of the Development from the Fertilized Egg Till the End of Metamorphosis. New York: Garland Publishing. [Google Scholar]
- Olson D. J., Hulstrand A. M. and Houston D. W. (2012). Maternal mRNA knock-down studies: antisense experiments using the host-transfer technique in Xenopus laevis and Xenopus tropicalis. Methods Mol. Biol. 917, 167-182. 10.1007/978-1-61779-992-1_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A. and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281-2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratzan W., Falco R., Salanga C., Salanga M. and Horb M. E. (2016). Generation of a Xenopus laevis F1 albino J strain by genome editing and oocyte host-transfer. Dev. Biol. 426, 188-193. 10.1016/j.ydbio.2016.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Session A. M., Uno Y., Kwon T., Chapman J. A., Toyoda A., Takahashi S., Fukui A., Hikosaka A., Suzuki A., Kondo M. et al. (2016). Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538, 336-343. 10.1038/nature19840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z., Wang F., Cui Y., Liu Z., Guo X., Zhang Y., Deng Y., Zhao H. and Chen Y. (2015). Heritable CRISPR/Cas9-mediated targeted integration in Xenopus tropicalis. FASEB J. 29, 4914-4923. 10.1096/fj.15-273425 [DOI] [PubMed] [Google Scholar]
- Shi Z., Tian D., Xin H., Lian J., Guo X. and Chen Y. (2017). Targeted integration of genes in Xenopus tropicalis. Genesis 55, doi: 10.1002/dvg.23006 10.1002/dvg.23006 [DOI] [PubMed] [Google Scholar]
- Shuttleworth J., Matthews G., Dale L., Baker C. and Colman A. (1988). Antisense oligodeoxyribonucleotide-directed cleavage of maternal mRNA in Xenopus oocytes and embryos. Gene 72, 267-275. 10.1016/0378-1119(88)90152-7 [DOI] [PubMed] [Google Scholar]
- Song J., Yang D., Xu J., Zhu T., Chen Y. E. and Zhang J. (2016). RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 7, 10548 10.1038/ncomms10548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truett G. E., Heeger P., Mynatt R. L., Truett A. A., Walker J. A. and Warman M. L. (2000). Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 29, 52, 54. [DOI] [PubMed] [Google Scholar]
- Wang S., Cha S.-W., Zorn A. M. and Wylie C. (2013). Par6b regulates the dynamics of apicobasal polarity during development of the stratified Xenopus epidermis. PLoS ONE 8, e76854 10.1371/journal.pone.0076854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Shi Z., Cui Y., Guo X., Shi Y.-B. and Chen Y. (2015). Targeted gene disruption in Xenopus laevis using CRISPR/Cas9. Cell Biosci. 5, 15 10.1186/s13578-015-0006-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W., Chen S., Yin H., Tammela T., Papagiannakopoulos T., Joshi N. S., Cai W., Yang G., Bronson R., Crowley D. G. et al. (2014). CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380-384. 10.1038/nature13589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Wang H., Shivalila C. S., Cheng A. W., Shi L. and Jaenisch R. (2013). One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370-1379. 10.1016/j.cell.2013.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Yang J. L., Byrne S., Pan J. and Church G. M. (2014). CRISPR/Cas9-directed genome editing of cultured cells. Curr. Protoc. Mol. Biol. 107, 31.1.1-31.1.17. 10.1002/0471142727.mb3101s107 [DOI] [PubMed] [Google Scholar]
- Yu Z., Chen H., Liu J., Zhang H., Yan Y., Zhu N., Guo Y., Yang B., Chang Y., Dai F. et al. (2014). Various applications of TALEN- and CRISPR/Cas9-mediated homologous recombination to modify the Drosophila genome. Biol. Open 3, 271-280. 10.1242/bio.20147682 [DOI] [PMC free article] [PubMed] [Google Scholar]