

Abstract
Pemphigus and bullous pemphigoid are autoantibody-mediated blistering skin diseases. In pemphigus, keratinocytes in epidermis and mucous membranes lose cell-cell adhesion, and in pemphigoid, the basal keratinocytes lose adhesion to the basement membrane. Pemphigus lesions are mediated directly by the autoantibodies, whereas the autoantibodies in pemphigoid fix complement and mediate inflammation. In both diseases, the autoantigens have been cloned and characterized; pemphigus antigens are desmogleins (cell adhesion molecules in desmosomes), and pemphigoid antigens are found in hemidesmosomes (which mediate adhesion to the basement membrane). This knowledge has enabled diagnostic testing for these diseases by enzyme-linked immunosorbent assays and dissection of various pathophysiological mechanisms, including direct inhibition of cell adhesion, antibody-induced internalization of antigen, and cell signaling. Understanding these mechanisms of disease has led to rational targeted therapeutic strategies.
Keywords: autoimmune, autoantibody, blistering, skin
Introduction
The terms pemphigus and pemphigoid refer to two prototypical autoimmune blistering skin diseases. Both are mediated by autoantibodies, but their mechanisms of pathophysiology and their pathologies are different.
There are two major types of pemphigus, pemphigus vulgaris (PV) and pemphigus foliaceus (PF). The rarer subgroups of pemphigus-like diseases, such as paraneoplastic pemphigus and IgA pemphigus, are beyond the scope of this review. We also discuss the major type of pemphigoid, bullous pemphigoid (BP), but not the rarer subtypes of subepidermal blistering skin diseases such as pemphigoid (or herpes) gestationis, mucous membrane pemphigoid and ocular pemphigoid, epidermolysis bullosa acquisita, and linear IgA disease.
Epidemiology in Brief
The figures for age-adjusted mortality per one million persons in the United States from 1999 to 2009 for BP and pemphigus as major or contributing causes of mortality were 0.3–1 (821–3268 deaths) and 0.2–0.6 (568–1856 deaths), respectively (1). BP occurs mainly in the elderly, with a median age at presentation in the UK of 80 years and with twice as high a risk of death compared with age-matched controls, whereas the median age of onset of PV is 71 (with a broad spread), and the risk of death is three times that of controls (2). The incidence of these diseases depends on the population. In the UK, the incidence (per 100,000 persons per year) of BP is 46 among those over 90 years old but only 1.5 among those 50–59 years old. The incidence of PV is 0.7 for the total UK population but 1.6 for the population of Jerusalem (3). There is an endemic form of PF (i.e., fogo selvagem) in rural areas of Brazil that has a prevalence of 3.4% on certain Amerindian reservations and an incidence of 1–4 cases per 1,200 persons per year (4). This high prevalence and incidence are thought to be due to an environmental factor, possibly related to the bite of a sand fly (5).
Pemphigus: Clinical Presentation and Histological and Immunohistological Findings
In PV, oral mucous membranes are usually affected with or without skin involvement, whereas in PF, only the skin is affected (6, 7). Flaccid blisters—which may occur anywhere on the skin surface—are the primary lesion of pemphigus. In PV, these blisters break easily and result in erosions that have a tendency to spread at their periphery and become large (7) (Figure 1a). The Nikolsky sign, mechanical induction of such erosions by the shearing of normal-appearing epidermis with sideward friction, is positive in active disease. Erosions, rather than intact blisters, are seen in mucous membranes, and even on skin only crusted erosions may be present. In PF, the blister is even more fragile than in PV, and therefore skin lesions usually present as scaly-crusted small erosions, because the blisters break when they are quite small (Figure 1b). The Nikolsky sign is positive in PF patients with active disease.
Figure 1.
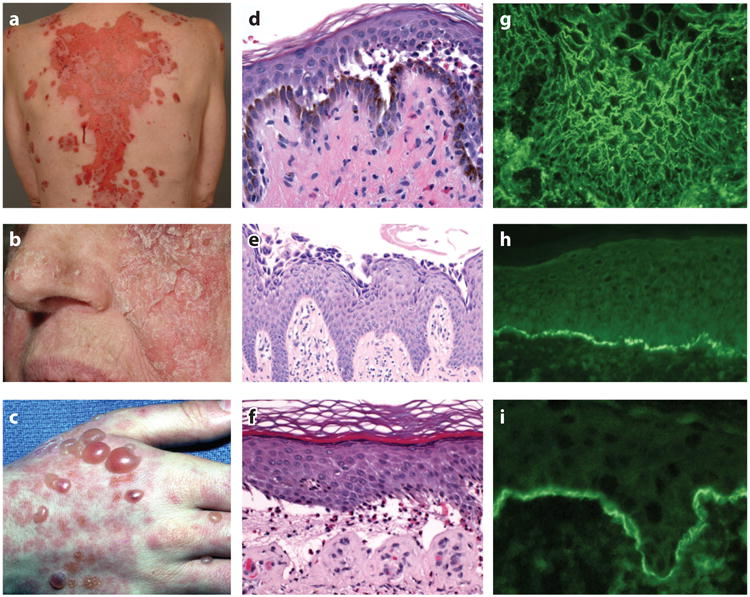
Clinical, histological, and immunopathological findings in pemphigus and pemphigoid. Clinical images of (a) PV with large erosions, (b) PF with scaly crusted lesions, and (c) BP with tense blisters. (d) Histology of PV shows suprabasilar blister, (e) PF shows acantholysis in the granular layer of the epidermis, and (f) BP shows a subepidermal blister with eosinophils. Indirect immunofluorescence of (g) PV on monkey esophagus and (h) BP on normal human skin shows presence of circulating IgG binding cell surface and epidermal basement membrane, respectively. (i) Direct immunofluorescence of perilesional skin in a BP patient shows C3 at the epidermal basement membrane. Abbreviations: BP, bullous pemphigoid; C3, third component of complement; IgG, immunoglobulin G; PF, pemphigus foliaceus; PV, pemphigus vulgaris.
Histologically, pemphigus is characterized by loss of keratinocyte cell-cell adhesion, a process called acantholysis. In PV, acantholysis is present in the basal cell layer and the cells directly above it, forming a suprabasilar blister (Figure 1d). Often the basal keratinocytes lose cell-cell (but not cell–basement membrane) adhesion and assume a cuboidal to rectangular shape, forming a so-called row of tombstones appearance. In PF, acantholysis occurs in the upper epidermis, below the stratum corneum, within or immediately beneath the granular layer (Figure 1e) (6).
The immunohistological hallmark of pemphigus is the detection of cell surface–bound immunoglobulin G (IgG) within the epidermis. All pemphigus patients have such a positive finding in direct immunofluorescence tests of perilesional skin, and (depending on the substrate used) more than 80% of pemphigus patients have circulating IgG directed against keratinocyte cell surfaces (Figure 1g). Usually it is not possible to differentiate PV and PF based on the pattern of immunofluorescence alone (7).
Pemphigus Pathophysiology
Autoantibodies in Pemphigus Are Directed Against Desmogleins
Immunochemical and molecular cloning techniques have shown that the antigens in pemphigus are desmogleins (Dsgs), transmembrane glycoproteins of desmosomes that confer cell-cell adhesion within the epidermis (Figure 2).
Figure 2.

Schematics of the intercellular keratinocyte desmosome and the basal keratinocyte hemidesmosome at the basement membrane. Dsgs, the targets of pemphigus antibodies, mediate cell-cell contact by trans- and cis-adhesion. Adhesion is also provided by Dscs, which, like Dsgs, are in the cadherin adhesion molecule supergene family. Intracellular proteins of desmosomes, such as PG, PKP-1, and DP, link Dsgs and Dscs to KIFs. The hemidesmosome links basal keratinocytes to the epidermal basement membrane zone. BP180 and BP230 are the molecules targeted by autoantibodies in BP patients. BP230 and plectin link the hemidesmosome to KIFs. DP, BP230, and plectin, all of which link the KIFs to the desmosome or hemidesmosome, are in the same gene family of plakins. Abbreviations: BP, bullous pemphigoid; Col, collagen; DP, desmoplakin; Dsc, desmocollin; Dsg, desmoglein; KIF, keratin intermediate filament; Lam332, laminin 332; LD, lamina densa; LL, lamina lucida; PG, plakoglobin; PKP-1, plakophilin 1; SLD, sublamina densa.
Autoantibodies from sporadic and endemic PF patients bound an antigen of ∼160 kDa that was identified as desmoglein 1 (Dsg1) (8, 9). Interestingly, most PF antibodies bound to a calcium-sensitive epitope on Dsg1; chelation of calcium abrogated reactivity with Dsg1 in those sera (10). By demonstrating that both PF and PV sera coimmunoprecipitated an 85-kDa polypeptide—which was the armadillo family adherens junction molecule plakoglobin (PG)—researchers established a close relation between the PF and PV antigen complexes as desmosomal components (11). Molecular cloning, by screening of a keratinocyte cDNA expression library with a PV serum, indicated that the PV antigen was also a desmoglein, now called Dsg3 (12). Further study of the molecular structure and immunoreactivity of the five ectodomains of the PV antigen defined the major pathogenic epitope as being on the amino-terminal extracellular domain, a region important for homophilic adhesion of cadherins (13). Comparable observations were made during studies on Dsg1 (14).
The pivotal role of the pemphigus antigens for keratinocyte adhesion has been shown by genetic deletion of the PV antigen, Dsg3, in mice (15) and enzymatic inactivation of the PF antigen, Dsg1, by exfoliative toxin A (produced by Staphylococcus aureus) in mice and humans (16). The deletion or inactivation of these antigens causes the same clinical and histological pathology as seen in patients with PV and PF and in passive transfer of Dsg3- and Dsg1-specific autoantibodies to neonatal mice or to organ culture of normal human skin.
The discovery of desmogleins as the autoantigens in almost all pemphigus patients has permitted development of enzyme-linked immunosorbent assays (ELISA) for diagnosis (17). This testing also allows for monitoring and, in part, prediction of disease activity through autoantibody levels (18, 19).
Although Dsgs are the main autoantigens targeted by autoantibodies in the vast majority of pemphigus patients, antibodies against other desmosomal cadherins (e.g., desmocollins) and even classical cadherins may rarely be found in disease (20–25).
Autoantibodies Alone Can Cause Disease
Passive transfer experiments in mice have confirmed the pathogenicity of circulating IgG antibodies from pemphigus patients, which result in dose-dependent disease (26, 27) and point to a predominance of the IgG4 subclass in PF patients' sera and in disease induction among mice (28). Predominance of IgG4 autoantibodies has been observed in PV patients as well (29, 30).
Of note, disease can be induced in mice with bivalent F(ab')2 and monovalent Fab' fragments purified from PF patients' sera (31) and in human skin organ culture with recombinantly produced monovalent single-chain variable fragments cloned from both PF and PV patients (32–34), demonstrating that Fc-mediated effects are dispensable in pemphigus pathophysiology, and that antibodies directly mediate acantholysis.
The observation of neonatal PV in babies born to mothers with PV corresponds to a passive transfer experiment in humans (35). Due to a different distribution pattern of Dsg3 and Dsg1 in neonatal epidermis, Dsg3 antibodies alone (from a mother with mucosal PV) can lead to skin blistering in the newborn (see Desmoglein Compensation Explains Localization of Blisters subsection, below). Cases of neonatal PF have also been reported but are unusual owing to desmoglein compensation(again, see below)(36.
Desmoglein Compensation Explains Localization of Blisters
Dissection of the serum autoantibody profiles in pemphigus patients led to the observation that PV patients show a dual status of serum antibodies. In most PV patients with disease localized to mucous membranes, only Dsg3-specific serum antibodies are found, whereas most patients with mucocutaneous disease have antibodies against both Dsg3 and Dsg1. In contrast, PF sera demonstrate only Dsg1 reactivity (29, 37).
These autoantibody profiles, combined with the determination of the normal tissue distributions of Dsg3 and Dsg1, led to the desmoglein compensation model to explain the distinct histological sites of blister formation in mucosal PV, mucocutaneous PV, and PF (38–40) (Figure 3). Basically, this model posits that anti-Dsg1 or -Dsg3 antibodies inactivate only their specific Dsg. If both Dsg1 and Dsg3 are present at any level of the epidermis and only one of them is inactivated, then the other will compensate and provide adhesion; however, if only one desmoglein is present at a particular level of epidermis and it is inactivated, then acantholysis will occur. Figure 3 illustrates how this model explains the blister localization in PV and PF. In addition, the model explains why neonatal PF is only rarely seen: because neonatal human skin shows expression patterns of Dsgs comparable to those seen in adult mucosa, where Dsg3 expressed throughout the epidermis protects from blistering due to anti-Dsg1 PF antibodies (41).
Figure 3.

Desmoglein compensation in pemphigus. Triangles show the usual localization of Dsg1 (green) and Dsg3 (yellow) in the epidermis (skin) and mucous membrane. Triangle width indicates the relative amount of Dsg present at each cell level. Loss of color in a triangle represents loss of function of that particular Dsg due to the presence of αDsg1 or αDsg3. In any area in which Dsg1 or Dsg3 function has been inactivated and the other Dsg is not present to compensate, a blister (shown as loss of cell-cell adhesion) occurs. Abbreviations: αDsg1, anti-Dsg1 antibodies; αDsg3, anti-Dsg3 antibodies; Dsg, desmoglein.
Experimental proof for this model comes from multiple studies. Passive transfer of PF anti-Dsg1 antibodies to neonatal mice causes blisters in the superficial epidermis (just as in patients) because that level normally contains only Dsg1 without Dsg3, so inactivation of Dsg1 causes a superficial epidermal blister; however, the deep epidermis expresses Dsg3, which maintains adhesion at that level. Forced expression of Dsg3 in the superficial epidermis of transgenic mice protects them from PF antibody–induced blistering (41). Another genetic mouse model examined PV blisters from passive transfer to neonatal mice. Normally, Dsg3−/− (knockout) neonatal mice do not show skin blisters because Dsg1 is present throughout the epidermis to compensate for genetic loss of Dsg3; however, transfer of anti-Dsg1 antibodies to these mice results in a severe PV-like blistering (40) because loss of both Dsg3 (genetically) and Dsg1 (by autoantibody) produces symptoms similar to those of PV patients who have cutaneous blisters caused by anti-Dsg1 and -Dsg3 antibodies. Finally, telogen hair in mice is anchored by adhesion provided by Dsg3. Accordingly, the telogen hair is shed early in Dsg3 −/− mice, leading to alopecia; however, forced genetic expression of Dsg1 in the telogen club prevents or delays this loss (42). All these studies indicate that one desmoglein may compensate, at least somewhat, for loss of another.
Autoantibodies Directly Interfere with Cell Adhesion
Abundant data suggest that some pemphigus antibodies directly interfere with cell adhesion. Monovalent Fab' antibodies from pemphigus patients—as well as single-chain variable fragments cloned from PV and PF patients, which do not possess the ability to cross-link antigens—can cause pemphigus in mice and human skin organ culture (see Autoantibodies Alone Can Cause Disease subsection, above) (32, 33). Mechanistically, some PV autoantibodies have been shown to interfere directly with homophilic trans-interaction of Dsg3 molecules (presumably through steric hindrance of Dsg adhesion), as shown by atomic force microscopy and in keratinocyte culture without reliance on transduction of signaling (43, 44). Further evidence for a direct inhibition of adhesion comes from epitope mapping studies with PV and PF autoantibodies on domain-swapped and point-mutated Dsg1/Dsg3 molecules; here, the authors were able to show that most of the dominant epitopes bound by pemphigus sera mapped to the amino-terminal ectodomains of Dsg1 and Dsg3, which are, in analogy to classical cadherins, critical for adhesion (45). In addition, the majority of pathogenic pemphigus sera target regions of the mature (as opposed to precursor) Dsgs (46). Adhesion ability of cadherins—and, by implication, desmogleins—is thought to be unmasked by this proteolytic cleavage (47); therefore, targeting of the mature, processed Dsg implies targeting of the adhesion domain. Extending previous findings that interruption of trans-adhesion of Dsgs is critical for disease induction, researchers recently demonstrated that patients may harbor pathogenic antibodies that target the cis-adhesive interface within the amino-terminal extracellular domain (48). This type of cis-adhesion of Dsg is also thought to be important in strengthening cell-cell adhesion through clustering of the desmoglein in the desmosome. Finally, pemphigus antibody binding to desmogleins is primarily dependent on their normal, calcium-stabilized conformation (10, 45, 49), as is the adhesive property of cadherins, suggesting again that pemphigus antibodies bind important adhesive domains.
Autoantibodies Cluster and/or Internalize Desmogleins and Deplete Them in the Desmosome
In addition to the direct steric hindrance model detailed above, other models explaining loss of cell-cell adhesion have been proposed. These models most likely are not mutually exclusive but may all contribute to pemphigus pathophysiology.
The model discussed here has been called the Dsg nonassembly depletion hypothesis (50, 51). This model attributes loss of cell adhesion to the ability of multivalent pemphigus anti-Dsg antibodies to crosslink and cluster Dsgs. This crosslinking results in internalization of the nonjunctional desmoglein and prevents newly synthesized Dsg from being incorporated into the desmosome. Ultimately, the desmosome is depleted of Dsg and fails to provide adhesion. This model is supported by multiple lines of evidence. In patients with PV and PF, clustering of Dsg3 and Dsg1 by anti-Dsg3 and anti-Dsg1 autoantibodies, respectively, is seen (50, 51). Similar results of clustering and Dsg depletion in desmosomes have been shown in cell culture (52–54). Interestingly, even monoclonal, monovalent, pathogenic anti-Dsg3 PV antibodies can deplete Dsg3 incorporation into newly formed desmosomes (55). Further evidence for this model is that forced increased expression of Dsg3 by adenovirus delivery can counteract the effect of Dsg3 loss in the desmosome and prevent acantholysis (53).
Autoantibodies Cause Cell Signaling That Contributes to Loss of Cell Adhesion
Early evidence implicating intracellular signaling mechanisms in induction of acantholysis came from the observation that polyclonal PV IgG causes retraction of keratin intermediate filaments (presumably contributing to intercellular detachment) in cultured keratinocytes from wild-type mice but not from PG-deficient mice (56). Further studies then showed that PG is a suppressor of c-Myc expression and that PV antibodies trigger c-Myc upregulation by depletion of PG together with Dsg3. Increased c-Myc then leads to cell proliferation and weakened cell-cell adhesion; furthermore, pharmacological inhibition of c-Myc inhibits the ability of PV antibodies to cause acantholysis in mice (57, 58).
Among other cascades of signaling, the p38MAPK signaling pathway has been extensively studied in pemphigus pathophysiology (59–62). HSP27 and p38MAPK, members of this pathway, have been shown to be phosphorylated upon incubation of human keratinocyte cell cultures with PV IgG (59, 60) and to be linked to Dsg3 internalization (61). Pharmacological inhibition of p38MAPK and its downstream targets blocked blister formation in vivo when studied in the passive transfer mouse model for PV and PF (62–64). This signaling cascade was also shown to be active in PV and PF patients' skin (65). Finally, epidermal growth factor receptor (EGFR) signaling was activated by PV antibody addition to human keratinocytes and was shown to be downstream of p38. Further implicating EGFR signaling in pemphigus, pharmacological inhibition of such signaling prevented blister formation induced by PV IgG in the passive transfer mouse model (66).
The issue of whether p38 activation is a primary event causing acantholysis or is secondary to initial loss of cell adhesion has not been fully resolved. Work using monoclonal PV antibodies, as opposed to polyclonal PV IgG, indicated that p38MAPK is not required for loss of intercellular adhesion but that it may augment endocytosis of Dsg3 and thus blistering in PV (67). In an attempt to integrate all existing data, recent research has clarified that (a) steric hindrance (see Autoantibodies Directly Interfere with Cell Adhesion subsection, above) and (b) Dsg3 clustering, depletion (see Autoantibodies Cluster and/or Internalize Desmogleins and Deplete Them in the Desmosome subsection, above), and signaling are separate events. Monoclonal pathogenic antibodies can cause loss of intercellular adhesion through steric hindrance and do not rely on p38MAPK signaling, whereas polyclonal PV IgG causes Dsg3 clustering and endocytosis via a p38MAPK-dependent pathway (44). Finally, p38MAPK signaling may contribute to spontaneous blistering in pemphigus but not to fragile, Nikolsky-positive skin (62).
Contribution of T Cells to Pemphigus Pathology
Human leukocyte antigen restriction in pemphigus
There is a strong association between certain human leukocyte antigen (HLA) class II alleles and susceptibility to PV. A higher prevalence of PV in certain ethnic groups (e.g., Ashkenazi Jews) is consistent with the increased prevalence of such alleles in those groups. Early studies demonstrated association between HLA-DRB1*0402 and PV in Jewish patients (68), and between HLA-DRB1*14/DQB1*0503 and PV in non-Jewish patients (69, 70). A more recent meta-analysis of the association between HLA-DRB1 and PV indicated that the alleles DRB1*04, DRB1*08, and DRB1*14 may serve as susceptibility factors, whereas DRB1*03, DRB1*07, and DRB1*15 were inversely associated with PV (71). These data suggest that patients who develop the disease must have HLA class II molecules capable of presenting Dsg3 peptides to T cells.
Presentation of Dsg peptides to T cells
CD4+ T cells from patients with PV proliferate when antigenic peptides of the extracellular domain of Dsg are presented to them via autologous antigen-presenting cells, with a restriction to the HLA class II alleles DRB1*1401 and DRB1*0402 (72). Dsg3-specific T cells were demonstrated to be present in both PV patients and HLA-concordant healthy individuals, with the former exhibiting mainly a TH2 polarization of those cells and the latter an exclusive TH1 response (73). Analysis of the peptides that these Dsg3-specific T cells recognize showed that they derive mostly from the amino-terminal extracellular domain of Dsg3 and share anchor residues at relative positions 1, 4, and 6 (position 4 being positively charged and critical for binding to the negatively charged p4 binding pocket of DRB1*0402) (74). The relevance of the restriction of the Dsg3-specific CD4+ T cell response to HLA-DRB1*0402 for human disease pathophysiology was demonstrated in vivo by immunization of mice transgenic for HLA-DRB1*0402 with such peptides, in which case anti-Dsg3 pathogenic antibodies were produced; however, such peptides do not produce autoantibodies in HLA-DRB1*0401-transgenic mice, and Dsg3 peptides that do not bind the HLA-DRB1*0402 pocket also do not produce autoantibodies in HLA-DRB1*0402-transgenic mice (75). Finally, CD4+CD25+ regulatory T cells have been implicated in maintaining peripheral tolerance to Dsg3 in mice, and in healthy human carriers of the PV-associated HLA-DRB1*0402 and DQB1*0503 alleles, through the suppression of CD4+ effector T cells (76, 77). Central mechanisms of T cell tolerance induction within the thymus have been described for both Dsg3 and Dsg1, but their exact contributions to health and disease are subject to further investigation (78, 79).
Characterization of Pemphigus Antibodies
Cloning of the B Cell Anti-Desmoglein Repertoire in Pemphigus
To better understand the genetics and the precise function of human pemphigus antibodies, researchers have cloned anti-Dsg B cell repertoires from PV and PF patients to generate and analyze monoclonal antibodies (mAbs).
Antibody phage display (APD) is a powerful high-throughput molecular tool to screen for and isolate antigen-specific mAbs. APD was used to detect anti-Dsg clonal lines [each share a common heavy chain complementarity-determining region 3 (H-CDR3)] from two PV and two PF patients (32–34, 80–82). Seven clonal antibody lines were isolated from each PV patient. Eleven clones were taken from one PF patient and six clones from the other. In all cases, at least one pathogenic clone (i.e., the mAb derived from the clone caused acantholysis in neonatal mice and/or human skin organ culture) was identified, but most anti-Dsg antibodies were nonpathogenic. Most monoclonal antibodies from the PV patients bound only Dsg3, but some bound both Dsg3 and Dsg1. All monoclonal antibodies from the PF patients bound only Dsg1; however, some bound only the intracellular, immature precursor Dsg1, which is not expressed on the keratinocyte cell surface. Interestingly, people without pemphigus also have B cells that code antibodies against precursor Dsg1, but they lack antibodies against the mature cell surface protein, suggesting that B cell tolerance in normal people is only for the exposed, mature Dsg1 (80). Pemphigus sera taken from multiple PV and PF patients blocked binding of these PV and PF mAbs, respectively, demonstrating that the cloned antibodies bind epitopes at, or near, those defined by patients' autoantibodies, and suggesting that idiotypes on pemphigus antibodies may be shared across patients (32, 33). Indeed, a consensus amino acid motif of D/E-X-X-X-W (D/E-acidic amino acid, W-tryptophan) was identified in the H-CDR3 of six pathogenic mAbs and the murine pathogenic PV mAb AK23, with the tryptophan residue thought to be critical for disruption of homophilic Dsg-mediated adhesion (34). These same studies also suggested that pathogenicity and Dsg binding mainly sort with the variable heavy (VH) and not the variable light (VL) region.
Heterohybridoma studies in pemphigus patients have also been used to analyze the pemphigus antibody repertoire. Seventy-three anti-Dsg hybridomas were obtained from six PV patients, most of them IgM+, in one study; IgG4 was not seen (83). More than 90% of the clones showed bispecificity toward both Dsg3 and Dsg1; a more restricted VH gene use in the IgG compared with the IgM compartment; a more promiscuous use of VL genes when compared with VH genes in autoantibodies (again arguing for a major role of the heavy chain over the light chain in antigen binding); and extensive somatic mutations, indicating affinity maturation (83). Analogous findings resulted from a study of nine patients with the endemic form of PF (84).
Another extensive study used EBV-transformed B cells to clone 15 Dsg3-specific IgG antibodies from two PV patients (48). Of these, three were pathogenic in a keratinocyte culture dissociation assay. Mapping the epitopes of pathogenic antibodies indicated that they disrupted Dsg3 cis (as opposed to trans) interactions. In addition, the autoantibodies were highly mutated, and when they reverted back to germline, they lost their Dsg3-binding ability, suggesting that autoreactivity develops through somatic mutation.
Combining APD and heterohybridoma methods for genetic analyses of Dsg3-specific B cells from four PV patients led to the discovery that usage of the variable heavy chain gene VH1-46 is favored in anti-Dsg3 B cells (81). Although anti-Dsg3 B cells used various VH genes for the autoantibodies, VH1-46 was the only VH gene identified in anti-Dsg3 B cells across all four patients, as well as in a mouse monoclonal pathogenic anti-Dsg3 PV antibody. This study went on to show that the VH1-46-encoded anti-Dsg3 mAbs (as opposed to the anti-Dsg3 mAbs encoded by other VH genes) require few to no somatic mutations to bind to Dsg3 and identified acidic amino acid residues in H-CDRs (not just H-CDR3) that are critical for Dsg3 binding. The authors speculate that VH1-46-containing antibodies may be important early in the development of pemphigus autoimmunity.
Clonal Anti-Desmoglein B Cell Persistence Over Time
Assessment of H-CDR3 length distributions (also known as immunoscope analyses) in pemphigus patients with active disease has indicated clonal expansions of B cells in active disease (85, 86). Complete remission in one of these patients (induced by anti-CD20 B cell ablation) led to normalization of CDR3 lengths toward a Gaussian distribution, indicating reestablishment of a more normal B cell repertoire. In a patient with relapse after remission, new clonal expansions were seen, and in a patient with incomplete remission, the original clonal expansion was maintained. These studies suggest that there are specific clones of anti-Dsg B cells in pemphigus that may be eliminated with adequate treatment, but they did not trace ablation, persistence, or reexpression with relapse of particular antigen-specific B cells (as identified by their H-CDR3 amino acid sequences) over time to answer the question of whether the same pathological clones persist (or recur in relapse) or new clonal lines appear in the circulation of patients who relapse. We studied this question by longitudinally cloning anti-Dsg3 IgG+ B cells from two PV patients with APD over 14 patient years. We found that nontolerant anti-Dsg3 B cell lineages persist in patients who relapse, even after periods of complete remission off therapy and, in one patient, multiple courses of rituximab (an antibody that ablates CD20+ B cells) (82). Interestingly, in two patients who had long-standing complete (clinical and serological) remissions off therapy, we could not detect any anti-Dsg3 IgG+ B cell clones anymore, which is consistent with the determination by Colliou et al. (85) that B cells with anti-Dsg3 B cell receptors were difficult to find in patients in remission. We interpreted these findings to suggest that, at least in the patients we studied, B cell loss of tolerance to Dsg3 may be a time-limited event that allows clones of anti-Dsg B cells to escape tolerance and proliferate in the periphery, but that this defect in tolerance for newly formed B cells does not persist; therefore, if therapy can eliminate all nontolerant anti-Dsg3 B cell clones, new ones will not escape tolerance when the B cell repertoire is reestablished from stem cells in the bone marrow; however, if all the anti-Dsg3 B cells in a patient are not eliminated by therapy, they will ultimately proliferate and, probably through differentiation into short-term plasmablasts, cause increased anti-Dsg antibody production and disease relapse. In other words, we can think about curing pemphigus as we do cancer: If we get rid of all the abnormal cells (neoplastic in cancer, nontolerant B cells in pemphigus), we can cure disease.
Therapy of Pemphigus Based on The Understanding of Disease Pathogenesis
Prednisone
Prednisone and other corticosteroids have been the mainstay of pemphigus therapy since their discovery. They are effective quite quickly after systemic therapy (within days) as well as when injected locally. Clearly, they are not effective in this short time or locally through decreasing circulating pathogenic anti-Dsg antibodies. They may be therapeutic, however, because they increase the synthesis of desmogleins in keratinocytes (87). Such increased synthesis may overcome the ability of the anti-Dsg antibodies to internalize newly synthesized desmogleins and ultimately deplete Dsg in the desmosome (see Autoantibodies Cluster and/or Internalize Desmogleins and Deplete Them in the Desmosome subsection, above), as has been shown in cell culture (53). Another possibility, in terms of the Dsg compensation model for adhesion (again, see above), is that corticosteroids and other transcription factor–modulating agents increase synthesis of Dsg isoforms or other cell adhesion molecules that are not targeted by pemphigus antibodies. These adhesion molecules then compensate in function for the loss of the Dsg targeted by the pemphigus antibodies.
General Immunosuppression
Additional immunosuppressive agents used in pemphigus, such as azathioprine and mycophenolate mofetil, are probably effective through general immunosuppression, which decreases antibody production (88, 89). Although these drugs are the mainstay of current clinical practice for most patients, the general immunosuppression they produce is not targeted to the autoantibody-producing cells, and although they spare corticosteroids, they have their own plethora of potential side effects.
Depletion and Increased Catabolism of Autoantibodies
Plasmapheresis and immunoadsorption may be highly effective tools to quickly reduce excessive serum levels of pathogenic antibodies, but they remain adjuvant strategies of treatment that need to be combined with systemic immunosuppression to prevent a rebound of newly synthesized antibodies (90, 91). Intravenous immunoglobulin (IVIg) decreases levels of pathogenic anti-Dsg antibodies too quickly to be explained by a suppression of the synthesis of autoantibodies; instead, the mode of action is thought to be by induction of a general increase in the catabolism of antibodies, including pathogenic antibodies (92). IVIg has been shown in studies with PV and PF patients to be therapeutically effective (93, 94), but as monotherapy may not lead to long-term remission.
Rituximab (Anti-CD20 Antibody)
Anti-CD20 antibody administration leads to a depletion of CD20+ peripheral B cells lasting for at least half a year, after which reconstitution of the B cell repertoire with naïve and transitional B cells derived from the stem cell pool is observed (85, 86). Rituximab is now used mainly as a second-line treatment, although studies for its use as a first-line therapy are underway. It is sometimes combined with IVIg or immunoadsorption. Rituximab induces clinical remission in 50% or more of patients, but relapses frequently occur, necessitating additional cycles of its use (91, 95, 96). In one study, median time to relapse was 15 months after a first cycle of rituximab (97). In one of the best case series (85), about 50% of all pemphigus patients treated with one or more cycles of rituximab went into complete remission off all therapy; however, all of five patients in whom rituximab was used as first-line therapy went into complete remission off all therapy. These data suggest that rituximab may be more effective if used early in disease, perhaps because it is more likely to destroy all nontolerant anti-Dsg B cell clones. Another large single-center study showed that treating pemphigus early in the disease course led to better remission rates (98).
Of note, total IgG levels and IgG titers against immunogens such as tetanus toxoid or pneumococcal capsular polysaccharides, which are synthesized by CD20-negative long-lived plasma cells, do not change after rituximab administration—as opposed to Dsg-specific IgG levels, which, as measured by ELISA assays, drop markedly over several months. These observations suggest that anti-Dsg IgG is synthesized by short-lived plasmablasts that are continually formed from members of the CD20+ memory B cell pool (99). Once that precursor pool is eliminated by rituximab, the short-lived plasmablasts are not renewed, and the pemphigus antibody titer falls through normal catabolism of IgG.
Unique Approaches to Therapy Based on Modulation of Adhesion and Signaling
Innovative approaches to treatment have been suggested recently based on knowledge gained from cell biological studies of desmosomal adhesion and signaling in pemphigus (see Pemphigus Pathophysiology section, above). A tandem peptide that targeted the trans-adhesive interfaces of Dsgs to crosslink them stabilized adhesion and inhibited skin blistering and activation of the p38MAPK pathway by PV-IgG (100). Another study successfully manipulated the expression of an intracellular armadillo protein, plakophilin-1, that links, in concert with PG and desmoplakin (DP), desmosomal cadherins to the keratin intermediate filaments of keratinocytes. Overexpression of plakophilin-1 resulted in hyperadhesive desmosomes that were significantly less prone to PV-IgG-mediated pathology (101). Similarly, introducing a point mutation (S2849G) into DP led to inhibition of both Dsg3 depletion from the cell surface and keratin filament retraction caused by PV-IgG (102), by preventing protein kinase C–dependent phosphorylation of DP at that specific site. Because the protein kinase C inhibitor Bim-X has the same inhibiting effects, this compound may serve as a new pharmacological tool in pemphigus. Finally, as discussed above (again, see Pemphigus Pathophysiology section), inhibition of various signaling pathways implicated in pemphigus antibody–induced acantholysis is a potential therapeutic approach (62).
Bullous Pemphigoid: Clinical Presentation and Histological and Immunohistological Findings
BP is characterized clinically by tense bullae that arise on normal or erythematous skin and that are not as fragile as those seen in pemphigus (6, 103) (Figure 1c). The Nikolsky sign is usually negative. The disease is often associated with pruritus that may precede blistering by months. Oral mucous membrane erosions may be present in a minority of patients.
Subepidermal blisters with inflammation are the hallmark of BP histology (6, 103) (Figure 1f). Almost always there is an inflammatory cell infiltrate in the superficial dermis containing eosinophils; eosinophils are often seen in the blister cavity and at the intact basement membrane zone (BMZ), where they may degranulate before blister formation. Early prebullous, urticarial-type lesions may show eosinophilic spongiosis. Direct immunofluorescence (DIF) of perilesional skin shows linear deposits of the third component of complement (C3) (Figure 1i) and, in most cases, IgG. In fact, if DIF is negative for C3, the diagnosis of BP is doubtful. Immunohistochemical staining of formalin-fixed biopsies for complement proteins C3 (C3d) and C4d can be used to support a diagnosis of BP in case only paraffin-embedded, and not fresh frozen, tissue is available (104, 105). Indirect immunofluorescence shows circulating IgG that binds the epidermal basement membrane in most cases (Figure 1h).
Bullous Pemphigoid Pathophysiology
Figure 4 summarizes the most salient points of the pathophysiology of BP, discussed below.
Figure 4.

Pathophysiological pathways in bullous pemphigoid. Many of these pathways have been demonstrated in mouse models. Although neutrophils are critical for subepidermal blisters in the mouse, in humans the infiltrate is usually eosinophil rich, with many fewer neutrophils. Abbreviations: BMZ, basement membrane zone; BP, bullous pemphigoid; EC, ectodomain; Ig, immunoglobulin; MC, mast cell; NE, neutrophil elastase.
Bullous Pemphigoid Antigens in the Hemidesmosome
Immunoelectron microscopy localized the antigens targeted in BP to the epidermal hemidesmosome, distributed in both intra- and extracellular sites (106, 107) (Figure 2). Immunoprecipitation with BP sera of extracts of cultured keratinocytes identified a protein of ∼230 kDa (now called the BP antigen 1, BPAG1/BPAG1e, or BP230 antigen) (108). Researchers determined the nucleotide coding sequence of BP230 by screening a cDNA expression library, which led to the discovery that BP230 represented the first hemidesmosomal member of a new family of cell adhesion junction plaque proteins, known as plakins, involved in intermediate filament organization (109–112). BP230 was localized to the inner dense plaque of the hemidesmosome (113), where it is thought to anchor keratin filaments. Alternatively spliced gene products of the BP230 antigen, termed BPAG1a (or BPAG1n) and BPAG1b, are found in neurons and striated muscle, respectively, where they also stabilize the cytoskeleton (112, 114). The association of BP with dementia, cerebrovascular disease, and neurological disease (115, 116) may be related to these antigens, but exactly how is not evident at this time.
Immunoblotting and immunoprecipitation indicated that circulating autoantibodies from BP patients also react with a second epidermal protein of ∼ 180 kDa, termed BP180 (BPAG2, collagen XVII), shown to be distinct from BP230 with regard to its coding sequence, antigenic epitopes, and transmembrane localization within hemidesmosomes (117, 118). BP180 has a type II orientation (i.e., the N terminus is located within the cell, and the C terminus externally) and consists of a long C-terminal extracellular region with 15 collagenous domains, interspersed with 16 noncollagenous (NC) segments. The major epitopes bound by BP autoantibodies are localized in the NC16A domain, which is just distal to the cell membrane (119).
BP180, being accessible to autoantibodies—as opposed to the intracellular BP230—is critical for disease induction by autoantibodies, as demonstrated in animal and cell culture models of disease (see below). In this regard, disease activity correlates well with the titers of anti-BP180 antibodies measured by ELISA (120).
Antibodies against both BP180 and BP230, as measured by ELISA, are used for the diagnosis of bullous pemphigoid (121–123).
Immunoglobulin G–Induced Inflammation at the Epidermal Basement Membrane Causes a Subepidermal Blister
By DIF, complement factor C3 has been found in virtually all patients at the epidermal BMZ, implying that complement-dependent pathways may lead to inflammation and clinical disease. To dissect the effector phase of BP, various mouse models have been developed.
Rabbit anti-mouse BP180 NC14A (corresponding to human NC16A) antibodies injected into neonatal mice bound to the dermoepidermal junction and initiated, via activation of complement, mast cell degranulation, attraction and activation of neutrophils, degradation of BP180, and subsequent subepidermal blister formation (124). This cascade did not take place in mice unable to activate the classical complement pathway (C4−/− mice), and was delayed in mice deficient in alternative pathway factors (e.g., factor B in Fb−/− mice); furthermore, C5aR−/− mice were resistant to anti-BP180-induced blistering despite C5a deposits at the dermoepidermal junction, because C5aR-expressing mast cells are crucial for disease progression (125). After degranulation of mast cells, neutrophils are attracted to the BMZ and activated to release potentially tissue-destructive enzymes such as neutrophil elastase and matrix metalloproteinase-9. This neutrophil activation is dependent on binding of FcγRIII by the Fc domain of BP IgG and, accordingly, was inhibited in FcγRIII−/− mice (126); furthermore, in a genetic mouse model in which human BP180 was substituted for mouse BP180 (127, 128), use of anti-BP180 F(ab')2 fragments (which lack the Fc domain) cloned from a BP patient were able to bind the BMZ, block binding of the full-length anti-BP180 IgG with its Fc fragment, and thereby prevent disease (129).
Finally, activated neutrophils secrete neutrophil elastase, which may play a major role in cleaving BP180, whose cleaved fragments may serve as a neutrophil chemoattractant, thereby amplifying the pathological cascade (130).
However, in spite of these elegant murine studies in which neutrophils figure prominently, it should be noted that in BP patients, eosinophils, not neutrophils, are the major cells seen in the histology of the blister and are also increased in the blood (see Bullous Pemphigoid: Clinical Presentation and Histological and Immunohistological Findings section, above, and Immunoglobulin E and Eosinophils: Contribution to Disease subsection, below).
Immunoglobulin G Autoantibodies May Cause BP180 Depletion in the Hemidesmosome and Direct Subepidermal Blister Formation
Although animal models and observations in BP patients suggest that antibody-induced inflammation contributes to blister formation, BP-IgG has also been shown, under certain circumstances, to contribute directly to subepidermal blister formation without requiring activation of complement. BP-IgG (as well as BP-IgE) can cause internalization of BP180, probably via macropinocytosis, from the basal cell membranes of basal cells in cultured human keratinocytes and human skin organ cultures, reducing the adhesive strength of these cells to the basement membrane (131–133). Interestingly, keratinocytes secrete IL-6 and IL-8 cytokines upon BP-IgG/E binding, potentially attracting neutrophils independently of complement (132, 134). Further supporting data stem from an in vivo study using BP180-humanized mice deficient for complement factor C3 (135); here the authors showed that deposition of autoantibodies and subsequent internalization of BP antigen suffice to induce blister formation.
Regarding the pathophysiological role of complement, authors have reported both its necessity (see Immunoglobulin G–Induced Inflammation at the Epidermal Basement Membrane Causes a Subepidermal Blister subsection, above, and 129, 136) and its dispensability (see above and 135, 137) in various models of BP. Putting these data together, especially taking into account the findings of complement fixation at the basement membrane in BP patients, we conclude that complement, if not absolutely necessary for blister formation, at a minimum amplifies skin pathology.
Immunoglobulin E and Eosinophils: Contribution to Disease
About 50% of BP patients have blood eosinophilia (138) and about 70% have elevated serum immunoglobulin E (IgE) (139). In addition, more than 70% have serum IgE against BP180 (140, 141). This IgE is directed against both the NC16A and non-NC16A domains of BP180, as well as against the BP230 antigen (140, 142). Furthermore, degranulation of eosinophils at the epidermal BMZ takes place early in BP and is most pronounced in early urticarial and erythematous lesions (143). These data indicate a TH2-polarized autoimmune response in BP.
The anti-BP180 IgE present in most patients is bound not to the BMZ in vivo (as visualized by DIF), but to mast cells in the reticular and papillary dermis (141). Furthermore, circulating blood basophils show a significant release of histamine upon stimulation with BP180, indicating the presence of anti-BP180 IgE on their surfaces. Histological studies have positioned mast cell activation upstream of that of eosinophils by showing that the latter are attracted to the site of mast cell degranulation (144). Like mast cells and basophils, eosinophils were recently shown to express the high-affinity IgE receptor FcεRI, which provides an explanation of how eosinophils may be activated in BP—namely by binding anti-BP180 IgE that is crosslinked by BP180 or its degradation products (143, 145).
Direct proof for the pathogenicity of BP-IgE derives from studies of human skin xenografts to mice (146). Injections of affinity-purified human BP-IgE into the grafts (which contain mast cells) produced urticarial plaques and mast cell degranulation within minutes, resulting in microscopic subepidermal blisters. Similar results were obtained from a human skin xenograft mouse model in which a murine IgE monoclonal antibody against a shed ectodomain of BP180 was subcutaneously injected (147).
Therapy of Bullous Pemphigoid Based on The Understanding of Disease Pathogenesis
Current treatment strategies for BP include general immunosuppression by corticosteroids and adjunct corticosteroid-sparing immunomodulatory drugs. These approaches probably nonspecifically decrease autoantibody production.
In treatment-resistant cases, rituximab has been used successfully (148). Rituximab decreased anti-BP180 and anti-BP230 IgG titers within one month, whereas levels of total IgG and anti-varicella-zoster-IgG did not change (148), indicating, as discussed with regard to pemphigus [see Rituximab (Anti-CD20 Antibody) subsection, above], that the autoantibodies are probably derived from short-lived plasma cells. Interestingly, anti-BP180 IgE antibodies declined less than anti-BP180 IgG during treatment and follow-up, potentially making patients with IgE-predominant BP (e.g., urticarial BP) more difficult to treat (148).
Because many BP patients have anti-BP180 IgE antibodies that contribute to the pathophysiology of their disease, treatment with omalizumab, a humanized monoclonal antibody that blocks IgE from binding to its receptors, may be effective therapy, especially in those patients who show urticarial skin lesions, high IgE levels, eosinophilia, and resistance to standard regimens of therapy (149).
Finally, the complement cascade would be a logical target to treat in BP patients; however, although complement-targeted therapy is being developed (150), it has not been well evaluated in BP.
Acknowledgments
This review was supported by grants from the DFG (HA6736/1-1 and 2-1) and the Section of Medicine at the University of Luebeck (J03-2015) to C.M.H. and by grant R01-AR052672 and P30-AR057217 from the NIAMS to J.R.S.
Footnotes
Disclosure Statement: The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Literature Cited
- 1.Lott JP, Gross CP. Mortality from nonneoplastic skin disease in the United States. J Am Acad Dermatol. 2014;70:47–54. doi: 10.1016/j.jaad.2013.09.039. [DOI] [PubMed] [Google Scholar]
- 2.Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJ, West J. Bullous pemphigoid and pemphigus vulgaris—incidence and mortality in the UK: population based cohort study. BMJ. 2008;337:a180. doi: 10.1136/bmj.a180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pisanti S, Sharav Y, Kaufman E, Posner LN. Pemphigus vulgaris: incidence in Jews of different ethnic groups, according to age, sex, and initial lesion. Oral Surg Oral Med Oral Pathol. 1974;38:382–87. doi: 10.1016/0030-4220(74)90365-x. [DOI] [PubMed] [Google Scholar]
- 4.Aoki V, Millikan RC, Rivitti EA, Hans-Filho G, Eaton DP, et al. Environmental risk factors in endemic pemphigus foliaceus (fogo selvagem) J Investig Dermatol Symp Proc. 2004;9:34–40. doi: 10.1111/j.1087-0024.2004.00833.x. [DOI] [PubMed] [Google Scholar]
- 5.Qian Y, Jeong JS, Maldonado M, Valenzuela JG, Gomes R, et al. Cutting edge: Brazilian pemphigus foliaceus anti-desmoglein 1 autoantibodies cross-react with sand fly salivary LJM11 antigen. J Immunol. 2012;189:1535–39. doi: 10.4049/jimmunol.1200842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lever WF. Pemphigus. Medicine (Baltimore) 1953;32:1–123. doi: 10.1097/00005792-195302000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Payne AS, Stanley JR. Pemphigus. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, editors. In Fitzpatrick's Dermatology in General Medicine. New York: McGraw Hill Medical: 2012. pp. 586–99. [Google Scholar]
- 8.Koulu L, Kusumi A, Steinberg MS, Klaus-Kovtun V, Stanley JR. Human autoantibodies against a desmosomal core protein in pemphigus foliaceus. J Exp Med. 1984;160:1509–18. doi: 10.1084/jem.160.5.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stanley JR, Koulu L, Klaus-Kovtun V, Steinberg MS. A monoclonal antibody to the desmosomal glycoprotein desmoglein I binds the same polypeptide as human autoantibodies in pemphigus foliaceus. J Immunol. 1986;136:1227–30. [PubMed] [Google Scholar]
- 10.Eyre RW, Stanley JR. Human autoantibodies against a desmosomal protein complex with a calcium-sensitive epitope are characteristic of pemphigus foliaceus patients. J Exp Med. 1987;165:1719–24. doi: 10.1084/jem.165.6.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korman NJ, Eyre RW, Klaus-Kovtun V, Stanley JR. Demonstration of an adhering-junction molecule (plakoglobin) in the autoantigens of pemphigus foliaceus and pemphigus vulgaris. N Engl J Med. 1989;321:631–35. doi: 10.1056/NEJM198909073211002. [DOI] [PubMed] [Google Scholar]
- 12.Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67:869–77. doi: 10.1016/0092-8674(91)90360-b. [DOI] [PubMed] [Google Scholar]
- 13.Amagai M, Karpati S, Prussick R, Klaus-Kovtun V, Stanley JR. Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J Clin Investig. 1992;90:919–26. doi: 10.1172/JCI115968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kowalczyk AP, Anderson JE, Borgwardt JE, Hashimoto T, Stanley JR, Green KJ. Pemphigus sera recognize conformationally sensitive epitopes in the amino-terminal region of desmoglein-1 (Dsg1) J Investig Dermatol. 1995;105:147–52. doi: 10.1111/1523-1747.ep12316680. [DOI] [PubMed] [Google Scholar]
- 15.Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, et al. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. 1997;137:1091–102. doi: 10.1083/jcb.137.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amagai M, Matsuyoshi N, Wang ZH, Andl C, Stanley JR. Toxin in bullous impetigo and staphy-lococcal scalded-skin syndrome targets desmoglein 1. Nat Med. 2000;6:1275–77. doi: 10.1038/81385. [DOI] [PubMed] [Google Scholar]
- 17.Ishii K, Amagai M, Hall RP, Hashimoto T, Takayanagi A, et al. Characterization of autoantibodies in pemphigus using antigen-specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010–17. [PubMed] [Google Scholar]
- 18.Cheng SW, Kobayashi M, Kinoshita-Kuroda K, Tanikawa A, Amagai M, Nishikawa T. Monitoring disease activity in pemphigus with enzyme-linked immunosorbent assay using recombinant desmogleins 1 and 3. Br J Dermatol. 2002;147:261–65. doi: 10.1046/j.1365-2133.2002.04838.x. [DOI] [PubMed] [Google Scholar]
- 19.Abasq C, Mouquet H, Gilbert D, Tron F, Grassi V, et al. ELISA testing of anti–desmoglein 1 and 3 antibodies in the management of pemphigus. Arch Dermatol. 2009;145:529–35. doi: 10.1001/archdermatol.2009.9. [DOI] [PubMed] [Google Scholar]
- 20.Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, et al. Autoimmunity to desmocollin 3 in pemphigus vulgaris. Am J Pathol. 2010;177:2724–30. doi: 10.2353/ajpath.2010.100483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozlowska A, Hashimoto T, Jarzabek-Chorzelska M, Amagai A, Nagata Y, et al. Pemphigus herpetiformis with IgA and IgG antibodies to desmoglein 1 and IgG antibodies to desmocollin 3. J Am Acad Dermatol. 2003;48:117–22. doi: 10.1067/mjd.2003.23. [DOI] [PubMed] [Google Scholar]
- 22.Rafei D, Muller R, Ishii N, Llamazares M, Hashimoto T, et al. IgG autoantibodies against desmo-collin 3 in pemphigus sera induce loss of keratinocyte adhesion. Am J Pathol. 2011;178:718–23. doi: 10.1016/j.ajpath.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flores G, Culton DA, Prisayanh P, Qaqish BF, James K, et al. IgG autoantibody response against keratinocyte cadherins in endemic pemphigus foliaceus (fogo selvagem) J Investig Dermatol. 2012;132:2573–80. doi: 10.1038/jid.2012.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hisamatsu Y, Amagai M, Garrod DR, Kanzaki T, Hashimoto T. The detection of IgG and IgA autoantibodies to desmocollins 1–3 by enzyme-linked immunosorbent assays using baculovirus-expressed proteins, in atypical pemphigus but not in typical pemphigus. Br J Dermatol. 2004;151:73–83. doi: 10.1111/j.1365-2133.2004.05995.x. [DOI] [PubMed] [Google Scholar]
- 25.Evangelista F, Dasher DA, Diaz LA, Prisayanh PS, Li N. E-cadherin is an additional immunological target for pemphigus autoantibodies. J Investig Dermatol. 2008;128:1710–18. doi: 10.1038/sj.jid.5701260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med. 1982;306:1189–96. doi: 10.1056/NEJM198205203062001. [DOI] [PubMed] [Google Scholar]
- 27.Roscoe JT, Diaz L, Sampaio SA, Castro RM, Labib RS, et al. Brazilian pemphigus foliaceus autoantibodies are pathogenic to BALB/c mice by passive transfer. J Investig Dermatol. 1985;85:538–41. doi: 10.1111/1523-1747.ep12277362. [DOI] [PubMed] [Google Scholar]
- 28.Rock B, Martins CR, Theofilopoulos AN, Balderas RS, et al. The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem) N Engl J Med. 1989;320:1463–69. doi: 10.1056/NEJM198906013202206. [DOI] [PubMed] [Google Scholar]
- 29.Ding X, Aoki V, Mascaro JM, Jr, Lopez-Swiderski A, Diaz LA, Fairley JA. Mucosal and mucocutaneous (generalized) pemphigus vulgaris show distinct autoantibody profiles. J Investig Dermatol. 1997;109:592–96. doi: 10.1111/1523-1747.ep12337524. [DOI] [PubMed] [Google Scholar]
- 30.Funakoshi T, Lunardon L, Ellebrecht CT, Nagler AR, O'Leary CE, Payne AS. Enrichment of total serum IgG4 in patients with pemphigus. Br J Dermatol. 2012;167:1245–53. doi: 10.1111/j.1365-2133.2012.11144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rock B, Labib RS, Diaz LA. Monovalent Fab' immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal BALB/c mice. J Clin Investig. 1990;85:296–99. doi: 10.1172/JCI114426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishii K, Lin C, Siegel DL, Stanley JR. Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Investig Dermatol. 2008;128:939–48. doi: 10.1038/sj.jid.5701132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Payne AS, Ishii K, Kacir S, Lin C, Li H, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Investig. 2005;115:888–99. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamagami J, Payne AS, Kacir S, Ishii K, Siegel DL, Stanley JR. Homologous regions of autoantibody heavy chain complementarity-determining region 3 (H-CDR3) in patients with pemphigus cause pathogenicity. J Clin Investig. 2010;120:4111–17. doi: 10.1172/JCI44425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kardos M, Levine D, Gurcan HM, Ahmed RA. Pemphigus vulgaris in pregnancy: analysis of current data on the management and outcomes. Obstet Gynecol Surv. 2009;64:739–49. doi: 10.1097/OGX.0b013e3181bea089. [DOI] [PubMed] [Google Scholar]
- 36.Avalos-Diaz E, Olague-Marchan M, Lopez-Swiderski A, Herrera-Esparza R, Diaz LA. Transplacental passage of maternal pemphigus foliaceus autoantibodies induces neonatal pemphigus. J Am Acad Dermatol. 2000;43:1130–34. doi: 10.1067/mjd.2000.110400. [DOI] [PubMed] [Google Scholar]
- 37.Amagai M, Tsunoda K, Zillikens D, Nagai T, Nishikawa T. The clinical phenotype of pemphigus is defined by the anti-desmoglein autoantibody profile. J Am Acad Dermatol. 1999;40:167–70. doi: 10.1016/s0190-9622(99)70183-0. [DOI] [PubMed] [Google Scholar]
- 38.Amagai M, Koch PJ, Nishikawa T, Stanley JR. Pemphigus vulgaris antigen (desmoglein 3) is localized in the lower epidermis, the site of blister formation in patients. J Investig Dermatol. 1996;106:351–55. doi: 10.1111/1523-1747.ep12343081. [DOI] [PubMed] [Google Scholar]
- 39.Shirakata Y, Amagai M, Hanakawa Y, Nishikawa T, Hashimoto K. Lack of mucosal involvement in pemphigus foliaceus may be due to low expression of desmoglein 1. J Investig Dermatol. 1998;110:76–78. doi: 10.1046/j.1523-1747.1998.00085.x. [DOI] [PubMed] [Google Scholar]
- 40.Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanation for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Investig. 1999;103:461–68. doi: 10.1172/JCI5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H, Wang ZH, Yan A, Lyle S, Fakharzadeh S, et al. Protection of neonates against pemphigus foliaceus by desmoglein 3. N Engl J Med. 2000;343:31–35. doi: 10.1056/NEJM200007063430105. [DOI] [PubMed] [Google Scholar]
- 42.Hanakawa Y, Matsuyoshi N, Stanley JR. Expression of desmoglein 1 compensates for genetic loss of desmoglein 3 in keratinocyte adhesion. J Investig Dermatol. 2002;119:27–31. doi: 10.1046/j.1523-1747.2002.01780.x. [DOI] [PubMed] [Google Scholar]
- 43.Heupel WM, Zillikens D, Drenckhahn D, Waschke J. Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol. 2008;181:1825–34. doi: 10.4049/jimmunol.181.3.1825. [DOI] [PubMed] [Google Scholar]
- 44.Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLOS ONE. 2012;7:e50696. doi: 10.1371/journal.pone.0050696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sekiguchi M, Futei Y, Fujii Y, Iwasaki T, Nishikawa T, Amagai M. Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J Immunol. 2001;167:5439–48. doi: 10.4049/jimmunol.167.9.5439. [DOI] [PubMed] [Google Scholar]
- 46.Yokouchi M, Saleh MA, Kuroda K, Hachiya T, Stanley JR, et al. Pathogenic epitopes of autoantibodies in pemphigus reside in the amino-terminal adhesive region of desmogleins which are unmasked by proteolytic processing of prosequence. J Investig Dermatol. 2009;129:2156–66. doi: 10.1038/jid.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozawa M, Kemler R. Correct proteolytic cleavage is required for the cell adhesive function of uvomorulin. J Cell Biol. 1990;111:1645–50. doi: 10.1083/jcb.111.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Zenzo G, Di Lullo G, Corti D, Calabresi V, Sinistro A, et al. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Investig. 2012;122:3781–90. doi: 10.1172/JCI64413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamiya K, Aoyama Y, Shirafuji Y, Hamada T, Morizane S, et al. A higher correlation of the antibody activities against the calcium-dependent epitopes of desmoglein 3 quantified by ethylenediamine-tetraacetic acid-treated enzyme-linked immunosorbent assay with clinical disease activities of pemphigus vulgaris. J Dermatol Sci. 2013;70:190–95. doi: 10.1016/j.jdermsci.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 50.Oktarina DA, van der Wier G, Diercks GF, Jonkman MF, Pas HH. IgG-induced clustering of desmogleins 1 and 3 in skin of patients with pemphigus fits with the desmoglein nonassembly depletion hypothesis. Br J Dermatol. 2011;165:552–62. doi: 10.1111/j.1365-2133.2011.10463.x. [DOI] [PubMed] [Google Scholar]
- 51.van der Wier G, Pas HH, Kramer D, Diercks GF, Jonkman MF. Smaller desmosomes are seen in the skin of pemphigus patients with anti-desmoglein 1 antibodies but not in patients with anti-desmoglein 3 antibodies. J Investig Dermatol. 2014;134:2287–90. doi: 10.1038/jid.2014.140. [DOI] [PubMed] [Google Scholar]
- 52.Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Investig Dermatol. 1999;112:67–71. doi: 10.1046/j.1523-1747.1999.00463.x. [DOI] [PubMed] [Google Scholar]
- 53.Jennings JM, Tucker DK, Kottke MD, Saito M, Delva E, et al. Desmosome disassembly in response to pemphigus vulgaris IgG occurs in distinct phases and can be reversed by expression of exogenous Dsg3. J Investig Dermatol. 2011;131:706–18. doi: 10.1038/jid.2010.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stahley SN, Saito M, Faundez V, Koval M, Mattheyses AL, Kowalczyk AP. Desmosome assembly and disassembly are membrane raft-dependent. PLOS ONE. 2014;9:e87809. doi: 10.1371/journal.pone.0087809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mao X, Choi EJ, Payne AS. Disruption of desmosome assembly by monovalent human pemphigus vulgaris monoclonal antibodies. J Investig Dermatol. 2009;129:908–18. doi: 10.1038/jid.2008.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, et al. A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol. 2001;153:823–34. doi: 10.1083/jcb.153.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williamson L, Raess NA, Caldelari R, Zakher A, de Bruin A, et al. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. 2006;25:3298–309. doi: 10.1038/sj.emboj.7601224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williamson L, Hunziker T, Suter MM, Muller EJ. Nuclear c-Myc: a molecular marker for early stage pemphigus vulgaris. J Investig Dermatol. 2007;127:1549–55. doi: 10.1038/sj.jid.5700735. [DOI] [PubMed] [Google Scholar]
- 59.Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, et al. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem. 2005;280:23778–84. doi: 10.1074/jbc.M501365200. [DOI] [PubMed] [Google Scholar]
- 60.Rubenstein DS, Diaz LA. Pemphigus antibody induced phosphorylation of keratinocyte proteins. Autoimmunity. 2006;39:577–86. doi: 10.1080/08916930600971885. [DOI] [PubMed] [Google Scholar]
- 61.Jolly PS, Berkowitz P, Bektas M, Lee HE, Chua M, et al. p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J Biol Chem. 2010;285:8936–41. doi: 10.1074/jbc.M109.087999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mao X, Li H, Sano Y, Gaestel M, Park JM, Payne AS. MAPKAP kinase 2 (MK2)-dependent and -independent models of blister formation in pemphigus vulgaris. J Investig Dermatol. 2014;134:68–76. doi: 10.1038/jid.2013.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. PNAS. 2006;103:12855–60. doi: 10.1073/pnas.0602973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berkowitz P, Chua M, Liu Z, Diaz LA, Rubenstein DS. Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering via p38 mitogen-activated protein kinase-dependent signaling in the skin. Am J Pathol. 2008;173:1628–36. doi: 10.2353/ajpath.2008.080391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berkowitz P, Diaz LA, Hall RP, Rubenstein DS. Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin. J Investig Dermatol. 2008;128:738–40. doi: 10.1038/sj.jid.5701080. [DOI] [PubMed] [Google Scholar]
- 66.Bektas M, Jolly PS, Berkowitz P, Amagai M, Rubenstein DS. A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J Biol Chem. 2013;288:9447–56. doi: 10.1074/jbc.M112.438010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mao X, Sano Y, Park JM, Payne AS. p38 MAPK activation is downstream of the loss of intercellular adhesion in pemphigus vulgaris. J Biol Chem. 2011;286:1283–91. doi: 10.1074/jbc.M110.172874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ahmed AR, Yunis EJ, Khatri K, Wagner R, Notani G, et al. Major histocompatibility complex haplotype studies in Ashkenazi Jewish patients with pemphigus vulgaris. PNAS. 1990;87:7658–62. doi: 10.1073/pnas.87.19.7658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ahmed AR, Wagner R, Khatri K, Notani G, Awdeh Z, et al. Major histocompatibility complex haplotypes and class II genes in non-Jewish patients with pemphigus vulgaris. PNAS. 1991;88:5056–60. doi: 10.1073/pnas.88.11.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sinha AA, Brautbar C, Szafer F, Friedmann A, Tzfoni E, et al. A newly characterized HLA DQ beta allele associated with pemphigus vulgaris. Science. 1988;239:1026–29. doi: 10.1126/science.2894075. [DOI] [PubMed] [Google Scholar]
- 71.Yan L, Wang JM, Zeng K. Association between HLA-DRB1 polymorphisms and pemphigus vulgaris: a meta-analysis. Br J Dermatol. 2012;167:768–77. doi: 10.1111/j.1365-2133.2012.11040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin MS, Swartz SJ, Lopez A, Ding X, Fernandez-Vina MA, etal Development and characterization of desmoglein-3 specific T cells from patients with pemphigus vulgaris. J Clin Investig. 1997;99:31–40. doi: 10.1172/JCI119130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Veldman C, Stauber A, Wassmuth R, Uter W, Schuler G, Hertl M. Dichotomy of autoreactive Th1 and Th2 cell responses to desmoglein 3 in patients with pemphigus vulgaris (PV) and healthy carriers of PV-associated HLA class II alleles. J Immunol. 2004;170:635–42. doi: 10.4049/jimmunol.170.1.635. [DOI] [PubMed] [Google Scholar]
- 74.Veldman CM, Gebhard KL, Uter W, Wassmuth R, Grotzinger J, et al. T cell recognition of desmoglein 3 peptides in patients with pemphigus vulgaris and healthy individuals. J Immunol. 2004;172:3883–92. doi: 10.4049/jimmunol.172.6.3883. [DOI] [PubMed] [Google Scholar]
- 75.Eming R, Hennerici T, Backlund J, Feliciani C, Visconti KC, et al. Pathogenic IgG antibodies against desmoglein 3 in pemphigus vulgaris are regulated by HLA-DRB1*04:02-restricted T cells. J Immunol. 2014;193:4391–99. doi: 10.4049/jimmunol.1401081. [DOI] [PubMed] [Google Scholar]
- 76.Veldman C, Höhne A, Dieckmann D, Schuler G, Hertl M. Type I regulatory T cells specific for desmoglein 3 are more frequently detected in healthy individuals than in patients with pemphigus vulgaris. J Immunol. 2004;172:6468–75. doi: 10.4049/jimmunol.172.10.6468. [DOI] [PubMed] [Google Scholar]
- 77.Yokoyama T, Matsuda S, Takae Y, Wada N, Nishikawa T, et al. Antigen-independent development of Foxp3+ regulatory T cells suppressing autoantibody production in experimental pemphigus vulgaris. Int Immunol. 2011;23:365–73. doi: 10.1093/intimm/dxr020. [DOI] [PubMed] [Google Scholar]
- 78.Wada N, Nishifuji K, Yamada T, Kudoh J, Shimizu N, et al. Aire-dependent thymic expression of desmoglein 3, the autoantigen in pemphigus vulgaris, and its role in T-cell tolerance. J Investig Dermatol. 2011;131:410–17. doi: 10.1038/jid.2010.330. [DOI] [PubMed] [Google Scholar]
- 79.Mouquet H, Berrih-Aknin S, Bismuth J, Joly P, Gilbert D, Tron F. Expression of pemphigus-autoantigen desmoglein 1 in human thymus. Tissue Antigens. 2008;71:464–70. doi: 10.1111/j.1399-0039.2008.01020.x. [DOI] [PubMed] [Google Scholar]
- 80.Yamagami J, Kacir S, Ishii K, Payne AS, Siegel DL, Stanley JR. Antibodies to the desmoglein 1 precursor proprotein but not to the mature cell surface protein cloned from individuals without pemphigus. J Immunol. 2009;183:5615–21. doi: 10.4049/jimmunol.0901691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cho MJ, Lo AS, Mao X, Nagler AR, Ellebrecht CT, etal Shared VH1-46 gene usage by pemphigus vulgaris autoantibodies indicates common humoral immune responses among patients. Nat Commun. 2014;5:4167. doi: 10.1038/ncomms5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hammers CM, Chen J, Lin C, Kacir S, Siegel DL, et al. Persistence of anti-desmoglein 3 IgG(+) B-cell clones in pemphigus patients over years. J Investig Dermatol. 2015;135:742–49. doi: 10.1038/jid.2014.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qian Y, Diaz LA, Ye J, Clarke SH. Dissecting the anti-desmoglein autoreactive B cell repertoire in pemphigus vulgaris patients. J Immunol. 2007;178:5982–90. doi: 10.4049/jimmunol.178.9.5982. [DOI] [PubMed] [Google Scholar]
- 84.Qian Y, Clarke SH, Aoki V, Hans-Filhio G, Rivitti EA, Diaz LA. Antigen selection of anti-DSG1 autoantibodies during and before the onset of endemic pemphigus foliaceus. J Investig Dermatol. 2009;129:2823–34. doi: 10.1038/jid.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Colliou N, Picard D, Caillot F, Calbo S, Le Corre S, et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Sci Transl Med. 2013;5:175ra30. doi: 10.1126/scitranslmed.3005166. [DOI] [PubMed] [Google Scholar]
- 86.Mouquet H, Musette P, Gougeon ML, Jacquot S, Lemercier B, et al. B-cell depletion immunotherapy in pemphigus: effects on cellular and humoral immune responses. J Investig Dermatol. 2008;128:2859–69. doi: 10.1038/jid.2008.178. [DOI] [PubMed] [Google Scholar]
- 87.Nguyen VT, Arredondo J, Chernyavsky AI, Kitajima Y, Pittelkow M, Grando SA. Pemphigus vulgaris IgG and methylprednisolone exhibit reciprocal effects on keratinocytes. J Biol Chem. 2004;279:2135–46. doi: 10.1074/jbc.M309000200. [DOI] [PubMed] [Google Scholar]
- 88.Hammers CM, Lunardon L, Schmidt E, Zillikens D. Contemporary management of pemphigus. Expert Opin Orphan Drugs. 2013;1:295–314. [Google Scholar]
- 89.Almugairen N, Hospital V, Bedane C, Duvert-Lehembre S, Picard D, et al. Assessment of the rate of long-term complete remission off therapy in patients with pemphigus treated with different regimens including medium- and high-dose corticosteroids. J Am Acad Dermatol. 2013;69:583–88. doi: 10.1016/j.jaad.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 90.Turner MS, Sutton D, Sauder DN. The use of plasmapheresis and immunosuppression in the treatment of pemphigus vulgaris. J Am Acad Dermatol. 2000;43:1058–64. doi: 10.1067/mjd.2000.109297. [DOI] [PubMed] [Google Scholar]
- 91.Kasperkiewicz M, Shimanovich I, Meier M, Schumacher N, Westermann L, et al. Treatment of severe pemphigus with a combination of immunoadsorption, rituximab, pulsed dexamethasone and azathioprine/mycophenolate mofetil: a pilot study of 23 patients. Br J Dermatol. 2012;166:154–60. doi: 10.1111/j.1365-2133.2011.10585.x. [DOI] [PubMed] [Google Scholar]
- 92.Czernik A, Beutner EH, Bystryn JC. Intravenous immunoglobulin selectively decreases circulating autoantibodies in pemphigus. J Am Acad Dermatol. 2008;58:796–801. doi: 10.1016/j.jaad.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 93.Amagai M, Ikeda S, Shimizu H, Iizuka H, Hanada K, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol. 2009;60:595–603. doi: 10.1016/j.jaad.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 94.Ahmed AR, Sami N. Intravenous immunoglobulin therapy for patients with pemphigus foliaceus unresponsive to conventional therapy. J Am Acad Dermatol. 2002;46:42–49. doi: 10.1067/mjd.2002.116338. [DOI] [PubMed] [Google Scholar]
- 95.Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immune globulin. N Engl J Med. 2006;355:1772–79. doi: 10.1056/NEJMoa062930. [DOI] [PubMed] [Google Scholar]
- 96.Joly P, Mouquet H, Roujeau JC, D'Incan M, Gilbert D, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357:545–52. doi: 10.1056/NEJMoa067752. [DOI] [PubMed] [Google Scholar]
- 97.Heelan K, Al-Mohammedi F, Smith MJ, Knowles S, Lansang P, et al. Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703–8. doi: 10.1001/jamadermatol.2013.6739. [DOI] [PubMed] [Google Scholar]
- 98.Lunardon L, Tsai KJ, Propert KJ, Fett N, Stanley JR, et al. Adjuvant rituximab therapy of pemphigus: a single-center experience with 31 patients. Arch Dermatol. 2012;148:1031–36. doi: 10.1001/archdermatol.2012.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. J Investig Dermatol. 2008;128:2850–58. doi: 10.1038/jid.2008.172. [DOI] [PubMed] [Google Scholar]
- 100.Spindler V, Rotzer V, Dehner C, Kempf B, Gliem M, et al. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Investig. 2013;123:800–11. doi: 10.1172/JCI60139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tucker DK, Stahley SN, Kowalczyk AP. Plakophilin-1 protects keratinocytes from pemphigus vulgaris IgG by forming calcium-independent desmosomes. J Investig Dermatol. 2014;134:1033–43. doi: 10.1038/jid.2013.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dehner C, Rotzer V, Waschke J, Spindler V. A desmoplakin point mutation with enhanced keratin association ameliorates pemphigus vulgaris autoantibody-mediated loss of cell cohesion. Am J Pathol. 2014;184:2528–36. doi: 10.1016/j.ajpath.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 103.Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320–32. doi: 10.1016/S0140-6736(12)61140-4. [DOI] [PubMed] [Google Scholar]
- 104.Pfaltz K, Mertz K, Rose C, Scheidegger P, Pfaltz M, Kempf W. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol. 2010;37:654–58. doi: 10.1111/j.1600-0560.2009.01450.x. [DOI] [PubMed] [Google Scholar]
- 105.Kwon EJ, Ntiamoah P, Shulman KJ. The utility of C4d immunohistochemistry on formalin-fixed paraffin-embedded tissue in the distinction of polymorphic eruption of pregnancy from pemphigoid gestationis. Am J Dermatopathol. 2013;35:787–91. doi: 10.1097/DAD.0b013e3182a6b6cc. [DOI] [PubMed] [Google Scholar]
- 106.Mutasim DF, Morrison LH, Takahashi Y, Labib RS, Skouge J, et al. Definition of bullous pemphigoid antibody binding to intracellular and extracellular antigen associated with hemidesmosomes. J Investig Dermatol. 1989;92:225–30. doi: 10.1111/1523-1747.ep12276753. [DOI] [PubMed] [Google Scholar]
- 107.Shimizu H, McDonald JN, Kennedy AR, Eady RAJ. Demonstration of intra- and extracellular localization of bullous pemphigoid antigen using cryofixation and freeze substitution for postembedding immunoelectron microscopy. Arch Dermatol Res. 1989;281:443–48. doi: 10.1007/BF00510078. [DOI] [PubMed] [Google Scholar]
- 108.Stanley JR, Hawley-Nelson P, Yuspa SH, Shevach EM, Katz SI. Characterization of bullous pemphigoid antigen: a unique basement membrane protein of stratified squamous epithelia. Cell. 1981;24:897–903. doi: 10.1016/0092-8674(81)90115-x. [DOI] [PubMed] [Google Scholar]
- 109.Stanley JR, Tanaka T, Mueller S, Klaus-Kovtun V, Roop D. Isolation of cDNA for bullous pemphigoid antigen by use of patients' autoantibodies. J Clin Investig. 1988;82:1864–70. doi: 10.1172/JCI113803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tanaka T, Parry DAD, Klaus-Kovtun V, Steinert PM, Stanley JR. Comparison of molecularly cloned bullous pemphigoid antigen to desmoplakin I confirms that they define a new family of cell adhesion junction plaque proteins. J Biol Chem. 1991;266:12555–59. [PubMed] [Google Scholar]
- 111.Green KJ, Virata MLA, Elgart GW, Stanley JR, Parry DAD. Comparative structural analysis of desmoplakin, bullous pemphigoid antigen and plectin: members of a new gene family involved in organization of intermediate filments. Int J Biol Macrobiol. 1992;14:145–53. doi: 10.1016/s0141-8130(05)80004-2. [DOI] [PubMed] [Google Scholar]
- 112.Bouameur JE, Favre B, Borradori L. Plakins, a versatile family of cytolinkers: roles in skin integrity and in human diseases. J Investig Dermatol. 2014;134:885–94. doi: 10.1038/jid.2013.498. [DOI] [PubMed] [Google Scholar]
- 113.Tanaka T, Korman NJ, Shimizu H, Eady RAJ, Klaus-Kovtun V, et al. Production of rabbit antibodies against carboxy-terminal epitopes encoded by bullous pemphigoid cDNA. J Investig Dermatol. 1990;94:617–23. doi: 10.1111/1523-1747.ep12876200. [DOI] [PubMed] [Google Scholar]
- 114.Yang Y, Dowling J, Yu QC, Kouklis P, Cleveland DW, Fuchs E. An essential cytoskeletal linker protein connecting actin microfilaments to intermediate filaments. Cell. 1996;86:655–65. doi: 10.1016/s0092-8674(00)80138-5. [DOI] [PubMed] [Google Scholar]
- 115.Taghipour K, Chi CC, Vincent A, Groves RW, Venning V, Wojnarowska F. The association of bullous pemphigoid with cerebrovascular disease and dementia: a case-control study. Arch Dermatol. 2010;146:1251–54. doi: 10.1001/archdermatol.2010.322. [DOI] [PubMed] [Google Scholar]
- 116.Langan SM, Groves RW, West J. The relationship between neurological disease and bullous pemphigoid: a population-based case-control study. J Investig Dermatol. 2011;131:631–36. doi: 10.1038/jid.2010.357. [DOI] [PubMed] [Google Scholar]
- 117.Diaz L, Ratrie H, Saunders WS, Futamura S, Squiquera HL, et al. Isolation of a human epidermal cDNA corresponding to the 180-kD autoantigen recognized by bullous pemphigoid and herpes gestationis sera. J Clin Investig. 1990;86:1088–94. doi: 10.1172/JCI114812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Giudice GJ, Emery DJ, Diaz LA. Cloning and primary structural analysis of the bullous pemphigoid autoantigen BP180. J Investig Dermatol. 1992;99:243–50. doi: 10.1111/1523-1747.ep12616580. [DOI] [PubMed] [Google Scholar]
- 119.Zillikens D, Rose PA, Balding SD, Liu Z, Olague-Marchan M, et al. Tight clustering of extracellular BP180 epitopes recognized by bullous pemphigoid autoantibodies. J Investig Dermatol. 1997;109:573–79. doi: 10.1111/1523-1747.ep12337492. [DOI] [PubMed] [Google Scholar]
- 120.Schmidt E, Obe K, Brocker EB, Zillikens D. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol. 2000;136:174–78. doi: 10.1001/archderm.136.2.174. [DOI] [PubMed] [Google Scholar]
- 121.Roussel A, Benichou J, Randriamanantany ZA, Gilbert D, Drenovska K, et al. Enzyme-linked immunosorbent assay for the combination of bullous pemphigoid antigens 1 and 2 in the diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:293–98. doi: 10.1001/archdermatol.2011.21. [DOI] [PubMed] [Google Scholar]
- 122.Thoma-Uszynski S, Uter W, Schwietzke S, Hofmann SC, Hunziker T, et al. BP230- and BP180-specific auto-antibodies in bullous pemphigoid. J Investig Dermatol. 2004;122:1413–22. doi: 10.1111/j.0022-202X.2004.22603.x. [DOI] [PubMed] [Google Scholar]
- 123.Sakuma-Oyama Y, Powell AM, Oyama N, Albert S, Bhogal BS, Black MM. Evaluation of a BP180-NC16a enzyme-linked immunosorbent assay in the initial diagnosis of bullous pemphigoid. Br J Dermatol. 2004;151:126–31. doi: 10.1111/j.1365-2133.2004.06082.x. [DOI] [PubMed] [Google Scholar]
- 124.Nelson KC, Zhao M, Schroeder PR, Li N, Wetsel RA, et al. Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Investig. 2006;116:2892–900. doi: 10.1172/JCI17891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Heimbach L, Li Z, Berkowitz P, Zhao M, Li N, et al. The C5A receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem. 2011;286:15003–9. doi: 10.1074/jbc.M111.221036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhao M, Trimbeger ME, Li N, Diaz LA, Shapiro SD, Liu Z. Role of FcRs in animal model of autoimmune bullous pemphigoid. J Immunol. 2006;177:3398–405. doi: 10.4049/jimmunol.177.5.3398. [DOI] [PubMed] [Google Scholar]
- 127.Olasz EB, Roh J, Yee CL, Arita K, Akiyama M, et al. Human bullous pemphigoid antigen 2 transgenic skin elicits specific IgG in wild-type mice. J Investig Dermatol. 2007;127:2807–17. doi: 10.1038/sj.jid.5700970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nishie W, Sawamura D, Natsuga K, Shinkuma S, Goto M, et al. A novel humanized neonatal autoimmune blistering skin disease model induced by maternally transferred antibodies. J Immunol. 2009;183:4088–93. doi: 10.4049/jimmunol.0800389. [DOI] [PubMed] [Google Scholar]
- 129.Wang G, Ujiie H, Shibaki A, Nishie W, Tateishi Y, et al. Blockade of autoantibody-initiated tissue damage by using recombinant fab antibody fragments against pathogenic autoantigen. Am J Pathol. 2010;176:914–25. doi: 10.2353/ajpath.2010.090744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lin L, Betsuyaku T, Heimbach L, Li N, Rubenstein D, et al. Neutrophil elastase cleaves the murine hemidesmosomal protein BP180/type XVII collagen and generates degradation products that modulate experimental bullous pemphigoid. Matrix Biol. 2012;31:38–44. doi: 10.1016/j.matbio.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Iwata H, Kamio N, Aoyama Y, Yamamoto Y, Hirako Y, et al. IgG from patients with bullous pemphigoid depletes cultured keratinocytes of the 180-kDa bullous pemphigoid antigen (type XVII collagen) and weakens cell attachment. J Investig Dermatol. 2009;129:919–26. doi: 10.1038/jid.2008.305. [DOI] [PubMed] [Google Scholar]
- 132.Messingham KN, Srikantha R, DeGueme AM, Fairley JA. FcR-independent effects of IgE and IgG autoantibodies in bullous pemphigoid. J Immunol. 2011;187:553–60. doi: 10.4049/jimmunol.1001753. [DOI] [PubMed] [Google Scholar]
- 133.Hiroyasu S, Ozawa T, Kobayashi H, Ishii M, Aoyama Y, et al. Bullous pemphigoid IgG induces BP180 internalization via a macropinocytic pathway. Am J Pathol. 2013;182:828–40. doi: 10.1016/j.ajpath.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nishie W. Update on the pathogenesis of bullous pemphigoid: an autoantibody-mediated blistering disease targeting collagen XVII. J Dermatol Sci. 2014;73:179–86. doi: 10.1016/j.jdermsci.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 135.Ujiie H, Sasaoka T, Izumi K, Nishie W, Shinkuma S, et al. Bullous pemphigoid autoantibodies directly induce blister formation without complement activation. J Immunol. 2014;193:4415–28. doi: 10.4049/jimmunol.1400095. [DOI] [PubMed] [Google Scholar]
- 136.Li Q, Ujiie H, Shibaki A, Wang G, Moriuchi R, et al. Human IgG1 monoclonal antibody against human collagen 17 noncollagenous 16A domain induces blisters via complement activation in experimental bullous pemphigoid model. J Immunol. 2010;185:7746–55. doi: 10.4049/jimmunol.1000667. [DOI] [PubMed] [Google Scholar]
- 137.Natsuga K, Nishie W, Shinkuma S, Ujiie H, Nishimura M, et al. Antibodies to pathogenic epitopes on type XVII collagen cause skin fragility in a complement-dependent and -independent manner. J Immunol. 2012;188:5792–99. doi: 10.4049/jimmunol.1003402. [DOI] [PubMed] [Google Scholar]
- 138.Bushkell LL, Jordon RE. Bullous pemphigoid: a cause of peripheral blood eosinophilia. J Am Acad Dermatol. 1983;8:648–51. doi: 10.1016/s0190-9622(83)70073-3. [DOI] [PubMed] [Google Scholar]
- 139.Arbesman CE, Wypych JI, Reisman RE, Beutner EH. IgE levels in sera of patients with pemphigus or bullous pemphigoid. Arch Dermatol. 1974;110:378–81. [PubMed] [Google Scholar]
- 140.Fairley JA, Fu CL, Giudice GJ. Mapping the binding sites of anti-BP180 immunoglobulin E autoantibodies in bullous pemphigoid. J Investig Dermatol. 2005;125:467–72. doi: 10.1111/j.0022-202X.2005.23853.x. [DOI] [PubMed] [Google Scholar]
- 141.Dimson OG, Giudice GJ, Fu CL, Van den Bergh F, Warren SJ, et al. Identification of a potential effector function for IgE autoantibodies in the organ-specific autoimmune disease bullous pemphigoid. J Investig Dermatol. 2003;120:784–88. doi: 10.1046/j.1523-1747.2003.12146.x. [DOI] [PubMed] [Google Scholar]
- 142.Delaporte E, Dubost-Brama A, Ghohestani R, Nicolas JF, Neyrinck JL, et al. IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol. 1996;157:3642–47. [PubMed] [Google Scholar]
- 143.Borrego L, Maynard B, Peterson EA, George T, Iglesias L, et al. Deposition of eosinophil granule proteins precedes blister formation in bullous pemphigoid. Comparison with neutrophil and mast cell granule proteins. Am J Pathol. 1996;148:897–909. [PMC free article] [PubMed] [Google Scholar]
- 144.Dvorak AM, Mihm MC, Jr, Osage JE, Kwan TH, Austen KF, Wintroub BU. Bullous pemphigoid, an ultrastructural study of the inflammatory response: eosinophil, basophil and mast cell granule changes in multiple biopsies from one patient. J Investig Dermatol. 1982;78:91–101. doi: 10.1111/1523-1747.ep12505711. [DOI] [PubMed] [Google Scholar]
- 145.Messingham KN, Holahan HM, Frydman AS, Fullenkamp C, Srikantha R, Fairley JA. Human eosinophils express the high affinity IgE receptor, FcεRI, in bullous pemphigoid. PLOS ONE. 2014;9:e107725. doi: 10.1371/journal.pone.0107725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fairley JA, Burnett CT, Fu CL, Larson DL, Fleming MG, Giudice GJ. A pathogenic role for IgE in autoimmunity: bullous pemphigoid IgE reproduces the early phase of lesion development in human skin grafted to nu/nu mice. J Investig Dermatol. 2007;127:2605–11. doi: 10.1038/sj.jid.5700958. [DOI] [PubMed] [Google Scholar]
- 147.Zone JJ, Taylor T, Hull C, Schmidt L, Meyer L. IgE basement membrane zone antibodies induce eosinophil infiltration and histological blisters in engrafted human skin on SCID mice. J Investig Dermatol. 2007;127:1167–74. doi: 10.1038/sj.jid.5700681. [DOI] [PubMed] [Google Scholar]
- 148.Hall RP, III, Streilein RD, Hannah DL, McNair PD, Fairley JA, et al. Association of serum B-cell activating factor level and proportion of memory and transitional B cells with clinical response after rituximab treatment of bullous pemphigoid patients. J Investig Dermatol. 2013;133:2786–88. doi: 10.1038/jid.2013.236. [DOI] [PubMed] [Google Scholar]
- 149.Yu KK, Crew AB, Messingham KA, Fairley JA, Woodley DT. Omalizumab therapy for bullous pemphigoid. J Am Acad Dermatol. 2014;71:468–74. doi: 10.1016/j.jaad.2014.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25:1265–75. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]