

Abstract
Background
Pre-implantation embryos exhibit sexual dimorphisms in both primates and rodents. To determine whether these differences reflected sex-biased expression patterns, we generated transcriptome profiles for six 40,XX, six 40,XY, and two 39,X mouse embryonic stem (ES) cells by RNA sequencing.
Results
We found hundreds of coding and non-coding RNAs that were differentially expressed between male and female cells. Surprisingly, the majority of these were autosomal and included RNA encoding transcription and epigenetic and chromatin remodeling factors. We showed differential Prdm14-responsive enhancer activity in male and female cells, correlating with the sex-specific levels of Prdm14 expression. This is the first time sex-specific enhancer activity in ES cells has been reported. Evaluation of X-linked gene expression patterns between our XX and XY lines revealed four distinct categories: (1) genes showing 2-fold greater expression in the female cells; (2) a set of genes with expression levels well above 2-fold in female cells; (3) genes with equivalent RNA levels in male and female cells; and strikingly, (4) a small number of genes with higher expression in the XY lines. Further evaluation of autosomal gene expression revealed differential expression of imprinted loci, despite appropriate parent-of-origin patterns. The 39,X lines aligned closely with the XY cells and provided insights into potential regulation of genes associated with Turner syndrome in humans. Moreover, inclusion of the 39,X lines permitted three-way comparisons, delineating X and Y chromosome-dependent patterns.
Conclusions
Overall, our results support the role of the sex chromosomes in establishing sex-specific networks early in embryonic development and provide insights into effects of sex chromosome aneuploidies originating at those stages.
Electronic supplementary material
The online version of this article (doi:10.1186/s13293-017-0150-x) contains supplementary material, which is available to authorized users.
Keywords: Sex chromosomes, Transcriptome, Embryonic stem cells, Transcription factors, Epigenetic enzymes
Background
The variation between males and females constitutes the largest phenotypic dimorphism in any given species [1]. In humans, this variation accounts for differences seen in the risk, incidence, and response to treatment for a plethora of diseases [2–4]. Examples include autoimmune diseases [5, 6], cancer susceptibility [7, 8], infectious disease incidence [9, 10], and cardiovascular disease presentation and risk [11, 12]. There is evidence showing transcriptional and epigenomic variability between men and women [13, 14]. The role of these differences in disease presentation and phenotype is unclear, but there is mounting evidence that gene regulation is linked to disease [15, 16], suggesting that the impact of sex on gene expression and epigenetic features is likely to be important [16, 17]. Sex hormones partially mediate sexual dimorphisms after gonadogenesis [18], but their action does not fully explain them [19].
The X and Y chromosomes encode multiple factors that have an important role in determining sexual biases in all tissues. This has been fully established in the four-core genotypes mouse model, in which the sex chromosome complement is uncoupled from gonadal sex [20, 21]. In this model, sexually dimorphic neural and behavioral traits were identified in adult mice and defects in neural tube closure at 10.5 days post coitum (dpc) were evident [21]. Differences in regulation of autosomal genes in male and female adult tissues were also shown in this system [22].
How soon do sexual dimorphisms appear in development? Differences between males and females start immediately after fertilization in both humans and mice. Male pre-implantation embryos develop more rapidly than female embryos, i.e., before gonadal sex differentiation, and, in mice, this difference persists at least up to day 9.5 of gestation [23]. Newborn males have a higher mortality rate than females [24]. Expression differences arising from the X and Y chromosomes directly or indirectly have been documented in mouse and humans [25–29]. The sex chromosome complement alone drives epigenome-wide differences in male and female murine embryonic stem (ES) cells [30–32]. These effects may have a lasting impact on developmental trajectories and disease risk [33, 34], even if transcriptional differences are not immediate.
We hypothesize that some sexual dimorphisms can be traced back to early embryogenesis because the presence of the sex chromosomes primes the genome differentially, leaving an epigenetic registry of sexual identity [35]. As a result, dosage differences, especially of transcription factors (TFs) and epigenetic complexes, can affect regulatory networks and determine differences in downstream developmental events. It has been shown that differential epigenetic marks can be latent, with effects on gene expression in later development, as tissue-specific signals become available.
It is widely accepted that various mouse strains are phenotypically different [36, 37]. Significant gene content variation exists between inbred mouse strains, including large deletions and amplifications [38], with the potential for biases in allelic gene expression [39]. To investigate the role of the sex chromosome complement on early transcriptional biases, we derived a panel of male and female hybrid mouse ES cell lines from reciprocal crosses between the C57BL/6 and CAST/EiJ mice and performed a genome-wide RNA expression comparison of 40,XX, 40,XY, and 39,X lines. This experimental design allowed us to gauge the impact of strain effects and focus on sex-specific expression. We found a substantial number of coding and non-coding RNAs that are differentially expressed between male and female ES cells, including RNAs encoding critical transcription factors and proteins involved in DNA methylation, histone modifications, and chromatin remodeling. Gene ontology analysis shows that male- and female-enriched RNAs code for proteins involved in distinct pathways, with male cells exhibiting a transcriptome more poised for differentiation than female cells. In addition, by comparing wild-type to 39,X cell lines, we identified genes dependent on either the X or the Y chromosome.
Methods
The experimental strategy is outlined in Additional file 1: Figure S1.
Embryonic stem cell derivation and culture
F1 hybrid blastocysts from natural matings were obtained at embryonic day 3.5 from reciprocal crosses of mouse substrains C57BL/6J and CAST/EiJ, denoted B and C hereafter; cell lines are designated as BC or CB, with the maternal line symbolized first. Blastocysts were cultured individually on mouse embryonic fibroblasts (MEFs) in gelatin-coated tissue culture plates and used to establish single, independent embryonic stem cell lines. Each line was maintained in ES cell culture medium (DMEM, 15% FCS, 1 mM sodium pyruvate, 2 mM L-glutamine, 1% nonessential amino acids, 0.1 mM 2-mercaptoethanol, and 1000 U/ml mouse leukemia inhibitory factor (LIF)) in 5% CO2 at 37 °C. Blastocysts hatched from the zona pellucida and adhered to the feeder layer after 2 days. After 4–6 days, the inner cell mass outgrowth was passaged using trypsin.
All mice were housed at the Fels Institute for Cancer Research and Molecular Biology animal facility. When required, euthanasia was performed by CO2 inhalation and animal sacrifice for blastocyst isolation was done by cervical dislocation. Animal studies for this project have been approved by the relevant institutional review board (protocol number 4472).
Sex determination
ES cell lines were sexed by the presence or absence of Sry and a restriction digest of Xist using AflIII which selectively digests the C57BL/6 allele at rs225909365. After polyacrylamide gel electrophoresis (PAGE), resulting bands were quantified by densitometry analysis (GelAnalyzer2010 software) to confirm the presence of the maternal and paternal X chromosomes in female cells.
Karyotyping
Sequenced lines were karyotyped at or near the passage from which RNA was obtained for library preparation to ensure chromosomal stability. Metaphase spreads were prepared according to previously published protocols [40]. A 100× oil immersion scope was used to obtained images of the spreads. Counting of > 100 cells was performed by separate individuals and compared for accuracy. The XY and XX cell lines possessed the diploid complement of 40 chromosomes in 85–90% of spreads. The two XO lines showed 39 chromosomes in 87–90% of spreads.
RNA sequencing and Transcriptome analysis
Male, female, and aneuploid ES cells are denoted XY, XX, and XO hereafter for brevity. Fourteen (six XY, six XX, and two XO) early passage independent cell lines were used for RNA sequencing. Cell morphology and growth rates were consistent across lines at the time of collection. RNA isolation was performed using Roche’s High Pure RNA Isolation kit for the undifferentiated lines following two consecutive 60-min MEF-depletion steps. Isolated RNA was treated with TURBO™ DNase following the kit instructions from Ambion. Bioanalyzer electrophoresis as well a negative reverse-transcriptase and PCR (RT-PCR) were performed to ensure integrity and purity of the isolated RNA and absence of DNA contamination. One microgram of RNA from each line was used to generate a library according to Illumina TruSeq® Stranded Total RNA sample preparation guide. Lines were prepared in batches of six and then pooled and submitted for sequencing through a single flow cell at Fox Chase Cancer Center Sequencing Core using HiSeq 2500 single end reads of 50 bp. One to two percent PhiX was spiked in at the time of sequencing as an additional quality check. To identify differences in gene expression, sequenced reads were aligned to mm9 genome assembly using TopHat 2 software suite [41]. To determine differential gene expression, we used the Cufflinks software suite [42]. We used change p < 0.01 after correction for multiple testing (false discovery rate, FDR) for significantly differentially expressed genes. We used R software with Cummerbund packages for downstream analysis and visualization of the RNA-seq output including but not limited to hierarchical clustering and principal component analysis. For the heat maps, we isolated the most stably expressed genes by selecting the genes for which expression levels fell within the first quartile of mean normalized standard deviation between biological replicates. Assessment of pluripotency was performed using genes curated from published interactions, namely PluriNetwork [43] and PluriNet82 [44] with the addition of dynamically expressed genes. Cytoscape [45] was used for network visualization. Functional annotation was performed using DAVID (https://david.ncifcrf.gov/home.jsp) with a gene list cutoff of FDR < 0.05 [46, 47]. Visualization of functional output and gene ontology terms was generated through GO-plot using modified Fisher exact p value cutoff of < 0.1 [48].
Quantitative PCR (qPCR) validation
Genes of interest showing differential expression were verified. The analysis was performed on cDNA generated using SuperScript™ II from RNA from multiple lines, including but not limited to the ones involved in the initial sequencing set. Relative gene expression was assessed using PowerUp SYBR Green Master Mix from Thermo Fisher and normalized to β-actin on Applied Biosystems StepOnePlus Real-Time PCR System. Some genes that showed no statistically significant difference in the training set were also tested to further confirm the validity of the RNA sequencing results (Additional file 2: Table S1).
Luciferase assays
The reporter plasmids with enhancers responsive to Prdm14 and Trim24 cloned into a pGL3-Oct4 promoter vector (Promega) were generously provided by Richard A. Young [49]. Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommendations. Testing was performed using three biological replicates from each cell line (XX, XY, and XO). ES cells were seeded onto a 12-well plate and then transfected with 800 ng of the reporter plasmid with or without the enhancer and 16 ng of the Renilla luciferase reporter (Promega) for 24 h at 37 °C. Firefly and Renilla activity were measured according to the instructions for Dual-Luciferase Reporter Assay System using a Glomax® Multi-Detection System (Promega). The relative luciferase activity was calculated by dividing Firefly luciferase by Renilla luciferase activity. Sequences were modified using primers designed on NEBaseChanger v1.2.5, and a protocol for mutagenesis using Q5® Hot Start High-Fidelity reaction components followed by digested with DpnI and subsequent ligation and transformation into competent bacteria. Successful deletion or scrambling of the Prdm14-responsive motifs was verified by sequencing (Eurofins Genomics) and alignment to plasmids with Geneious version 6 (http://www.geneious.com) [50].
Long non-coding (lnc)RNA interrogation
To evaluate differential long non-coding RNA expression between the male and female ES cells, sequences were aligned to mm10 and transcripts were identified with the NONCODE database (https://www.ncbi.nlm.nih.gov/pubmed/26586799). The lncRNA genes that overlapped with coding genes and were expressed from same DNA strand were filtered out from downstream analysis.
Transcription factor motif analysis
The differentially expressed genes between male and female cell lines (FDR ≤ 0.05) were used for de novo motif discovery with parameters set for 5000–1000 bp upstream of the transcriptional start site in HOMER (http://homer.ucsd.edu/homer/) (Additional file 3: Table S2).
Allele-specific RT-PCR
After reverse transcription, the Cdkn1c transcript was amplified using Ruby Taq Master Mix (Affymetrix - #71191). Following PCR, restriction digest was performed with a Taq I (New England Biolabs) and products were run on 7% PAGE [51]. The primers for Cdkn1c were 5′-CGGACGATGGAAGAACTCTGG-3′ and 5′-TATACCTTGGGACCAGCGTACTCC-3′.
Results
Sex-specific expression in ES cells
To determine whether differential expression occurs during early development, we used mouse ES cells cultured in serum and LIF, denoted as serum/LIF, as a model for peri-implantation embryos. This medium was preferred because culture of ES cells with inhibitors of Mek1/2 and Gsk3β in conjunction with LIF (2i/LIF) leads to widespread reduction in DNA methylation, including erosion of methylation of imprinting control regions (ICRs), compromising the ability of the cells to contribute to development [30, 52–55]. We derived stable male (XY), female (XX), and 39,X (XO) ES cell lines from blastocysts originated by reciprocal crosses between C57BL/6 (B) and CAST/EiJ (C) mice (designated as BC and CB, with maternal strain indicated first; male cells, BCM or CBM; and female cells, BCF or CBF). We analyzed the transcriptomes by RNA sequencing (RNA-seq) from three early passage cell lines of each cross and sex and two early passage XO cell lines derived from a BC cross (denoted BCO). We detected 14,766 transcripts and compared the data to quantitatively assess the differentially expressed genes. Alignment and transcript coverage was similar between all samples assayed.
We first compared transcriptomes between sexes within each cross to determine sex-specific differentially expressed genes. The ES cell lines showed hierarchical clustering by sex chromosome complement (Fig. 1a, b). Evaluation of the chromosomal distribution of the differentially expressed genes shows that the differences between XY and XX lines are primarily derived from expression originating from autosomal regions (Fig. 1c). The XO lines (BCO) more closely resembled the XY lines (BCM) than the XX lines (BCF) of the same cross, which is illustrated in the principal component analysis generated using differentially expressed genes at a FDR < 0.01 (Fig. 1d).
Fig. 1.
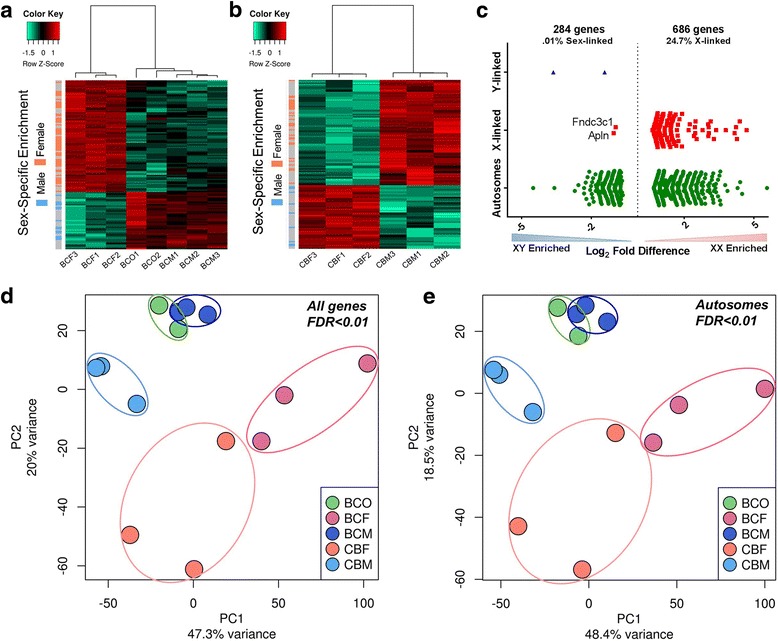
Identification and biological replication of sexually dimorphic genes in mouse ES cells. a, b Transcripts were defined as dimorphic from RNA-seq data using a genome-wide corrected FDR < 0.01 and standard deviation between biological replicates of less than 10%. Genes with sex-specific biases independent of strain are indicated on the y-axis. Sexually dimorphic expression in BC ES cells (a) showed clustering of the two XO lines with the three XY lines, separately and distinctly from the three XX lines. CB ES cells (b) showed clustering by sex chromosome complement with the three XY grouped together separately from the three XX lines. c Chromosomal distribution of sexually dimorphic genes when comparing all six XX and six XY ES cells at FDR < 0.01. d Principal component analysis (PCA) of differentially expressed genes at FDR < 0.01 from RNA -seq of ES cells shows distinct clustering of each cell line with close association of lines of the same strain and sex, with the exception of the two XO lines which highly overlap with the three XY lines of the same cross (BC). e Exclusion of sex chromosome-linked expression in the PCA at FDR < 0.01 shows maintenance of segregation of the ES lines based on strain and sex affirming that autosomal expression alone accounts for these biases
Because close to 25% of the differential gene expression originated from the sex chromosomes, we performed a principal component analysis excluding sex-linked genes at a FDR < 0.01 to evaluate if the expression levels from the sex chromosomes alone were driving the difference seen in the hierarchical clustering. Interestingly, autosomal gene expression differences alone are enough to segregate the ES cells by sex, with Fig. 1e showing segregation by cross and then by sex chromosome composition with the CBM and BCF lines showing the largest degree of separation. This result illustrates that the sex chromosome composition influences autosomal gene regulation and expression in a differential and detectable manner.
A limitation of some mouse studies is the use of a single inbred mouse line to identify sex-specific effects. Polymorphisms are capable of skewing gene expression and as there is mounting consensus for the use of multiparent strains to more closely approximate human diseases [56], we compared all XY to all XX ES lines. At FDR < 0.05, we detected over 1500 genes differentially expressed between XY and XX lines (Fig. 2b). Over a thousand genes were more highly expressed in XX cell lines. XY lines were enriched for epithelial development and differentiation as well as cell-cell junctions (Fig. 2a). Interestingly, genes with higher expression in female ES cells were enriched for terms associated with cell cycle, meiosis, nuclear chromosome, and microtubule organization (Fig. 2c). These results suggest that male cells are more poised for lineage determination even before differentiation induction. It also supports the hypothesis put forth previously that the double X chromosome dosage stabilizes pluripotency [27].
Fig. 2.
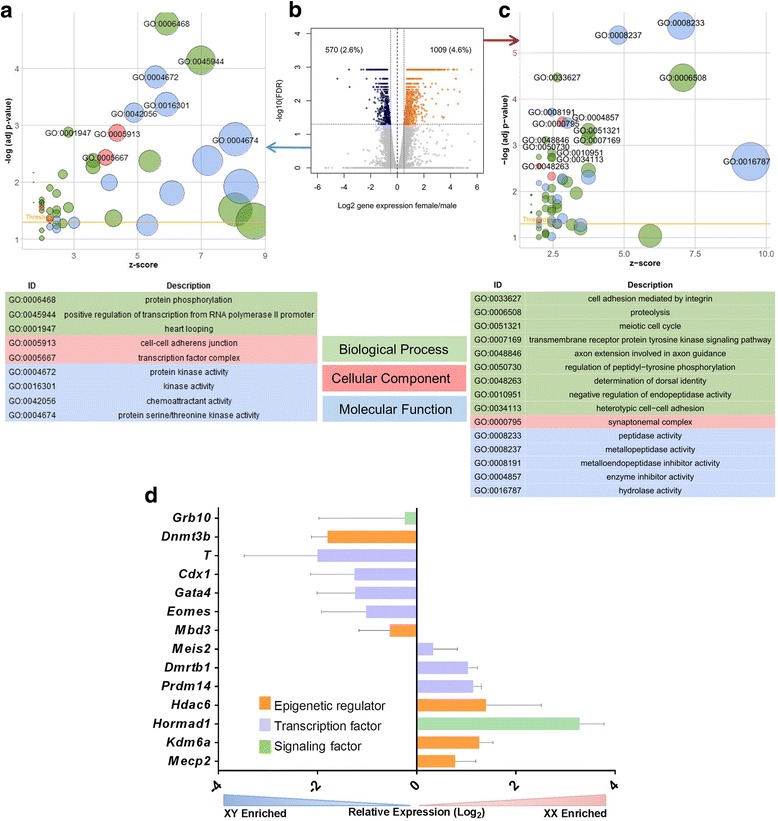
Gene ontology of differentially expressed genes between XY and XX ES cells. a, c Upregulated biological process, molecular function and cellular components between XY (a) and XX (c) ES cells. The XY lines had increased expression of genes related to early development (heart looping) and kinase activity. The XX lines showed increased expression of meiotic cell cycle-associated genes and peptidase activity. x-axis: z-score of associated terms, which indicates degree of upregulation; y-axis: significance of term. b Volcano plot showing selection of the differentially expressed genes between XY and XX lines at FDR < 0.05 (blue dots, higher in males; orange dots, higher in females). d qPCR validation of genes detected in the RNA sequencing dataset illustrating a sex-biased expression pattern of transcription factors, epigenetic enzymes and key signaling factors
To trace regulatory differences in XX and XY cells, we focused on transcription factors and epigenetic and chromatin remodeling factors (TFs and ERFs, respectively). At FDR < 0.01, 49 TFs were enriched in XX cells, including 13 X-linked factors: Klf8, Hmgb3, Rhox1, Rhox9, Elf4, Zfp182, Zfp449, and Aff2, among others. Autosomal TFs enriched in XX cells included Dmrtb1, Meis2, Prdm14, and interestingly, 11 genes of the Hox (homeobox) family. ERFs more highly expressed in XX cells (22) included Mecp2, Apobec2, Parp3, and Kdm6a (previously reported [26]) (Additional file 4: Table S3).
In XY cells, 59 TFs had significantly higher levels compared to XX cells. These included Eomes, Prdm6, Gata4, Mef2b, and Ybx2. XY cells had higher RNA levels of 20 ERFs, including Dnmt3a, Dnmt3b, Hdac5, Mbd3, Cxxc1, and Uty (Kdm6c) (Additional file 4: Table S3). We evaluated expression of a subset of genes of interest including TFs, ERFs, and signaling molecules with qPCR assays, to confirm the RNA sequencing data (Fig. 2d). These genes were selected based on their associations with key cellular pathways and processes.
Regulatory network for pluripotency in XX and XY ES cell lines
The XY ES cell lines exhibited enrichment for pathways associated with epithelial development and morphogenesis and seemed more poised for differentiation. We assessed the transcriptomes of all XX and XY ES cell lines to confirm that they maintained expression of the genes commonly associated with the minimal pluripotency network [57, 58]. All of the pluripotency-associated transcription factors (TFs) were expressed. However, two of them were differentially expressed between male and female cells, with Tcf3 expressed at higher levels in the males and Zfx higher in the females (Fig. 3a). As an X-linked gene, Zfx was expected to be 2-fold higher in female cells. Although the Y chromosome homolog, Zfy, could compensate this imbalance, it was not expressed in male cells. We investigated the regions upstream of Zfx and Tcf3 to see if they are regulated by TFs that are also differentially expressed by combining the histone modification profiles typical of enhancers from ENCODE data at the USCS genome browser (http://genome.ucsc.edu/) [59–63] and the conservation by comparative genomics from the dCODE website (http://www.dcode.org/) [64, 65]. The most likely enhancer for Zfx has binding sites for Mef2 TFs, one of which (Mef2b) was expressed at higher levels in female cell lines (Additional file 5: Figure S2A). A candidate enhancer for Tcf3 has binding sites for Gata4, present at higher levels in the male cells (Additional file 5: Figure S2B).
Fig. 3.
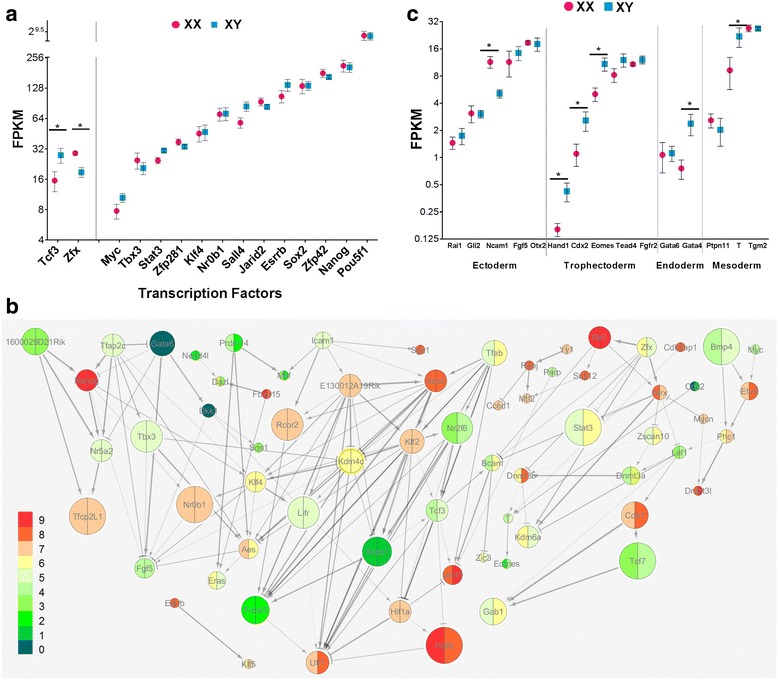
Pluripotency and lineage-specific factors in XX and XY ES cells. a Fragments per kilobase per million (FPKM) values from RNA sequencing of six XX and six XY ES cells showing expression of 15 of the minimal network of pluripotency transcription factors. Two transcription factors were differentially expressed between the XX and XY cell lines: Tcf3 was 1.8-fold greater in XY, and Zfx was 1.5-fold higher in XX. *FDR < 0.05. b In the expanded pluripotency network, there are detectable and distinct differences in expression of key genes between XX and XY cells. Node size reflects the clustering coefficient and line thickness denotes the statistical significance of the interaction with arrows indicating a positive interaction and T bars indicating inhibition. Colors represent relative expression levels in RPKM for XX (left) and XY ES cells (right). c Differences in expression of lineage-specific markers. Note that expression levels were lower compared to levels of pluripotency-associated genes. The ectodermal lineage-specific marker Ncam1 was 2-fold higher in XX lines. The XY cells were enriched for Hand1, Cdx2, Eomes, Gata4, and T when compared to the XX lines with fold change of 2.6, 2.5, 2.4, 3.7, and 2.5, respectively. *FDR < 0.03
Because we saw differences in the minimal network, we decided to assess additional genes curated from published interactions, namely PluriNetwork [43] and PluriNet82 [44]. Focusing on dynamically expressed genes and the core regulators of Nanog and Sox2, we generated a network of 90 genes. From this set of genes, we performed pathway analysis using the RNA-seq data generated from all of the XX and XY ES cell lines and found statistically significant interactions involving 69 of the genes, FDR < 0.05 (Fig. 3b).
Differential expression of Prdm14 in ES cell lines leads to differential enhancer activity
Prdm14 plays a key role in pluripotency [66, 67], as well as in X chromosome reactivation in reprogramming [68]. We observed that Prdm14 had significantly higher expression levels in XX ES cells (Fig. 2d). Prdm14 generally recognizes DNA motifs distant from transcriptional start sites, overlapping with candidate regulatory sequences [69], including enhancer clusters that drive high-level expression of genes that are key regulators of cell identity [49, 70]. To see if higher Prdm14 levels correlated with expression of its targets, we checked the overlap with pre-existing data on Prdm14-regulated genes [67, 71]. Indeed, genes repressed by Prdm14 such as Dnmt3b, Cdkn1c, FoxA2, and Gata4 exhibited lower expression in XX cells, whereas activated genes, such as Prdm14 itself, Meis2, Dazl, Mitf, and Zeb1 were more highly expressed. A substantial number of genes from the knockdown experiments, however, did not correlate with the higher Prdm14 levels, possibly because of the differences in culture media. Binding data from tagged Prdm14 ChIP-seq experiments, however, confirmed that Prdm14 is present in the vicinity of Dnmt3a, Dnmt3b, Gata4, and Meis2 [67].
To assess if Prdm14 dosage differences in XX and XY ES cells correlated with differential enhancer activity, we performed luciferase assays in three biological replicates of XX and XY ES cells with vectors containing known enhancers responsive to Prdm14 and Trim24 [49] (Fig. 4a). Luciferase activity of the plasmid containing the Prdm14-responsive enhancer was higher in the XX ES cells (Fig. 4b), highlighting that there are sex-specific differences in autosomal gene regulation very early in development. This is the first report of such biased enhancer response. Using the consensus binding motif derived from ChIP studies [67] and an algorithm for binding potential (Geneious version 10.0.2, http://www.geneious.com) [50], we identified and tested three potential Prdm14 motifs in the enhancer. Deletion and scrambling of one of these motifs (Fig. 4c) greatly reduced luciferase activity and abolished the sex-specific differences. Trim24 was expressed similarly between the XX and XY ES cell lines and served as a negative control. Enhancer activity in XO ES cells mimicked the XY cells. These results suggest that, in addition to chromosomal environment, TF dosage contributes to the expression outcome.
Fig. 4.
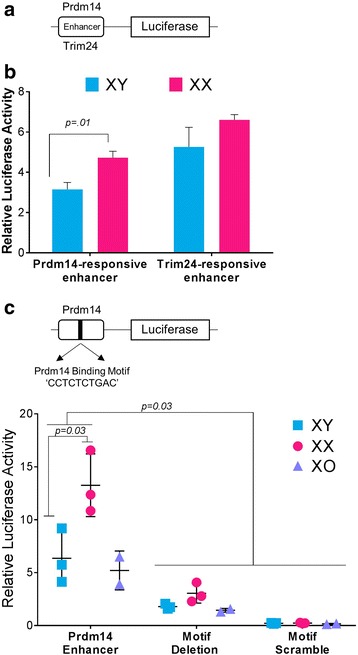
Prdm14-associated enhancer response correlates with Prdm14 dosage. a Schematic of vectors used in luciferase assays. b XX and XY ES cells were each transfected with vectors containing either a Prdm14- or Trim24-responsive enhancer. Luciferase activity was higher in XX ES cells for the Prdm14-responsive enhancer. Trim-24 mRNA levels were equal in XX and XY ES cells and the Trim24-responsive enhancer showed no difference in enhancer activity. Error bars represent SEM. Samples were normalized to an empty vector control. c Top, schematic of the luciferase plasmid and the binding motif for Prdm14. Both the deletion and scrambling of the motif reduced the sex-specific differences and decreased overall luciferase activity in all biological replicates
Differentiation markers in XX and XY cells
The variation in expression of pluripotency-associated genes prompted us to look at markers of differentiation. We evaluated the data using the 15 lineage-associated markers derived from the ESCAPE database [72] and found that XX lines were enriched for the ectodermal lineage marker Ncam1 (2-fold), while the XY and XO lines were enriched for trophectoderm markers Hand1, Eomes and Cdx2, endodermal marker Gata4, and the mesodermal marker T (Brachyury)(FDR < 0.04) (Fig. 3c).
We considered whether genes were differentially expressed in male and female cells because the XX cells are slightly delayed, as reported in a previous study [27]. If the observed differences were due to an offset in developmental stage, we would expect genes more highly expressed in male cells to be more typical of differentiated ES cells. Inspection of published data on ES cell differentiation into cardiomyocytes [73] showed that this was not the case. For example, Pou3f1, Cdx1, Mycn, Mef2b, and Dnmt3b, among others, are higher in male cells, but downregulated upon differentiation. Thus, higher expression levels of these genes are specific to undifferentiated XY ES cells. If the expression pattern in female cells is due to developmental delay, genes expressed more highly relative to XY cells would be downregulated upon differentiation. On the contrary, genes expressed more highly in females, such as Meis2, Zeb1, Apobec2, and Mecp2, were upregulated during differentiation.
Furthermore, we compared our data to the list of genes with delayed XX expression obtained by microarray analysis in one male and one female ES cell line upon differentiation into embryoid bodies [27]. Only 104 (8%) out of 1284 genes from that list overlapped with our list of RNAs enriched in XX cells. These data confirm that the vast majority of expression biases we observed are not due to stage-specific patterns but are bona fide sexual dimorphisms. Our results also suggest that analysis of six cell lines for each sex by RNA sequencing is more powerful and sensitive, allowing identification of a greater number of differentially expressed genes.
X chromosome-linked expression
X chromosome inactivation (XCI) in females is a crucial event during mammalian development [58, 74]. In ES cell lines maintained in serum/LIF conditions, a minimum of 94% of the cells consistently show two active X chromosomes [27, 75]. Thus, many X-linked genes are expected to be expressed 2-fold higher in female than in male cells. Not surprisingly, 25% of female-enriched genes were on the X chromosome. Unexpectedly, however, close to 35% of all the X-linked genes with higher expression in XX cells showed more than a 2-fold difference. One of these, Srpx, which was 5.6-fold higher in the XX cells, is a tumor suppressor gene that is downregulated in a number of human malignancies, including prostate, colorectal, and neuroendocrine cancers [76, 77]. Other X-linked genes with RNA levels more than 2-fold higher in female cells were Rhox1, Gm9, and a large cohort of genes from the Xlr family associated with immune-related processes. These findings suggest that there is an additional layer of regulation directing the expression of these genes. Interestingly, Xlr3c and Xlr3a are clustered, as are Rhox1 and Gm9, which may indicate common regulatory mechanisms (Additional file 6: Table S4).
Also surprising was the finding that close to 50% of all X-linked genes expressed in ES cells had equivalent levels in both male and female cell lines, before X chromosome inactivation had occurred.
Two X-linked genes were expressed at higher levels in the male cells, with FDR < 0.01 (Fig. 1c). One of these, Apln, has highly conserved regions upstream of the promoter with chromatin features consistent with enhancer activity. Analysis of these sequences yielded motifs for TFs Hand1 and Lef1, both of which were expressed more highly in male ES cells (Additional file 5: Figure S2B).
In mice, 3% of genes escape X chromosome inactivation (XCI) and remain active, depending on the tissue [78, 79]. We asked if those genes are more highly expressed than other X-linked genes in XX lines than in XY lines before ES cell differentiation, perhaps counteracting the chromosome-wide silencing mechanism. We did not find that to be the case, suggesting that, if those genes are pre-marked for later escape, other mechanisms are involved (Fig. 5a; Additional file 7: Table S5). Whether these genes maintain comparable levels to their XY counterparts after XCI is still under debate [80–82]. Escapees are also not overexpressed in XX versus XO ES cells (Fig. 5b).
Fig. 5.
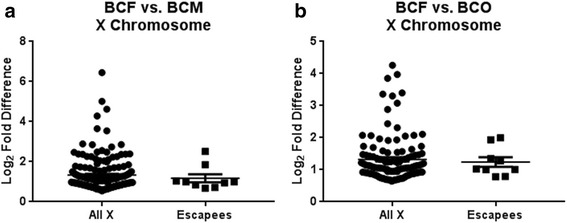
Expression of known X chromosome escapees compared to the expression of other X-linked genes that showed enrichment in XX ES lines using a Mann-Whitney test. a Comparison of expression levels of genes escaping XCI between BCF and BCM lines shows no statistical difference (p = 0.6420). b Expression levels of X-linked genes was compared between BCF and BCO, showing no statistical difference (p = 0.9747)
Identification of promoter signatures for differentially expressed genes
The promoter architecture of XX and XY differentially expressed genes were analyzed for enrichment of TF motifs from the transcriptional start site (TSS) − 1500 to + 500 and − 5000 to + 500 bp (Additional file 3: Table S2). For genes exhibiting higher expression in XX ES cells, motifs for Arid5a, Nkx2–5, HoxA2, Smad3, Tead2, HoxA5, Mecom, and Zfx, among others, were enriched (p ≤ 10− 8). In fact, RNA levels for HoxA2, HoxA5, Mecom, and Zfx were higher in XX cells. For genes with higher expression in male cells, motifs for Runx1, Foxq1, Hic1, Prrx2, and Tcf3, among others, were more prominent (p ≤ 10− 8). However, only Tcf3 was higher in XY cells. Many TFs have overlapping and redundant binding potential, so combinatorial effects as well as accessibility could be contributing to the sex-biased expression patterns.
Detection and differential expression of long non-coding RNAs
The RNA-seq data was assessed for differences in expression of lncRNAs. Comparison between all samples assayed showed the highest degree of variation between BCF and CBM, which coincides with the variation seen in coding genes (Fig. 6a). Comparison of lncRNA expression showed segregation first by direction of cross followed closely by sex chromosome complement (FDR < 0.01) (Fig. 6b, c). As with coding genes, the XO cell lines (BCO) were more similar to XY lines (BCM) of the same cross type, with only 10 lncRNAs showing differential expression (Fig. 6d). Of note, we detected more differences between BCM and BCF (412 lncRNAs) than between CBM and CBF (173 lncRNAs) (Fig. 5a).
Fig. 6.
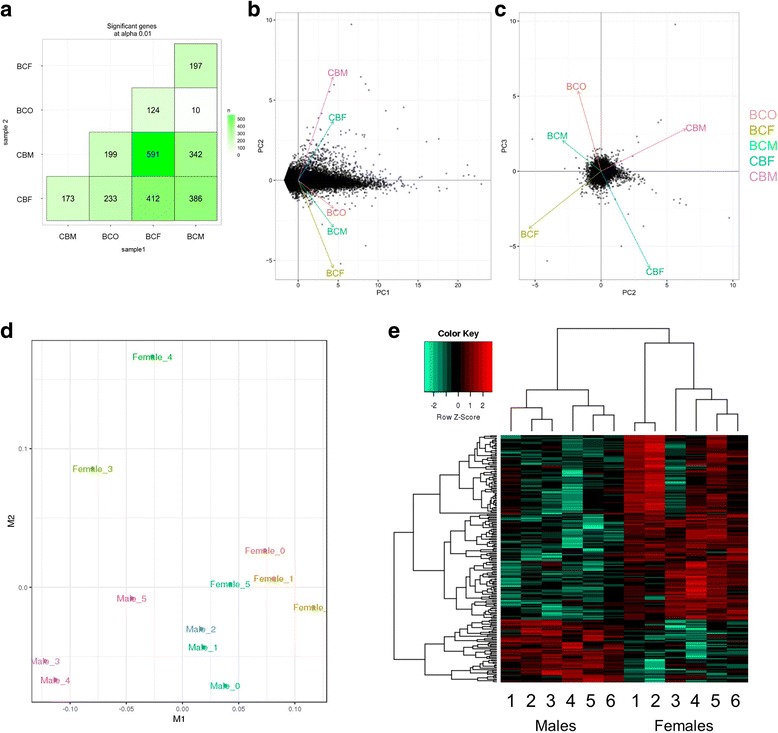
Identification and biological replication of sexually dimorphic lncRNAs identified in ES cells. Transcripts were defined as dimorphic from RNA-seq data using a genome-wide FDR (alpha) < 0.01. a Sexually dimorphic expression between all conditions tested. The highest degree of variation was seen between BCF and CBM which 591 differentially expressed lncRNAs. b Principal component analysis of lncRNAs shows segregation of cells based on strain of origin. c Plotting principal component 2 against component 3 shows segregation based on sex chromosome complement with BCO aligning more closely to BCM than to BCF, corresponding to the analysis performed using coding genes. d Multidimensional scaling plot showing clustering of the XY and XX lines. M1 corresponds to strain of origin, with M2 showing separation based on sex chromosome composition. e Heatmap showing dimorphic lncRNA expression and clustering of samples based on sex chromosome composition (FDR < 0.01)
To assess sex-specific expression of lncRNAs, we compared all six XY to all six XX ES cell lines. Over 200 lncRNAs were differentially expressed (FDR < 0.05). As with the coding genes, we saw separation based on sex chromosome composition (Fig. 6e).
Imprinted loci: expression and regulation
Unexpectedly, some imprinted genes exhibited higher expression in the male cell lines (Fig. 7) including the maternally expressed Cdkn1c, Phlda2, and Slc22a18. To see whether this was due to loss of imprinting, we performed allelic expression assays and observed monoallelic expression from the appropriate parent of origin allele for the BC cells, i.e., C57BL/6 allele, and maternally biased expression for the CB cells (Additional file 8: Figure S3). Loss of methylation is unlikely to occur in male cells, since they are more highly methylated than XX ES cells [83]. In the case of Cdkn1c and Phlda2, the higher expression could be due to the male cells being slightly ahead of the female cells in the lineage determination path, since both of these genes are greatly induced upon differentiation, but this is not the case for Slc22a18. Because these genes are clustered, a more likely explanation is that they are subject to an additional regulatory mechanism active on the maternal allele, directly or indirectly dependent on sex chromosome composition. Grb10 RNA levels, on the other hand, were only higher in the male cells of the CB cross. Strain differences present an opportunity to discover the as yet unknown enhancers directing expression of these genes during development.
Fig. 7.
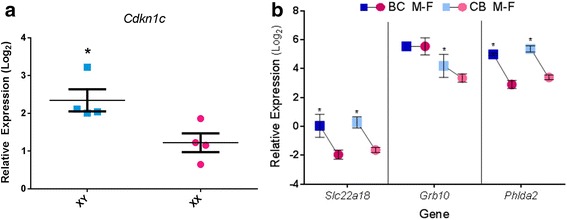
Imprinted gene expression. a Differential expression of imprinted gene Cdkn1c validated in four XX and XY lines. Error bars represent SEM. b Relative expression levels three additional imprinted genes. Comparisons are within each cross. Expression levels of these imprinted loci were generally greater in males than in females. Asterisk denotes significance (FDR < 0.01)
Comparisons with currently published datasets
We compared our results with a recent publication that analyzed expression differences between male and female ES cells across different developmental stages using single-cell RNA sequencing (scRNA-seq) [75]. We used homologous datasets, i.e., cells cultured in serum/LIF and the comparable cross, i.e., C57BL/6 × CAST/EiJ, because only this unidirectional cross was analyzed in that study. Due to the inherent noise and abundance of zero entries within the data matrix, a common occurrence within scRNA-seq analyses [84, 85], we compared the concordance of direction of gene expression between the two sexes in both datasets with a sign test. Comparison of the differentially expressed genes within our dataset at FDR < 0.05 using an exact binomial test showed a high degree of correlation at a p value of 2.2e− 16 and a 95% confidence interval (CI) of 0.58 to 0.60. To more closely examine the correlation, we compared expression of the 14 genes assessed by qPCR (Fig. 2d) with the single-cell data (Additional file 9: Figure S4) and found a high degree of correlation r = 0.78 (p = 0.001). We also performed a sign test on this subset of genes revealing a strong concordance of expression with a p value of 0.002 and 95% CI of 0.66 to 0.99. Of note, the sign test for Prdm14 agreed with our data, confirming that Prdm14 expression shows a sex-specific bias.
Single-cell RNA-seq [75] was also performed on sexed post-implantation embryos (5.5 dpc). We analyzed the published dataset to determine which genes had a sex-dependent bias (p < 0.01). We then compared with our ES cell data from the comparable cross to see if there were genes that maintained their sex biases. The overlap in female-biased genes between the two datasets was significant by hypergeometric testing (73 genes, p = 0.007), but only 10 male-biased genes overlapped (Additional file 10: Table S6). Among them were the Y-linked genes Ddx3y and Uty and interestingly, Dnmt3b. This shows that female ES cells and post-implantation embryos are more similar than the male counterparts, suggesting that the female 5.5 dpc embryos are still delayed at that time point.
Sex chromosome-dependent expression
We analyzed two XO cell lines that arose spontaneously during ES cell line establishment, both of which were derived from a BC cross (designated BCO). Both aneuploid cell lines expressed all the genes associated with the minimal pluripotency network [57, 58]. BCO lines were most similar to BCM cell lines, with only 116 genes showing statistically significant differences in expression (FDR < 0.05). As was the case for XY cells, XO lines had higher expression of Tcf3 than XX cells, but in addition, they had higher levels of Sall4 and Esrrb.
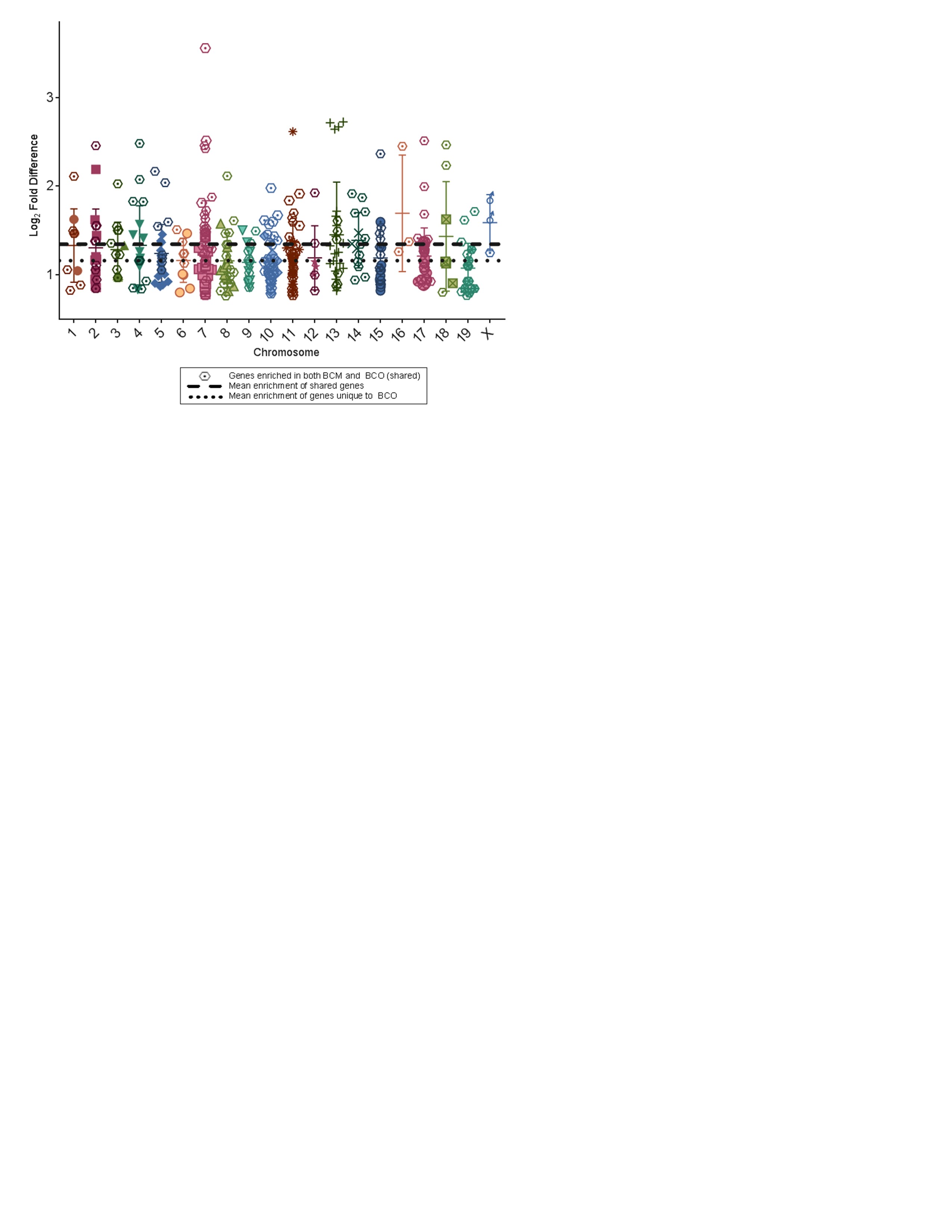
To identify if similar regulatory mechanisms were contributing to the differential expression in BCM and BCO when compared to BCF, we plotted the chromosomal distribution of the shared and unique enriched sets of genes for the male and XO cell lines (Additional file 11: Figure S5). The average proportion of overlap of enriched genes per chromosome relative to the female cells was 48%, with several chromosomes showing greater than 63% overlap, notably chromosome 1, chromosome 14, and chromosome 16. We quantified the variation in expression based on whether the gene was uniquely enriched in BCO or shared with BCM relative to BCF. The genes common to both sets showed statistically higher enrichment than those that did not (p = 7.3e− 7), suggesting that the lack of a second X chromosome is the predominant driver of expression differences in this model.
To better understand the impact of the sex chromosome aneuploidy, we did two-way comparisons between XX, XY, and XO cell lines and identified differentially expressed genes that were exclusive to each pair (Fig. 8a). Eight hundred twenty-four genes were upregulated in XX relative to both XO and XY cells: 129 of these are X-linked. Two hundred thirty-five genes were more highly expressed exclusively in XX relative to XO cells, suggesting RNA levels directly or indirectly dependent on the presence of the two X chromosomes. We expected most of these to be X-linked, but only 45 (20%) are on the X chromosome, highlighting that the bulk of the transcriptional differences are downstream of the sex chromosomes and reflect their regulatory effect on the autosomes. Eight TFs and four ERFs are among the differentially expressed genes, as well as eight genes associated with Turner syndrome (PROCR, NR3C1, INSR, APOB, BMP15, F8, IL6, and WAS) [86–88] and two genes that are haploinsufficient in humans (RAD50 and SLC33A1) [88]. Neither the Turner-associated nor the haploinsufficient genes are overexpressed in male versus female ES cells, indicating that they are specific for the aneuploidy. Additionally, two imprinted genes, the maternally expressed Rian and Grb10 had higher levels in XX versus XO lines.
Fig. 8.
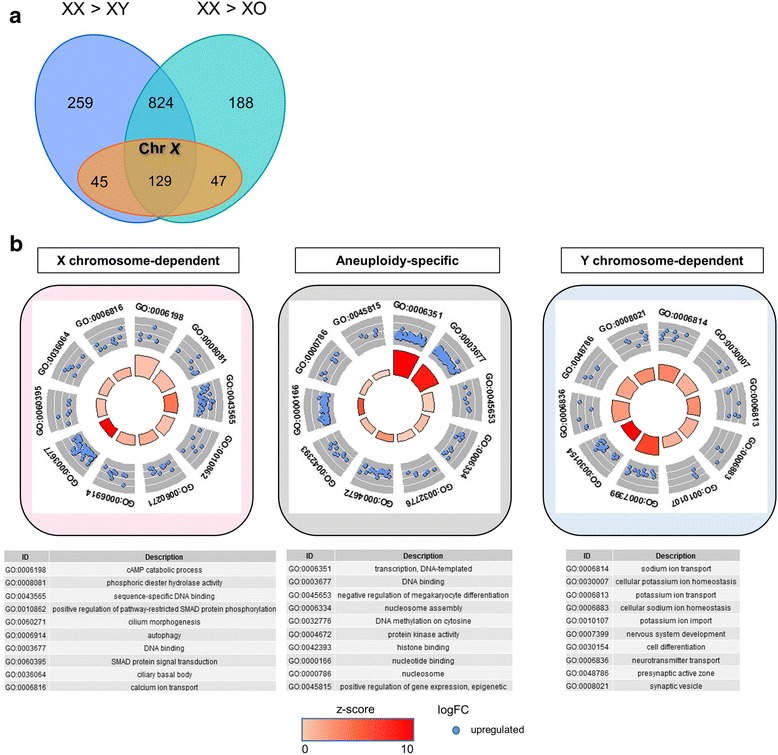
Differential gene expression analysis across karyotypes. a Expression comparisons between XX, XY, and XO ES cell lines, showing the extent of shared differences in pair-wise comparisons for X-linked and autosomal genes. b Gene ontology of genes showing X chromosome dependency, Y chromosome dependency, and aneuploidy-specific effects. Genes showing X chromosome dependency, aneuploidy-specific effects, or Y chromosome-dependency fall within unique ontology terms. Visualization performed using the GO-plot package in R. The outer circle shows a scatter plot of the log-fold change for the assigned genes within each distinct gene ontology term. Selection for enrichment resulted in blue dots only, which indicate upregulation. The inner circle is a bar plot with the height of the bar illustrating the p value of the term and the color representing the z-score, which evaluates the direction of the fold change of each of the genes within the assigned term and provides a relative scale for enrichment
X chromosome dosage also exerts strong repressive effects: hundreds of genes were upregulated in XO cells relative to XX lines. Four hundred thirty-four genes were more highly expressed in XO uniquely versus XX cell lines. Fifty-five TFs and 25 ERFs were among them. These genes showed enrichment for 50 ontology terms at FDR < 0.05 which included regulation of biosynthetic processes as well as terms associated with epigenetic regulation involving nucleosomes, chromatin, and DNA binding (Fig. 8b). Included in this group are four genes commonly associated with Turner syndrome (WNT4, MTHFR, FGFR3, and MEN1) [88]. The Turner-associated genes are not higher in XY versus XX cells, indicating that X chromosome dosage alone does not explain their derepression.
XO cell lines had very similar expression profiles to XY cells. Consistent with this, only 15 genes were more highly expressed in the XY cells, one mapping to the Y chromosome (Eif2s3y). Based on our data, we infer that genes that were higher in XY than in both XX and XO cells (294 genes) are dependent on the presence of the Y chromosome.
Discussion
Sex determination in mammals is classically defined as the developmental decision of whether the gonad becomes a testis or an ovary [89, 90]. There is long-standing evidence, however, that phenotypic differences between males and females precede the formation of the gonads [25, 35].
The findings presented in this study demonstrate that sexually biased genes expressed in embryonic stem cells integrate distinct regulatory networks very early in development. Transcription factors (TFs) and epigenetic and remodeling complexes (ERFs) are among the hundreds of differentially expressed genes, including TFs belonging to the pluripotency network. We found that an enhancer responsive to Prdm14 is more active in female ES cells, corresponding to the higher Prdm14 levels. We also detected more than 300 differentially expressed lncRNAs. Only a small fraction of differences can be accounted for by a developmental delay imposed on female cells by the need to inactivate an X chromosome. We also analyzed genes that were differentially expressed between 40,XX and 39,X ES cells and found multiple differences, some of which may account for phenotypes observed in Turner syndrome. These findings advance on previous work [26–28] by defining a signature for mouse ES cells in conditions that mimic the peri-implantation stage, by analyzing six cell lines for each sex and by highlighting the differences with monosomic X cell lines. In addition, we detect genotype-sex interactions which will provide a rich resource for understanding the effects of genomic variation and the need for canalizing the developmental processes essential for both sexes [91].
Sex-specific expression in mouse ES cells
We chose the ES cell serum/LIF system and analyzed 14 independent cell lines to robustly detect sex-specific differences. Because the ES cells in serum/LIF represent a less “naïve” state, with higher methylation levels, we reasoned that the transcriptomes from male and female cells would better reflect epigenetic differences dependent on the sex chromosome complement. In fact, we found many differentially expressed genes, including TFs belonging to the pluripotency network.
Together with results from previous reports, our data support the hypothesis that the sex chromosomes affect autosomal gene expression and epigenetic features, which can determine the sex-specific transcriptome later in development. In ES cells, the sex chromosome complement can influence gene expression before and after differentiation due to (1) gene activity from the Y chromosome in males and dosage differences in X-linked genes before X inactivation in females; (2) imprinted X-linked genes, active from the paternal, but not maternal, X chromosomes; (3) genes that escape XCI and are not dosage compensated when ES cells are differentiated; and (4) the process of XCI itself, which can substantially alter the availability and composition of epigenetic complexes in female relative to male cells. In addition, there is evidence that XCI per se delays the differentiation of female ES cells [27]. We suggest that this delay opens a window of opportunity for establishing epigenetic modifications unique to the female epigenome.
Dosage of transcription factor Prdm14 correlates with enhancer activity
In the light of the hypothesis, we focused on differentially expressed TFs and ERFs because they have the potential to imprint the epigenetic landscape and affect subsequent transcriptional activity. Because TFs often act synergistically or in multimers, variations in their dosage can quantitatively fine-tune the response of their target genes and can even expand the range of their targets. In addition, many epigenetic modifications are established by protein complexes and dosage differences in individual components can alter their stoichiometry or composition [92, 93]. In fact, our results indicate that the dosage of Prdm14 partially determines the activity of a developmentally regulated enhancer. This sequence, responsive to Oct4, Nanog, and Prdm14, induces higher transcriptional activity in the cells that have higher Prdm14 levels, i.e., the female cells, even though Oct4 and Nanog are not differentially expressed. As biological validation of regulatory elements progresses, it will be interesting to test the response of other sequences to the TF sex-specific biases.
Prdm14 can both activate and repress gene expression depending, at least partially, on its partner proteins [66]. Some targets of Prdm14 have been identified by siRNA knockdown [67] and gene knockout [71]. Higher levels of Prdm14 in XX ES cells correlates with repression of Dnmt3b, Cdkn1c, FoxA2, and Gata4 and activation of Dazl, Mitf, and Zeb1, indicating that dosage is a factor in regulation of some of its targets. Indeed, our luciferase assays identified a motif responsive to Prdm14 dosage.
Not all genes exhibited the differences in response to Prdm14 predicted by the knockdown studies. Clearly, the relationship between levels of a TF and transcriptional outcome in vivo is not linear, and other factors, such as accessibility, motif numbers or combinations, are involved. Variations in the actual sequence of the motif might also contribute to the response to Prdm14.
X-linked genes can be divided into four categories according to their expression levels
The X chromosome is the major contributor to female-enriched gene expression in the ES cells. Of the X-linked RNAs enriched in XX ES cells, there are two categories: one includes those expressed at a 2-fold level compared to the male cells, correlating with the dosage of the X chromosome. The other category encompasses X-linked genes that are upregulated more than 2-fold in the female cells. This indicates that these genes are influenced by an additional layer of regulation. For example, they could be responding to transcription factors that are more active in the female cells. Another possibility is that these genes are expressed more highly from one of the parental X chromosomes because it inherited a more open chromatin at its corresponding regulatory element. Yet a further explanation is that one of the two alleles of these genes is more highly expressed because of regulatory polymorphisms. Further studies to delineate the strain-specific transcriptional differences will help to answer this question.
The third, and most striking category, consists of X-linked genes that are expressed equally between male and female ES cells. Fifty percent of all expressed X-linked genes are in this group. This is not unique to our cells but is also seen in recent scRNA-seq data [75]. Considering that these genes are already dosage compensated, it will be interesting to determine the consequences of XCI on their relative levels.
A very limited number of X-linked genes constitute a fourth category. These are genes more highly expressed in male than in female ES cells, despite the presence of only one X chromosome. These may be responding directly or indirectly to genes expressed from the Y chromosome. Further studies will shed light on the mechanisms involved in this expression pattern.
Differentiation markers are sex-specific in ES cells cultured in serum/LIF
A previous study comparing the transcriptomes of ES cells cultured in serum/LIF versus 2i found that whereas the pluripotency genes were expressed at similar levels in both conditions, cells in serum/LIF exhibited higher expression of differentiation markers [94]. This is because these cells are in a metastable condition, balanced between pluripotency and lineage determination [30]. Interestingly, our data show that ES cells express certain differentiation markers, but they also highlight that male and female cell lines exhibit distinct profiles. It will be exciting to elucidate how the sex-specific regulatory networks connect to the dimorphisms in lineage determining molecules.
Comparison with single-cell RNA-seq and human data
Our data correlated well with a comparable dataset generated from single-cell RNA-seq [75]. Many differences were observed, however, especially for genes expressed at very low levels. Because transcription occurs in bursts, single-cell expression analysis more closely models the dynamics of RNA pol II activity in each individual cell [95–99]. Master transcriptional factors, however, are generally expressed at low levels. Thus, to determine the presence or absence of a TF, and especially to compare between different cells or tissues, we suggest that bulk RNA extraction and sequencing gives a more robust picture of the expression profile.
Sex biases in gene expression have been reported in human ES cells with microarrays [29]. However, undifferentiated human female ES cells have already undergone XCI and show little overlap with our datasets. Whether this is because of the X chromosome status or species-specific developmental differences (or both) is not clear, but it emphasizes that comparisons between ES cells of divergent species will be necessary.
Levels of some imprinted genes are sex-specific
We revealed a layer of regulation of imprinted genes beyond the differential DNA methylation that maintains monoallelic expression. Even though only the maternal alleles of Cdkn1c, Phlda2, and Slc22a18 were active, they expressed higher RNA levels in the XY cells than in the XX cells, an observation that will be followed up to determine the additional factors regulating these genes.
Sex chromosome-dependent transcriptional regulation
A previous report comparing global expression analysis in four somatic tissues of adult 40,XX and 39,X by microarray had identified genes overexpressed in XX females and revealed transcriptional changes in autosomal genes in response to X chromosome dosage [100]. Comparisons between XX, XY, and XO ES cells allowed us to stratify the transcripts with expression dependent on the presence or absence of the X and Y chromosomes in early development. Genes with sex biases in somatic tissues do not overlap with our data, likely due to the difference in transcriptomes between embryonic and differentiated cells. The XO ES cells are much more similar to XY cells, supporting the previously posited hypothesis that the presence of two X chromosomes delays differentiation due to the need for XCI [27]. Intriguingly, the XO ES cells also exhibited down-regulation of two imprinted genes, the maternally expressed Grb10 and Rian genes.
We also found evidence that the absence of two X chromosomes leads to overexpression of genes associated with Turner syndrome, including Fgfr3 and Wnt4 [88]. However, there are differences between 39,X mice and Turner syndrome patients, with mice presenting a milder phenotype [101]. A recent comparative study of DNA methylation and gene expression differences between 46,XX and 45,X females performed in leukocytes showed no overlap with the differentially expressed genes in our dataset [102], likely because of species differences and because none of the reported genes are expressed during embryogenesis.
Many of the regulatory elements that direct tissue-specific expression have a “poised” chromatin status in early development, i.e., they exhibit both activating and repressing chromatin marks that are “resolved” upon differentiation in one way or another [103]. Thus, epigenetic marks can be latent, with the potential to result in transcriptional outcomes only later in the life of the organism. The same scenario holds for TFs that work in concert to activate gene expression, where one serves as a placeholder until a partner or co-factor appears later on [104]. Evidence is accumulating that disruptions in embryonic stages can have consequences that only become apparent in adults [105, 106]. Integration of our results with previously reported datasets provides insights into how sex-specific differences appear and are translated into later developmental events [107, 108]. In addition, our data will contribute to understanding how male and female embryos differ in their response to the environment, with implications for the impact of maternal behaviors on fetal development.
Conclusions
Sex biases in gene expression lead to critical, health-related sexual dimorphisms that are widely appreciated but still understudied. We show here that differential expression patterns are established in early embryogenesis, before hormonal influence is unleashed. At the level of pathway analysis, these expression differences integrate distinct networks and are dependent directly or indirectly on the sex chromosome complement. Thus, substantial contributions to sex-related differences occur prior to and possibly upstream of gonadal sex determination [109].
Our datasets from XO ES cell lines are an important platform for understanding the impact of sex chromosome aneuploidies on pre-implantation embryogenesis and lineage determination. They will also contribute to refining the direct and indirect mechanisms by which the sex chromosomes interact with the autosomal component of the genome.
Additional files
{kind=link}
Derivation of mouse ES cell lines. F1 hybrid blastocysts were obtained at embryonic day 3.5 from reciprocal crosses of mouse substrains C57BL/6 and CAST/EIJ (designated as B and C, respectively). Blastocysts were cultured individually and used to establish independent cell lines.
Real-time PCR primers.
Transcription factor motifs enriched in promoters of differentially expressed genes in female and male ES cells.
Catalog of transcription factors (TFs) and epigenetic and remodeling factors (ERFs) expressed differentially in female and male ES cells.
{kind=link}
Identification of candidate regulatory elements. (A) Top, graphic display of the conservation profiles for regions upstream of Zfx from Dcode.org [64, 110]. The base genome is mouse. Evolutionarily conserved regions (ECRs) of a minimum of 100 bp conserved above 70% sequence identity are displayed as red (intergenic) peaks, with the x-axis representing positions in the base genome and the y-axis representing percentage identity between the base and the aligned genomes. Predicted transcription factor motifs are depicted as colored bars. Arrowhead points to predicted motif of TF expressed more highly in female ES cells. Bottom, UCSC genome browser view of the same regions including histone modifications from ENCODE data in mouse ES cells (http://genome.ucsc.edu, NCBI37/mm9). (B) Conservation analysis, TF motif prediction and UCSC browser view as in (A) for the Tcf3 gene. Arrowhead points to predicted motif of TF expressed more highly in male ES cells. (C) Conservation analysis, TF motif prediction and UCSC browser view as in (A) for the Apln gene, with arrowheads indicating motifs predicted to bind TFs more highly expressed in male ES cells.
Expression in undifferentiated murine embryonic stem (ES) cells of genes that escape X chromosome inactivation (XCI) after differentiation (BC cell lines).
Examples of genes expressed in undifferentiated ES cells of genes that do not escape XCI (BC cell lines).
{kind=link}
Allele-specific expression analysis for imprinted gene Cdkn1c. RT-PCR was followed by allele-specific restriction digest of the Cdkn1c coding sequence and polyacrylamide gel analysis. A single nucleotide polymorphism in the M. castaneus allele generates a restriction site for aTaqI. Two different F1 hybrid ES cell lines each derived from reciprocal crosses of C57BL/6 (B) and CAST/EIJ (C) mice exhibited monoallelic (BxC) and biased expression (CxB) from the maternal allele. The first lane is the marker, the last two lanes are digested controls from PCR products from B and C genomic DNA.
{kind=link}
Comparison of expression differences for 14 selected genes from our dataset with a single-cell (sc) RNA-seq study [75] shows a high degree of correlation. Individual plots of expression from the scRNA-seq analysis shows the variability within the assay and the abundance of zero readouts for some genes. To avoid this confounder, a sign test was performed using a binomial exact test which affirmed the sex-specific biases seen within our dataset.
Genes with a sex-specific bias that overlap between ES cells and 5.5 dpc embryos.
{kind=link}
Chromosomal distribution of genes enriched in BCO when compared to BCF ES cells. BCO ES cell lines had enrichment of 423 genes, 49% of which overlapped with genes enriched in BCM relative to BCF (FDR < 0.01). The genes common to both BCO and BCM had statistically higher enrichment relative to BCF, averaging 2.55- versus 2.23-fold (students t-test, two tailed p < 0.001). Error bars denote standard deviation.
Acknowledgements
We thank Gwendolyn Gallicio for the experimental assistance and Joanne Thorvaldsen for the helpful discussions.
Funding
This work was supported by the National Institutes of Health [R01 GM093066] and by a Temple University Bridge Grant.
Availability of data and materials
RNA sequencing data has been deposited: GEO accession: GSE90516
Abbreviations
- BCM, BCF
F1 hybrid male and female ES cell lines resulting from a C57BL/6 female × CAST/EiJ male cross
- BCO
F1 hybrid XO ES cell line from the same cross
- CBM, CBF
F1 hybrid male and female ES cell lines resulting from a CAST/EiJ female × C57BL/6 male cross
- CI
Confidence interval
- ERFs
Epigenetic and remodeling factors
- ES
Embryonic stem
- FDR
False discovery rate
- LIF
Leukemia inhibitory factor
- lncRNAs
Long non-coding RNAs
- MEFs
Mouse embryonic fibroblasts
- PAGE
Polyacrylamide gel electrophoresis
- TFs
Transcription factors
- XCI
X chromosome inactivation
Authors’ contributions
RJW and NE conceived and designed the experiments. RJW generated the data and all the figures. RJW and NE contributed to the data analysis, interpretation, and manuscript writing. BMS and JMH contributed to the collection of data. JJ contributed to the experimental design, data analysis, and interpretation. JM contributed bioinformatic support, data analysis, and interpretation. All authors gave final approval of the published version. All authors are accountable for all aspects of the work.
Ethics approval
All work was carried out in accordance with protocols approved by the Animal Care and Use Committee at Temple University and in conformance with principles enunciated in the NIH Guide for the care and use of laboratory animals.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13293-017-0150-x) contains supplementary material, which is available to authorized users.
References
- 1.Parsch J, Ellegren H. The evolutionary causes and consequences of sex-biased gene expression. Nat Rev Genet. 2013;14(2):83–87. doi: 10.1038/nrg3376. [DOI] [PubMed] [Google Scholar]
- 2.Klein SL, Schiebinger L, Stefanick ML, Cahill L, Danska J, de Vries GJ, Kibbe MR, McCarthy MM, Mogil JS, Woodruff TK, et al. Opinion: sex inclusion in basic research drives discovery. Proc Natl Acad Sci U S A. 2015;112(17):5257–5258. doi: 10.1073/pnas.1502843112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold AP. Promoting the understanding of sex differences to enhance equity and excellence in biomedical science. Biol Sex Differ. 2010;1(1):1. doi: 10.1186/2042-6410-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morrow EH. The evolution of sex differences in disease. Biology Sex Differences. 2015;6 [DOI] [PMC free article] [PubMed]
- 5.Hayter SM, Cook MC. Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun Rev. 2012;11(10):754–765. doi: 10.1016/j.autrev.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016;16(10):626–638. doi: 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- 7.Cook MB, Dawsey SM, Freedman ND, Inskip PD, Wichner SM, Quraishi SM, Devesa SS, McGlynn KA. Sex disparities in cancer incidence by period and age. Cancer Epidemiol Biomark Prev. 2009;18(4):1174–1182. doi: 10.1158/1055-9965.EPI-08-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh SK, Lupo PJ, Scheurer ME, Saxena A, Kennedy AE, Ibrahimou B, Barbieri MA, Mills KI, McCauley JL, Okcu MF, et al. A childhood acute lymphoblastic leukemia genome-wide association study identifies novel sex-specific risk variants. Medicine (Baltimore) 2016;95(46):e5300. doi: 10.1097/MD.0000000000005300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muenchhoff M, Goulder PJ. Sex differences in pediatric infectious diseases. J Infect Dis. 2014;209(Suppl 3):S120–S126. doi: 10.1093/infdis/jiu232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fish EN. The X-files in immunity: sex-based differences predispose immune responses. Nat Rev Immunol. 2008;8(9):737–744. doi: 10.1038/nri2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.den Ruijter HM, Haitjema S, Asselbergs FW, Pasterkamp G. Sex matters to the heart: a special issue dedicated to the impact of sex related differences of cardiovascular diseases. Atherosclerosis. 2015;241(1):205–207. doi: 10.1016/j.atherosclerosis.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 12.Winham SJ, de Andrade M, Miller VM. Genetics of cardiovascular disease: importance of sex and ethnicity. Atherosclerosis. 2015;241(1):219–228. doi: 10.1016/j.atherosclerosis.2015.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singmann P, Shem-Tov D, Wahl S, Grallert H, Fiorito G, Shin SY, Schramm K, Wolf P, Kunze S, Baran Y, et al. Characterization of whole-genome autosomal differences of DNA methylation between men and women. Epigenetics Chromatin. 2015;8:43. doi: 10.1186/s13072-015-0035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isensee J, Ruiz Noppinger P. Sexually dimorphic gene expression in mammalian somatic tissue. Gender Med. 2007;(4 Suppl B):S75–95. [DOI] [PubMed]
- 15.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152(6):1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kukurba KR, Parsana P, Balliu B, Smith KS, Zappala Z, Knowles DA, Fave MJ, Davis JR, Li X, Zhu X, et al. Impact of the X chromosome and sex on regulatory variation. Genome Res. 2016; [DOI] [PMC free article] [PubMed]
- 17.Rigby N, Kulathinal RJ. Genetic architecture of sexual dimorphism in humans. J Cell Physiol. 2015;230(10):2304–2310. doi: 10.1002/jcp.24979. [DOI] [PubMed] [Google Scholar]
- 18.Williams TM, Carroll SB. Genetic and molecular insights into the development and evolution of sexual dimorphism. Nat Rev Genet. 2009;10(11):797–804. doi: 10.1038/nrg2687. [DOI] [PubMed] [Google Scholar]
- 19.Erickson RP. Does sex determination start at conception? BioEssays. 1997;19(11):1027–1032. doi: 10.1002/bies.950191113. [DOI] [PubMed] [Google Scholar]
- 20.Arnold AP, Chen X. What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Front Neuroendocrinol. 2009;30(1):1–9. doi: 10.1016/j.yfrne.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Vries GJ, Rissman EF, Simerly RB, Yang LY, Scordalakes EM, Auger CJ, Swain A, Lovell-Badge R, Burgoyne PS, Arnold AP. A model system for study of sex chromosome effects on sexually dimorphic neural and behavioral traits. J Neurosci. 2002;22(20):9005–9014. doi: 10.1523/JNEUROSCI.22-20-09005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wijchers PJ, Yandim C, Panousopoulou E, Ahmad M, Harker N, Saveliev A, Burgoyne PS, Festenstein R. Sexual dimorphism in mammalian autosomal gene regulation is determined not only by Sry but by sex chromosome complement as well. Dev Cell. 2010;19(3):477–484. doi: 10.1016/j.devcel.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Seller MJ. Neural-tube defects and sex-ratios. Am J Med Genet. 1987;26(3):699–707. doi: 10.1002/ajmg.1320260325. [DOI] [PubMed] [Google Scholar]
- 24.Migeon BR. Why females are mosaics, X-chromosome inactivation, and sex differences in disease. Gender Med. 2007;4(2):97–105. doi: 10.1016/S1550-8579(07)80024-6. [DOI] [PubMed] [Google Scholar]
- 25.Burgoyne PS, Thornhill AR, Boudrean SK, Darling SM, Bishop CE, Evans EP. The genetic basis of XX-XY differences present before gonadal sex differentiation in the mouse. Philos Trans R Soc Lond Ser B Biol Sci. 1995;350(1333):253–260. doi: 10.1098/rstb.1995.0159. [DOI] [PubMed] [Google Scholar]
- 26.Berletch JB, Deng X, Nguyen DK, Disteche CM. Female bias in Rhox6 and 9 regulation by the histone demethylase KDM6A. PLoS Genet. 2013;9(5):e1003489. doi: 10.1371/journal.pgen.1003489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schulz EG, Meisig J, Nakamura T, Okamoto I, Sieber A, Picard C, Borensztein M, Saitou M, Bluthgen N, Heard E. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell. 2014;14(2):203–216. doi: 10.1016/j.stem.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 28.Lowe R, Gemma C, Rakyan VK, Holland ML. Sexually dimorphic gene expression emerges with embryonic genome activation and is dynamic throughout development. BMC Genomics. 2015;16:295. doi: 10.1186/s12864-015-1506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ronen D, Benvenisty N. Sex-dependent gene expression in human pluripotent stem cells. Cell Rep. 2014;8(4):923–932. doi: 10.1016/j.celrep.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 30.Habibi E, Brinkman AB, Arand J, Kroeze LI, Kerstens HH, Matarese F, Lepikhov K, Gut M, Brun-Heath I, Hubner NC, et al. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell. 2013;13(3):360–369. doi: 10.1016/j.stem.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 31.Zvetkova I, Apedaile A, Ramsahoye B, Mermoud JE, Crompton LA, John R, Feil R, Brockdorff N. Global hypomethylation of the genome in XX embryonic stem cells. Nat Genet. 2005;37(11):1274–1279. doi: 10.1038/ng1663. [DOI] [PubMed] [Google Scholar]
- 32.Bramble MS, Roach L, Lipson A, Vashist N, Eskin A, Ngun T, Gosschalk JE, Klein S, Barseghyan H, Arboleda VA, et al. Sex-specific effects of testosterone on the sexually dimorphic transcriptome and epigenome of embryonic neural stem/progenitor cells. Sci Rep. 2016;6:36916. doi: 10.1038/srep36916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aiken CE, Ozanne SE. Sex differences in developmental programming models. Reproduction. 2013;145(1):R1–13. doi: 10.1530/REP-11-0489. [DOI] [PubMed] [Google Scholar]
- 34.Donjacour A, Liu X, Lin W, Simbulan R, Rinaudo PF. In vitro fertilization affects growth and glucose metabolism in a sex-specific manner in an outbred mouse model. Biol Reprod. 2014;90(4):80. doi: 10.1095/biolreprod.113.113134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnold AP, Reue K, Eghbali M, Vilain E, Chen X, Ghahramani N, Itoh Y, Li J, Link JC, Ngun T, et al. The importance of having two X chromosomes. Philos Trans R Soc Lond Ser B Biol Sci. 2016;371(1688):20150113. doi: 10.1098/rstb.2015.0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grubb SC, Bult CJ, Bogue MA. Mouse phenome database. Nucleic Acids Res. 2014;42(Database issue):D825–D834. doi: 10.1093/nar/gkt1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svenson KL, Von Smith R, Magnani PA, Suetin HR, Paigen B, Naggert JK, Li R, Churchill GA, Peters LL. Multiple trait measurements in 43 inbred mouse strains capture the phenotypic diversity characteristic of human populations. J Appl Physiol (1985) 2007;102(6):2369–2378. doi: 10.1152/japplphysiol.01077.2006. [DOI] [PubMed] [Google Scholar]
- 38.Szatkiewicz JP, Beane GL, Ding Y, Hutchins L, Pardo-Manuel de Villena F, Churchill GA. An imputed genotype resource for the laboratory mouse. Mamm Genome. 2008;19(3):199–208. doi: 10.1007/s00335-008-9098-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crowley JJ, Zhabotynsky V, Sun W, Huang S, Pakatci IK, Kim Y, Wang JR, Morgan AP, Calaway JD, Aylor DL, et al. Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance. Nat Genet. 2015;47(4):353–360. doi: 10.1038/ng.3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagy A, Gertsenstein M, Vintersten K, Behringer R. Counting chromosomes in embryonic stem (ES) cells. Cold Spring Harb Protoc. 2009;2009(6):pdb prot4404. doi: 10.1101/pdb.prot4404. [DOI] [PubMed] [Google Scholar]
- 41.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Som A, Harder C, Greber B, Siatkowski M, Paudel Y, Warsow G, Cap C, Scholer H, Fuellen G. The PluriNetWork: an electronic representation of the network underlying pluripotency in mouse, and its applications. PLoS One. 2010;5(12):e15165. doi: 10.1371/journal.pone.0015165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boroviak T, Loos R, Lombard P, Okahara J, Behr R, Sasaki E, Nichols J, Smith A, Bertone P. Lineage-specific profiling delineates the emergence and progression of naive Pluripotency in mammalian embryogenesis. Dev Cell. 2015;35(3):366–382. doi: 10.1016/j.devcel.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smoot M, Ono K, Ideker T, Maere S. PiNGO: a Cytoscape plugin to find candidate genes in biological networks. Bioinformatics. 2011;27(7):1030–1031. doi: 10.1093/bioinformatics/btr045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang da W, Sherman BT, Zheng X, Yang J, Imamichi T, Stephens R, Lempicki RA. Extracting biological meaning from large gene lists with DAVID. Curr Protoc Bioinformatics. 2009;Chapter 13:Unit 13 11. doi: 10.1002/0471250953.bi1311s27. [DOI] [PubMed] [Google Scholar]
- 47.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walter W, Sanchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31(17):2912–2914. doi: 10.1093/bioinformatics/btv300. [DOI] [PubMed] [Google Scholar]
- 49.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schultz BM, Gallicio GA, Cesaroni M, Lupey LN, Engel N. Enhancers compete with a long non-coding RNA for regulation of the Kcnq1 domain. Nucleic Acids Res. 2015;43(2):745–759. doi: 10.1093/nar/gku1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.von Meyenn F, Iurlaro M, Habibi E, Liu NQ, Salehzadeh-Yazdi A, Santos F, Petrini E, Milagre I, Yu M, Xie Z, et al. Impairment of DNA methylation maintenance is the main cause of global demethylation in naive embryonic stem cells. Mol Cell. 2016;62(6):848–861. doi: 10.1016/j.molcel.2016.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hackett JA, Dietmann S, Murakami K, Down TA, Leitch HG, Surani MA. Synergistic mechanisms of DNA demethylation during transition to ground-state pluripotency. Stem Cell Reports. 2013;1(6):518–531. doi: 10.1016/j.stemcr.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ficz G, Hore TA, Santos F, Lee HJ, Dean W, Arand J, Krueger F, Oxley D, Paul YL, Walter J, et al. FGF signaling inhibition in ESCs drives rapid genome-wide demethylation to the epigenetic ground state of pluripotency. Cell Stem Cell. 2013;13(3):351–359. doi: 10.1016/j.stem.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yagi M, Kishigami S, Tanaka A, Semi K, Mizutani E, Wakayama S, Wakayama T, Yamamoto T, Yamada Y. Derivation of ground-state female ES cells maintaining gamete-derived DNA methylation. Nature. 2017;548:224–227. [DOI] [PubMed]
- 56.Srivastava A, Morgan AP, Najarian ML, Sarsani VK, Sigmon JS, Shorter JR, Kashfeen A, McMullan RC, Williams LH, Giusti-Rodriguez P, et al. Genomes of the mouse collaborative cross. Genetics. 2017;206(2):537–556. doi: 10.1534/genetics.116.198838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang HM, Liu T, Liu CJ, Song S, Zhang X, Liu W, Jia H, Xue Y, Guo AY. AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 2015;43(Database issue):D76–D81. doi: 10.1093/nar/gku887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Froberg JE, Yang L, Lee JT. Guided by RNAs: X-inactivation as a model for lncRNA function. J Mol Biol. 2013;425(19):3698–3706. doi: 10.1016/j.jmb.2013.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raney BJ, Dreszer TR, Barber GP, Clawson H, Fujita PA, Wang T, Nguyen N, Paten B, Zweig AS, Karolchik D, et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC genome browser. Bioinformatics. 2014;30(7):1003–1005. doi: 10.1093/bioinformatics/btt637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. ENCODE data in the UCSC genome browser: year 5 update. Nucleic Acids Res. 2013;41(Database issue):D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457(7231):854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010; [DOI] [PMC free article] [PubMed]
- 64.Loots GG, Ovcharenko I. Dcode.Org anthology of comparative genomic tools. Nucleic Acids Res. 2005;33(Web Server issue):W56–W64. doi: 10.1093/nar/gki355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32(Web Server issue):W280–W286. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakaki F, Saitou M. PRDM14: a unique regulator for pluripotency and epigenetic reprogramming. Trends Biochem Sci. 2014;39(6):289–298. doi: 10.1016/j.tibs.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 67.Ma Z, Swigut T, Valouev A, Rada-Iglesias A, Wysocka J. Sequence-specific regulator Prdm14 safeguards mouse ESCs from entering extraembryonic endoderm fates. Nat Struct Mol Biol. 2011;18(2):120–127. doi: 10.1038/nsmb.2000. [DOI] [PubMed] [Google Scholar]
- 68.Payer B, Rosenberg M, Yamaji M, Yabuta Y, Koyanagi-Aoi M, Hayashi K, Yamanaka S, Saitou M, Lee JT. Tsix RNA and the germline factor, PRDM14, link X reactivation and stem cell reprogramming. Mol Cell. 2013;52(6):805–818. doi: 10.1016/j.molcel.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Verburg PE, Tucker G, Scheil W, Erwich JJ, Dekker GA, Roberts CT. Sexual dimorphism in adverse pregnancy outcomes—a retrospective Australian population study 1981-2011. PLoS One. 2016;11(7):e0158807. doi: 10.1371/journal.pone.0158807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamaji M, Ueda J, Hayashi K, Ohta H, Yabuta Y, Kurimoto K, Nakato R, Yamada Y, Shirahige K, Saitou M. PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell. 2013;12(3):368–382. doi: 10.1016/j.stem.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 72.Xu H, Baroukh C, Dannenfelser R, Chen EY, Tan CM, Kou Y, Kim YE, Lemischka IR, Ma'ayan A. ESCAPE: database for integrating high-content published data collected from human and mouse embryonic stem cells. Database (Oxford) 2013;2013:bat045. doi: 10.1093/database/bat045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bulger M. Maps for changing landscapes: viewing epigenomic signatures through differentiation. Cell Stem Cell. 2012;11(5):581–582. doi: 10.1016/j.stem.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 74.Chaligne R, Heard E. X-chromosome inactivation in development and cancer. FEBS Lett. 2014;588(15):2514–2522. doi: 10.1016/j.febslet.2014.06.023. [DOI] [PubMed] [Google Scholar]
- 75.Chen G, Schell JP, Benitez JA, Petropoulos S, Yilmaz M, Reinius B, Alekseenko Z, Shi L, Hedlund E, Lanner F, et al. Single-cell analyses of X chromosome inactivation dynamics and pluripotency during differentiation. Genome Res. 2016; [DOI] [PMC free article] [PubMed]
- 76.Shimakage M, Kodama K, Kawahara K, Kim CJ, Ikeda Y, Yutsudo M, Inoue H. Downregulation of drs tumor suppressor gene in highly malignant human pulmonary neuroendocrine tumors. Oncol Rep. 2009;21(6):1367–1372. doi: 10.3892/or_00000362. [DOI] [PubMed] [Google Scholar]
- 77.Kim CJ, Shimakage M, Kushima R, Mukaisho K, Shinka T, Okada Y, Inoue H. Down-regulation of drs mRNA in human prostate carcinomas. Hum Pathol. 2003;34(7):654–657. doi: 10.1016/S0046-8177(03)00240-5. [DOI] [PubMed] [Google Scholar]
- 78.Berletch JB, Ma W, Yang F, Shendure J, Noble WS, Disteche CM, Deng X. Escape from X inactivation varies in mouse tissues. PLoS Genet. 2015;11(3):e1005079. doi: 10.1371/journal.pgen.1005079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Balaton BP, Brown CJ. Escape artists of the X chromosome. Trends Genet. 2016;32(6):348–359. doi: 10.1016/j.tig.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 80.Disteche CM. Dosage compensation of the sex chromosomes and autosomes. Semin Cell Dev Biol. 2016;56:9–18. doi: 10.1016/j.semcdb.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lin F, Xing K, Zhang J, He X. Expression reduction in mammalian X chromosome evolution refutes Ohno's hypothesis of dosage compensation. Proc Natl Acad Sci U S A. 2012;109(29):11752–11757. doi: 10.1073/pnas.1201816109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pessia E, Makino T, Bailly-Bechet M, McLysaght A, Marais GA. Mammalian X chromosome inactivation evolved as a dosage-compensation mechanism for dosage-sensitive genes on the X chromosome. Proc Natl Acad Sci U S A. 2012;109(14):5346–5351. doi: 10.1073/pnas.1116763109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schulz R, Proudhon C, Bestor TH, Woodfine K, Lin CS, Lin SP, Prissette M, Oakey RJ, Bourc'his D. The parental non-equivalence of imprinting control regions during mammalian development and evolution. PLoS Genet. 2010;6(11):e1001214. doi: 10.1371/journal.pgen.1001214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vallejos CA, Risso D, Scialdone A, Dudoit S, Marioni JC. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nat Methods. 2017;14(6):565–571. doi: 10.1038/nmeth.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yuan GC, Cai L, Elowitz M, Enver T, Fan G, Guo G, Irizarry R, Kharchenko P, Kim J, Orkin S, et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017;18(1):84. doi: 10.1186/s13059-017-1218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Trovo de Marqui AB. Turner syndrome and genetic polymorphism: a systematic review. Rev Paul Pediatr. 2015;33(3):364–371. doi: 10.1016/j.rpped.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhong Q, Layman LC. Genetic considerations in the patient with turner syndrome—45,X with or without mosaicism. Fertil Steril. 2012;98(4):775–779. doi: 10.1016/j.fertnstert.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.OMIM. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University & National Center for Biotechnology Information, National Library of Medicine, http://www.ncbi.nlm.nih.gov/omim. Accessed 11 Jan 2016.
- 89.Lin YT, Capel B. Cell fate commitment during mammalian sex determination. Curr Opin Genet Dev. 2015;32:144–152. doi: 10.1016/j.gde.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Koopman P. The curious world of Gonadal development in mammals. Curr Top Dev Biol. 2016;116:537–545. doi: 10.1016/bs.ctdb.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 91.Ingleby FC, Flis I, Morrow EH: Sex-biased gene expression and sexual conflict throughout development. Csh Perspect Biol. 2015;7(1):1–7. [DOI] [PMC free article] [PubMed]
- 92.Veitia RA, Birchler JA. Dominance and gene dosage balance in health and disease: why levels matter! J Pathol. 2010;220(2):174–185. doi: 10.1002/path.2623. [DOI] [PubMed] [Google Scholar]
- 93.Birchler JA, Riddle NC, Auger DL, Veitia RA. Dosage balance in gene regulation: biological implications. Trends Genet. 2005;21(4):219–226. doi: 10.1016/j.tig.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 94.Marks H, Kalkan T, Menafra R, Denissov S, Jones K, Hofemeister H, Nichols J, Kranz A, Stewart AF, Smith A, et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell. 2012;149(3):590–604. doi: 10.1016/j.cell.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135(2):216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dar RD, Razooky BS, Singh A, Trimeloni TV, McCollum JM, Cox CD, Simpson ML, Weinberger LS. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc Natl Acad Sci U S A. 2012;109(43):17454–17459. doi: 10.1073/pnas.1213530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Molina N, Suter DM, Cannavo R, Zoller B, Gotic I, Naef F. Stimulus-induced modulation of transcriptional bursting in a single mammalian gene. Proc Natl Acad Sci U S A. 2013;110(51):20563–20568. doi: 10.1073/pnas.1312310110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tantale K, Mueller F, Kozulic-Pirher A, Lesne A, Victor JM, Robert MC, Capozi S, Chouaib R, Backer V, Mateos-Langerak J, et al. A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nat Commun. 2016;7:12248. doi: 10.1038/ncomms12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chubb JR. Gene regulation: stable noise. Curr Biol. 2016;26(2):R61–R64. doi: 10.1016/j.cub.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 100.Lopes AM, Burgoyne PS, Ojarikre A, Bauer J, Sargent CA, Amorim A, Affara NA. Transcriptional changes in response to X chromosome dosage in the mouse: implications for X inactivation and the molecular basis of turner syndrome. BMC Genomics. 2010;11:82. doi: 10.1186/1471-2164-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lynn PM, Davies W. The 39,XO mouse as a model for the neurobiology of turner syndrome and sex-biased neuropsychiatric disorders. Behav Brain Res. 2007;179(2):173–182. doi: 10.1016/j.bbr.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 102.Trolle C, Nielsen MM, Skakkebaek A, Lamy P, Vang S, Hedegaard J, Nordentoft I, Orntoft TF, Pedersen JS, Gravholt CH. Widespread DNA hypomethylation and differential gene expression in turner syndrome. Sci Rep. 2016;6:34220. doi: 10.1038/srep34220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu J, Watts JA, Pope SD, Gadue P, Kamps M, Plath K, Zaret KS, Smale ST. Transcriptional competence and the active marking of tissue-specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes Dev. 2009;23(24):2824–2838. doi: 10.1101/gad.1861209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261(5):412–417. doi: 10.1111/j.1365-2796.2007.01809.x. [DOI] [PubMed] [Google Scholar]
- 106.Gabory A, Attig L, Junien C. Sexual dimorphism in environmental epigenetic programming. Mol Cell Endocrinol. 2009;304(1–2):8–18. doi: 10.1016/j.mce.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 107.Dipietro JA, Voegtline KM. The gestational Foundation of sex Differences in development and vulnerability. Neuroscience. 2017;342:4–20. doi: 10.1016/j.neuroscience.2015.07.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sundrani DP, Roy SS, Jadhav AT, Joshi SR. Sex-specific differences and developmental programming for diseases in later life. Reprod Fertil Dev. 2017;(Epub ahead of print); PMID:28380326. doi:10.1071/RD16265. [DOI] [PubMed]
- 109.Arnold AP, Chen X, Itoh Y. What a difference an X or Y makes: sex chromosomes, gene dose, and epigenetics in sexual differentiation. Handb Exp Pharmacol. 2012;214:67–88. doi: 10.1007/978-3-642-30726-3_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Loots GG. Genomic identification of regulatory elements by evolutionary sequence comparison and functional analysis. Adv Genet. 2008;61:269–293. doi: 10.1016/S0065-2660(07)00010-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Derivation of mouse ES cell lines. F1 hybrid blastocysts were obtained at embryonic day 3.5 from reciprocal crosses of mouse substrains C57BL/6 and CAST/EIJ (designated as B and C, respectively). Blastocysts were cultured individually and used to establish independent cell lines.
Real-time PCR primers.
Transcription factor motifs enriched in promoters of differentially expressed genes in female and male ES cells.
Catalog of transcription factors (TFs) and epigenetic and remodeling factors (ERFs) expressed differentially in female and male ES cells.
Identification of candidate regulatory elements. (A) Top, graphic display of the conservation profiles for regions upstream of Zfx from Dcode.org [64, 110]. The base genome is mouse. Evolutionarily conserved regions (ECRs) of a minimum of 100 bp conserved above 70% sequence identity are displayed as red (intergenic) peaks, with the x-axis representing positions in the base genome and the y-axis representing percentage identity between the base and the aligned genomes. Predicted transcription factor motifs are depicted as colored bars. Arrowhead points to predicted motif of TF expressed more highly in female ES cells. Bottom, UCSC genome browser view of the same regions including histone modifications from ENCODE data in mouse ES cells (http://genome.ucsc.edu, NCBI37/mm9). (B) Conservation analysis, TF motif prediction and UCSC browser view as in (A) for the Tcf3 gene. Arrowhead points to predicted motif of TF expressed more highly in male ES cells. (C) Conservation analysis, TF motif prediction and UCSC browser view as in (A) for the Apln gene, with arrowheads indicating motifs predicted to bind TFs more highly expressed in male ES cells.
Expression in undifferentiated murine embryonic stem (ES) cells of genes that escape X chromosome inactivation (XCI) after differentiation (BC cell lines).
Examples of genes expressed in undifferentiated ES cells of genes that do not escape XCI (BC cell lines).
Allele-specific expression analysis for imprinted gene Cdkn1c. RT-PCR was followed by allele-specific restriction digest of the Cdkn1c coding sequence and polyacrylamide gel analysis. A single nucleotide polymorphism in the M. castaneus allele generates a restriction site for aTaqI. Two different F1 hybrid ES cell lines each derived from reciprocal crosses of C57BL/6 (B) and CAST/EIJ (C) mice exhibited monoallelic (BxC) and biased expression (CxB) from the maternal allele. The first lane is the marker, the last two lanes are digested controls from PCR products from B and C genomic DNA.
Comparison of expression differences for 14 selected genes from our dataset with a single-cell (sc) RNA-seq study [75] shows a high degree of correlation. Individual plots of expression from the scRNA-seq analysis shows the variability within the assay and the abundance of zero readouts for some genes. To avoid this confounder, a sign test was performed using a binomial exact test which affirmed the sex-specific biases seen within our dataset.
Genes with a sex-specific bias that overlap between ES cells and 5.5 dpc embryos.
Chromosomal distribution of genes enriched in BCO when compared to BCF ES cells. BCO ES cell lines had enrichment of 423 genes, 49% of which overlapped with genes enriched in BCM relative to BCF (FDR < 0.01). The genes common to both BCO and BCM had statistically higher enrichment relative to BCF, averaging 2.55- versus 2.23-fold (students t-test, two tailed p < 0.001). Error bars denote standard deviation.
Data Availability Statement
RNA sequencing data has been deposited: GEO accession: GSE90516