

Abstract
Balance between cholinergic and dopaminergic signaling is central to striatal control of movement and cognition. In dystonia, a common disorder of movement, anticholinergic therapy is often beneficial. This observation suggests there is a pathological increase in cholinergic tone, yet direct confirmation is lacking. In DYT1, an early-onset genetic form of dystonia caused by a mutation in the protein torsinA (TorA), the suspected heightened cholinergic tone is commonly attributed to faulty dopamine D2 receptor (D2R) signaling where D2R agonists cause excitation of striatal cholinergic interneurons (ChIs), rather than the normal inhibition of firing observed in wild-type animals, an effect known as “paradoxical excitation”. Here, we provide for the first time direct measurement of elevated striatal extracellular acetylcholine (ACh) in a knock-in mouse model of human DYT1 dystonia (TorAΔE/+ mice), confirming a striatal hypercholinergic state. We hypothesized that this elevated extracellular ACh might cause chronic over-activation of muscarinic acetylcholine receptors (mAChRs) and disrupt normal D2R function due to their shared coupling to Gi/o-proteins. We tested this concept in vitro first using a broad-spectrum mAChR antagonist, and then using a M2/M4 mAChR selective antagonist to specifically target mAChRs expressed by ChIs. Remarkably, we found that mAChR inhibition reverses the D2R–mediated paradoxical excitation of ChIs recorded in slices from TorAΔE/+ mice to a typical inhibitory response. Furthermore, we recapitulated the paradoxical D2R excitation of ChIs in striatal slices from wild type mice within minutes by simply increasing cholinergic tone through pharmacological inhibition of acetylcholinesterase (AChE) or by prolonged agonist activation of mAChRs. Collectively, these results show that enhanced mAChR tone itself is sufficient to rapidly reverse the polarity of D2R regulation of ChI excitability, correcting the previous notion that the D2R mediated paradoxical ChI excitation causes the hypercholinergic state in dystonia. Further, using a combination of genetic and pharmacological approaches, we found evidence that this switch in D2R polarity results from a change in coupling from the preferred Gi/o pathway to non-canonical β-arrestin signaling. These results highlight the need to fully understand how the mutation in TorA leads to pathologically heightened extracellular ACh. Furthermore the discovery of this novel ACh-dopamine interaction and the participation of β-arrestin in regulation of cholinergic interneurons is likely important for other basal ganglia disorders characterized by perturbation of ACh-dopamine balance, including Parkinson and Huntington diseases, l-DOPA-induced dyskinesia and schizophrenia.
Keywords: Dystonia, DYT1, dorsal striatum, cholinergic interneurons, GPCRs, Dopamine-Acetylcholine interaction, β-arrestin, D2 receptors, muscarinic receptors
Graphical abstract
Introduction
Striatal control of motor and cognitive functions relies on proper balance between cholinergic and dopaminergic modulation of synaptic circuits. This balance is ensured by a complex, close interaction between cholinergic interneurons (ChIs) and midbrain dopaminergic neurons (Stoof et al., 1992a; Pisani et al., 2007; Aosaki et al., 2010). These two neuron populations exhibit coincident physiological changes in their activity during behavior (Morris et al., 2004; Joshua et al., 2008) and exert reciprocal control on the release of their neurotransmitters (Stoof et al., 1992a; DeBoer and Abercrombie, 1996; Pisani et al., 2003; Rice and Cragg, 2004; Cragg, 2006; Threlfell et al., 2012). A particularly important site of functional interaction in striatum is at autonomous pacemaker ChIs, the main source of striatal acetylcholine (ACh). ChIs express high levels of dopamine D2Rs and M2/M4 mACh autoreceptors (Yan and Surmeier, 1996; Straub et al., 2014; Wieland et al., 2014). D2Rs and M2/M4 mAChRs reduce tonic firing of ChIs, and subsequent ACh release, through shared coupling to Gi/o signaling, which diminishes the opening of Cav2 Ca2+ channels in response to membrane depolarization (Yan and Surmeier, 1996; Yan et al., 1997; Calabresi et al., 1998; Maurice et al., 2004; Ding et al., 2006; Bonsi et al., 2008; Zhao et al., 2016). In vivo microdialysis (Drukarch et al., 1990; Stoof et al., 1992b; DeBoer and Abercrombie, 1996) and electrophysiological (Yan et al., 1997) studies revealed that the ability of the D2R agonist quinpirole to decrease ACh release and to reduce Ca2+ currents in ChIs is limited by increasing levels of striatal ACh, suggesting that dopamine modulation of striatal ACh release is influenced by striatal cholinergic tone, perhaps through functional interactions between D2R and M2/M4 mACh autoreceptors. Defining this interaction will have significant impact on understanding the most common basal ganglia motor disorders which are often associated with alterations in the normal balance between dopaminergic and cholinergic systems (Pisani et al., 2007; Aosaki et al., 2010; Bonsi et al., 2011; Benarroch, 2012). Dystonia, a common motor disorder characterized by abnormal muscle contractions, is of particular relevance in this context because of abundant evidence for alterations in striatal cholinergic-dopaminergic balance (Breakefield et al., 2008; Eskow Jaunarajs et al., 2015).
DYT1 dystonia, the most common early-onset inherited form of isolated dystonia, is caused by a three-nucleotide (GAG) deletion in the TOR1A gene, leading to loss of a glutamate residue near the C-terminus of the encoded protein torsinA, ΔE-TorA (Ozelius et al., 1999). TorA belongs to the AAA+ (ATPase associated with diverse cellular activities) family of chaperones and is involved in trafficking of proteins between the nucleus and endoplasmic reticulum. How these cell biological properties relate to neural system disturbances seen in dystonia is not well understood (Breakefield et al., 2008).
Mounting evidence suggests imbalanced cholinergic transmission plays a pivotal role in the pathophysiology of DYT1 dystonia (Eskow Jaunarajs et al., 2015). Clinically, anticholinergic drugs (e.g., trihexyphenidyl) are the most effective treatments available for DYT1 and other dystonias (Jankovic, 2013). Across several rodent models based on mutant TorA, increased cholinergic signaling underlies impaired bidirectional corticostriatal synaptic plasticity, and comparable abnormalities in neural plasticity of motor circuits are found in human dystonia (Martella et al., 2009; Quartarone and Hallett, 2013; Martella et al., 2014; Calabresi et al., 2016). A consistent finding in all DYT1 rodent models examined so far is a “paradoxical excitation” of ChIs in response to D2R activation (Pisani et al., 2006; Martella et al., 2009; Sciamanna et al., 2012). This finding has led to the prevailing hypothesis that faulty D2R signaling causes the hypercholinergic state, with downstream effects on synaptic plasticity at corticostriatal synapses (Martella et al., 2009; Maltese et al., 2014; Martella et al., 2014; Calabresi et al., 2016). These clinical and experimental data predict that there should be elevated striatal cholinergic tone in DYT1 dystonia, but to date, ACh levels have not been measured directly in vivo in preclinical dystonia models or human dystonia.
Here, we provide direct evidence of elevated striatal cholinergic tone in a DYT1 knock-in mouse model (TorAΔE/+ mice) and challenge the view that D2R signaling causes the hypercholinergic state. In fact, the reverse is true: we find that the strength of the cholinergic tone dictates the polarity of D2R modulation of ChI excitability. Moreover, our data support the interpretation that, as a result of the hypercholinergic state, a sustained activation of mACh autoreceptors on ChIs drives a shift from canonical D2R–Gi/o coupling, which mediates the inhibition of ChI firing (Yan et al., 1997), to non-canonical D2R-β-arrestin signaling which triggers the aberrant increase in ChI firing. These findings not only provide a new perspective on DYT1 dystonia pathophysiology, but also have relevance to broader aspects of G-protein-coupled receptor signaling in conditions of dopamine/ACh imbalance found in a wide range of neurological disorders, including Parkinson’s disease, Huntington’s disease, l-DOPA induced dyskinesia and schizophrenia.
Materials and Methods
All experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham.
Animal Model
Heterozygous DYT1 mutant knock-in mice, TorA ΔE /+ (Goodchild et al., 2005) were maintained congenically by breeding with C57BL/6J mice from Jackson Laboratories (Bar Harbor, ME, USA). Mice were housed with a 12 h light/dark cycle. Food and water were provided ad libitum. Tail DNA was genotyped using a primer pair to detect the 34 base pair loxP site in the DYT1 mutant (forward primer, AGTCTGTGGCTGGCTCTCCC; reverse primer, CCTCAGGCTGCTCACAACCAC). PCR products were run using a 2% agarose gel. Male mutant (TorAΔE /+) and littermate controls (TorA+/+) were evaluated between 4–6 months of age. The generation of β-Arr2 homozygous KO mice has been previously described (Bohn et al., 1999). These mice have been backcrossed more than 12 generations to and maintained on the C57BL/6 background.
In vivo microdialysis
Male TorA+/+ and TorAΔE /+ littermates were anesthetized with isoflurane (1–3%) and placed in a stereotaxic apparatus. Unilateral microdialysis cannulae (CMA Microdialysis, Stockholm, Sweden) were implanted vertically above the striatum (anterior +0.6; lateral +1.9; ventral −1.6mm from bregma, according to the coordinates of (Paxinos et al., 1985). Two anchor screws (CMA Microdialysis) were placed behind the cannula and then fixed to the skull with dental cement. Following surgery, buprenorphine (0.03mg/kg, i.p.) was injected for pain relief. Three to four days following surgery, dummy cannulae were removed and microdialysis probes were inserted (CMA7: 2 mm, CMA Microdialysis). Mice were habituated to the microdialysis environment for 3 h while the probes were perfused with artificial cerebrospinal fluid (aCSF; 127.6mM NaCl, 4.02mM KCl, 750uM NaH2PO4, 2.1mM Na2HPO4, 2.00mM MgCl2, 1.71mM CaCl2; pH 7.4) at a constant rate of 2 µL/min for the duration of the experiment. In order to determine basal levels of ACh, three dialysate samples were collected every 20 min for one h following habituation. Following baseline, 10, 100 and 500 nM neostigmine bromide (Neo) were added to aCSF and samples were collected every 20 min for 1 h per dose in an escalating dose design for a total of 3 h. At the end of sampling, mice were killed and brains were removed and post-fixed with 4% paraformaldehyde. Fixed brains were then sectioned using a freezing microtome (Leica SM 200R, Buffalo Grove, IL) and cresyl violet staining was used to determine accurate probe placement. All microdialysis samples were analyzed for ACh using HPLC-ED (Eicom HTEC-500 with Eicom Autosampler INSIGHT, San Diego, CA) with an enzyme reactor (Eicom, AC-ENZYM II 1.0 × 4.0mm) and a platinum electrode (Eicom, WE-PT; +450 mV vs Ag/AgCl). The mobile phase consisted of 50 mM potassium bicarbonate, 134 uM ethylenediaminetetraacetic acid, and 1.22 mM decanesulfonate (pH 8.5) and was delivered at a rate of 150 µL/min. Microdialysate sample chromatograms were analyzed based on established concentration curves for ACh (1–100 nM). The limit of detection was ~10 fM.
Slice preparation
Male TorAΔE /+, their wild-type littermates, TorA+/+ and β-Arr2 KO mice of 3–4 months of age were deeply anesthetized with isoflurane and perfused transcardially with ice-cold aCSF, bubbled with 95% O2-5% CO2, and containing (in mM): 2.5 KCl, 126 NaCl, 26 NaHCO3, 1.25 Na2HPO4, 2 CaCl2, 2 MgSO4, and 10 glucose. The brain was removed, blocked in the coronal plane, and sectioned at a thickness of 280 µm in ice-cold aCSF. Slices were then submerged in aCSF bubbled with 95% O2-5% CO2, and stored at room temperature (RT) for at least 1 h prior to recording.
Electrophysiology
The slices were transferred to a recording chamber mounted on an Olympus BX50WI upright, fixed-stage microscope and perfused (2ml/min) with oxygenated aCSF at RT. A 40× / 0.9 N.A. water-immersion objective was used to examine the slice, cholinergic interneurons were visualized on a monitor using a differential interference contrast optical system combined with an infrared filter, and a monochrome CCD camera (RC300, MTI). Electrophysiological recordings were obtained with an Axopatch 200B amplifier (Axon Instruments, Sunnyvale, CA), using borosilicate glass pipette pulled on a P97 puller (Sutter Instruments, Novato, CA). Patch pipette resistance was typically 2.8–3 MΩ when filled with aCSF. ChI spontaneous firing was recorded at RT (22–25°C) in the cell-attached configuration (seal resistance: 200 MΩ − > 5 GΩ), voltage-clamp mode, at a command potential at which the amplifier current, I amp, is at 0 pA. Signals were digitized at 100 kHz and logged onto a personal computer with the Clampex 10.5 software (Molecular Devices, Sunnyvale, CA).
Data analysis and statistics
Data were analyzed using pClampfit 10.5 (Molecular Device) and Prism 6.0e (GraphPad). Numerical data are presented as means ± SEM; n values represent independent observations. Basal ACh data were analyzed using an independent two-sample t-test. Neo dose response data were analyzed by repeated-measures 2-way ANOVA. Post-hoc tests were completed using Tukey’s multiple comparisons tests. The evaluation of statistical difference for electrophysiological experiments was performed using Wilcoxon signed rank test (matched pairs) or repeated measurement (RM) oneway ANOVA and post-hoc Bonferroni’s test for multiple comparisons. Two-sample Kolmogorov– Smirnov (K–S) test was used to compare ISI cumulative distribution functions pre and post treatments and RM 2-way ANOVA to compare treatment effects between genotypes.
Drug source and handling
All drugs were obtained from Sigma Aldrich (St. Louis, MO) and TOCRIS Bioscience (UK). Drugs were applied by reverse microdialysis for in vivo microdialysis, or bath perfusion for slice electrophysiology.
Results and Discussion
Extracellular striatal acetylcholine is elevated in TorAΔE/+ mice
We used in vivo microdialysis to investigate extracellular ACh in striatum in awake behaving TorAΔE/+ and TorA+/+ mice. We found that TorAΔE/+ mice have approximately 16-fold higher baseline levels of striatal ACh compared to littermate TorA+/+ mice (Fig. 1A, TorA+/+: 0.08 ng/10 µL, n=4; TorAΔE/+: 1.42 ng/10 µL, n=5; t4.122=4.451, p< 0.05). The addition of neostigmine (Neo), an AChE inhibitor, to the microdialysis perfusate led to a concentration dependent increase in measured extracellular levels of ACh in both TorA+/+ and TorAΔE/+ mice, but extracellular ACh levels remained increased in TorAΔE/+ compared to TorA+/+ mice. These differences were statistically significant at each concentration examined except 10nM Neo, where the difference between genotypes was smaller and did not reach statistical significance. (Fig. 1B, TorA+/+: 0 nM=0.08, 10 nM=43.8, 100 nM=146, 500 nM=194 ng/10 µL; TorAΔE/+: 0 nM=1.42, 10 nM=63.4, 100 nM=340, 500 nM=398 ng/10 µL; Fgenotype(1,7)=3.79, ns; Fdose(3,21)=18.8, p< 0.05; Finteraction(3,21)=2.72, ns). This finding represents the first direct measurement of elevated basal ACh levels in the striatum of a DYT1 mouse model, implicating pathologically increased cholinergic tone.
Figure 1. Basal ACh measured in vivo is elevated in DYT1 mouse model.

(A) TorAΔE /+ mice show siginificantly increased basal extracellular ACh levels; *p< 0.05. (B) Dose-related ACh increase in both TorA+/+ and TorAΔE /+ animals with neostigmine bromide in aCSF; *p< 0.05 ANOVA with Tukey’s post-hoc test for individual concentrations. (C) Diagram of probe placements in striatum (CPu).
Cholinergic interneurons from TorAΔE /+ mice show paradoxical excitation in response to D2R activation but have a normal response to mAChR activation
The pathological increase in striatal cholinergic tone in rodent models of DYT1 has been proposed to be a consequence of the paradoxical excitation of ChI in response to D2R activation. Indeed, we confirmed that ChIs from TorAΔE /+ mice show paradoxical excitation in response to D2R stimulation. We performed in vitro cell-attached current-clamp recordings of ChI spontaneous firing in acute striatal slices. Using videomicroscopy, all recorded cells exhibited typical morphological features of ChIs, such as a large polygonal cell body (>15 microns) and two-three major dendritic branches (Fig. 2A); the presence of spontaneous activity characteristic of ChIs confirmed the morphological identification. In slices from TorA+/+ mice, the D2R agonist quinpirole (Quin; 10 µM, 28 min) elicited depression of ChI firing rates as observed previously (Maurice et al., 2004) (Pisani et al., 2006), Fig. 2B1, baseline: 2 ± 0.5 Hz; 20 min in Quin: 1.1 ± 0.2 Hz, n=9, p< 0.05). Also consistent with previous reports (Pisani et al., 2006; Martella et al., 2009; Sciamanna et al., 2012), a paradoxical increase in firing rate of ChIs was found in TorAΔE/+ mice during bath application of Quin (Fig. 2C1, baseline: 1.8 ± 0.3 Hz; 20 min in Quin: 2.6 ± 0.5 Hz, n=11, p< 0.05).
Figure 2. Paradoxical excitation by D2R activation in TorAΔE /+ striatal ChIs and inhibitory modulation by M2/M4 mACh autoreceptors.

(A) Photomicrograph of a patch clamp recording of a striatal ChI in an acute brain slice. (B1) In TorA+/+ mice, spontaneous activity of ChIs in cell-attached recordings is significantly decreased by D2R activation with quinpirole (Quin 10 µM, blue, n=9 cells, 3 animals * p< 0.05, **p< 0.01, Wilcoxon matched pairs signed rank test) and (B2) by mAChR activation with oxotremorine (Oxo 1µM, red, n=7 cells, 3 animals * p< 0.05, Wilcoxon matched pairs signed rank test). (C1) In TorAΔE /+ mice, D2R activation induces a paradoxical excitation of ChI firing rate (Quin 10 µM, n=11 cells, 5 animals * p< 0.05, Wilcoxon matched pairs signed rank test) while Oxo (C2) remains inhibitory on ChI firing rate (1µM, red, n=9 cells, 5 animals **p< 0.01, Wilcoxon matched pairs signed rank test). The effects are shown as a time course of the firing rate (symbols are population means of averaged 2 min bins ± SEM), and as a summary bar chart of the % of change at 10 and 20 min after agonist application. Voltage traces from cell-attached somatic recordings of ChIs are shown; scale bars apply to all traces.
Next, we confirmed that stimulation of postsynaptic M2/M4 mAChRs using the non-selective mAChR agonist oxotremorine (Oxo) significantly depressed the ChI firing rate in both TorA+/+ mice (Fig. 2B2, baseline: 3.8 ± 0.9 Hz; 20 min in Oxo: 0.8 ± 0.4 Hz, n=7, p< 0.05) and in TorAΔE/+ mice (Fig. 2C2, baseline: 5.4 ± 1.1 Hz; 20 min in Oxo: 1.6 ± 0.6 Hz, n=9, p< 0.01) with no statistical difference between genotypes (Fgenotype(1–14)= 0.47, p= 0.5). This result shows that, despite the heightened basal ACh levels measured using microdialysis in TorAΔE/+ mice, postsynaptic mAChRs in vitro are not desensitized, remain functional, and can decrease firing rate following agonist activation. The long-lasting effects of both Quin and Oxo, as shown in time course plots, suggest either that there is no receptor desensitization during a 28 min time window of continuous activation, or that receptor activation initiates long-lasting effects unrelated to the continuous presence of agonist.
Pharmacological antagonism of mAChRs reverses D2R–mediated paradoxical excitation of ChIs in TorAΔE /+ mice
The elevated extracellular ACh levels in TorAΔE/+ mice (Fig. 1) show that in these animals there is a pathologically increased cholinergic tone. Because M2/M4 mAChRs and D2Rs share coupling to Gi/o signaling which mediates the decrease in ChI firing rate and ACh release in physiological conditions (Drukarch et al., 1990; Stoof et al., 1992b; DeBoer and Abercrombie, 1996; (Yan et al., 1997), we considered the possibility that chronic activation of mAChRs due to the elevated ACh levels could be causally related to the aberrant D2R mediated increase in ChI firing rate. Precedent for this view exists, as previous work has shown that elevated ACh tone, through activation of M2/M4 mACh autoreceptors, can occlude D2R–coupling to Gi/o signaling (Drukarch et al., 1990; Yan et al., 1997).
To examine the possibility of a functional interaction between M2/M4 mAChR and D2R activation, we asked whether pharmacological inhibition of mAChRs could reverse the paradoxical D2R mediated increase in firing rate of ChIs in slices from TorAΔE /+ mice. We first tested the effect of Quin after a 15 min bath application of the non-selective mAChR antagonist scopolamine (Scop, 10 µM). Interestingly, in the presence of Scop, the excitatory effect of Quin on firing rate was significantly reduced, although the effect was not reversed to the normal inhibition of firing (Fig. 3A1–3, TorAΔE /+ baseline: 2.4 ± 0.6 Hz; 20 min in Quin: 3.1 ± 0.8 Hz, p= 0.61; Fig 3A5, CVbaseline=0.35, CVQuin=0.35, p= 0.67, n=10). Surprisingly, despite the elevated striatal ACh tone measured in vivo, we did not find a statistically significant effect of Scop on the baseline firing frequency of ChIs, as shown by the null effects on firing rate time course (Fig. 3A1, firing rate 15 min in Scop: 2.3 ± 0.8 Hz, n=10, p> 0.99), as well as on the cumulative distribution of the interspike interval (ISI) (Fig. 3A4) and coefficient of variation (CV) of the ISI (Fig. 3A5, CVScop=0.39, p= 0.38). This result, together with the evidence that activation of mAChRs (Oxo) produces the same inhibitory effect on ChI firing in TorAΔE /+ and TorA+/+ mice (Fig. 2B2–C2), suggests that mAChRs are not tonically active in ChIs from TorAΔE /+ slices, likely because the heightened cholinergic tone in vivo is not maintained in ex vivo brain preparations. Importantly, the ability of short-term (15min) pharmacological mAChR inhibition to eliminate the D2R paradoxical excitation of ChI in TorAΔE /+ slices suggests that the mAChR effect on D2R signaling must be initiated in vivo due to the increased cholinergic tone and persists in vitro, even in the absence of the maintained heightened cholinergic tone (Fig. 3A).
Figure 3. Antagonizing mAChRs reverses the D2R mediated paradoxical excitation of ChIs in TorAΔE/+ mice.

(A) Potential paradoxical D2R–mediated excitation using Quin (10µM, blue) was tested after 15 min bath application of the broad-spectrum mAChR antagonist scopolamine (Scop 10 µM, red, n=10 cells, 4 animals p= 0.61 RM one-way ANOVA) or in slices preincubated for at least 1 h before recording in (B) scopolamine (Scop, red, n=8 cells, 4 animals * p< 0.05, Wilcoxon matched pairs signed rank test) and (C) in M2/M4 mAChR selective antagonist (AF DX 384, 300nM, red, n=8 cells, 4 animals * p< 0.05, Wilcoxon matched pairs signed rank test). (1) Time course of spontaneous firing rate change, (2) summary bar chart, (3) example traces, (4) cumulative inter-spike interval (ISI) distribution and (5) changes of the median CV of ISI in ChI from TorAΔE /+ slices.
We next tested whether longer-term pharmacological mAChR inhibition (at least 1 h) can fully reverse the D2R–mediated paradoxical excitation in ChIs from TorAΔE /+ to the typical inhibition of firing. We preincubated TorAΔE /+ striatal slices in Scop for at least 1 h prior to measuring modulation of ChI spontaneous activity by the D2R agonist, Quin. Remarkably, mAChR antagonism now completely reversed the paradoxical excitation of ChIs following D2R activation, restoring the D2R mediated inhibition of firing typically observed in control animals. Specifically, in the presence of Scop (10µM), Quin reduced the firing rate of ChIs (Fig. 3 B1–3, TorAΔE /+ baseline: 4.5 ± 0.8 Hz; 20 min in Quin: 3.07 ± 0.5 Hz, n=8, p< 0.05) and regularity of discharge as shown by the increase in the CV (Fig. 3B5, CVbaseline=0.18, CVQuin=0.21, p< 0.01); accordingly, the cumulative distribution of the ISI from ChIs recorded in the Scop treated slices was shifted to the right of the ISI distribution from ChIs recorded in untreated TorAΔE /+ striatal slices (Fig. 3B4, p< 0.0001). This rightward shift is indicative of a slowed firing rate, mimicking the normal physiological response of D2R activation observed in TorA+/+ mice (Fig. 2B1).
Next, to confirm that this “rescue” is a consequence of inhibition of overactivated M2/M4 mAChRs, which are expressed by ChIs, we preincubated TorAΔE/+ slices in the M2/M4 selective antagonist AF-DX 384 (300 nM) for at least 1 h. Again, the paradoxical D2R–mediated increase in firing was prevented, although the normalization of the inhibitory effect was not observed as in Scop (Fig. 3C1–3, TorAΔE/+ baseline: 3.8 ± 0.9 Hz; 20 min in Quin: 3.2 ± 0.95 Hz, n= 8, p= 0.25). However, the discharge pattern became less regular and the CV increased as with Scop (Fig. 3C3 and 3C5, CVbaseline=0.31, CVQuin=0.48, p< 0.05), and there was a significant rightward shift in the cumulative distribution of the ISI from ChIs recorded in AF-DX 384 treated slices compared to non-treated (Fig. 3C4, p< 0.0006). It is important to mention that AF-DX 384 at the concentrations studied was not able to induce a complete normalization of D2R–mediated inhibition, likely because M2/M4 mAChRs are not fully blocked by these drug concentrations. At higher concentrations this agent loses M2/M4 mAChR selectivity (Dorje et al., 1991; Bonsi et al., 2008). Collectively, these new findings suggest that heightened activation of mAChRs, likely the M2/M4 mAChRs expressed by ChIs, is responsible for the D2R–mediated paradoxical excitation of ChI in DYT1 dystonia.
Over-activation of mAChRs in wild-type TorA+/+ mice drives D2R paradoxical excitation of ChIs
The results above suggest that in striatal ChIs from TorAΔE /+ mice, in vivo chronic activation of M2/M4 mAChRs switches D2R signaling, an effect that outlasts agonist activation of mAChRs and has a slow reversal in the presence of the receptor antagonist (Fig 3). If this is the case, rather than the TorA mutation itself directly causing the aberrant D2R response, we should be able to induce D2R–mediated paradoxical excitation in wild-type TorA+/+ mice simply by mimicking the elevated extracellular ACh that occurs in TorAΔE/+ mice. To test this hypothesis, we first used the AChE inhibitor, neostigmine (Neo, 100nM), to elevate ACh levels in striatal slices from TorA+/+ mice, and asked if subsequent D2R activation with Quin could enhance ChI firing frequency. As expected, the firing frequency of ChIs decreased in the presence of Neo, confirming an increase in extracellular ACh that activates M2/M4 mAChR autoreceptors (Fig. 4A1–3, TorA+/+ baseline: 2.02 ± 0.2 Hz; 10 min in Neo: 1.1 ± 0.2 Hz, n=10, p< 0.03). The decreased ChI firing frequency is clearly seen in the statistically significant rightward shift of the ISI cumulative distribution (Fig. 4A4, p< 0.0001) and the increased irregularity of discharge (Fig. 4A3) is shown by the significant increase of the median CV of the ISI frequency distribution histogram (Fig. 4A5, CVbaseline= 0.31; CVNeo= 1.01, n=10, p< 0.004). During the pharmacologically induced hypercholinergic state, D2R activation with Quin now causes an increase in the firing frequency, reversing the mAChR-mediated depression and mimicking the paradoxical excitation that occurs in TorAΔE/+ mice and other rodent models of DYT1 dystonia (Fig. 4A1–3, firing rate 20 min in Quin: 1.9 ± 0.4 Hz n=10, p> 0.9). The cumulative distributions of ISI shifted back to the left (Fig. 4A4), indicating an increase in the firing rate, and the median CV decreased to levels not significantly different from baseline (Fig. 4A5, CVQuin=0.43, p= 0.2).
Figure 4. Increased ACh levels in TorA+/+ mice drive the D2R paradoxical excitation of ChI firing through mAChR activation.

(A) Increase in ChI firing rate induced by the D2R agonist Quin (10 µM) in the presence of the AChE inhibitor Neo (100nM). n=10, 4 animals * p< 0.05, **p< 0.005, RM one-way ANOVA) and (B) in the presence of a mAChR agonist (Oxo 1µM, n=10 cells, 5 animals * p< 0.05, RM one-way ANOVA). (C) Quin does not induce the paradoxical excitation of ChI firing in the presence of Oxo and the D2Rs antagonist sulpiride (3 µM, n=11, 4 animals **p< 0.005, RM one-way ANOVA). (1) Time course of firing rate change; symbols represent mean ± SEM, (2) summary bar chart of % of change, (3) example traces of spontaneous firing, (4) inter-spike intervals (ISI) cumulative distribution and (5) box plot summary of the change in the coefficient of variation (CV) of the ISI frequency histograms.
In order to more specifically test the involvement of mAChR activation, and to exclude the possibility of involvement of nicotinic cholinergic receptors in the switch in polarity of the D2R response, we used the nonselective mAChR agonist Oxo, (1µM). Oxo itself, like Neo, induced a depression of ChI firing frequency, likely through M2/M4 mAChR activation (Fig. 4B1–3, TorA+/+ baseline: 5.7 ± 1.3 Hz; 10 min in Oxo: 2.7 ± 0.9 Hz, n=8, p= 0.0009; 4B4, 4B3 and 4B5, CVbaseline= 0.28; CVOxo= 0.94, n=8, p= 0.03). Again, within minutes of D2R activation with Quin, the depressed firing frequency was reversed, bringing it back to pre-Oxo baseline values (Fig. 4B1–3, firing rate 20 min in Quin: 5.5 ± 1.3 Hz, n=8, p> 0.9; 4B4 and 4B5, CVQuin= 0.38 p< 0.1). The increased excitability induced by Quin in the presence of Oxo was prevented by the D2R antagonist, sulpiride, confirming that the increased firing frequency was indeed mediated by D2Rs. The mean firing rate was reduced (Fig. 4C1–3, TorA+/+ baseline: 4.2 ± 0.7 Hz; 10 min in Oxo+Sulp: 2.06 ± 0.3 Hz, p= 0.011; 20 min in Quin: 2.4 ± 0.4 Hz, n= 10 p= 0.015) and the left-shifted ISI cumulative distribution (Fig. 4C4, p< 0.0001) and the increased CV (Fig. 4C5, CVbaseline=0.21, CVoxo+sulp=0.59, CVQuin=0.52, p= 0.04) remained significantly different from baseline. These results show that in TorA+/+ mice, mAChR activation is sufficient to reverse the polarity of D2R modulation of ChI firing frequency with an onset occurring within minutes.
Given the above results, we then asked if the ability of enhanced cholinergic tone to switch the polarity of D2R modulation of ChI firing rate persists beyond the period of mAChR activation with agonist. To address this possibility, we attempted to reproduce the experimental conditions in TorAΔE/+ mice, where the heightened extracellular ACh levels occurring in vivo gets “washed-out” in the in vitro slice, yet the D2R–mediated paradoxical excitation persists. To this end, we preincubated slices from TorA+/+ mice in Oxo for at least 1h, to mimic the chronic mAChR activation that occurs in vivo in TorAΔE/+ mice and then washed out the mAChR agonist for up to 4 h prior to testing whether D2R activation with Quin still induces an increase in ChI firing frequency (Fig. 5A). Interestingly, we found that, regardless of the duration of Oxo washout (WO) time, Quin increased the firing rate of ChIs (Fig. 5A–B, TorA+/+ baseline: 1.69 ± 0.9 Hz; 10 min in Quin after up to 4h WO of Oxo: 3.1 ± 1.1 Hz, n=5, p= 0.01). Surprisingly, the Quin-induced increase of firing frequency also outlasted exposure to the agonist, since the firing rate remained elevated for at least 16 min of Quin washout, as shown by the time course (Fig. 5 A–C) and right-shift of the ISI cumulative distribution (Fig. 5D p< 0.0001), with non significant changes of the CV of the ISI indicating that the regularity of spikes did not change (Fig. 5E, CVbaseline=0.64, CVQuin=0.6, CVQuinWO=0.51, p= 0.2 RM one-way ANOVA). Collectively, these results indicate that a temporary increase in cholinergic tone is sufficient to cause a long-lasting switch in the polarity of D2R modulation of ChI firing frequency. In fact, up to 4 h after removal of the mAChR agonist, activation of D2Rs continues to elicit an increase in ChI firing frequency, recapitulating the D2R mediated paradoxical excitation of ChIs in slices from TorAΔE/+ mice.
Figure 5. The cholinergic-induced switch in D2R signaling persists after washout of mAChR agonist.

The excitatory effect of Quin on TorA+/+ ChI firing induced by pretreatment with Oxo is shown in (A) time course of firing rate changes (n= 5, 3 animals, *p< 0.01, **p< 0.001 RM one-way ANOVA), (B) box plot summary, for each interneuron is indicated the duration (h) of Oxo washout (WO, red) before treatment with Quin, (C) example traces of somatic firing, (D) ISI cumulative distribution and (E) median CV of ISI.
A G-protein preferring D2R agonist does not elicit paradoxical ChI excitation
How can changes in mAChR activity influence the polarity of the D2R modulation on excitability? Since both D2R and mACh autoreceptors inhibit ChI spontaneous firing through a similar membrane-delimited, Gi/o/Gβγ signaling cascade (Yan and Surmeier, 1996; Yan et al., 1997), the D2R–mediated paradoxical increase of ChI activity induced by a hypercholinergic state must be mediated by the engagement of an alternative signaling pathway. Many GPCRs are able to transduce signals through more than one intracellular pathway. A growing body of evidence has shown that D2R signaling can occur through a non-canonical G protein-independent pathway involving recruitment of β-arrestin-2 (Beaulieu et al., 2005; Beaulieu et al., 2007a; Beaulieu et al., 2007b; Masri et al., 2008; Urs et al., 2012). This discovery led to the development of new functionally selective ligands, which preferentially engage either canonical or non-canonical D2R pathways (Allen et al., 2011; Free et al., 2014). We decided to use one of these new drugs, MLS1547, a G protein-biased D2R ligand with an antagonist effect on β-arrestin recruitment (Free et al., 2014), to explore the differential role of G protein and β-arrestin signaling pathways in mediating the D2R paradoxical excitation of ChIs.
In recordings of ChIs in slices from TorA+/+ mice, MLS1547 (10 µM, 28min) induced a long-lasting decrease of firing rate (Fig. 6A1–3, TorA+/+ baseline: 2.8 ± 0.8 Hz; 20 min in MLS1547: 1.6 ± 0.6 Hz, n=7, p< 0.05), accompanied by a statistically significant leftward shift in the ISI cumulative distribution (Fig. 6A4, p< 0.0001) and increase in CV (Fig. 6A5, CVbaseline=0.42, CVMLS=0.61, p< 0.05). These results suggest that the MLS1547 inhibitory effect is mediated by D2R activation, through the G protein signaling (Yan et al., 1997). Indeed, the MLS1547-induced depression of firing rate is prevented by the D2R antagonist, sulpiride (Sulp, 10µM, Fig. 6B1–5, TorA+/+ baseline: 3.4 ± 1.4 Hz, 8min in Sulp: 3.5 ± 1.5 Hz, 14min in MLS1547: 3.6 ± 1.5 Hz, n=7, p= 0.65). Interestingly, MLS1547, unlike Quin, did not induce paradoxical excitation of ChIs during tonic activation of mAChRs in the presence of Oxo. The rate and regularity of pacemaking were depressed by Oxo and did not further change in MLS1547 (Fig. 6C1–3, TorA+/+ baseline: 4.6 ± 0.8 Hz, 10 min in Oxo: 1.8 ± 0.6 Hz, p= 0.009; 20 min in MLS1547: 1.6 ± 0.9 Hz, n=8, p= 0.04; Fig. 6C5, CVbaseline=0.2, CVoxo=0.72, p= 0.03; CVMLS=1.15, p= 0.01). Furthermore, a statistical comparison between the time courses of the effect on firing rate of Oxo alone versus MLS+Oxo showed no significant difference (Fcell(1–11)=1.37, p= 0.2), suggesting that they are acting through a common Gi/o pathway, thus, mAChR activation by Oxo occludes further depression by MLS1547. These results suggest that D2R activation of G-protein signaling alone is insufficient to drive the paradoxical response of ChI.
Figure 6. G protein-biased D2R agonist fails to induce a paradoxical increase of ChI firing rate.

(A) Inhibitory effect of a G-preferred D2Rs agonist MLS1547 (10 µM, green) on ChI spontaneous firing rate in TorA+/+ mice. (1) Time course, (2) summary bar chart, (3) example traces of spontaneous firing, (4) cumulative distribution of ISI and (5) box plot of changes in CV of ISI, n=7 cells, 2 animals * p< 0.05, Wilcoxon matched pairs signed rank test. (B) MLS1547 did not induce an inhibitory response in ChIs in the presence of sulpiride (Sulp 10, µM, blue) as shown by (1) time course of firing rate change, (2) summary bar chart, (3) example traces of spontaneous firing (4) cumulative distribution of ISI and (5) CV of ISI before and after MLS1547 in the presence of Sulp, n=7 cells, 3 animals, RM one-way ANOVA). (C) MLS1547 did not change the inhibitory response induced by Oxo on ChIs firing rate (Oxo 1, µM, red) as shown by (1) time course of firing rate change, (2) summary bar chart, (3) example traces of spontaneous firing, (4) cumulative distribution of ISI and (5) CV of ISI before and after MLS1547 in the presence of Oxo, n=8 cells, 4 animals *p< 0.01, **p< 0.001 RM one-way ANOVA).
Recruitment of β-arrestin signaling at D2Rs is required for the ChI paradoxical excitatory response
To test the hypothesis that β-arrestin signaling is involved in the D2R–mediated increase in firing rate of ChIs, we used a functionally selective D2R agonist with opposite pharmacology of MLS1547. These ligands have recently been developed and characterized based on the scaffold of the unbiased D2R ligand aripiprazole and they do not elicit any Gi/o agonist activity downstream of D2Rs but have partial agonist activity mediated through β-arrestin recruitment (Allen et al., 2011; Chen et al., 2012). Recently, the β-arrestin-biased D2R ligand UNC9994 has been reported to enhance the firing of parvalbumin-positive (PV) fast-spiking interneurons (FSIs) of the prefrontal cortex (PFC), suggesting that D2R-βarrestin agonism drives cortical FSIs excitability (Urs et al., 2016). We tested UNC9994 in our system with the intent of verifying the idea that β-arrestin recruitment by D2R activation drives the increase of striatal ChI firing as well. When tested in wild-type mice, UNC9994 had no significant effect on the spontaneous striatal ChI firing rate at baseline (Supplementary Fig. S1A1) or after activation of mAChRs with Oxo (Fig. S1A2). As a positive control, we tested the effect of UNC9994 on PV-FSIs excitability in prefrontal cortex using PV/tdTomato mice and reproduced the excitatory effect described by Urs and colleagues (Urs et al., 2016) (Fig. S1B1), while it totally lacked efficacy on striatal ChIs in the same mouse (Fig. S1B2). Importantly, Urs and colleagues also reported that UNC9994 does not reverse the inhibitory effect of Quin in D2R–expressing projection neurons (MSNs) in striatum. They suggest that the same pharmacological agent UNC9994 can act as a D2R-βarrestin agonist on cortical FSIs but as a D2R-β-arrestin antagonist in striatal D2 MSNs, due to regional differences in expression patterns of transducer proteins such GRKs, and β-arrestins (Urs et al., 2016; Urs et al., 2017). Our data suggest that the same may be true of striatal ChIs, and that UNC9994 and other currently available drugs are not suitable for a direct test of the hypothesized role of β-arrestin signaling in driving D2R–mediated excitatory responses of striatal ChI.
Since the pharmacological approach proved to be inadequate to verify the role of β-arrestin, we used a genetic approach and tested the effect of Quin on ChI firing rate in striatal slices from mice lacking the β-arrestin2 gene (β-Arr2 KO). In β-Arr2 KO mice, activation of D2Rs with Quin under basal conditions tended to reduce the ChI spontaneous discharge rate, although the effect was small and the group data did not reach statistical significance (data not shown: β-Arr2 KO baseline: 3.04 ± 0.9 Hz; 20 min in Quin: 2.6 ± 0.5 Hz p= 0.64, n=8 cells, 5 animals). In the presence of Oxo, which in TorA+/+ ChIs leads to the D2R paradoxical excitation, bath application of Quin was ineffective in reversing the Oxo-induced depression of firing rate and regularity of discharge in β-Arr2 KO ChIs (Fig. 7A1–3, β-Arr2 KO baseline: 7.5 ± 0.5 Hz, 12 min in Oxo: 4.5 ± 0.8 Hz, p= 0.02, 20 min in Quin: 4.1 ± 0.8 Hz, p= 0.009, n=8; Fig. 7A4, p< 0.0001 and Fig. 7A5, CVbaseline=0.17, CVoxo=0.32, CVQuin=0.37, p< 0.001). This evidence supports the idea that the recruitment of β-arrestin signaling is required in order to produce the D2R–mediated paradoxical excitation of ChIs during activation of mAChRs.
Figure 7. Recruitment of β-arrestin signaling pathway is required for the D2R–mediated paradoxical excitatation of ChIs.

(A) In β-Arr2 KO mice, in the presence of Oxo (1µM,red), which drives the D2R excitatory effect in wild-type mice, the effect of Quin (10µM, blue) is shown as (1) time course of firing rate change (2) summary bar chart of % of change, (3) example traces, (4) cumulative distribution of ISI and (5) changes in the CV of ISI. Note the lack of D2R–mediated paradoxical excitation, n=8 cells, 4 animals * p< 0.05, **p< 0.005 RM one-way ANOVA. (B) In TorAΔE /+ ChIs, the inhibitory effect induced by a G-biased D2R agonist (MLS1547 10µM, green) on (1) time course of firing rate, (2) summary bar chart of % of change, (3) example traces of spontaneous firing, (4) cumulative distribution of ISI and (5) changes of CV of ISI (n=6 cells, 4 animals * p< 0.05, Wilcoxon matched pairs signed rank test) suggests the involvement of β-arrestin signaling in the D2R–mediated paradoxical excitation.
The next pertinent question brings us back to DYT1 mice, where the Quin-induced paradoxical excitation of ChIs was first discovered. We then asked whether, in the TorAΔE /+ DYT1 mouse, a paradoxical excitation can be elicited using the G-protein biased D2R agonist, MLS1547, in place of the unbiased agonist Quin. We bath applied MLS1547 during ChI recordings in TorAΔE /+ slices and found that the paradoxical excitation was absent, and only an inhibitory response was observed, likely mediated by the D2R–G-protein coupled pathway. The mean firing rate was reduced significantly (Fig. 7B1–3, TorAΔE /+ baseline: 4.9 ± 1.4 Hz, 20 min in MLS1547: 2.6 ± 1.4 Hz, p= 0.03, n=6,), the ISI cumulative distribution shifted to the left (Fig. 7B4, p< 0.0001) and the CV rose from CVbaseline=0.19 to CVMLS=0.48, but did not reach statistical significance (Fig. 7B5, p= This suggests that in ChIs from TorAΔE /+ mice a highly efficacious G-biased D2R agonist can still put D2Rs in a conformation suitable for Gi/o activation, allowing them to act as inhibitors of ChI spontaneous firing rate. Importantly, the evidence that MLS1547, which stimulates G protein–based signaling downstream of D2R activation while simultaneously inhibiting signaling through the β-arrestin pathway, prevents the paradoxical increase in excitability of TorAΔE /+ ChI suggests that the β-arrestin pathway may be involved in mediating the increase of ChI excitability, since Gi/o stimulation alone is insufficient to drive the paradoxical excitation.
Although the mechanism by which the activation of mAChRs leads to the shift in biased signaling at D2Rs is at present unclear, one possibility is that there is a direct receptor-receptor interaction similar to what has been described for adenosine 2A receptor and D2R (A2A)-D2R heterodimers. Previous studies have shown that A2A agonist-activated allosteric interaction can induce a D2R conformational change, which switches D2Rs binding from Gi/o to β-arrestin. Thus, D2R–Gi/o signaling is reduced and signaling via β-arrestin dominates (Borroto-Escuela et al., 2011; Borroto-Escuela et al., 2016). Our experimental evidence suggests that a similar mechanism may be mediating interactions of M2/M4 mAChRs and D2Rs found on striatal ChIs (Fig. 8). However, since there is no direct evidence that such mAChR/D2R heterodimers exist, an alternative possibility could be that interactions occur at the level of the signaling pathways. This interpretation is supported by evidence that there is a dramatic decline in the ability of striatal D2Rs to activate their cognate Gi/o proteins in transgenic DYT1 mice (Napolitano et al., 2010).
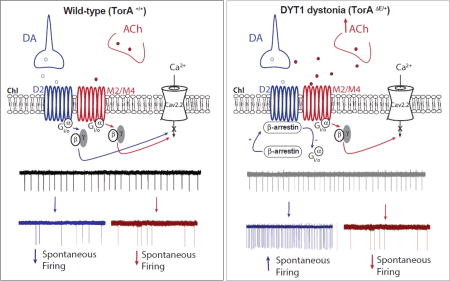
Figure 8. The extracellular ACh level influences the ChI response to dopamine D2R activation.

In TorAΔE/+ mice, an increase in striatal cholinergic tone and ChI muscarinic M2/M4 autoreceptor activation switch the coupling of D2Rs from Gi/o (left panel) towards a non-canonical β-arrestin signaling pathway (right panel). This in turn enhances ChIs spontaneous firing, reversing the physiological inhibitory modulation of dopamine D2Rs on this class of interneurons.
Conclusion
In this study, we demonstrate: 1) there is an intrinsic hypercholinergic state in the TorAΔE/+ mouse model of DYT1dystonia; and 2) that the ΔE-TorA mutation is not directly responsible for the paradoxical effect of D2R stimulation on firing rate in TorAΔE/+ mice. Furthermore, we identify a novel interaction between muscarinic AChRs and dopaminergic D2Rs in modulating striatal ChI excitability.
The hypercholinergic state was established through direct measurement of extracellular striatal ACh, a 16-fold increase was found in TorAΔE/+ mice. The cause of the “D2R paradoxical excitation” of ChI spontaneous firing and the novel interaction of mAChRs and D2Rs were revealed by several findings. First, pharmacological block of M2/M4 mACh autoreceptors on ChIs of TorAΔE/+ mice prevented the paradoxical excitatory effect of D2R activation on ChI firing which is typically found in these animals. Second, in wild-type TorA+/+ mice, increasing the extracellular ACh level or directly activating mAChRs can within minutes reverse the polarity of D2R signaling, converting the normal D2R–mediated inhibition of ChI firing into a “paradoxical” stimulation, mimicking the paradoxical excitation of ChIs observed in the DYT1 knock-in TorAΔE/+ mouse and other rodent models of DYT1 dystonia (Eskow Jaunarajs et al., 2015). Interestingly, while the cholinergic-induced switch in D2R signaling is rapid in onset, as shown by experiments in wild-type mice where the D2R–mediated paradoxical increase in ChI firing occurs after only a few minutes of mAChR activation, it is very slow to reverse, requiring at least 1 h of mAChR pharmacological antagonism to reverse D2R excitation into inhibition. Furthermore, once this change is induced by temporary activation of mAChRs, it can persist hours later following mAChR agonist washout. This observation reveals that a form of “signaling” plasticity exists where prior activation of one GPCR can directly impact signaling of another, similarly coupled GPCR, rendering a response in the opposite direction.
Third, we found that in mice with genetic deletion of β-arrestin, the D2R paradoxical response was absent even when mAChRs were pharmacologically activated. Furthermore, MLS1547, a G-protein biased D2R agonist that does not engage β-arrestin signaling, was never able to excite ChI firing either in TorAΔE/+ mice with intrinsic hypercholinergic tone or in wild-type mice with artificially enhanced cholinergic activity.
Our findings support the view that the hypercholinergic state is the primary defect in the TorAΔE /+ mouse model of human DYT1 dystonia. What causes the increase in ACh in this animal model remains elusive, but our findings warrant further attention on the potential roles of TorA in regulating ACh release (Torres et al., 2004; Kakazu et al., 2012a; Kakazu et al., 2012b).
Functional significance
In most physiological conditions we would expect that low cholinergic tone would prevail and would be reinforced by the inhibitory effects of D2R activation on ChIs, since the ambient dopamine tone is likely sufficient to activate high-affinity D2R signaling. In the presence of pathologically high cholinergic tone, instead, the gain of excitatory D2R function on ChIs could further amplify the hypercholinergic state by promoting further ACh release. The situation in DYT1 animals, however, may be more complicated as the heightened extracellular ACh will likely cause desensitization of presynaptic nAChRs on dopaminergic terminals, leading to decreased tonic extracellular dopamine levels and increased phasic dopamine release during burst of dopaminergic neurons activity (Exley and Cragg, 2008). This idea is supported by reports showing enhanced responsiveness of striatal dopamine release in a dystonia mouse model to phasic versus tonic stimulation (Bao et al., 2010).
With this in mind, we suggest that the excitatory switch we observed in D2Rs as a consequence of the elevated ACh tone might be most functionally important during episodic elevations in dopamine activity occurring in associative and motor learning situations, rather than in basal dopaminergic states. D2R dysregulation of signaling may cause aberrant responses of ChIs during episodic elevations of dopamine leading to impaired synaptic plasticity that supports long-term changes in motor function and contributing to the pathophysiology of dystonia. Interventions targeted at both the primary defect in hypercholinergic tone and the downstream alterations in striatal signaling may be useful in modulating or reversing these abnormal motor functions.
Supplementary Material
Somatic cell-attached recording of ChI spontaneous firing change induced by D2R–β-arrestin biased agonist UNC9994 (10µM in 10−4 DMSO+20% v/v HBC). Non statistically significant changes were found either on (A1) basal firing rate (baseline: 2.7 ± 1.03 Hz, 20 min in UNC9994: 2.8 ± 0.93 Hz, n= 6 cells, 2 animals, p= 0.56) nor (A2) in the presence of mAChR agonist Oxo (baseline 2.8 ± 0.6 Hz, 10min in Oxo: 1.5 ± 0.5 Hz, 20min in UNC9994: 1.2 ± 0.4 Hz, n= 7cells, 4 animals, p= 0.12, RM one-way ANOVA). Different effects of UNC9994 on firing rate evoked by +200pA current steps in representative whole cell, current-clamp recordings from (B1) a parvalbumin-positive (PV) fast spiking interneuron (FSI) of prefrontal cortex and (B2) a striatal cholinergic interneuron from a mouse that expresses tdTomato in PV expressing interneurons. The pipette solution used for whole cell experiments consisted of (in mM): Kgluconate (120), EGTA (0.6) MgCl2 (5), ATPNa (2), GTP-Na (0.3) and HEPES (40).
Box plot summary of the mean with SEM of baseline firing rates of ChI from TorA+/+(circle, 4.07 ± 0.4 Hz, n= 40), TorAΔE/+ (square, 3.64 ± 0.4 Hz, n= 26) and β-Arr2 KO (triangle, 4.45 ± 0.5 Hz, n= 25) shows that there is no statistical difference between genotypes F(2,88)= 0.58, p= 0.56, one-way ANOVA.
Highlights.
In a mouse model of DYT1 dystonia there is enhanced striatal cholinergic tone
Cholinergic tone modulates the polarity of D2R signaling in cholinergic neurons
Elevated cholinergic tone, as found in DYT1, causes D2R “paradoxical excitation”
Muscarinic antagonists restore the normal inhibitory effect of D2R signaling
Changes in D2R polarity arises from a shift from Gi/o to β-arrestin pathways
Acknowledgments
We thank Allie J. Widman for intellectual contributions, Dr. Lynn Dobrunz for providing PV-Cre/tdTomato mice, Dr. Jian Jin at the University of North Carolina Chapel Hill for the UNC9994 compound, and Dr. Antonio Pisani for comments on the manuscript.
Funding sources: This work was supported by National Institute of Health [P01 NS087997, 2015–16] and Johnson Family Research Acceleration Fund for Dystonia [2015–16].
Abbreviations
- ACh
acetylcholine
- AChE
acetylcholinesterase
- ChI
cholinergic interneurons
- D2R
dopamine D2 receptors
- mAChR
muscarinic receptors
- Oxo
oxotremorine
- Quin
quinpirole
- Neo
neostigmine
- TorA
torsinA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: MS, LLM and DGS designed the experiments and wrote the manuscript; MS performed and analyzed electrophysiology experiments; CNZ and KLJ performed and analyzed microdialysis experiments; QW provided significant intellectual input on experimental design and data interpretation.
References
- Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, Jensen NH, Che X, Bai X, Frye SV, Wetsel WC, Caron MG, Javitch JA, Roth BL, Jin J. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aosaki T, Miura M, Suzuki T, Nishimura K, Masuda M. Acetylcholine-dopamine balance hypothesis in the striatum: an update. Geriatrics & gerontology international. 2010;10(Suppl 1):S148–157. doi: 10.1111/j.1447-0594.2010.00588.x. [DOI] [PubMed] [Google Scholar]
- Bao L, Patel JC, Walker RH, Shashidharan P, Rice ME. Dysregulation of striatal dopamine release in a mouse model of dystonia. Journal of neurochemistry. 2010;114:1781–1791. doi: 10.1111/j.1471-4159.2010.06890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR, Caron MG. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends in pharmacological sciences. 2007a;28:166–172. doi: 10.1016/j.tips.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Tirotta E, Sotnikova TD, Masri B, Salahpour A, Gainetdinov RR, Borrelli E, Caron MG. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007b;27:881–885. doi: 10.1523/JNEUROSCI.5074-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE. Effects of acetylcholine in the striatum. Recent insights and therapeutic implications. Neurology. 2012;79:274–281. doi: 10.1212/WNL.0b013e31825fe154. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science (New York, NY) 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bonsi P, Martella G, Cuomo D, Platania P, Sciamanna G, Bernardi G, Wess J, Pisani A. Loss of muscarinic autoreceptor function impairs long-term depression but not long-term potentiation in the striatum. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:6258–6263. doi: 10.1523/JNEUROSCI.1678-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsi P, Cuomo D, Martella G, Madeo G, Schirinzi T, Puglisi F, Ponterio G, Pisani A. Centrality of striatal cholinergic transmission in Basal Ganglia function. Frontiers in neuroanatomy. 2011;5:6. doi: 10.3389/fnana.2011.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroto-Escuela DO, Romero-Fernandez W, Tarakanov AO, Ciruela F, Agnati LF, Fuxe K. On the existence of a possible A2A–D2-beta-Arrestin2 complex: A2A agonist modulation of D2 agonist-induced beta-arrestin2 recruitment. Journal of molecular biology. 2011;406:687–699. doi: 10.1016/j.jmb.2011.01.022. [DOI] [PubMed] [Google Scholar]
- Borroto-Escuela DO, Pintsuk J, Schafer T, Friedland K, Ferraro L, Tanganelli S, Liu F, Fuxe K. Multiple D2 heteroreceptor complexes: new targets for treatment of schizophrenia. Therapeutic advances in psychopharmacology. 2016;6:77–94. doi: 10.1177/2045125316637570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nature reviews Neuroscience. 2008;9:222–234. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Sancesario G, North RA, Bernardi G. Muscarinic IPSPs in rat striatal cholinergic interneurones. The Journal of physiology. 1998;510(Pt 2):421–427. doi: 10.1111/j.1469-7793.1998.421bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Rothwell J, Ghiglieri V, Obeso JA, Picconi B. Hyperkinetic disorders and loss of synaptic downscaling. Nature neuroscience. 2016;19:868–875. doi: 10.1038/nn.4306. [DOI] [PubMed] [Google Scholar]
- Chen HT, Ruan NY, Chen JC, Lin TY. Dopamine D2 receptor-mediated Akt/PKB signalling: initiation by the D2S receptor and role in quinpirole-induced behavioural activation. ASN neuro. 2012;4:371–382. doi: 10.1042/AN20120013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ. Meaningful silences: how dopamine listens to the ACh pause. Trends in neurosciences. 2006;29:125–131. doi: 10.1016/j.tins.2006.01.003. [DOI] [PubMed] [Google Scholar]
- DeBoer P, Abercrombie ED. Physiological release of striatal acetylcholine in vivo: modulation by D1 and D2 dopamine receptor subtypes. The Journal of pharmacology and experimental therapeutics. 1996;277:775–783. [PubMed] [Google Scholar]
- Ding J, Guzman JN, Tkatch T, Chen S, Goldberg JA, Ebert PJ, Levitt P, Wilson CJ, Hamm HE, Surmeier DJ. RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nature neuroscience. 2006;9:832–842. doi: 10.1038/nn1700. [DOI] [PubMed] [Google Scholar]
- Dorje F, Wess J, Lambrecht G, Tacke R, Mutschler E, Brann MR. Antagonist binding profiles of five cloned human muscarinic receptor subtypes. The Journal of pharmacology and experimental therapeutics. 1991;256:727–733. [PubMed] [Google Scholar]
- Drukarch B, Schepens E, Stoof JC. Muscarinic receptor activation attenuates D2 dopamine receptor mediated inhibition of acetylcholine release in rat striatum: indications for a common signal transduction pathway. Neuroscience. 1990;37:1–9. doi: 10.1016/0306-4522(90)90186-8. [DOI] [PubMed] [Google Scholar]
- Eskow Jaunarajs KL, Bonsi P, Chesselet MF, Standaert DG, Pisani A. Striatal cholinergic dysfunction as a unifying theme in the pathophysiology of dystonia. Progress in neurobiology. 2015;127–128:91–107. doi: 10.1016/j.pneurobio.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R, Cragg SJ. Presynaptic nicotinic receptors: a dynamic and diverse cholinergic filter of striatal dopamine neurotransmission. British journal of pharmacology. 2008;153(Suppl 1):S283–297. doi: 10.1038/sj.bjp.0707510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Free RB, et al. Discovery and characterization of a G protein-biased agonist that inhibits beta-arrestin recruitment to the D2 dopamine receptor. Molecular pharmacology. 2014;86:96–105. doi: 10.1124/mol.113.090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Jankovic J. Medical treatment of dystonia. Movement disorders : official journal of the Movement Disorder Society. 2013;28:1001–1012. doi: 10.1002/mds.25552. [DOI] [PubMed] [Google Scholar]
- Joshua M, Adler A, Mitelman R, Vaadia E, Bergman H. Midbrain dopaminergic neurons and striatal cholinergic interneurons encode the difference between reward and aversive events at different epochs of probabilistic classical conditioning trials. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:11673–11684. doi: 10.1523/JNEUROSCI.3839-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakazu Y, Koh JY, Ho KW, Gonzalez-Alegre P, Harata NC. Synaptic vesicle recycling is enhanced by torsinA that harbors the DYT1 dystonia mutation. Synapse (New York, NY) 2012a;66:453–464. doi: 10.1002/syn.21534. [DOI] [PubMed] [Google Scholar]
- Kakazu Y, Koh JY, Iwabuchi S, Gonzalez-Alegre P, Harata NC. Miniature release events of glutamate from hippocampal neurons are influenced by the dystonia-associated protein torsinA. Synapse (New York, NY) 2012b;66:807–822. doi: 10.1002/syn.21571. [DOI] [PubMed] [Google Scholar]
- Maltese M, Martella G, Madeo G, Fagiolo I, Tassone A, Ponterio G, Sciamanna G, Burbaud P, Conn PJ, Bonsi P, Pisani A. Anticholinergic drugs rescue synaptic plasticity in DYT1 dystonia: role of M1 muscarinic receptors. Movement disorders : official journal of the Movement Disorder Society. 2014;29:1655–1665. doi: 10.1002/mds.26009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella G, Maltese M, Nistico R, Schirinzi T, Madeo G, Sciamanna G, Ponterio G, Tassone A, Mandolesi G, Vanni V, Pignatelli M, Bonsi P, Pisani A. Regional specificity of synaptic plasticity deficits in a knock-in mouse model of DYT1 dystonia. Neurobiology of disease. 2014;65:124–132. doi: 10.1016/j.nbd.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Martella G, Tassone A, Sciamanna G, Platania P, Cuomo D, Viscomi MT, Bonsi P, Cacci E, Biagioni S, Usiello A, Bernardi G, Sharma N, Standaert DG, Pisani A. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain : a journal of neurology. 2009;132:2336–2349. doi: 10.1093/brain/awp194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, Caron MG. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13656–13661. doi: 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Mercer J, Chan CS, Hernandez-Lopez S, Held J, Tkatch T, Surmeier DJ. D2 dopamine receptor-mediated modulation of voltage-dependent Na+ channels reduces autonomous activity in striatal cholinergic interneurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:10289–10301. doi: 10.1523/JNEUROSCI.2155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H. Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron. 2004;43:133–143. doi: 10.1016/j.neuron.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Napolitano F, Pasqualetti M, Usiello A, Santini E, Pacini G, Sciamanna G, Errico F, Tassone A, Di Dato V, Martella G, Cuomo D, Fisone G, Bernardi G, Mandolesi G, Mercuri NB, Standaert DG, Pisani A. Dopamine D2 receptor dysfunction is rescued by adenosine A2A receptor antagonism in a model of DYT1 dystonia. Neurobiology of disease. 2010;38:434–445. doi: 10.1016/j.nbd.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Page CE, Klein C, Hewett JW, Mineta M, Leung J, Shalish C, Bressman SB, de Leon D, Brin MF, Fahn S, Corey DP, Breakefield XO. The TOR1A (DYT1) gene family and its role in early onset torsion dystonia. Genomics. 1999;62:377–384. doi: 10.1006/geno.1999.6039. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C, Pennisi M, Topple A. Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. Journal of neuroscience methods. 1985;13:139–143. doi: 10.1016/0165-0270(85)90026-3. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends in neurosciences. 2007;30:545–553. doi: 10.1016/j.tins.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bonsi P, Centonze D, Gubellini P, Bernardi G, Calabresi P. Targeting striatal cholinergic interneurons in Parkinson’s disease: focus on metabotropic glutamate receptors. Neuropharmacology. 2003;45:45–56. doi: 10.1016/s0028-3908(03)00137-0. [DOI] [PubMed] [Google Scholar]
- Pisani A, Martella G, Tscherter A, Bonsi P, Sharma N, Bernardi G, Standaert DG. Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiology of disease. 2006;24:318–325. doi: 10.1016/j.nbd.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Quartarone A, Hallett M. Emerging concepts in the physiological basis of dystonia. Movement disorders : official journal of the Movement Disorder Society. 2013;28:958–967. doi: 10.1002/mds.25532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. Nicotine amplifies reward-related dopamine signals in striatum. Nature neuroscience. 2004;7:583–584. doi: 10.1038/nn1244. [DOI] [PubMed] [Google Scholar]
- Sciamanna G, Hollis R, Ball C, Martella G, Tassone A, Marshall A, Parsons D, Li X, Yokoi F, Zhang L, Li Y, Pisani A, Standaert DG. Cholinergic dysregulation produced by selective inactivation of the dystonia-associated protein torsinA. Neurobiology of disease. 2012;47:416–427. doi: 10.1016/j.nbd.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoof JC, Drukarch B, de Boer P, Westerink BH. In vitro and in vivo acetylcholine release from rat striatum as a functional paradigm of signal transduction via a D-2 dopamine receptor. Neurochemistry international. 1992a;20(Suppl):201S–205S. doi: 10.1016/0197-0186(92)90239-n. [DOI] [PubMed] [Google Scholar]
- Stoof JC, Drukarch B, de Boer P, Westerink BH, Groenewegen HJ. Regulation of the activity of striatal cholinergic neurons by dopamine. Neuroscience. 1992b;47:755–770. doi: 10.1016/0306-4522(92)90027-y. [DOI] [PubMed] [Google Scholar]
- Straub C, Tritsch NX, Hagan NA, Gu C, Sabatini BL. Multiphasic modulation of cholinergic interneurons by nigrostriatal afferents. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:8557–8569. doi: 10.1523/JNEUROSCI.0589-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Torres GE, Sweeney AL, Beaulieu JM, Shashidharan P, Caron MG. Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated DeltaE-torsinA mutant. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15650–15655. doi: 10.1073/pnas.0308088101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs NM, Peterson SM, Caron MG. New Concepts in Dopamine D2 Receptor Biased Signaling and Implications for Schizophrenia Therapy. Biological psychiatry. 2017;81:78–85. doi: 10.1016/j.biopsych.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs NM, Snyder JC, Jacobsen JP, Peterson SM, Caron MG. Deletion of GSK3beta in D2R–expressing neurons reveals distinct roles for beta-arrestin signaling in antipsychotic and lithium action. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:20732–20737. doi: 10.1073/pnas.1215489109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs NM, Gee SM, Pack TF, McCorvy JD, Evron T, Snyder JC, Yang X, Rodriguiz RM, Borrelli E, Wetsel WC, Jin J, Roth BL, O’Donnell P, Caron MG. Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E8178–E8186. doi: 10.1073/pnas.1614347113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland S, Du D, Oswald MJ, Parlato R, Kohr G, Kelsch W. Phasic dopaminergic activity exerts fast control of cholinergic interneuron firing via sequential NMDA, D2, and D1 receptor activation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:11549–11559. doi: 10.1523/JNEUROSCI.1175-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ. Muscarinic (m2/m4) receptors reduce N- and P-type Ca2+ currents in rat neostriatal cholinergic interneurons through a fast, membrane-delimited, G-protein pathway. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16:2592–2604. doi: 10.1523/JNEUROSCI.16-08-02592.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Song WJ, Surmeier J. D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein-kinase-C-insensitive pathway. Journal of neurophysiology. 1997;77:1003–1015. doi: 10.1152/jn.1997.77.2.1003. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Zhang K, Liu X, Yan H, Ma X, Zhang S, Zheng J, Wang L, Wei X. Involvement of HCN Channel in Muscarinic Inhibitory Action on Tonic Firing of Dorsolateral Striatal Cholinergic Interneurons. Frontiers in cellular neuroscience. 2016;10:71. doi: 10.3389/fncel.2016.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Somatic cell-attached recording of ChI spontaneous firing change induced by D2R–β-arrestin biased agonist UNC9994 (10µM in 10−4 DMSO+20% v/v HBC). Non statistically significant changes were found either on (A1) basal firing rate (baseline: 2.7 ± 1.03 Hz, 20 min in UNC9994: 2.8 ± 0.93 Hz, n= 6 cells, 2 animals, p= 0.56) nor (A2) in the presence of mAChR agonist Oxo (baseline 2.8 ± 0.6 Hz, 10min in Oxo: 1.5 ± 0.5 Hz, 20min in UNC9994: 1.2 ± 0.4 Hz, n= 7cells, 4 animals, p= 0.12, RM one-way ANOVA). Different effects of UNC9994 on firing rate evoked by +200pA current steps in representative whole cell, current-clamp recordings from (B1) a parvalbumin-positive (PV) fast spiking interneuron (FSI) of prefrontal cortex and (B2) a striatal cholinergic interneuron from a mouse that expresses tdTomato in PV expressing interneurons. The pipette solution used for whole cell experiments consisted of (in mM): Kgluconate (120), EGTA (0.6) MgCl2 (5), ATPNa (2), GTP-Na (0.3) and HEPES (40).
Box plot summary of the mean with SEM of baseline firing rates of ChI from TorA+/+(circle, 4.07 ± 0.4 Hz, n= 40), TorAΔE/+ (square, 3.64 ± 0.4 Hz, n= 26) and β-Arr2 KO (triangle, 4.45 ± 0.5 Hz, n= 25) shows that there is no statistical difference between genotypes F(2,88)= 0.58, p= 0.56, one-way ANOVA.