

Abstract
Prostate-specific membrane antigen (PSMA) is highly expressed in prostate cancer cells and nonprostatic solid tumor neovasculature and is a target for anticancer imaging and therapeutic agents. PSMA acts as a glutamate carboxypeptidase (GCPII) on small molecule substrates, including folate, the anticancer drug methotrexate, and the neuropeptide N-acetyl-l-aspartyl-l-glutamate. Here we present the 3.5-Å crystal structure of the PSMA ectodomain, which reveals a homodimer with structural similarity to transferrin receptor, a receptor for iron-loaded transferrin that lacks protease activity. Unlike transferrin receptor, the protease domain of PSMA contains a binuclear zinc site, catalytic residues, and a proposed substrate-binding arginine patch. Elucidation of the PSMA structure combined with docking studies and a proposed catalytic mechanism provides insight into the recognition of inhibitors and the natural substrate N-acetyl-l-aspartyl-l-glutamate. The PSMA structure will facilitate development of chemotherapeutics, cancer-imaging agents, and agents for treatment of neurological disorders.
Keywords: crystallography, folate hydrolase, glutamate carboxypeptidase II, Naaladase I
Prostate-specific membrane antigen (PSMA), also known as glutamate carboxypeptidase II (EC 3.4.17.21), N-acetyl-α-linked acidic dipeptidase I [Naaladase (NLD) I], or folate hydrolase, is a 750-residue type II transmembrane glycoprotein that is highly expressed in prostate cancer cells and in nonprostatic solid tumor neovasculature (1) and expressed at lower levels in other tissues (2, 3), including healthy prostate, kidney, liver, small intestine, and brain (reviewed in refs. 4 and 5). PSMA expression increases with prostate disease progression and metastasis (6), thus PSMA is the target of a Food and Drug Administration-approved prostate cancer-imaging agent, the 111In-labeled 7E11 monoclonal antibody (Prostascint, Cytogen, Princeton, NJ), which binds an intracellular epitope exposed upon cell death. The extracellular domain of PSMA is targeted by monoclonal antibodies such as J591, which is in clinical trials for specific delivery of imaging agents or therapeutics to cells overexpressing PSMA (7).
In addition to its role as a tumor marker, PSMA contains a binuclear zinc site and is active as a glutamate carboxypeptidase, catalyzing the hydrolytic cleavage of α- or γ-linked glutamates from peptides or small molecules (8). Its substrates include poly-γ-glutamated folates, which are essential nutrients, and the poly-γ-glutamated form of the anticancer drug methotrexate (Fig. 1A) (9), in which case cleavage renders it less efficacious. The enzymatic activity of PSMA can be exploited for the design of prodrugs, in which an inactive glutamated form of the drug is selectively cleaved and thereby activated only at cells that express PSMA (10).
Fig. 1.

Structural comparison of PSMA and related proteins. The common fold of the protease or protease-like domain consists of a central seven- to eight-stranded mixed β-sheet and 6-11 flanking α-helices. Ribbon diagrams of side and top view of PSMA (A), side and top view of TfR1 (PDB ID code 1CX8) (B), AAP (PDB ID code 1RTQ) (C), CPG2 (PDB ID code 1CG2) (D). The protease (or protease-like) domains are shown in blue, the apical domains of PSMA and TfR1 in green, the helical domains in red, and the CPG2 dimerization domain in pink. Water molecules are red, zinc ions are orange, zinc-binding residues are yellow sticks, and carbohydrate residues are shown in a color-coded ball-and-stick representation. Representative substrates are shown below the appropriate molecule. R1 = OH or NH2, and R2 = H or CH3 for folate and methotrexate, respectively. Note: A mono-γ-glutamated folate is shown for clarity; in poly-γ-glutamated folate/methotrexate, the additional glutamates (up to seven) are attached to the glutamate shown via a γ-peptide linkage.
PSMA also cleaves and inactivates the abundant neuropeptide N-acetyl-l-aspartyl-l-glutamate (α-NAAG) (11) (Fig. 1 A), which is an inhibitor of the NMDA ionotropic receptor and an agonist of the type II metabotropic glutamate receptor subtype 3 (12). A breakdown of the regulation of glutamatergic neurotransmission by α-NAAG is implicated in schizophrenia, seizure disorders, Alzheimer's disease, Huntington's disease, and amyotrophic lateral sclerosis (13). Thus, inhibition of PSMA potentially confers neuroprotection both by reducing glutamate and increasing α-NAAG. Indeed, the subnanomolar inhibitor 2-(phosphonomethyl) pentanedioc acid has been shown to provide neuroprotection in cell culture and/or animal models of ischemia, diabetic neuropathy, drug abuse, chronic pain, and amyotrophic lateral sclerosis (14).
PSMA is a member of a superfamily of zinc-dependent exopeptidases, which include carboxypeptidases with a mononuclear zinc active site (e.g., carboxypeptidase A) (15) and carboxy- and aminopeptidases with a binuclear zinc active site [e.g., carboxypeptidase G2 (CPG2) (16), peptidases T and V (PepT and PepV) (17, 18), Streptomyces griseus aminopeptidase (Sgap) (19), and Aeromonas proteolytica aminopeptidase (AAP) (20)]. In addition to a limited region of homology with these soluble single-domain (e.g., AAP, Fig. 1C) (19, 20) or double-domain (e.g., CPG2, Fig. 1D) (16-18) zinc-dependent exopeptidases, the entire sequence of PSMA is homologous to at least four other human proteins: NLDL (expressed in ileum; 35% identity) (2, 21), NLD2 (expressed in ovary, testis, and brain; 67% identity) (2, 21, 22), transferrin receptor (TfR) 1 (TfR1; expressed in most cell types; 26% identity) (23) (Fig. 1B), and TfR2 (expressed predominantly in liver; 28% identity) (24). Of these homologs, only the 3D structure of TfR1 is available; it is a type II transmembrane glycoprotein with three extracellular domains: the protease-like, apical, and helical domains (29%, 25%, and 23% identical to PSMA, respectively) (23). The helical and apical domains have no counterparts in the single- and double-domain zinc-dependent exopeptidases (Fig. 1). TfR1 binds the hemochromatosis protein HFE and iron-loaded transferrin at overlapping binding sites (25). Both TfR1 and PSMA are internalized into endosomes via cytoplasmic YXRF and MXXXL motifs, respectively (26, 27). Whereas TfR1 delivers iron-loaded transferrin and HFE to cells, PSMA may internalize folate or other ligands, a property that can be exploited for targeted delivery of chemotherapeutics. Indeed, the binding of PSMA to the monoclonal antibody J591 increases its internalization rate and potential uptake of tethered ligands (28).
Here we present the 3.5-Å crystal structure of the PSMA ectodomain (residues 44-750), which corresponds approximately to the alternatively spliced PSM′ protein expressed in the cytosol (residues 60-750) (29). The structure reveals a two-fold symmetric homodimer with overall similarity to TfR1, but PSMA has a more compact dimer interface and an enzymatically active protease domain. The active site of PSMA contains two zinc atoms coordinated by histidine and glutamate/aspartate residues and a water molecule, similar to active sites in Sgap, AAP, PepV, PepT, and CPG2. An arginine-rich patch in PSMA replaces the hydrophobic substrate-binding site of the aminopeptidases, consistent with their different substrate preferences. Substrate docking and sequence alignments suggest that the arginine patch stabilizes the binding of a C-terminal glutamate-containing substrate.
Materials and Methods
For additional experimental details, see the supporting information, which is published on the PNAS web site.
Expression and Purification of Soluble Human PSMA. The cDNA encoding full-length human PSMA was kindly provided by Joseph Neale and Tomas Bzdega (Georgetown University, Washington, DC). The extracellular portion of PSMA (residues 44-750) was subcloned into a baculovirus transfer vector (pAcGP67A; BD Biosciences) containing a hydrophobic secretion signal, Factor Xa cleavage site, and N-terminal 6x-His affinity tag. Supernatants from High 5 cells infected with recombinant virus were concentrated and exchanged into 50 mM Tris, pH 7.4/150 mM NaCl. PSMA was purified at yields of ≈10 milligrams of protein per liter of infected cell supernatant by using Ni-NTA affinity (Qiagen, Chatsworth, CA) and Superdex 200 16/60 (Amersham Pharmacia Biosciences) size-exclusion chromatography. The enzymatic activity of the purified PSMA ectodomain was within the previously reported range for cleavage of α-NAAG by blood plasma glutamate carboxypeptidase II (2.5-4.5 nanomoles glutamate produced per minute per milligram of enzyme) (30).
Crystallization, Data Collection, and Processing. Crystals of PSMA were obtained at 4°C in 2 days by vapor diffusion from hanging drops containing 0.8 μl of protein (at ≈10 mg/ml in 20 mM Tris, pH 7.5) and 0.8 μl of reservoir solution [18% polyethylene glycol (PEG) 3350/0.2 M sodium thiocyanate]. Crystals were transferred serially through increasing concentrations of glycerol and PEG 3350 to a final concentration of 0.2 M sodium thiocyanate/28% PEG 3350/20% glycerol for cryopreservation. Data were collected at 100 K from two cryopreserved crystals at beamline 8.2.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory (Berkeley, CA) and beamline 11-1 at Stanford Synchrotron and Radiation Laboratory (SSRL; Stanford, CA). A fluorescence energy scan collected at SSRL demonstrated that the PSMA crystals contained zinc, although no exogenous zinc was added during purification or crystallization of the protein. Data were collected at 1.2818 Å, the K-edge of zinc, to aid in the location of the zinc ions. Data processing and scaling were carried out by using the HKL package (Table 1) (31). Crystals were indexed in spacegroup P21 with unit cell dimensions a = 75.8 Å, b = 159.5 Å, and c = 134.4 Å; β = 93.2° and two PSMA dimers per asymmetric unit. Reflections were initially visible to ≈2.7 Å, but the crystals decayed rapidly in the beam, and data from both crystals were combined to obtain a complete dataset to 3.5 Å. Higher-resolution data could not be obtained, because the P21 crystals were difficult to reproduce due to complications from a P212121 crystal form that diffracted to low resolution and had variable unit cell edges. Thus only 2 of ≈300 crystals screened provided useful diffraction data.
Table 1. Data collection and refinement statistics for PSMA.
| Space group | P21 |
|---|---|
| Unit cell (a, b, c, β) | (74.9 Å, 157.8 Å, 133.8 Å, 93.2°) |
| Molecules per asymmetric unit | Two dimers |
| Resolution (outer shell) | 30-3.5 Å (3.62-3.5) |
| Theoretical reflections in resolution range | 77,001 |
| Working set | 70,431 (91.5%) |
| Test set | 3,671 (4.8%) |
| Completeness (outer shell)* | 99% (98.1) |
| Rmerge (outer shell)† | 14% (43.2) |
| I/σ (outer shell) | 15.4 (5.9) |
| Rcryst‡, Rfree§ | 25.2%, 28.4% |
| Average B factor (main chain, side chain) | 66 Å2 (54, 72) |
| non-H atoms per asymmetric unit | 22,876 |
| Protein, Zn(II), carbohydrate, water | 22, 184; 8; 672; 12 |
| rms deviation from ideality: bond, angle | 0.009 Å, 1.47° |
| Non-Gly residues in Ramachandran plot | |
| Most-favored | 74.2% |
| Additionally allowed | 22.9% |
| Generously allowed | 2% |
| Disallowed | 0.8% |
Completeness indicates number of independent reflections/total theoretical number.
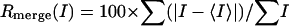 where I is the integrated intensity of a given reflection.
where I is the integrated intensity of a given reflection.
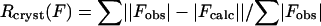 where |Fobs| and |Fcalc| are the observed and calculated structure-factor amplitudes for the HKL reflection.
where |Fobs| and |Fcalc| are the observed and calculated structure-factor amplitudes for the HKL reflection.
Rfree is calculated from reflections in a test set not included in the atomic refinement.
Structure Solution, Refinement, and Analysis. The structure was determined by molecular replacement by using the program amore (32) and a search model based on the structure of TfR1 [residues 122-197, 251-310, 328-511, and 531-750 of TfR1 from chains C and F in TfR1 (PDB ID code 1DE4), which is equivalent to PSMA residues 56-124, 197-269, 288-479, and 561-747]. Side chains for residues that differed between PSMA and TfR1 were truncated to alanine. A solution was obtained for data between 30 and 3.5 Å [Rfree = 52%, Rcryst = 51% after rigid body refinement with refmac5 (32), in which each domain was treated as a separate rigid body]. The model was built by using a combination of the programs o (33) and main (34) with 2Fobs-Fcalc annealed omit maps calculated by using the ccp4 program dm and density-modified four-fold noncrystallographic symmetry (NCS) averaged 2Fobs-Fcalc maps. Cycles of torsional, positional, and B group refinement (35) with a bulk solvent correction, four-fold NCS constraints, and a zinc patch were used to generate a final model with Rfree = 28.4% and Rcryst = 25.2% for data between 30 and 3.5 Å (all reflections with F >0). An anomalous difference Patterson map generated with ccp4 (32) revealed two zinc ions at each active site in the PSMA homodimer, both with full occupancy (as determined by refinement using cns), but experimental phases calculated using anomalous scattering data from these atoms did not improve the electron density maps. Density for a bridging ligand was modeled as a water, but a Tris molecule, as seen in an AAP structure (36), or another buffer component cannot be ruled out at this resolution. Carbohydrate sites in addition to the zinc ligands aided in the initial assignment of the amino acid register in regions where PSMA and TfR1 shared little or no sequence identity. The final model contains residues 56-750 of PSMA chains A-D, two zinc ions, 12 N-acetyl glucosamine residues, and three water molecules per monomer and has an overall B factor of 66 Å2. N-terminal residues 44-55 were not observed in the density.
Results and Discussion
Overall Structure of PSMA. The structure reveals a symmetric dimer with each polypeptide chain containing three domains analogous to the three TfR1 domains: a protease domain (residues 56-116 and 352-591), an apical domain (residues 117-351), and a helical domain (residues 592-750) (Fig. 1 and Table 1). A large cavity (≈1,100 Å2) at the interface between the three domains includes a binuclear zinc site (Fig. 2) and predominantly polar residues (66% of 70 residues) (supporting information). The observation of two zinc ions and conservation of many of the cavity-forming residues among PSMA orthologs and homologs (Table 2) identify the cavity as the substrate-binding site.
Fig. 2.
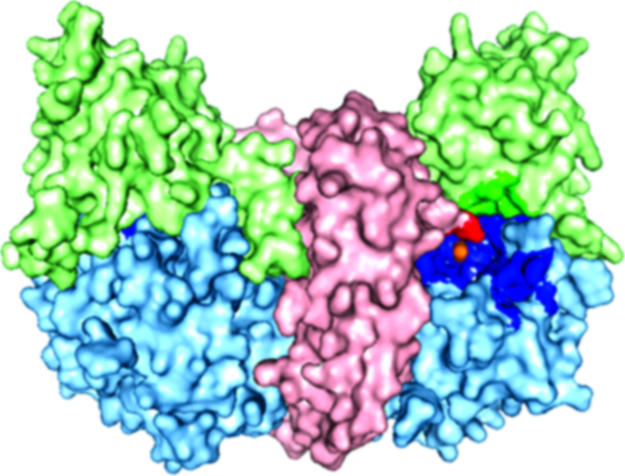
A surface rendering in which the helical domain is light red, the apical domain is light green, the protease domain is light blue, and zinc ions are orange. The residues lining the substrate-binding cavity are highlighted in a darker version of the color corresponding to the domain from which the residue originates.
Table 2. Residues within or in the vicinity of the PSMA active site.

* Residues are highlighted according to their proposed roles: yellow, zinc ligands; purple, substrate- or water-binding residues; blue, residues with structural roles; green, residues that stabilize arginine patch.
† Counterpart residues listed in single-letter code for 10 species of PSMA (Sus scrofa, Mus musculus, Rattus norvegicus, Danio rerio, Caenorhabditis briggsae, A. thaliana, Magnaporthe grisea, Aspergillus nidulans, Neurospora crassa, Gibberella zeae) and for NLD2, NLDL, Sgap, AAP, CPG2, pepT, pepV, and TfR1. Mut, mutant; ϕ, no detectable enzymatic activity; low, enzymatic activity too low for determination of kinetic parameters; ND, not determined or too low activity for determination of Vmax and Km.
‡ Additional mutagenesis results can be found in ref. 45.
The protease domain of PSMA resembles the protease and protease-like domains of bacterial binuclear zinc exopeptidases (Sgap, AAP, PepV, PepT, and CPG2) and TfR1, respectively (Fig. 1), and contains 11 α-helices, an eight-stranded β-sheet, and insertions (residues 485-487 and 502-510), which extend a loop joining the sixth and seventh β-strands and include a short α-helix. As in TfR1, the apical domain is inserted between strands 1 and 2 of the protease domain β-sheet. PepV, PepT, and CPG2 (16-18) (Fig. 1D) also contain an inserted domain, although for these proteins the insertions are between strands 6 and 7 of the central β-sheet, and each serves as a dimerization domain.
The apical domain of PSMA (also called the protease-associated domain) consists of two β-sheets, three α-helices, and one helical turn. This domain is also found in subtilases and trafficking receptors (37). In PSMA, it forms one side of the substrate-binding cavity. In contrast, Sgap and AAP lack an apical domain counterpart and have more open active sites (19, 20).
The helical domain of PSMA contains six α-helices and associated connecting regions, one of which (residues 687-704) is involved in forming the substrate-binding cavity. Another important function of the helical domain is in forming the dimer interface, which involves three helices (Fig. 1 A). Although PSMA can convert between a dimeric and monomeric form in vitro by addition of metal chelators or changes in salt concentration (38), only the dimeric form of PSMA has folate hydrolase and naaladase activity and is expressed on the surface of prostate cells (38). The dimer interface of PSMA is large, burying a total of ≈4,600 Å2, making it unlikely that an intact monomer would dissociate from PSMA without at least partial unfolding, perhaps rationalizing why PSMA monomers are inactive (38). Consistent with experiments demonstrating that deletion of the 15 C-terminal residues in PSMA abolishes enzymatic activity (39), the C-terminal residues 721-747 of PSMA form a helix at the dimer interface, suggesting that C-terminal truncation of the ectodomain disrupts PSMA dimerization.
Comparison with TfR1. Although the overall structure and domain organization of PSMA and TfR1 are similar (Fig. 1) (23, 40) (rms deviation of 2.1 Å for 847 of 1,270 Cα atoms in the homodimers), specific structural differences are relevant to their different functions. First, two large insertions in the apical domain of PSMA (residues 145-158 and 238-248) contribute to a large increase in buried surface area between the apical and protease domains (a total of ≈4,170 Å2), as compared with TfR1 (≈2,630 Å2). There is also a modest increase in buried surface area between the PSMA apical and helical domains (≈2,830 Å2), as compared with TfR1 (≈2,540 Å2). These additional interdomain contacts are involved in positioning the PSMA apical domain to form one side of the substrate-binding cavity. In addition, insertions in the PSMA helical and protease domains (residues 698-703 and 543-548) narrow its substrate-binding cavity, as compared with the analogous region of TfR1. Finally, PSMA does not bind HFE or transferrin (data not shown), and the residues involved in TfR1 binding to HFE or transferrin (40-42) are nonconservatively substituted in PSMA (supporting information).
Glycosylation of PSMA. Glycosylation of PSMA is required for secretion of the recombinant extracellular domain, membrane expression of full-length PSMA, and enzymatic activity (43, 44). The PSMA ectodomain contains nine potential N-linked carbohydrate sites, all of which have been shown to be glycosylated (43, 44). Seven of the glycosylation sites show carbohydrate density in 2Fobs-Fcalc maps (Asn-76, -121, -140, -153, -459, -476, and -638), but none of the glycosylation sites are close enough to the PSMA active site to directly contact a bound substrate. Therefore, loss of enzymatic activity resulting from prevention or removal of glycosylation (43, 44) likely results from indirect effects, such as misfolding or inhibition of dimerization. We note that the N-linked carbohydrates attached to Asn-638 are positioned such that they could be involved in intersubunit interactions, suggesting that their removal may affect the dimerization of PSMA, thereby resulting in the reduced enzymatic activity associated with PSMA monomers (38).
Antibody Epitopes. The structure of PSMA clarifies the epitopes recognized by monoclonal antibodies being studied for applications in cancer imaging and therapeutics (reviewed in ref. 4). The epitope for the J591 antibody that is in clinical trials has been mapped by using truncated PSMA constructs to residues 153-347 (7). The structure reveals that this region corresponds to the majority of the apical domain, which is distant from the membrane surface (Fig. 1 A) and hence generally exposed and accessible. It was previously suggested that the epitope for J591 is near Asn-476 or the carbohydrate normally attached to this residue, because a mutant at this site (N476A) was not detected by J591 (43). However, the PSMA structure shows that Asn-476 is distant from the apical domain and is instead located in the membrane-proximal region of the molecule, where it is less accessible to antibodies.
Active Site of PSMA. The structure of PSMA also provides insight into its catalytic mechanism and substrate specificity. Although zinc was not added to PSMA during purification or crystallization, the crystal structure reveals two zinc atoms separated by ≈3.2 Å. The zinc ions are each coordinated by three endogenous ligands: a histidine (His-553 or -377), a terminal aspartate (Asp-453) or glutamate (Glu-425), and a bridging aspartate (Asp-387) (Fig. 3). Additionally, a water ligand asymmetrically bridges the zinc ions. With the exception of Asp-453, these five endogenous zinc-binding residues are substituted in the protease-like domain of TfR1, which does not bind zinc or function as a peptidase (23). The zinc-coordinating residues were implicated in previous mutagenesis studies (45) and are conserved in PSMA orthologs from plants to mammals, in the aminopeptidases Sgap, AAP, PepV, and PepT, and include a single conservative substitution in the bacterial carboxypeptidase CPG2 (Table 2). The active site geometries of these enzymes are similar (Fig. 3C); e.g., the bridging ligand modeled as a water molecule in the PSMA active site is at a similar position as a water in CPG2 and a bound Tris molecule in AAP (16-20, 36). The water molecule in PSMA is likely to have significant hydroxide character, because a single zinc ion can lower the pKa of a water ligand from 15.7 to 9.0 (46), and the adjacent residue Glu-424 could act as an active site base to deprotonate the bound water. Consistent with a role in catalysis, Glu-424 is conserved in all species of PSMA, other naaladases, AAP, Sgap, PepV, PepT, and CPG2, and substitution of this residue leads to a reduced Vmax and increased Km in PSMA (45) (Table 2). The analogous residue in AAP is involved in the hydrolysis reaction and functions as an active site acid and base (47). Another residue that could potentially act as an active site base is Tyr-552, which is conserved in all PSMA and PSMA-related proteins except AAP and PepV, in which the analogous residue is an isoleucine or a methionine, respectively (Table 2). PSMA Tyr-552 is within 3.8 Å of the water ligand, suggesting involvement in catalysis, but its exact role remains to be elucidated.
Fig. 3.

The PSMA active site. (A) Stereoview of the PSMA active site. Zinc ions are orange spheres, and a water molecule is shown as a red sphere. Zinc-binding residues are yellow sticks, water- or substrate-binding ligands are purple sticks, and residues with structural roles are light blue sticks. (B) Stereoview of the PSMA model in the region of the PSMA active site superimposed on a 3.5-Å 2Fobs-Fcalc annealed omit electron density map contoured at 1σ (yellow) and 4σ (blue). The zinc ions and zinc-binding residues were omitted from the model before generation of the map. Some of the extra density corresponds to nonzinc-binding residues that were omitted from the figure for clarity. (C) Overlay of the active sites of PSMA (yellow), aminopeptidase AAP (green) (PDB ID code 1RTQ), and carboxypeptidase CPG2 (blue) (PDB ID code 1CG2) with the zinc ions as spheres and the zinc-binding residues in ball-and-stick representation.
Other residues near the zinc ligands (Asp-379, Glu-557, Pro-388, and Asn-519) are also conserved or conservatively substituted between PSMA orthologs and related binuclear zinc-dependent exopeptidases (Table 2). Substitutions of the strictly conserved Asp-379, which is within hydrogen-bonding distance of the zinc ligand His-377, abolish detectable enzymatic activity (Table 2). Glu-557 is within hydrogen-bonding distance of the backbone nitrogen of Asp-387 and is either a glutamate or aspartate in all PSMA orthologs and related binuclear zinc-dependent exopeptidases. Pro-388, which was modeled as a cis-peptide in the PSMA structure, occupies a position at which residues with a cis-peptide conformation are found in the mononuclear (48, 49) and binuclear (16-19, 36) zinc-dependent exopeptidases. This residue is broadly conserved across different species of PSMA, and alanine substitution results in an increased Km (Table 2). The substitution of Asn-519, which is within 3.8 Å of the active site water molecule, affects enzymatic activity (45), and this residue (or its serine or threonine counterpart in nonmammalian PSMAs and other related peptidases) could be involved in hydrogen bonding to the C or N terminus of the substrate (Table 2).
Substrate-Binding Cavity of PSMA. The substrate-binding cavity contains a patch of three arginines (Arg-463, -534, and -536), which are clustered within 4.5 Å of each other and located 6-12 Å from the nearest zinc ion. The arginines in the patch are stabilized by interactions with Glu-457, Asp-465, and Ser-454. The arginine patch is conserved in mammalian and zebrafish PSMA (Table 2) and may be involved in correctly orienting substrates for catalysis by interacting with the negatively charged C-terminal glutamate residue. The positively charged character of the cavity is in contrast to the hydrophobic substrate-binding cavities found in aminopeptidases, which accommodate substrates with apolar residues (50). Nonconservative substitutions of PSMA residues Arg-463 and -536 result in increased Km values, and mutagenesis of Ser-454 leads to a decrease in Vmax (45) (Table 2). Similarly, CPG2, which cleaves folic acid and methotrexate (51), also includes three arginines near its active site (CPG2 residues Arg-262, -288, and -324, which are 8-19 Å away from the nearest zinc ion) (16). A mutant at the nearest of these arginines (R324A) showed reduced cleavage of methotrexate (16). In PSMA, a fourth arginine, Arg-210 from the apical domain, which is 100% conserved in all PSMA orthologs and NLD2 and NLDL, is situated on the opposite side of the cavity and may also be involved in substrate binding.
An arginine-rich substrate-binding cavity is also observed in carboxypeptidase A (48), although this enzyme is not specific for C-terminal glutamate-containing substrates. In this case, the arginines are not clustered but rather are more linearly arranged with a distance of ≈9.0 Å between the two furthest arginines. The arginine that is closest to the active site zinc (≈4 Å) is proposed to stabilize the tetrahedral intermediate (52). The two other arginines are both ≈7 Å from the zinc ion but ≈9 Å from each other. Thus, although an arginine-lined substrate-binding cavity is a general feature of zinc carboxypeptidases that facilitates interactions with carboxyl groups, the clustering of the four arginines in the substrate-binding cavity of PSMA could provide additional specificity for substrates with C-terminal glutamate residues.
Conservation of all four arginine residues correlates with the ability of PSMA and NLD2 to cleave α-NAAG (2), consistent with the proposed role of these arginine residues in binding substrates containing C-terminal glutamates. NLDL and Arabidopsis thaliana PSMA (AMP1) (53) do not cleave α-NAAG, and they lack two or three of the four arginines, respectively (Table 2). In NLDL, only Arg-210 and -463 are present, and in AMP1, only Arg-210 is present. Threading of the sequences of NLDL and AMP1 onto the structure of human PSMA suggests that the NLDL and AMP1 substrate-binding cavities do not contain compensating arginine residues (data not shown) but rather have hydrophobic substrate-binding cavities, suggesting interactions with apolar substrates. Although the substrate for AMP1 has not been identified, plants with AMP1 mutations show changes in apical meristem size, suggesting the importance of this PSMA ortholog in plant development and a potential role for its substrate as a signaling compound (53).
Mechanistic Implications. Taken together with prior crystallographic and mechanistic studies of members of the zinc-dependent exopeptidase superfamily (16-19, 36, 47-49, 54, 55), we can use the structure and substrate docking results (supporting information) to propose a catalytic mechanism for the cleavage of α-NAAG by PSMA (Fig. 4) that is consistent with mutagenesis studies. In this mechanism, α-NAAG binds such that the glutamate side chain forms electrostatic interactions with the arginine patch of PSMA, and the oxygen from the carbonyl to be attacked and the C terminus bind to the zinc ions. The remainder of the substrate (e.g., N-acetyl-aspartate in α-NAAG) is accommodated by the substrate-binding cavity with specific interactions with Arg-210. Larger substrates [e.g., poly-γ-glutamated folates with up to four γ-linked glutamates (9)] require some structural rearrangements to avoid steric clashes with the pocket and could have an alternate mode of binding. Nevertheless, the proposed reaction mechanism is the same: binding of one water molecule to the zinc ions and deprotonation by Glu-424 to activate the water molecule for nucleophilic attack of the peptide bond of the bound substrate, leading to a tetrahedral intermediate, bond cleavage, and product dissociation. This mechanism is consistent with catalysis by other members of the zinc-dependent exopeptidase superfamily, in which each active site includes one water molecule that is activated for nucleophilic attack of the peptide bond through deprotonation by a nearby carboxylate (Glu-424 for PSMA) (16-19, 36, 47-49, 54). The active site architecture of PSMA and its implications for substrate binding and catalysis provide insight into the function of this important enzyme and a rationale for designing reagents for detection and treatment of cancer and/or neurological disorders.
Fig. 4.

A mechanistic scheme for α-NAAG (glutamate is red, and NAA is blue) binding and cleavage by PSMA (black). The zinc-binding PSMA residues are assumed to remain bound during the reaction mechanism and are omitted in the substrate-binding and cleavage steps for clarity. The C-terminal carboxyl group binds the zinc ions in a bridging fashion, and there are electrostatic interactions between the glutamate side chain and the arginine patch and the aspartate side chain and Arg-210. The zinc-bound water molecule is poised to attack the peptide bond of the bound substrate, leading to a tetrahedral intermediate, bond cleavage, and product dissociation. An analogous mechanism would be expected for the cleavage of α-NAAG by NLD2 (2).
Supplementary Material
Acknowledgments
We thank members of the Bjorkman laboratory for technical assistance and critical reading of the manuscript, Doug Rees and Michael Chan for helpful discussions, the staff at Stanford Synchrotron and Radiation Laboratory and Advanced Light Source for assistance with synchrotron data collection, Adam Paiz for help with crystallization, Peter Snow and Inder Nangiana of the California Institute of Technology Protein Expression Facility for protein expression, Gary Hathaway of the California Institute of Technology Protein/Peptide Microanalysis Facility for mass spectrometry and N-terminal sequencing, Jens Kaiser for assistance with main and the zinc patch, David Mathog of the California Institute of Technology Sequence and Structure Analysis Facility for help with sequence alignments, and Tomas Bzdega and Joseph Neale (Georgetown University, Washington, DC) for the PSMA gene. This work was supported by a Department of Defense Prostate Cancer Postdoctoral Award (to M.I.D.), an American Liver Foundation Postdoctoral Award (to M.I.D.), and the Howard Hughes Medical Institute (to P.J.B.).
Author contributions: M.I.D., M.J.B., and P.J.B. designed research; M.I.D. performed research; M.I.D., M.J.B., and P.J.B. analyzed data; M.I.D., M.J.B., and P.J.B. wrote the paper; and L.M.T. assisted with data collection and initial structure solution.
Abbreviations: PSMA, prostate-specific membrane antigen; AAP, A. proteolytica aminopeptidase; AMP1, A. thaliana PSMA; CPG2, carboxypeptidase G2; α-NAAG, N-acetyl-l-aspartyl-l-glutamate; NLD, Naaladase; PepT, peptidase T; PepV, peptidase V; Sgap, S. griseus aminopeptidase; TfR, transferrin receptor.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 1Z8L).
References
- 1.Ghosh, A. & Heston, W. D. W. (2004) J. Cell. Biochem. 91, 528-539. [DOI] [PubMed] [Google Scholar]
- 2.Pangalos, M. N., Neefs, J. M., Somers, M., Verhasselt, P., Bekkers, M., van der Helm, L., Fraiponts, E., Ashton, D. & Gordon, R. D. (1999) J. Biol. Chem. 274, 8470-8483. [DOI] [PubMed] [Google Scholar]
- 3.Berger, U. V., Luthi-Carter, R., Passani, L. A., Elkabes, S., Black, I., Konradi, C. & Coyle, J. T. (1999) J. Comp. Neurol. 415, 52-64. [DOI] [PubMed] [Google Scholar]
- 4.Gong, M. C., Chang, S. S., Sadelain, M., Bander, N. H. & Heston, W. D. W. (1999) Cancer Metastasis Rev. 18, 483-490. [DOI] [PubMed] [Google Scholar]
- 5.Elgamal, A. A., Holmes, E. H., Su, S. L., Tino, W. T., Simmons, S. J., Peterson, M., Greene, T. G., Boynton, A. L. & Murphy, G. P. (2000) Semin. Surg. Oncol. 18, 10-16. [DOI] [PubMed] [Google Scholar]
- 6.Murphy, G. P., Snow, P. B., Brandt, J., Elgamal, A. & Brawer, M. K. (2000) Prostate 42, 145-149. [DOI] [PubMed] [Google Scholar]
- 7.Bander, N. H., Nanus, D. M., Milowsky, M. I., Kostakoglu, L., Vallabahajosula, S. & Goldsmith, S. J. (2003) Semin. Oncol. 30, 667-676. [DOI] [PubMed] [Google Scholar]
- 8.Rawlings, N. D., O'Brien, E. A. & Barrett, A. J. (2002) Nucleic Acids Res. 30, 343-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinto, J. T., Suffoletto, B. P., Berzin, T. M., Qiao, C. H., Lin, S., Tong, W. P., May, F., Mukhrjee, B. & Heston, W. D. W. (1996) Clin. Cancer Res. 2, 1445-1451. [PubMed] [Google Scholar]
- 10.Denny, W. A. (2001) Eur. J. Med. Chem. 36, 577-595. [DOI] [PubMed] [Google Scholar]
- 11.Carter, R. E., Feldman, A. R. & Coyle, J. T. (1996) Proc. Natl. Acad. Sci. USA 93, 749-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neale, J. H., Bzdega, T. & Wroblewska, B. (2000) J. Neurochem. 2000, 443-452. [DOI] [PubMed] [Google Scholar]
- 13.Passani, L. A., Vonsattel, J. P. G., Carter, R. E. & Coyle, J. T. (1997) Mol. Chem. Neuropathol. 31, 97-118. [DOI] [PubMed] [Google Scholar]
- 14.Jackson, P. F. & Slusher, B. S. (2001) Curr. Med. Chem. 8, 949-957. [DOI] [PubMed] [Google Scholar]
- 15.Rees, D. C., Lewis, M. & Lipscomb, W. N. (1983) J. Mol. Biol. 168, 367-387. [DOI] [PubMed] [Google Scholar]
- 16.Rowsell, S., Pauptit, R. A., Tucker, A. D., Melton, R. G., Blow, D. M. & Brick, P. (1997) Structure (Cambridge, U.K.) 5, 337-347. [DOI] [PubMed] [Google Scholar]
- 17.Håkansson, K. & Miller, C. G. (2002) Eur. J. Biochem. 269, 443-450. [DOI] [PubMed] [Google Scholar]
- 18.Jozic, D., Bourenkow, G., Bartunik, H., Scholze, H., Dive, V., Henrich, B., Huber, R., Bode, W. & Maskos, K. (2002) Structure (Cambridge, U.K.) 10, 1097-1106. [DOI] [PubMed] [Google Scholar]
- 19.Greenblatt, H. M., Almog, O., Maras, B., Spungin-Bialik, A., Barra, D., Blumberg, S. & Shoham, G. (1997) J. Mol. Biol. 265, 620-636. [DOI] [PubMed] [Google Scholar]
- 20.Chevrier, B., D'Orchymont, H., Schalk, C., Tarnus, C. & Moras, D. (1996) Eur. J. Biochem. 237, 393-398. [DOI] [PubMed] [Google Scholar]
- 21.Shneider, B. L., Thevananther, S., Moyer, M. S., Walters, H. C., Rinaldo, P., Devarajan, P., Sun, A. Q., Dawson, P. A. & Ananthanarayanan, M. (1997) J. Biol. Chem. 272, 31006-31015. [DOI] [PubMed] [Google Scholar]
- 22.Bzdega, T., Crowe, S. L., Ramadan, E. R., Sciarretta, K. H., Olszewski, R. T., Ojeifo, O. A., Rafalski, V. A., Wroblewska, B. & Neale, J. H. (2004) J. Neurochem. 89, 627-635. [DOI] [PubMed] [Google Scholar]
- 23.Lawrence, C. M., Ray, S., Babyonyshev, M., Galluser, R., Borhani, D. W. & Harrison, S. C. (1999) Science 286, 779-782. [DOI] [PubMed] [Google Scholar]
- 24.Kawabata, H., Yang, R., Hirama, T., Vuong, P. T., Kawano, S., Gombart, A. F. & Koeffler, H. P. (1999) J. Biol. Chem. 274, 20826-20832. [DOI] [PubMed] [Google Scholar]
- 25.Giannetti, A. M., Snow, P. M., Zak, O. & Bjorkman, P. J. (2003) PLoS Biol. 1, 341-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Enns, C. A. (2002) in Molecular and Cellular Iron Transport, ed. Templeton, D. M. (Dekker, New York), pp. 71-94.
- 27.Rajasekaran, S. A., Anilkumar, G., Oshima, E., Bowie, J. U., Liu, H., Heston, W., Bander, N. H. & Rajasekaran, A. K. (2003) Mol. Biol. Cell. 14, 4835-4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu, H., Rajasekaran, A. K., Moy, P., Xia, Y., Kim, S., Navarro, V., Rahmati, R. & Bander, N. H. (1998) Cancer Res. 58, 4055-4060. [PubMed] [Google Scholar]
- 29.Grauer, L. S., Lawler, K. D., Marignac, J. L., Kumar, A., Goel, A. S. & Wolfert, R. L. (1998) Cancer Res. 58, 4787-4789. [PubMed] [Google Scholar]
- 30.Gingras, R., Richard, C., El-Alfy, M., Morales, C. R., Potier, M. & Pshezhetsky, A. V. (1999) J. Biol. Chem. 274, 11742-11750. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowksi, Z. & Minor, W. (1997) Methods Enzymol. 276, 307-326. [DOI] [PubMed] [Google Scholar]
- 32.Collaborative Computational Project (1994) Acta Crystallogr. D 50, 760-763.15299374 [Google Scholar]
- 33.Jones, T. A. & Kjeldgaard, M. (1997) Methods Enzymol. 277, 173-208. [DOI] [PubMed] [Google Scholar]
- 34.Turk, D. (1992) Ph.D. thesis (Technische Universität, Munich, Germany).
- 35.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 36.Desmarais, W. T., Bienvenue, D. L., Bzymek, K. P., Holz, R. C., Petsko, G. A. & Ringe, D. (2002) Structure (Cambridge, U.K.) 10, 1063-1072. [DOI] [PubMed] [Google Scholar]
- 37.Mahon, P. & Bateman, A. (2000) Protein Sci. 9, 1930-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schulke, N., Varlamova, O. A., Donovan, G. P., Ma, D., Gardner, J. P., Morrissey, D. M., Arrigale, R. R., Zhan, C., Chodera, A. J., Surowitz, K. G., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 12590-12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barinka, C., Mlcochova, P., Sacha, P., Hilgert, I., Majer, P., Slusher, B. S., Horejsi, V. & Konvalinka, J. (2004) Eur. J. Biochem. 271, 2782-2790. [DOI] [PubMed] [Google Scholar]
- 40.Bennett, M. J., Lebron, J. A. & Bjorkman, P. J. (2000) Nature 403, 46-53. [DOI] [PubMed] [Google Scholar]
- 41.Giannetti, A. M. & Bjorkman, P. J. (2004) J. Biol. Chem. 279, 25866-25875. [DOI] [PubMed] [Google Scholar]
- 42.Cheng, Y., Zak, O., Aisen, P., Harrison, S. C. & Walz, T. (2004) Cell 116, 565-576. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh, A. & Heston, W. D. W. (2003) Prostate 57, 140-151. [DOI] [PubMed] [Google Scholar]
- 44.Barinka, C., Sacha, P., Sklenar, J., Man, P., Bezouska, K., Slusher, B. S. & Konvalinka, J. (2004) Protein Sci. 13, 1627-1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Speno, H. S., Luthi-Carter, R., Macias, W. L., Valentine, S. L., Joshi, A. R. T. & Coyle, J. T. (1999) Mol. Pharmacol. 55, 179-185. [DOI] [PubMed] [Google Scholar]
- 46.Christianson, D. & Cox, D. (1999) Annu. Rev. Biochem. 68, 33-57. [DOI] [PubMed] [Google Scholar]
- 47.Bzymek, K. P. & Holz, R. C. (2004) J. Biol. Chem. 279, 31018-31025. [DOI] [PubMed] [Google Scholar]
- 48.Lipscomb, W. N., Hartsuck, J. A., Reeke, G. N., Quiocho, F. A., Bethge, P. H., Ludwig, M. L., Steitz, T. A., Muirhead, H. & Coppola, J. C. (1969) Brookhaven Symp. Biol. 21, 24-90. [PubMed] [Google Scholar]
- 49.Schmid, M. F. & Herriott, J. R. (1976) J. Mol. Biol. 103, 175-190. [DOI] [PubMed] [Google Scholar]
- 50.Gonzales, T. & Bobert-Baudouy, J. (1996) FEMS Microbiol. Rev. 18, 319-344. [DOI] [PubMed] [Google Scholar]
- 51.Goldman, P. & Levy, C. C. (1967) Proc. Natl. Acad. Sci. USA 58, 1299-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phillips, M. A., Fletterick, R. & Rutter, W. J. (1990) J. Biol. Chem. 265, 20692-20698. [PubMed] [Google Scholar]
- 53.Helliwell, C. A., Chin-Atkins, A. N., Wilson, I. W., Chapple, R., Dennis, E. S. & Chaudhury, A. (2001) Plant Cell 13, 2115-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Makarova, K. S. & Grishin, N. V. (1999) J. Mol. Biol. 292, 11-17. [DOI] [PubMed] [Google Scholar]
- 55.Holz, R. C. (2002) Coord. Chem. Rev. 232, 5-26. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}