

Abstract
Purpose
Liquid biopsies allow the tracking of clonal dynamics and detection of mutations during treatment.
Experimental design
We evaluated under blinded conditions the ability of cell free DNA (cfDNA) to detect RAS/BRAF mutations in the plasma of 42 metastatic colorectal cancer patients treated on a phase Ib/II trial of FOLFOX and dasatinib, with or without cetuximab.
Results
Prior to treatment, sequencing of archival tissue detected mutations in 25/42 patients (60%), while the cfDNA assay detected mutations in 37/42 patients (88%). Our cfDNA assay detected mutations with allele frequencies as low as 0.01%. After exposure to treatment, 41/42 patients (98%) had a cfDNA detected RAS/BRAF mutation. Of 21 patients followed with serial measurements who were RAS/BRAF mutant at baseline, 11 (52%) showed additional point mutation following treatment and 3 (14%) no longer had detectable levels of another mutant allele. Of RAS/BRAF wild type tumors at baseline, 4/5 (80%) showed additional point mutations. cfDNA quantitative measurements from this study closely mirrored changes in CEA and CT scan results, highlighting the importance of obtaining quantitative data beyond the mere presence of a mutation.
Conclusions
Our findings demonstrate the development of new RAS/BRAF mutations in patients regardless of whether they had pre-existing mutations in the pathway, demonstrating a convergent evolutionary pattern.
Keywords: circulating DNA, Heterogeneity, RAS, Resistance, Liquid biopsy
INTRODUCTION
Targeted therapies are rapidly being integrated into the treatment of patients with cancer. However, the acquisition of resistance to such treatments is observed in virtually all cases. This resistance may arise as de novo mutations or as the expansion of a sub-clonal population of cells with pre-existing resistance (1,2). Detecting these resistant sub-clones and monitoring clonal populations over time is difficult and requires the development of more sensitive assays that are also convenient and safe for patients to have serially performed. While the advancement of next generational sequencing (NGS) coupled with pre and post-treatment biopsies may provide snapshots of clonal evolution, they are often difficult to implement and do not provide a longitudinal assessment of heterogeneity (2–8) Not only do these biopsies not provide the ability to frequently assess heterogeneity and follow changes, they are also hindered by the fact that biopsies are inherently of a single location, often the safest lesion to biopsy, and this may not represent the entire clonal population of a tumor (8–10)
The ability to comprehensively and longitudinally characterize the clonal architecture of a tumor will have profound treatment implications. Early detection of acquired resistance may allow initiation of new therapies that suppress the expansion of clones that would otherwise result in disease progression. In addition, there may be clonal populations that were resistant to previous therapies that subsequently experience an extinction event, thus rendering the patient sensitive to re-use of a previous line of therapy and expanding potential treatment options. “Liquid biopsies” or cell free DNA (cfDNA) may better capture the complexity of tumor heterogeneity, while at the same time minimizing patient risks and associated costs (11,12). Recent studies have shown that liquid biopsies can be used to effectively genotype tumors (13,14) and monitor the emergence of resistant clones during the course of treatment (15–17)
Use of the EGFR directed monoclonal antibodies cetuximab and panitumumab in the treatment of metastatic colorectal cancer (mCRC) is limited to RAS wild type (WT) patients (18–20). Activating KRAS and NRAS (RAS) mutations in exons 2,3, and 4 of each gene have been shown to result in a lack of benefit from EGFR targeted therapy (21,22). Other genetic alterations associated with resistance to EGFR directed therapy include KRAS amplification (4), activating-BRAF mutations (4,23), activating-PIK3CA mutations and PTEN loss (4,24). Despite the knowledge of these mechanisms of resistance, there are still RAS WT patients without these other perturbations who display primary resistance to cetuximab/panitumumab (1,19). In addition, secondary resistance develops in all patients and is associated with the development of secondary KRAS, NRAS, EGFR or BRAF mutations (11,25,26). This demonstrated the ability to detect additional mutation of these secondary KRAS and NRAS mutations in the blood of patients receiving anti-EGFR therapies using a liquid biopsy approach and showed that resistant clones could be detected months before radiographic progression. Further reports have confirmed this technique and highlight the potential of cfDNA as a powerful way to longitudinally follow cancer patients (27,28). Diaz et al (15) described a mathematical modelling to suggest variants that lead to resistance are present prior to initiation of therapy. Thus, we can speculate that the mutations corresponding to those emerging clones may have initially been below the limit of detection of the platform before treatment. As a result, we will below refer to the “detection of additional mutation” rather than “developed” or “emerging mutation”.
In this study, we present Intplex, a novel multi-marker assay for the analysis of cfDNA and demonstrate its ability to sensitively detect the changing clonal dynamics of colorectal cancer. IntPlex is based on a refined allele specific competitive blocker q-PCR method specifically designed to improve quantification based on the structure and size of cfDNA (29–31) It enables simultaneous determination of five parameters: the total concentration of cfDNA, the presence of a point mutation, the mutant DNA concentration, the mutant allele fractions of cfDNA, and the cfDNA fragmentation index (32,33). Using IntPlex, we evaluated cfDNA in the plasma of 42 mCRC patients treated on a phase Ib/II trial of FOLFOX and dasatinib with or without cetuximab and compared our findings to the results obtained from the archival tissue-based assay, currently considered the standard of care. The use of serial plasma collections from some of the patients on this trial allowed evaluation of the clonal dynamics of RAS and BRAF during therapy. Here we present the evolutionary changes that occurred and compare our findings to standard clinical observation with radiographic imaging and carcinoembryonic antigen (CEA) of a select subset of patients.
Materials and Methods
Ethics statement, eligibility criteria, drug administration and study design, treatment are described in Parseghian et al (back to back report)
Study Design and treatment
We conducted a single-institution, open-label, investigator-initiated phase IB/II study in refractory metastatic colorectal cancer patients treated with modified FOLFOX6, cetuximab, and dasatinib, with dasatinib dose escalation by cohort. This study is presented in a companion publication (34). The regimen consisted of cetuximab (400 mg/m2 week 1 followed by weekly doses of 250mg/m2), oxaliplatin (85 mg/m2 q2weeks), bolus 5-FU (400 mg/m2 q2weeks) and leucovorin (400 mg/m2 q2weeks), followed by a 46-hour infusion of 5-FU (2400 mg/m2 q2weeks). Dasatinib was dosed orally daily in cohorts of 100mg/d, 150mg/d, and 200mg/d, administered without interruption. Dasatinib dose escalation was performed using a standard “3+3” design. Appropriate radiographic images were collected for all patients enrolled at baseline and at all post-baseline evaluations by the same radiological method, in order to assess response. All radiological tests that demonstrated tumor at baseline were repeated after every 4 cycles and at discontinuation of study treatment. For the Phase II study, patients deemed to be KRAS codon 12/13 wildtype based on archival tissue received the MTD from the Phase I study, while KRAS mutant patients received the same, without cetuximab.
Sample preparation
We retrospectively assessed archival tissue and plasma samples at baseline for mutational status, with repeat plasma samples occurring every 4 cycles prior to each restaging CT scan (2 to 6 hours after taking the daily dose of dasatinib). Investigators were blinded to the results of the clinical grade assay during the experiment. Plasma samples for cfDNA analysis were stored at −80°C and transferred between institutions on dry ice. The plasma isolation protocol we adapted from Chiu et al (35) as well as handling and storage conditions were previously described (36). Briefly, plasma was first centrifuged at 1600g at 4C° for 10 minutes and supernatant was centrifuged at 16,000g at 4°C for 10 minutes. Supernatant was immediately used for DNA extraction and stored at −20°C. Total cfDNA was extracted from 1mL of isolated plasma using the QIAmp DNA mini blood kit (Qiagen, CA) in accordance with the pre-analytical guidelines we previously described (36) in an elution volume of 130μL. DNA extracts were kept at −20°C until use or used immediately. In total, we analyzed 81 serial plasma samples from 46 mCRC patients (Supplemental Figure S1).
Tumor tissue collection/storage/analysis
DNA was extracted from paraffin-embedded formalin-fixed tumor tissue. Genomic analysis samples were evaluated by using a next generation sequencing (NGS) platform using the semiconductor-based Ion PGM NGS platform with Ampliseq Cancer Hot Spot Panel v2 in 46, 50 gene panels for the detection of frequently reported point mutations in human malignancies in CLIA-certified molecular diagnostics laboratory.
Intplex analysis of cfDNA
Intplex analysis was performed as previously described (31,32). Briefly, the qPCR assays were performed according the MIQE guidelines. Q-PCR amplifications were carried out at least in duplicate in a 25μL volume on a CFX96 instrument using the CFX manager software (Bio-Rad). Each PCR reaction was composed of 12.5 μL of IQ supermix Sybr Green (Bio-Rad), 2.5 μL of free water (Qiagen) or specific oligoblocker, 2.5μL of forward and reverse primers (0.3 pmol/mL) and 5μL of template. Thermal cycling comprised three repeated steps: a hot-start activation step at 95°C for 3 minutes, followed by 40 cycles of denaturation-amplification at 95°C for 10 seconds, then 60°C for 30 seconds. Melting curves were investigated by increasing the temperature from 60°C to 90°C with a plate reading every 0.2°C. Standard curve were performed for each run with a genomic extract of the DiFi cell line at 1.8 pg/μL of DNA. Each PCR run was carried out with no template control and positive control for each primer set. Positive controls were extracted from cell lines bearing KRAS/BRAF or NRAS point mutations or we purchased synthetic DNA (Horizon dx, UK. Cambridge). In each single run, negative and positive controls for each tested mutation were included and one standard curve was prepared. Validation of Q-PCR amplification was made by melt curve differentiation. ctDNA mutation testing was made here without any sensitivity cutoff while a threshold of >0.5% (mutant to WT ctDNA ratio) was used in our previous report (31). Consequently, ctDNA mutation testing here is much more sensitive (up to 500-fold) with a sensitivity ranging from 0.001% to 0.005% depending upon the mutations. When point mutations were found in only one of the two replicates, the point mutation was confirmed in triplicate.
Design of patient’s follow-up and detection of point mutations
Point mutations tested in this retrospective study with the IntPlex Q-PCR method are presented in the supplemental Table S1. Total circulating cfDNA quantification was carried out on all serial plasma samples. However, for patients with KRAS c12/13, NRAS, or BRAF V600E point mutations at baseline, only these mutations were assessed for on-treatment samples. Final plasma samples at disease progression for all patients were assessed for all mutations in KRAS (exon 2, 3, 4), NRAS (exon 2, 3, 4), and BRAF V600E. In the case of a patient having no KRAS/NRAS/BRAF point mutation at baseline, we also assessed KRAS codons 61, 146 and NRAS point mutations in all on-treatment samples. EGFR S492R mutation testing was performed only on patients with prior cetuximab therapy (37,38).
RESULTS
Prior to initiation of therapy, 25/42 patients (59.5%) had detectable KRAS 12/13, BRAFV600E or NRAS mutations in archival tumor tissue and 37/42 patients (88%) were found to have mutations in cfDNA (Figure 1A). Among the 25 patients with mutations in archival tissue, 24 (96%) also had mutations in cfDNA (Figure 1B). Considering archival tumor sequencing as the “gold standard”, the concordance between our cfDNA and tissue based methodologies for KRAS exon2 testing was 71%, while the specificity and sensitivity of the cfDNA assay were 48% and 95%, respectively (Supplemental Table S2A). For BRAFV600E testing the concordance between the two assays was 97%, while the specificity and sensitivity of the cfDNA was 97% and 100% respectively (Supplemental Table S2A), compared to the archival tissue based assessment. As can be seen in Table 1, the two cohorts were well matched with no difference in age, gender, grade, presence of an intact primary, number of metastatic sites, or previous therapies. Fourteen patients in Cohort 2 had received prior anti-EGFR therapy, while only 1 patient in Cohort 1 had received prior anti-EGFR therapy.
Figure 1. Comparison of mutational status of RAS/BRAF as determined by tumor tissue and plasma analysis at baseline (n=42).
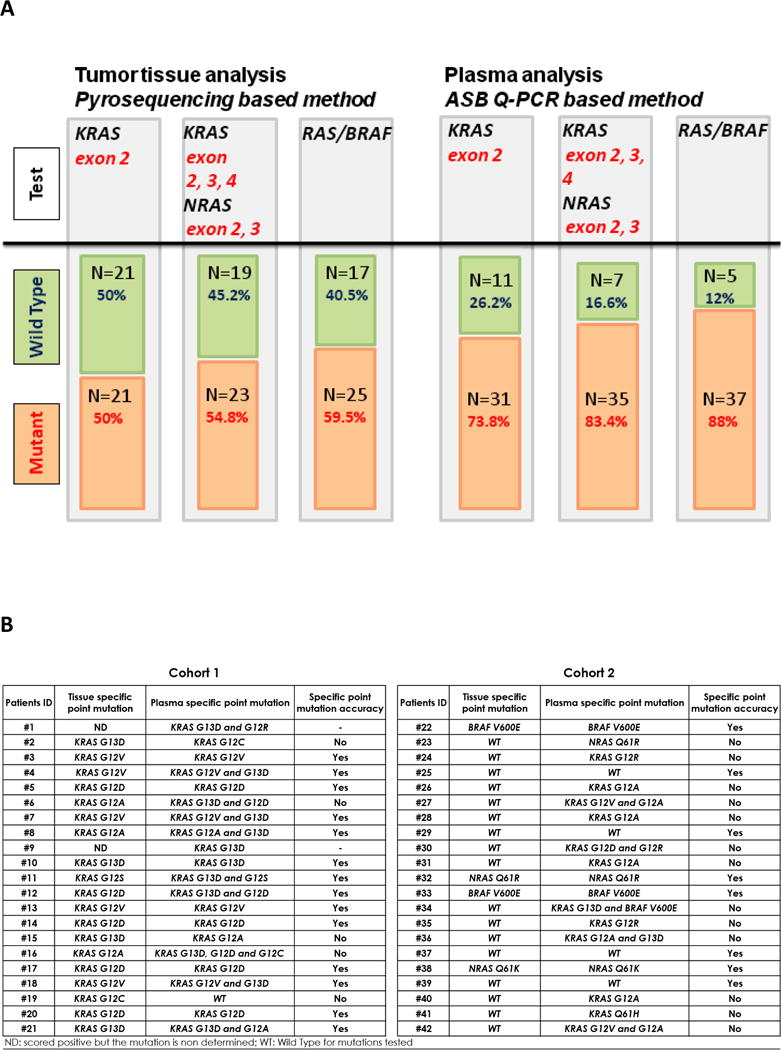
A) Plasma analysis was carried out in cohort 1 and 2 before initiation of treatments (baseline) according with targeted genes. B) Comparison of the type of point mutation as determined by tumor tissue and plasma analysis before initiation of treatments and for cohort 1 and 2. Extended RAS testing was only performed on plasma for patients who were KRAS exon 2 and BRAFV600E wild type on all assays. ND, scored positive but the mutation is non-determined; WT, wild type for mutations tested.
Table 1. Patient’s baseline clinicopathological characteristics.
FOLFOX/dasatinib treatment (cohort 1) and FOLFOX/dasatinib/cetuximab treatment (cohort 2).
|
|
|||
|---|---|---|---|
| Cohorts N (%) | |||
|
| |||
| Cohorts 1 + 2 | Cohort 1 | Cohort 2 | |
| N | 42 | 21 (50) | 21 (50) |
| Age | |||
| Median | 58.5 | 58 | 59 |
| Min–Max | 26–74 | 26–74 | 33–70 |
| Gender | |||
| Male | 27 (64.3) | 15 (35.7) | 12 (28.6) |
| Female | 15 (35.7) | 6 (14.3) | 9 (21.4) |
| Grade of tumour | |||
| Moderate | 34 (81) | 17 (40.5) | 17 (40.5) |
| Moderate–poor | 6 (14) | 3 (7) | 3 (7) |
| missing data | 2 (4.8) | 1 (2.4) | 1 (2.4) |
| Primary tumor in place | |||
| Yes | 21 (50) | 8 (19) | 13 (31) |
| No | 20 (47.6) | 13 (31) | 7 (16.6) |
| missing data | 1 (2.4) | 0 | 1 (2.4) |
| Delay between tumor tissue and blood collection | |||
| < 1 year | 3 (7) | 2 (4.8) | 1 (24) |
| [1 year – 2 years] | 15 (35.7) | 10 (23.8) | 5 (11.9) |
| > 2 years | 24 (57) | 9 (21.7) | 15 (35.7) |
| No. of metastatic sites | |||
| 1 | 26 (62) | 13 (31) | 13 (31) |
| >1 | 15 (36) | 8 (19) | 7 (17) |
| missing data | 1 (2.4) | 0 | 1 (2.4) |
| Previous chemotherapy | |||
| Oxaliplatin | 38 (91) | 20 (48) | 18 (43) |
| Irinotecan | 33 (78.6) | 1 6 (38.1) | 17 (40.5) |
| Any | 3 (7.1) | 1 (2.4) | 2 (4.8) |
| Previous targeted therapies | |||
| bevacizumab | 33 (78.6) | 19 (45.3) | 14 (33.3) |
| cetuximab | 14 (33.3) | 1 (2.4) | 13 (31) |
| panitumumab | 2 (4.8) | 0 | 2 (4.8) |
| Any | 3 (7.1) | 1 (2.4) | 2 (4.8) |
Mutation Analysis and cfDNA Assay Sensitivity
Cohort 1: Patients with previously documented RAS mutation and treated with FOLFOX plus dasatinib
Prior to treatment, 100% patients were found to have KRAS c12/13 point mutations in archival tissue, while 20/21 (95%) patients had cfDNA mutations detected (Figure 2A). No BRAFV600E mutations were detected at baseline in tissue or cfDNA. Compared to tissue detected mutations, 4/19 patients (21%) had different KRAS 12/13 point mutations noted in cfDNA while 7/14 patients (50%) with concordant results had a second mutation detected in cfDNA (Figure 1B). Of the 20 patients with cfDNA detected mutations, 50% had a second concurrent mutation while no patients with two concurrent mutations were detected in tissue (Figure 2A).
Figure 2. RAS/BRAF mutational status.

Comparison of RAS/BRAF mutational status as determined by tumor tissue and plasma analysis. A) Before initiation of treatment of cohorts 1 and 2 (baseline). B) Longitudinal plasma genotyping during treatment (n=26). C) Distribution of mutational status from cfDNA analysis of cohort 2 patients before and after EGFR directed therapy. Percentage of mutant patients as determined by tumor tissue analysis and plasma analysis before initiation and during FOLFOX/dasatinib + cetuximab treatment as determined by serial plasma analysis. Results are shown according the target genes. An, acquired n additional mutation; CO, co-occurring mutations from plasma analysis; WT, wild type for mutations tested; Single, one mutation was found in patient; Multiple, at least two mutations were found in patient.
Pre-treatment (FOLFOX/dasatinib) and post progression plasma was available for 11/21 patients (52%) who had completed between 4 and 8 cycles of therapy. New point mutations were found in 8/11 (73%) patients (Figure 2B), 6 of whom had additional KRAS mutations, 1 of whom developed 2 new KRAS mutations, and 3 of whom developed new BRAFV600E mutations. There was 1 patient with a baseline KRAS G12C mutation that was no longer detected after 4 cycles of therapy. Since all plasma were scored KRAS exon2/BRAF mutant at baseline or during treatment, no extended RAS mutation testing was performed in this cohort.
Cohort 2: Patients without a previously documented RAS mutation and treated with FOLFOX, dasatinib plus cetuximab
Eleven out of twenty-one patients (52%) were KRAS 12/13 mutant using our cfDNA analysis before treatment (Figure 2A), while 3 patients (14%) were BRAFV600E mutant using the cfDNA analysis. Archival testing detected only 2 (10%) patients with BRAFV600E mutations (Figure 2A). Concordance of BRAFV600E mutation results using archival tissue results as the “gold standard” was 95%, with specificity of 95% and sensitivity of 100% (Supplemental Table S2C). Extended RAS mutation testing was performed on cfDNA only in patients with WT KRAS 12/13 and BRAF. No KRAS exon 3 or 4 mutations were noted in archival tissue, whereas 1 patient had a KRAS Q61H mutation by cfDNA testing prior to treatment. Two patients were NRAS 61 mutant on tissue while 3 were NRAS 61 mutant in plasma (Figure 2A). The same NRAS exon 61 mutation was detected from tumor tissue and plasma. Eight out of twenty-one patients (38%) had concordant pre-treatment results by archival tissue and cfDNA analysis. Conversely, 13/21 (62%) were scored mutant only using plasma cfDNA analysis: 11 were KRAS 12/13 mutant, 1 was KRAS Q61H mutant, and another was NRAS Q61R mutant. In Cohort 2, 5/17 patients (29%) were found to have multiple concurrent RAS mutations by plasma analysis at baseline, while none had multiple mutations in the tissue based assay (Figure 2A). One patient was KRAS 12/13 and BRAFV600E mutant before initiation of treatment.
Pre-treatment and post progression plasma was available for 15/21 patients who had completed between 4 and 16 cycles of FOLFOX, dasatinib plus cetuximab therapy. Plasma analysis showed new point mutations in 6/15 patients (40%), including detection of additional point mutations in patients who were WT extended RAS/RAF before treatment (Figure 2B). Seven of fifteen patients (47%) had no molecular change during treatment, including only 1 patient who remained RAS/BRAF WT during treatment and never developed a documented resistance mechanism in the RAS/BRAF pathway. In 2/15 patients (13%) the cfDNA point mutation detected at baseline was no longer detected during treatment. Compared to exon 2 RAS testing, extended RAS in plasma decreased the number of WT patients from 48% to 29% at baseline, and 43% to 19% upon completion of treatment (Figure 2C). Our cfDNA assay re-classified even more patients to contain a mutation in the RAS/BRAF axis. Pre-treatment, 19% of patients were deemed to be extended RAS and BRAF V600E WT, while post treatment only 5% lacked a mutation. Three of fifteen (20%) patients with single baseline RAS mutations developed novel concurrent RAS mutations by the time of disease progression.
Combined Cohorts Assay Analysis
The median interval between tumor tissue and plasma collection was longer for Cohort 2 patients than for Cohort 1 (1074 days and 699 days, respectively; P-value, 0.038) (Supplemental Figure S2A). Delay between tissue/plasma collection was statistically different between patients with concordant and discordant data (692 versus 1074 days, respectively (P-value: 0.0163) (Supplemental Figure S2B). The median time between tumor tissue collection and enrollment on the clinical trial was 845 days in both arms compared to less than one day for plasma collection.
Eighty percent (4/5) of patients who were RAS/BRAF WT on both tissue and cfDNA assay became RAS or BRAF mutant during or at the end of the therapy. Only one patient (2%) among the 42 examined in the two arms was WT for both RAS/BRAF. No detection of EGFR S492R was found either before or during anti-EGFR targeted therapy in plasma of this patient.
Impact of cetuximab re-challenge on mutational status
Among patients examined in Cohort 2, 13 patients had previously received cetuximab in a prior line of therapy. Of these patients, 9/13 (69%) had a RAS mutation noted and another 1/13 (8%) had a BRAFV600E mutation at baseline. Thus, 77% of re-challenged patients previously determined to be WT by archival tissue analysis, were indeed mutant. Notably, 30% (3/10) of the re-challenged RAS mutant patients had multiple co-occurring RAS point mutations at baseline. All plasma samples originating from cetuximab re-challenged patients (n=13) were tested for the EGFR S492R point mutation. No patients carried EGFR S492R mutations before, during, or after FOLFOX, dasatinib plus cetuximab treatment.
Temporal trends in cfDNA concentration during therapy
Circulating WT KRAS/BRAF cfDNA concentration
The total concentration of WT cfDNA was determined by quantifying a wild type sequence of both KRAS and BRAF genes and is shown in Table 2A and 2B, for cohort 1 and 2 respectively. At baseline and as expected, data showed a linear positive relationship between the RefA values (ng/mL) of KRAS and BRAF wild type sequences (Supplemental Figure S3A) as previously observed (32). The KRAS:BRAF ratio was close to 1, both before and during treatment (Supplemental Figure S3B). KRAS WT concentration did not statistically differ between cohorts (P-value: 0.36) (Supplemental Figure S4), and in most patients KRAS WT concentration was higher at the end of treatment than at baseline (Figure 3A & 3B). Of patients with serial measurements, 4/11 (36%) and 12/15 patients (80%) showed an increase of 50% (1.5 fold) above their baseline value at the end of FOLFOX plus dasatinib or FOLFOX, dasatinib plus cetuximab therapy respectively (Figure 3). No significant difference was seen in total concentration of WT cfDNA at baseline between patients whose treatment was stopped before cycle 4 and those treated with more than 4 cycles (P- value: 0.76) (Supplemental Figure S5).
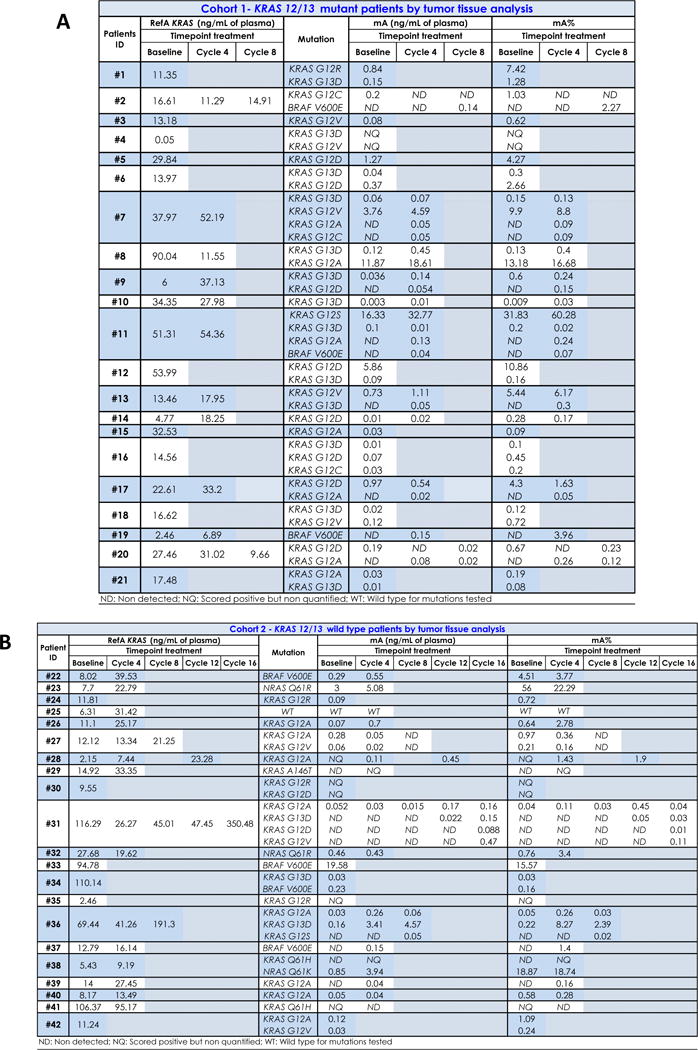
Table 2. Impact of treatment on total cfDNA and mutant RAS/BRAF cfDNA concentration and mutant RAS/BRAF allele frequency.
A) The total cfDNA concentration was determined by Intplex as described in materials and methods, for each patients of cohorts 1 before initiation of treatment up to progression and at the end of treatment. Also, the concentration of mutant cfDNA and mutation loads were determined by Intplex as described in materials and methods, for all point mutations found in each mutant patients of cohorts 1 before initiation of treatments up to the end of treatment. B) The total cfDNA concentration was determined by Intplex as described in materials and methods, for each patients of cohorts 2 before initiation of treatment up to progression and at the end of treatment. Also, the concentration of mutant cfDNA and mutation loads were determined by Intplex as described in materials and methods, for all point mutations found in each mutant patients of cohorts 2 before initiation of treatment up to the end of treatment. RefA, concentration of total cfDNA (ng/mL of plasma); mA, concentration of mutant cfDNA (ng/mL of plasma); mA%, mutation load.
|
Figure 3. Impact of treatment on the evolution of cfDNA concentration.
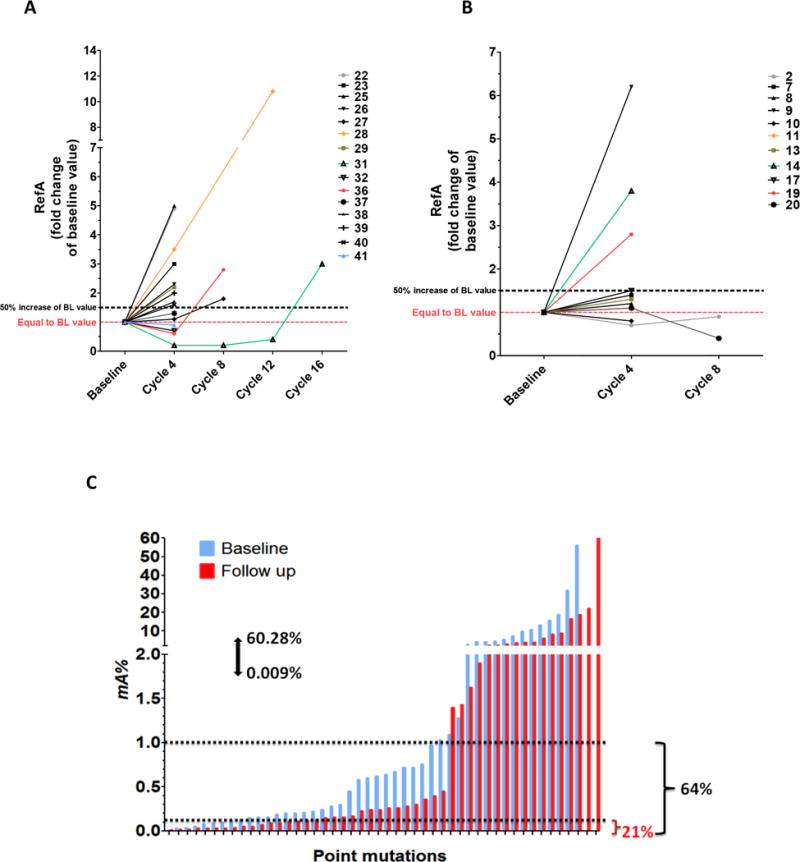
A) RefA values during Folfox/dasatinib treatment (Cohort 1, n=11), and B) during Folfox/dasatinib/cetuximab treatment (Cohort 2, n=15). RefA are expressed in fold change of baseline value. C) cfDNA mutant allele frequency of each detected mutant samples. mA% was determined by Intplex as described in materials and methods before (blue, baseline) and after treatment (red, follow-up) in both cohorts. mA% varies from 0.009% to 60.28%. 21% and 64% of all samples showed an mutant allele frequency below 0.1% and 1% (dotted lines), respectively. RefA, concentration of total cfDNA (ng/mL of plasma); BL, baseline; mA%, cfDNA mutation load.
Circulating mutant RAS/BRAF cfDNA concentration
Concentration of mutant cfDNA (Table 2A and 2B) did not differ at baseline when comparing cohorts 1 and 2 (P-value: 0.65) (Supplemental Figure S6). No significant difference was observed in the baseline concentration of mutant cfDNA between patients whose treatment was stopped before the fourth cycle and patients who continued more than four cycles without progression (P-value: 0.18) (Supplemental Figure S7). In both cohorts combined, 7/46 point mutations (15.2%) found at baseline had concentrations higher than 1 ng/mL of plasma, 19/46 mutations (41.3%) had concentrations less than 0.1 ng/mL, and 4/46 (8.7%) were noted at 0.01 ng/mL (Table 2A and 2B).
Mutant allele frequency
The mutant allele frequency represents the proportion of detected alleles that are mutant for a particular point mutation. The distribution of mutant allele fraction (Table 2A and 2B) for both cohorts highly varied before and during treatment, ranging from 0.009% to 60% (Figure 3C). Pre-treatment, 65% (30/46) of detected mutations were below an allele frequency of 1%, including 45.68% (21/46) below 0.5% and 15% (7/46) below 0.1% (Table 2A and 2C). At baseline, mutant allele fraction varied from 0.009% to 31.83%. In patients with concordant results between archival tissue and cfDNA, 56% (10/18) had a mutant allele fraction >1%, 33% (6/18) were between 0.01% and 1%, and 11% (2/18) were <0.01% (Supplemental Table S3). One mutation was detectable but not quantifiable by our Q-PCR assay, as its allele fraction was <0.01%.
During treatment 65% (31/48) of variants were found below 0.5% allele frequency, including 27% (13/48) below 0.1% (Table 2A and 2B). We noted that 83% (15/18) of new point mutations during treatment occur with a mutant allele fraction less than 0.5% and 50% (9/18) are first noted at fractions ≤0.1% (Supplemental Table S4). We observed no difference between mutant allele fraction at baseline between Cohorts 1 and 2 (P-value: 0.77) (Supplemental Figure S8).
Ability of cfDNA to monitor disease progression and track therapeutic resistance
We collected data on imaging CT-scan and CEA biomarker in most of the patients from baseline to stop of the treatment and compare them to mutant cfDNA parameters such as mA%, mA or refA (Supplemental Table S5). To illustrate the clinical application of circulating cfDNA to track detection of additional mutant subclones and assess disease response/progression, we present four example patients. Patient #8 was treated with FOLFOX plus dasatinib while patients #28, #31, and #36, were treated with FOLFOX, dasatinib plus cetuximab. Treatment of patients #8, #36, #28, and #31 was stopped at cycles 4, 8, 12, and 16, respectively (Figure 4A and 4B). RAS WT and mutant allele concentration and mutant allele fraction were compared to standard clinical assessment with CT-scan and CEA level. For all patients except #8, RAS WT concentration, RAS mutant concentration, and mutant allele fraction closely matched laboratory and radiologic indicators of disease status. For patient #36, cfDNA may have provided a signal of disease progression earlier than standard clinical assessments when a rising KRAS G12S clone was noted.
Figure 4. Quantitative cfDNA analysis in the course of treatments in comparison with serum CEA and CT scan measurements.
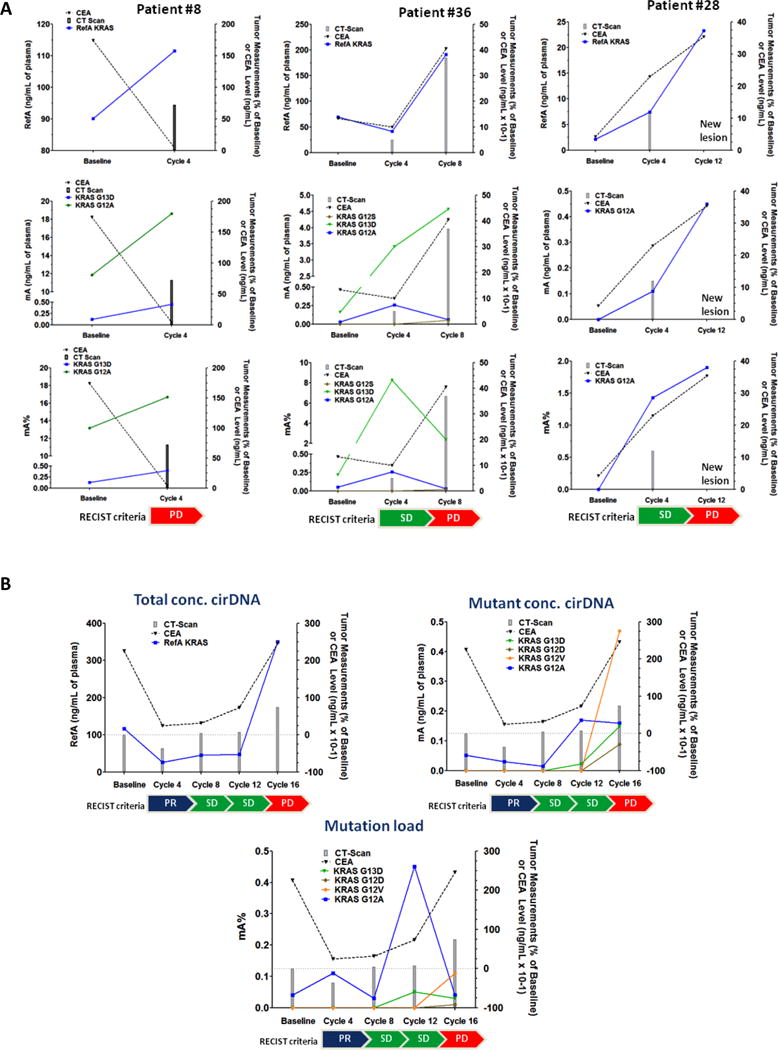
Patients #8, #28, #31 and #36 illustrate impact of treatment on those biomarkers. A) Patients #8, #28 and #36 are patients who responded to treatment with stable disease. B) patient #31 was the single patient in both cohort showing initial response to therapy (cetuximab). CEA, carcinoembryonic antigen (expressed in ng/mL); CT-scan, computed tomography scan (expressed as percentage change of baseline value) ; RefA, concentration of total cfDNA (ng/mL of plasma); mA, concentration of mutant cfDNA (ng/mL of plasma); mA%, mutation load; PR, Partial response; SD, Stable disease (RECIST criteria); PD, progressive disease (RECIST criteria).
Interestingly, differential evolution of mutant subclones may be occurring in patients #31 and #36 (Figure 4A and 4B), with some clones increasing in mutant allele frequency while others decrease over time. In patient #36 there was detection of additional KRAS G12S mutant subclone and a decrease of a KRAS G12A subclone at cycle 8. Similarly in patient #31 there are KRAS G12A and G12V clones that have inverse dynamics noted.
Discussion
Although monoclonal antibodies to EGFR improve mCRC patient survival, they are limited by primary and secondary resistance (39). Standard of care clinical biomarkers poorly track treatment resistance during early tumor progression. The selective pressure from targeted therapy combined with spatial and temporal clonal heterogeneity lead to a complex clinical challenge. Early detection of secondary resistance may impact clinical outcomes. Bertotti et al (40) characterized secondary resistance to EGFR blockade in mCRC and observed that alterations in KRAS, NRAS, and BRAF were important mechanisms of resistance. With two recent reports showing the ability of cfDNA to detect secondary resistance to anti-EGFR therapies with using liquid biopsy (15,17), this study further explored cfDNA and expanded upon work in the field by examining both the ability of cfDNA assays to assess for the presence of a mutation, and the ability of quantitative parameters associated with this assay such as RAS WT concentration, RAS mutant concentration, and mutant allele fraction, to impact outcome.
cfDNA Assay Performance
Approximately half (52%) of the patients from Cohort 2 who initially tested WT for KRAS exon 2 mutations from archival tumor tissue analysis were actually noted to have KRAS exon 2 mutations in their plasma, and even more patients were found to have extended RAS or BRAF mutations. While concordance between tissue and plasma appeared better for BRAFV600E mutations, discordance for KRAS exon 2 mutations was notable. Concordance between cfDNA and archival tissue seems to be lower in patients who do not have their primary tumor in situ at the point of study entry as well in case of long delay between tumor-tissue and blood collection (Supplementary Table S6). Our study suggests that cfDNA may be more sensitive than archival tumor tissue analysis in detecting mutations. We can speculate that discordance in 10 patients from cohort 2 might result from the fact that blood was collected following pre-study targeted therapy, causing selection of mutant clones potentially detectable by plasma analysis at baseline before study treatment of cohort 2. (Figure 2A). We recently reported similar findings in another prospective and blinded study where we found a much higher proportion of mCRC patients with RAS or BRAF mutations using a cfDNA assay compared to tissue (76% instead of 55%) – (41). Various factors may explain this discordance. Tissue biopsies provide a static snapshot of the tumor at the time of tissue collection and do not necessarily represent the entire tumor genome due to tumor heterogeneity and clonal evolution (9,10,42). Temporal clonal evolution (43) may also impact concordance as we noted that time between collection of archival tissue and blood draw differed significantly between those with concordant and discordant results (692 versus 1074 respectively; P-value: 0.016; Figure S11 and S12). This suggests that therapeutic pressures and tumor evolution over time make archival tissue less reliable for determining the genotype of a tumor and highlights the advantage of real-time genotyping with cfDNA.
There are only a few other prospective studies examining the concordance of RAS mutation in tissue and cfDNA under blinded conditions (44–46).
Many other retrospective or small studies are available in the literature (45,47). In these studies, specificity, sensitivity and overall concordance in comparison to archival tissue was highly variable (55%–97%, 26%–92%, 64%–96%, respectively), likely due to assay characteristics coupled with time between tissue and plasma collection. When using an ultrasensitive method (48) such as employed in this study, another group noted a significant level of discordance (15–25%), mainly associated with an increase in tumors classified as mutation positive by mutant cfDNA assessment. Our results are in agreement with Kuo et al. (49) who observed that 21% of patients who were wild type by archival tissue were actually cfDNA positive for a KRAS mutation for an overall concordance of 79%. Since detection of actionable mutations by plasma analysis is not limited by (i), intra-tumoral, (ii), inter-tumoral and (iii), temporal clonal heterogeneity, in contrast to tumor tissue analysis, cfDNA testing has clear advantages that may lead to the earlier and more sensitive detection of mutations. Tumor tissue in now challenged as the gold standard in screening for actionable mutations in oncology.
Sensitivity is a crucial requirement for analyzing cfDNA due to the potentially low mutant allele frequency noted during sampling. To address this, we developed IntPlex, an allele specific Q-PCR technique that uses several nested primer sets and takes into account the low fragment size of cfDNA (mean size of 100 bp) to optimize assay performance. IntPlex enables detection of variant alleles down to a sensitivity of 0.005% mutant to WT ratio that greatly surpasses conventional NGS-based methods (0.5%–2%) (4,50,51). Spiking experiments showed clearly single-copy detection in highly diluted PCR mixture under Poisson law distribution (Supplemental Figure S9). Using this technique one mutant fragment may be detected among 10 Billion cfDNA fragments providing a sensitivity level (0.005%, mutant/WT ratio) This targeted method is limited by the number of mutant fragments existing in the plasma, with a limit of 26 mutant cfDNA fragments/mL of plasma. Despite the fact that plasma for the present study was stored frozen for more than 4 years (4–6 years storage range) and the pre-analytical conditions were not optimal, IntPlex was still able to detect mutations at very low frequencies and performed well. Note, no KRAS exon 2 and BRAFV600E mutation could be detected in 29 certified healthy volunteers in an Ad-Hoc study of our previous report, regardless of the threshold of detection used (31).
There are some limitations to our assay. IntPlex enables the detection of only a restricted set of common mutations (i.e. known point mutations). Only 15 KRAS, 9 NRAS and one BRAF mutations were tested in this study, however these hotspot mutations represent more than 95–99%, 92–99% and 96–97% of all mutations found in these three genes in CRC patients, respectively.
Significance of low mutant allele frequency mutations
The impressive sensitivity of our assay and its ability to quantify mutations noted at as low as 0.01 ng/mL allowed the identification of many mutations with very small allelic fractions. The ability to confidently detect these low frequency mutations likely explains part of the discrepancy between the results from the initial clinical assay used for trial enrollment and our results. CLIA certified laboratory often have a mutant allele fraction limit of detection of 5%. Positive results below this threshold are often suppressed from clinicians because a false positive result due to sequencing errors cannot be ruled out. We retrospectively sequenced archival tissue with a high sensitivity assay to confirm mutations (Figure 1B), and note significant discordance regardless of whether a clinical grade or high-sensitivity research based platform is used (Figure 1).
We observed subclones with very low allele frequency (down to 0.009%) either at baseline or during the course of treatment (Supplemental figure S10). There were 30% of patients with a mutation at an allele frequency below 0.1% at baseline, and 20% of patients with a mutation noted below 0.1% during the course of treatment. These rare subclones may exist before treatment but be undetectable by clinical grade assays and subsequently emerge under anti-EGFR treatment. Secondary RAS mutations have been previously shown to develop in up to 60% of tumors progressing during anti-EGFR therapy and we noted that they could be found in all but 1 patient on this study, however many of these mutations may have been present yet undetectable at baseline. Retrospective studies have suggested that many of these mutations may be detected in cfDNA prior to initiation of therapy (13), while other work using mathematic models of tumor kinetics and growth has shown that these mutations must be present prior to initiation of therapy (15).
Since these low frequency mutations may result in treatment resistance, very sensitive methods to initially select patients for anti-EGFR therapies are needed. Tougeron et al (52) recently demonstrated that low-prevalence KRAS mutations detected by an advanced pyrosequencing method in tumors previously defined as KRAS wild type were associated with shorter PFS. Our data revealed that 52% (22/42) of patients had an allele fraction below 1%, 31% (13/42) were below 0.5% and 19% (8/42) were below 0.1% (Supplemental Table S7). These low allelic values may not perfectly correlate to the allele frequency that would be detected in a tumor. Plasma analysis relies on the total number of cfDNA fragments shed into the blood stream from normal and tumor cells. In contrast, tumor tissue analysis relies on the number of malignant cells in a biopsy and may be enriched by pathologist selection of section with higher tumor content. As a consequence, the value used to estimate cfDNA sensitivity does not correspond directly to that of tumor tissue analysis which precludes use of a universal sensitivity threshold for detection of mutations. We illustrated this by showing that in the 16% (4/25) of patients with cfDNA allele frequency below 0.5%, mutations could be readily detected in matched tumor tissue (supplemental table S8) (41).
Impact of therapy on clonal dynamics and development of additional RAS mutations
Following EGFR directed therapy, we noted that 98% of patients had at least one mutation in RAS or BRAF that likely contributed to treatment resistance. More interestingly, of the 21 patients defined as RAS/BRAF mutant at baseline, 11 (50%) showed additional point mutation that was not present at baseline while 3 (14%) lost previously detected mutations. There was even a single patient with a baseline KRAS G12A mutation that subsequently developed additional G12D, G12V, and G13D mutations after receiving 16 cycles of cetuximab containing therapy. These findings demonstrate that even if a single clonal population with a resistance mechanism is present in a tumor, treatment pressures may drive the development of additional alternative resistance mechanisms in a pattern of convergent evolution. These multiple convergent mutations were noted to develop whether patients received FOLFOX plus dasatinib or FOLFOX, dasatinib plus cetuximab. Another important clonal dynamic that we noted was that some RAS mutations appeared to be more strongly associated with secondary mutation than primary resistance. Our data shows that KRAS codon 61 was expressed more frequently in acquired resistance mutations (5.5%) than in the general mCRC population (1–4%) (Zhang,2011), suggesting that mutations at this codon might provide a selective growth advantage under cetuximab treatment. This was also observed for KRAS codon 146 mutations by Morelli et al (48). Our findings also reinforce the importance of BRAF mutations as mechanisms of resistance, as we demonstrated that 4/42 patients (10%) had additional BRAFV600E mutations during therapy.
cfDNA to follow disease progression
cfDNA quantitative measurements from this study closely mirrored changes in CEA and CT scan results as evaluated in 18 and 16 patients in cohort 1 and 2, respectively. We noted that both the qualitative detection of a mutation and quantitative information such as RAS mutant and WT cfDNA concentrations and mutant allele fraction appeared to be useful in following disease course. The level of RAS/BRAF mutations expressed as % of mutant cfDNA as well as ng/mL plasma of mutant cfDNA did not correlate with the treatment response highlighting the necessity of detecting low level and low frequency mutations. (Supplemental Table S9). The circulating WT cfDNA concentration appeared to closely approximate other clinical parameters, and it was not only the detection of a mutant cfDNA sample that was related to disease progression (Figure 4B). The patient in Figure 4B had an interesting molecular profile which demonstrated falling levels of RAS DNA, both WT and mutant during response to therapy. Subsequently, after 16 cycles of therapy this patient had developed 3 additional RAS mutations beyond the baseline KRAS G12A mutation. Mutant allele frequency of the single baseline mutation was very low (0.04%), and the subsequent new clones all remained subclonal with allelic frequencies ranging from 0.01%–0.11%, potentially explaining why the patient continued to respond for over several cycles. There was likely a large proportion of this tumor that lacked RAS mutations.
Siravegna et al (53) elegantly demonstrated the utility of following cfDNA to measure KRAS mutant subclones as a potential for re-using prior targeted therapies. Upon withdrawal of the selective pressure of anti-EGFR treatment, KRAS mutant clones declined (53). These patients subsequently regained sensitivity to anti-EGFR therapy. In a similar manner, following the patient in Figure 4B serially may allow continuation of therapy until a resistance clone becomes more dominant, highlighting the importance of obtaining quantitative data beyond the mere presence of a mutation (12,54). If therapy had been stopped at the first incidence of a new resistance mutation, that patient would have switched therapy 2 months earlier than was required clinically.
Limitations
While this study demonstrates the potential applications of highly sensitive cfDNA to follow tumor dynamics, it must be interpreted within the contexts of its limitations. The small sample size of our study makes it difficult to draw firm conclusions. Our assessment of mutation status was blinded and samples were prospectively collected, both features that add to the rigor of experimental design but do not obviate the need for larger trials for validation. We were also limited by the infrequent plasma samples that were available for cfDNA analysis, occurring at baseline and once every 4 cycles. While our study does not provide evidence that cfDNA detects mutant clones and progression earlier than clinical assessment, this is likely due to the fact that we only assessed cfDNA once every four cycles.
An important finding in the present study was the convergent evolution of clones with redundant resistance mechanisms in the EGFR pathway. We were only able to test three clinically relevant genes and did not test genes involved in EGFR downstream signaling pathways or the mTOR pathway. PIK3CA mutation, PTEN loss (55), EGFR mutations, as well as MET and ERBB2 amplifications may predict a lack of response to anti-EGFR agents. Thus, it would be interesting to see if there were redundant resistance pathways that also developed in these genes in addition to the multiple concurrent KRAS clones that were noted (40,48). Because EGFR S492R has been described as an alternate mechanism of resistance, we did test the 13 plasma samples of patients who had previously received anti-EGFR therapy for EGFR S492R mutations at baseline, however none of the 13 plasma samples were positive for this mutation.
Conclusion
With our assay we were able to uncover the development of secondary resistance mutations in RAS and BRAF in all but one out of 42 patients. A significant fraction of these mutations detected either before or in the course of treatment from plasma analysis harbor low mutant allele frequency in cfDNA. This shows the need of a ultra-sensitive method to detect additional point mutation when tracking secondary resistance. Periodic liquid biopsies require a simple blood draw, and if optimized for clinical use would optimize patient safety and convenience. We note that not only was the qualitative presence of a mutant clone important, but the additional quantitative measurements of WT KRAS concentration, mutant KRAS concentration, and mutant allele fraction may be useful parameters to follow. We also noted that many patients experienced convergent evolution that resulted in redundant mutations that would provide multiple mechanisms of resistance to EGFR directed therapy.
Supplementary Material
Statement of translational relevance.
Cell-free circulating DNA (cfDNA) provides a liquid biopsy alternative to tissue biopsies for monitoring cancer genetic changes over time. In this study, we used a recently developed multi-marker assay enabling a qualitative and a quantitative cfDNA analysis for tracking acquired resistance by studying the real-time clonal evolution of the tumor. CfDNA of refractory mCRC patients to anti-EGFR therapy may harbor mutations at very low frequency down to 0.01% before initiation or during treatment revealing the need of a high sensitive technique. CfDNA demonstrates convergent evolution and common resistance mechanisms during treatment of colorectal cancer. Serial analysis of cfDNA provides a unique opportunity to study the evolving genomic landscape of a cancer during therapy, and to identify the early emergence of treatment resistance and guide targeted therapeutic decisions.
Acknowledgments
The authors thank Thibault Mazard, Frederic Bibeau, Philippe Blache, Corinne Prevostel, Celine Gongora and Evelyne Crapez for helpful discussion, Vanessa Guillaumon for help in supporting this work, and Amaelle Otandault and Baptiste Auzemery for technical assistance. Alain R. Thierry is supported by INSERM. This study was supported by the SIRIC Montpellier Grant «INCa-DGOS-Inserm 6045». This work was supported by BMS and Roche France.
Footnotes
Financial support: This study was supported by the SIRIC Montpellier Grant «INCa-DGOS-Inserm 6045» and was supported by BMS and Roche France. Alain R. Thierry is supported by INSERM.
Conflict of interest: No conflict of interest.
References
- 1.Fakih M. Biologic therapies in colorectal cancer: indications and contraindications. Am Soc Clin Oncol Educ Book Am Soc Clin Oncol Meet. 2015:e197–206. doi: 10.14694/EdBook_AM.2015.35.e197. [DOI] [PubMed] [Google Scholar]
- 2.Li Z-Z, Wang F, Zhang Z-C, Wang F, Zhao Q, Zhang D-S, et al. Mutation profiling in chinese patients with metastatic colorectal cancer and its correlation with clinicopathological features and anti-EGFR treatment response. Oncotarget. 2016 May 10;7(19):28356–68. doi: 10.18632/oncotarget.8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanco-Calvo M, Concha Á, Figueroa A, Garrido F, Valladares-Ayerbes M. Colorectal Cancer Classification and Cell Heterogeneity: A Systems Oncology Approach. Int J Mol Sci. 2015 Jun 15;16(6):13610–32. doi: 10.3390/ijms160613610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haley L, Tseng L-H, Zheng G, Dudley J, Anderson DA, Azad NS, et al. Performance characteristics of next-generation sequencing in clinical mutation detection of colorectal cancers. Mod Pathol Off J U S Can Acad Pathol Inc. 2015 Oct;28(10):1390–9. doi: 10.1038/modpathol.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westwood M, van Asselt T, Ramaekers B, Whiting P, Joore M, Armstrong N, et al. KRAS mutation testing of tumours in adults with metastatic colorectal cancer: a systematic review and cost-effectiveness analysis. Health Technol Assess Winch Engl. 2014 Oct;18(62):1–132. doi: 10.3310/hta18620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laurent-Puig P, Pekin D, Normand C, Kotsopoulos SK, Nizard P, Perez-Toralla K, et al. Clinical relevance of KRAS-mutated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin Cancer Res Off J Am Assoc Cancer Res. 2015 Mar 1;21(5):1087–97. doi: 10.1158/1078-0432.CCR-14-0983. [DOI] [PubMed] [Google Scholar]
- 7.Ciardiello F, Normanno N, Maiello E, Martinelli E, Troiani T, Pisconti S, et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: findings from the CAPRI-GOIM trial. Ann Oncol Off J Eur Soc Med Oncol. 2014 Sep;25(9):1756–61. doi: 10.1093/annonc/mdu230. [DOI] [PubMed] [Google Scholar]
- 8.Caldas C. Cancer sequencing unravels clonal evolution. Nat Biotechnol. 2012 May 7;30(5):408–10. doi: 10.1038/nbt.2213. [DOI] [PubMed] [Google Scholar]
- 9.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012 Mar 8;366(10):883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012 Oct 1;72(19):4875–82. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol Off J Am Soc Clin Oncol. 2014 Feb 20;32(6):579–86. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016 Sep;35(3):347–76. doi: 10.1007/s10555-016-9629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014 Feb 19;6(224):224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newman AM, Bratman SV, To J, Wynne JF, Eclov NCW, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014 May;20(5):548–54. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012 Jun 28;486(7404):537–40. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murtaza M, Dawson S-J, Tsui DWY, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013 May 2;497(7447):108–12. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 17.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012 Jun 28;486(7404):532–6. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lièvre A, Bachet J-B, Le Corre D, Boige V, Landi B, Emile J-F, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006 Apr 15;66(8):3992–5. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 19.Van Cutsem E, Lenz H-J, Köhne C-H, Heinemann V, Tejpar S, Melezínek I, et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2015 Mar 1;33(7):692–700. doi: 10.1200/JCO.2014.59.4812. [DOI] [PubMed] [Google Scholar]
- 20.Allegra CJ, Rumble RB, Hamilton SR, Mangu PB, Roach N, Hantel A, et al. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J Clin Oncol Off J Am Soc Clin Oncol. 2016 Jan 10;34(2):179–85. doi: 10.1200/JCO.2015.63.9674. [DOI] [PubMed] [Google Scholar]
- 21.Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, Karapetis CS. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol Off J Eur Soc Med Oncol. 2015 Jan;26(1):13–21. doi: 10.1093/annonc/mdu378. [DOI] [PubMed] [Google Scholar]
- 22.Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol Off J Eur Soc Med Oncol. 2014 Jul;25(7):1346–55. doi: 10.1093/annonc/mdu141. [DOI] [PubMed] [Google Scholar]
- 23.Boissière-Michot F, Lopez-Crapez E, Frugier H, Berthe M-L, Ho-Pun-Cheung A, Assenat E, et al. KRAS genotyping in rectal adenocarcinoma specimens with low tumor cellularity after neoadjuvant treatment. Mod Pathol Off J U S Can Acad Pathol Inc. 2012 May;25(5):731–9. doi: 10.1038/modpathol.2011.210. [DOI] [PubMed] [Google Scholar]
- 24.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010 Aug;11(8):753–62. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015 Aug 26;7(302):302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 26.Reinert T, Schøler LV, Thomsen R, Tobiasen H, Vang S, Nordentoft I, et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut. 2016 Apr;65(4):625–34. doi: 10.1136/gutjnl-2014-308859. [DOI] [PubMed] [Google Scholar]
- 27.Douillard J-Y, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013 Sep 12;369(11):1023–34. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 28.Kim M-J, Lee HS, Kim JH, Kim YJ, Kwon JH, Lee J-O, et al. Different metastatic pattern according to the KRAS mutational status and site-specific discordance of KRAS status in patients with colorectal cancer. BMC Cancer. 2012 Aug 9;12:347. doi: 10.1186/1471-2407-12-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thierry AR, Mouliere F, Gongora C, Ollier J, Robert B, Ychou M, et al. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010 Oct;38(18):6159–75. doi: 10.1093/nar/gkq421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, et al. High fragmentation characterizes tumour-derived circulating DNA. PloS One. 2011;6(9):e23418. doi: 10.1371/journal.pone.0023418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014 Apr;20(4):430–5. doi: 10.1038/nm.3511. [DOI] [PubMed] [Google Scholar]
- 32.Mouliere F, El Messaoudi S, Pang D, Dritschilo A, Thierry AR. Multi-marker analysis of circulating cell-free DNA toward personalized medicine for colorectal cancer. Mol Oncol. 2014 Jul;8(5):927–41. doi: 10.1016/j.molonc.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El Messaoudi S, Mouliere F, Du Manoir S, Bascoul-Mollevi C, Gillet B, Nouaille M, et al. Circulating DNA as a Strong Multimarker Prognostic Tool for Metastatic Colorectal Cancer Patient Management Care. Clin Cancer Res Off J Am Assoc Cancer Res. 2016 Jun 15;22(12):3067–77. doi: 10.1158/1078-0432.CCR-15-0297. [DOI] [PubMed] [Google Scholar]
- 34.Parseghian C, Parikh NU, Wu JY, Jiang Z-Q, Henderson LD, Tian F, et al. Dual Inhibition of EGFR and c-Src by Cetuximab and Dasatinib Combined with FOLFOX Chemotherapy in Patients with Metastatic Colorectal Cancer. Clin Cancer Res Off J Am Assoc Cancer Res. doi: 10.1158/1078-0432.CCR-16-3138. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiu RW, Poon LL, Lau TK, Leung TN, Wong EM, Lo YM. Effects of blood-processing protocols on fetal and total DNA quantification in maternal plasma. Clin Chem. 2001 Sep;47(9):1607–13. [PubMed] [Google Scholar]
- 36.El Messaoudi S, Rolet F, Mouliere F, Thierry AR. Circulating cell free DNA: Preanalytical considerations. Clin Chim Acta Int J Clin Chem. 2013 Sep 23;424:222–30. doi: 10.1016/j.cca.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 37.Arena S, Bellosillo B, Siravegna G, Martínez A, Cañadas I, Lazzari L, et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2015 May 1;21(9):2157–66. doi: 10.1158/1078-0432.CCR-14-2821. [DOI] [PubMed] [Google Scholar]
- 38.Esposito C, Rachiglio AM, La Porta ML, Sacco A, Roma C, Iannaccone A, et al. The S492R EGFR ectodomain mutation is never detected in KRAS wild-type colorectal carcinoma before exposure to EGFR monoclonal antibodies. Cancer Biol Ther. 2013 Dec;14(12):1143–6. doi: 10.4161/cbt.26340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waring P, Tie J, Maru D, Karapetis CS. RAS Mutations as Predictive Biomarkers in Clinical Management of Metastatic Colorectal Cancer. Clin Colorectal Cancer. 2016 Jun;15(2):95–103. doi: 10.1016/j.clcc.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015 Oct 8;526(7572):263–7. doi: 10.1038/nature14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thierry AR, Messaoudi SE, Mollevi C, Pastor B, Sanchez C, Raoul J-L, et al. Abstract 2632: Clinical validation of circulating DNA analysis for the detection of point mutations and of the longitudinal metastatic colorectal patient follow up for detecting emergence of resistance to targeted therapy. Cancer Res. 2016 Jul 15;76(14 Supplement):2632–2632. [Google Scholar]
- 42.Ulz P, Auer M, Heitzer E. Detection of Circulating Tumor DNA in the Blood of Cancer Patients: An Important Tool in Cancer Chemoprevention. Methods Mol Biol Clifton NJ. 2016;1379:45–68. doi: 10.1007/978-1-4939-3191-0_5. [DOI] [PubMed] [Google Scholar]
- 43.Uchi R, Takahashi Y, Niida A, Shimamura T, Hirata H, Sugimachi K, et al. Integrated Multiregional Analysis Proposing a New Model of Colorectal Cancer Evolution. PLoS Genet. 2016 Feb;12(2):e1005778. doi: 10.1371/journal.pgen.1005778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sasaki Y, Akasu T, Saito N, Kojima H, Matsuda K, Nakamori S, et al. Prognostic and predictive value of extended RAS mutation and mismatch repair status in stage III colorectal cancer. Cancer Sci. 2016 Jul;107(7):1006–12. doi: 10.1111/cas.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yen L-C, Yeh Y-S, Chen C-W, Wang H-M, Tsai H-L, Lu C-Y, et al. Detection of KRAS oncogene in peripheral blood as a predictor of the response to cetuximab plus chemotherapy in patients with metastatic colorectal cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2009 Jul 1;15(13):4508–13. doi: 10.1158/1078-0432.CCR-08-3179. [DOI] [PubMed] [Google Scholar]
- 46.Spindler K-LG, Pallisgaard N, Appelt AL, Andersen RF, Schou JV, Nielsen D, et al. Clinical utility of KRAS status in circulating plasma DNA compared to archival tumour tissue from patients with metastatic colorectal cancer treated with anti-epidermal growth factor receptor therapy. Eur J Cancer Oxf Engl 1990. 2015 Nov;51(17):2678–85. doi: 10.1016/j.ejca.2015.06.118. [DOI] [PubMed] [Google Scholar]
- 47.Ryan BM, Lefort F, McManus R, Daly J, Keeling PWN, Weir DG, et al. A prospective study of circulating mutant KRAS2 in the serum of patients with colorectal neoplasia: strong prognostic indicator in postoperative follow up. Gut. 2003 Jan;52(1):101–8. doi: 10.1136/gut.52.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morelli MP, Overman MJ, Dasari A, Kazmi SMA, Mazard T, Vilar E, et al. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann Oncol Off J Eur Soc Med Oncol. 2015 Apr;26(4):731–6. doi: 10.1093/annonc/mdv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuo Y-B, Chen J-S, Fan C-W, Li Y-S, Chan E-C. Comparison of KRAS mutation analysis of primary tumors and matched circulating cell-free DNA in plasmas of patients with colorectal cancer. Clin Chim Acta Int J Clin Chem. 2014 Jun 10;433:284–9. doi: 10.1016/j.cca.2014.03.024. [DOI] [PubMed] [Google Scholar]
- 50.Altimari A, de Biase D, De Maglio G, Gruppioni E, Capizzi E, Degiovanni A, et al. 454 next generation-sequencing outperforms allele-specific PCR, Sanger sequencing, and pyrosequencing for routine KRAS mutation analysis of formalin-fixed, paraffin-embedded samples. OncoTargets Ther. 2013;6:1057–64. doi: 10.2147/OTT.S42369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao J, Wu H, Wang L, Zhang H, Duan H, Lu J, et al. Validation of targeted next-generation sequencing for RAS mutation detection in FFPE colorectal cancer tissues: comparison with Sanger sequencing and ARMS-Scorpion real-time PCR. BMJ Open. 2016 Jan 8;6(1):e009532. doi: 10.1136/bmjopen-2015-009532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tougeron D, Lecomte T, Pagès JC, Villalva C, Collin C, Ferru A, et al. Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Ann Oncol Off J Eur Soc Med Oncol. 2013 May;24(5):1267–73. doi: 10.1093/annonc/mds620. [DOI] [PubMed] [Google Scholar]
- 53.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015 Jul;21(7):827. doi: 10.1038/nm0715-827b. [DOI] [PubMed] [Google Scholar]
- 54.Perkins G, Yap TA, Pope L, Cassidy AM, Dukes JP, Riisnaes R, et al. Multi-purpose utility of circulating plasma DNA testing in patients with advanced cancers. PloS One. 2012;7(11):e47020. doi: 10.1371/journal.pone.0047020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol Stockh Swed. 2014 Jul;53(7):852–64. doi: 10.3109/0284186X.2014.895036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.