

Abstract
In vitro, EGFR inhibition, combined with the BRAF inhibitor vemurafenib, causes synergistic cytotoxicity for BRAFV600E metastatic colorectal cancer (mCRC), further augmented by irinotecan. The safety and efficacy of vemurafenib, irinotecan, and cetuximab in BRAF-mutated malignancies are not defined. In this 3+3 phase I study, patients with BRAFV600E advanced solid cancers received cetuximab and irinotecan with escalating doses of vemurafenib. Nineteen patients (18 with mCRC, 1 with appendiceal cancer) were enrolled. Three patients experienced dose-limiting toxicities. The maximum tolerated dose of vemurafenib was 960 mg twice daily. Six of 17 evaluable patients (35%) achieved a radiographic response by RECIST 1.1 criteria, consistent with in vivo models demonstrating tumor regressions with the triplet regimen. Median progression-free survival was 7.7 months. BRAFV600E cfDNA trends correlated with radiographic changes, and acquired mutations from cfDNA in genes reactivating MAPK signaling were observed at progression.
Keywords: BRAF, MAPK, cfDNA, metastasis, colorectal cancer
Introduction
BRAFV600E mutations are present in approximately 8% to 10% of patients with metastatic colorectal cancer(1–3) and are associated with poor survival(4–6). In patients with previously treated BRAF-mutated colorectal cancer, radiographic disease progression is often observed by the time of first restaging, the result of both rapid clinical deterioration and poor sensitivity to standard cytotoxic regimens(7). Therefore, these patients are in dire need of novel, effective therapies.
Vemurafenib is a small-molecule tyrosine kinase inhibitor specific to the ATP-binding domain of BRAF V600E(8). Inhibition of V600E-mutated BRAF by vemurafenib may lead to decreased activity of the mitogen-activated protein kinase (MAPK) pathway, and vemurafenib as a single agent has been active in multiple advanced malignancies with BRAFV600E mutations(9–11). However, in patients with metastatic BRAFV600E colorectal cancer, vemurafenib monotherapy has a response rate of only 5%(12). In preclinical models, blockade of BRAF V600E by vemurafenib generates a reflexive activation of EGFR, which can bypass BRAF and promote tumor progression via multiple downstream signaling pathways(13, 14). Dual blockade of BRAF by vemurafenib and EGFR by cetuximab in mice caused tumor regression not observed with both agent alone, and pilot studies of BRAF and EGFR inhibition have demonstrated evidence of clinical activity(15, 16).
Irinotecan combined with cetuximab is approved for metastatic colorectal cancer in patients with RAS wild-type tumors, and this combination has shown efficacy for patients who previously progressed on irinotecan monotherapy(17). Xenograft models derived from colorectal tumors of patients with BRAFV600E mutation have suggested that mice treated with vemurafenib, irinotecan, and cetuximab have better response rates and longer survival than mice treated with irinotecan and cetuximab or vemurafenib and cetuximab(18). In light of these findings, we performed a phase I study to determine the safety and activity of vemurafenib in combination with irinotecan and cetuximab in patients with refractory, advanced, BRAFV600E-mutated solid tumors.
Results
Patient Characteristics
Nineteen patients were enrolled and treated between June 2013 and May 2015. Demographic data are listed in Supplementary Table 1. The median number of prior therapies was 2 (range, 1–4). Fourteen patients (74%) had previously received irinotecan, 8 (42%) had previously received cetuximab, 1 had previously received a BRAF inhibitor, and 1 had previously received a MEK inhibitor. Genomic and pathologic features of 17 participants’ tumors are listed in Table 1. KRAS and NRAS mutations were not detected in pretreatment samples from any of these patients. A PIK3CA mutation was detected in only one of 17 tested patients. Of the 14 tumors in which tumor tissue was available for microsatellite testing to be performed, microsatellite instability was detected in 3 (21%).
Table 1.
Genomic profiling and microsatellite status for each patient on study according to treatment response.
| BRAF | KRAS | NRAS | PIK3CA | AKT | MSI-H | BEST RESPONSE | |
|---|---|---|---|---|---|---|---|
| 1 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | (Not tested) | SD |
| 2 | V600E | Wild-type | Wild-type | (Not tested) | (Not tested) | MSI-H | PR |
| 3 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | MSS | PR |
| 4 | V600E | Wild-type | Wild-type | (Not tested) | (Not tested) | MSS | SD |
| 5 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | MSS | PD |
| 6 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | MSS | PR |
| 7 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | MSS | SD |
| 8 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | MSS | SD |
| 9 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | MSI-H | PR |
| 10 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | MSS | N/A |
| 11 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | (Not tested) | SD |
| 12 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | MSS | SD |
| 13 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | (Not tested) | SD |
| 14 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | (Not tested) | PR |
| 15 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | (Not tested) | SD |
| 16 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | MSS | SD |
| 17 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | MSS | PR |
| 18 | V600E | Wild-type | Wild-type | Wild-type | Wild-type | MSS | SD |
| 19 | V600E | Wild-type | Wild-type | H1047R | Wild-type | MSI-H | PD |
Safety
Six patients were treated at dose level 1, six at dose level 2, and 7 at dose level 3. A dose-limiting toxic effect was observed in one patient at each dose level: arthralgia at dose level 1 and diarrhea at dose levels 2 and 3. The patient with arthralgia experienced resolution of symptoms after reduction of the vemurafenib dose. One of the patients with dose-limiting diarrhea ceased study participation, and the other experienced improvement of symptoms after reduction of the irinotecan dose.
Adverse events, according to each dose level, are listed in Table 2. The most common adverse events were fatigue (89% of patients), diarrhea (84%), nausea (79%), and rash (74%). The most common grade 3 adverse event across all dose levels, observed during cycles 2 (one patient), 5 (two patients), and 6, 7, and 8 (one patient each), was diarrhea (26%). Four patients (21%) developed grade 3 or 4 leukopenia, and one developed febrile neutropenia. No patient developed keratoacanthoma or cutaneous squamous cell carcinoma. On the basis of these findings, vemurafenib 960 mg by mouth twice daily was declared safe in combination with irinotecan and cetuximab every 14 days at the doses used in this study and will serve as the recommended dose for phase II studies.
Table 2.
Adverse events, according to dose level, associated with treatment with vemurafenib in combination with irinotecan and cetuximab.
| Total | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 1–2 | Grade 3–4 | Total | % |
|---|---|---|---|---|---|---|---|---|
| Alopecia | 3 | 1 | 1 | 4 | 1 | 5 | 26 | |
| Anemia | 7 | 6 | 2 | 13 | 2 | 15 | 79 | |
| Anorexia | 7 | 3 | 10 | 0 | 10 | 53 | ||
| Arthralgia | 5 | 1 | 2 | 6 | 2 | 8 | 42 | |
| Blurry vision | 1 | 1 | 0 | 1 | 5 | |||
| Constipation | 4 | 4 | 0 | 4 | 21 | |||
| Cramping | 1 | 1 | 0 | 1 | 5 | |||
| Diarrhea | 6 | 5 | 5 | 11 | 5 | 16 | 84 | |
| Dry heaves | 1 | 1 | 0 | 1 | 5 | |||
| Dry mouth | 1 | 1 | 0 | 1 | 5 | |||
| Dyspnea | 4 | 1 | 5 | 0 | 5 | 26 | ||
| Epistaxis | 1 | 1 | 0 | 1 | 5 | |||
| Fatigue | 9 | 6 | 2 | 15 | 2 | 17 | 89 | |
| Fever | 1 | 1 | 0 | 1 | 5 | |||
| Flushing | 1 | 1 | 0 | 1 | 5 | |||
| GERD | 1 | 1 0 | 1 5 | |||||
| HFS | 2 | 2 | 0 | 2 | 11 | |||
| HTN | 1 | 1 | 0 | 1 | 5 | |||
| Inc bilirubin | 1 | 1 | 0 | 1 | 5 | |||
| Inc LFT | 1 | 1 | 0 | 1 | 5 | |||
| Infection | 1 | 1 | 0 | 1 | 5 | |||
| Leukopenia | 2 | 3 | 3 | 1 | 5 | 4 | 9 | 47 |
| Low albumin | 1 | 1 | 0 | 1 | 5 | |||
| Low K | 1 | 1 | 0 | 1 | 5 | |||
| Low Mg | 2 | 2 | 0 | 2 | 11 | |||
| Mucositis | 4 | 1 | 5 | 0 | 5 | 26 | ||
| Myalgia | 9 | 2 | 11 | 0 | 11 | 58 | ||
| Nausea | 11 | 3 | 1 | 14 | 1 | 15 | 79 | |
| Neuropathy | 4 | 2 | 6 | 0 | 6 | 32 | ||
| Proteinuria | 1 | 1 | 0 | 1 | 5 | |||
| Pruritis | 1 | 1 | 0 | 1 | 5 | |||
| Rash | 12 | 1 | 1 | 13 | 1 | 14 | 74 | |
| Taste change | 1 | 1 | 0 | 1 | 5 | |||
| Taste change | 1 | 1 | 0 | 1 | 5 | |||
| Vomiting | 5 | 1 | 6 | 0 | 6 | 32 | ||
| Weight loss | 1 | 1 | 2 | 0 | 2 | 11 |
Efficacy
Two patients did not have restaging: one left the study because of dose-limiting diarrhea, and another withdrew consent because of arthralgia. Seventeen patients (16 with colorectal cancer, 1 with appendiceal cancer) underwent at least one restaging study following initiation of vemurafenib, irinotecan, and cetuximab. Among these 17 evaluable patients, six had a response (response rate, 35%; 95% confidence interval, 14% to 62%) (Figure 1A). All six of these patients had confirmed responses with at least one additional imaging study showing persistent response according to the RECIST 1.1 criteria (Figure 1B). Among the 17 evaluable patients, 15 (88%) had disease control, defined as stable disease or radiographic response. The lone patient with appendiceal cancer had disease progression by the time of the first restaging study. Patterns in disease control were distributed similarly across the three dose-level cohorts (Figure 1C).
Figure 1.
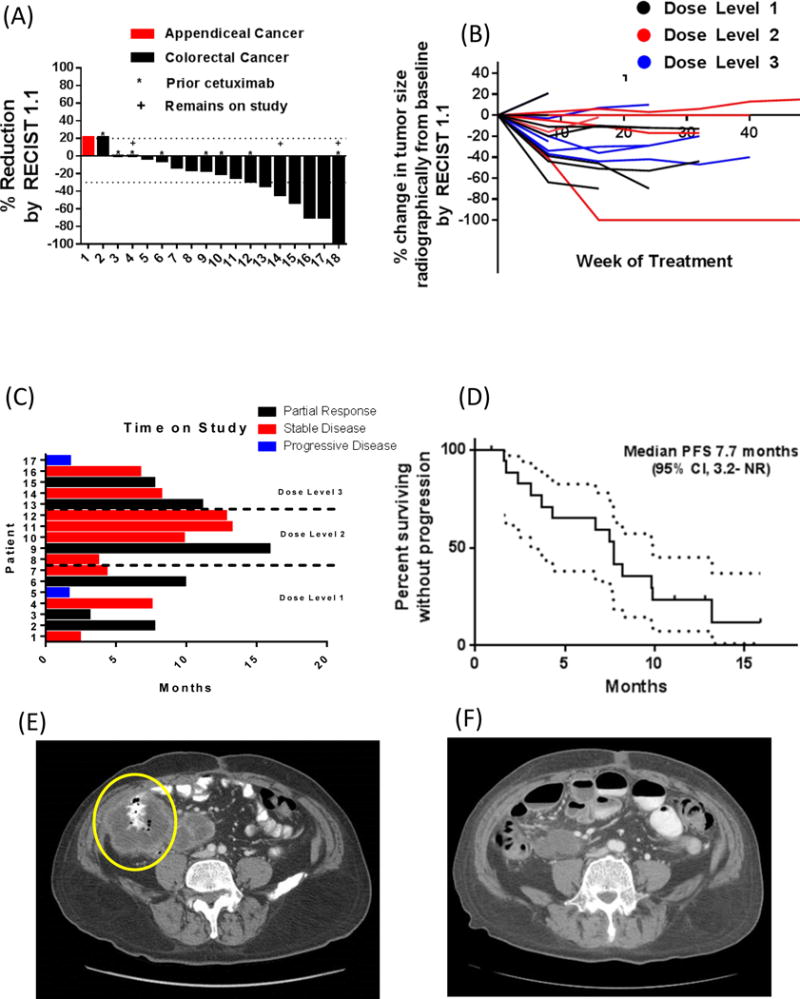
(A) Waterfall plot characterizing best response to treatment for each patient (B) Spider plot demonstrating response to treatment for each patient over time according to the dose level (C) Doses of treatment for each patient on study according to dose level (D) Kaplan-Meier plot for progression-free survival. Representative sequential imaging of a pretreatment abdominal tumor (E) with subsequent shrinkage upon treatment (F) are also shown.
The estimated median progression-free survival was 7.7 months (95% confidence interval, 3.1 months-NA) (Figure 1D). Radiographic responses, when present, lasted a median of 8.8 months before progression; an example of pre-treatment CT imaging studies of a tumor and repeat radiographic improvement of a responding patient is seen in Figures 1E and 1F, respectively. For the 17 evaluable patients, the median number of doses of treatment administered was 16 (interquartile range, 6 to 20). Three patients remain on study at the time of data cutoff. For the 14 patients in whom microsatellite testing was performed, there was no association of response with the presence of accompanying microsatellite instability (odds ratio [OR] 6.0, P=.20). Likewise, no association was detected between radiographic response and prior treatment with irinotecan (OR .60, P=.23) or with prior anti-EGFR therapy (OR = 1.2, P=.64).
cfDNA Analyses
Twelve patients had serial plasma samples available. Trends in cfDNA by both studies mirrored changes in radiographic tumor volume over the course of treatment. For two of these patients, serial plasma samples were analyzed across all time points on study via next-generation sequencing methods that incorporated all genes on the selected gene panel. Declines in BRAFV600E fraction paralleled declines in other truncal mutations like TP53 and SMAD4 (Supplemental Figure 1). A waterfall plot of changes in percentage of BRAF mutant allele at the time of first restaging (Figure 2A) appears similar to the waterfall plot detailing changes in radiographic tumor volume with treatment (Figure 1A). The magnitude of change in BRAFV600E allele from baseline correlated to radiographic response, as patients with partial responses were more likely to demonstrate a deeper reduction in BRAFV600E cfDNA fraction following one dose of treatment by either ddPCR or cfDNA NGS relative to those with stable or progressive disease at the time of first restaging (P<.01). To allow for a more direct comparison between radiographic measurements and changes in mutant cfDNA fraction to detect response to the triple combination, we plotted the change in volume per RECIST 1.1 measurement at the first restaging against the change in BRAFV600E mutant fraction from a blood specimen collected at the same time point (Figure 2B). All six patients with radiographic responses demonstrated near-complete reductions in BRAFV600E cfDNA allele fraction at the same time point. Two of the “non-responder” patients were found to have near-complete disappearances of mutant BRAFV600E just prior to their second dose of treatment (Figure 2A), and maintained these reductions even by the time of their first restaging. Both of these patients demonstrated radiographically stable disease while on study, with maximum reductions in tumor size of −13% and −17%.
Figure 2.
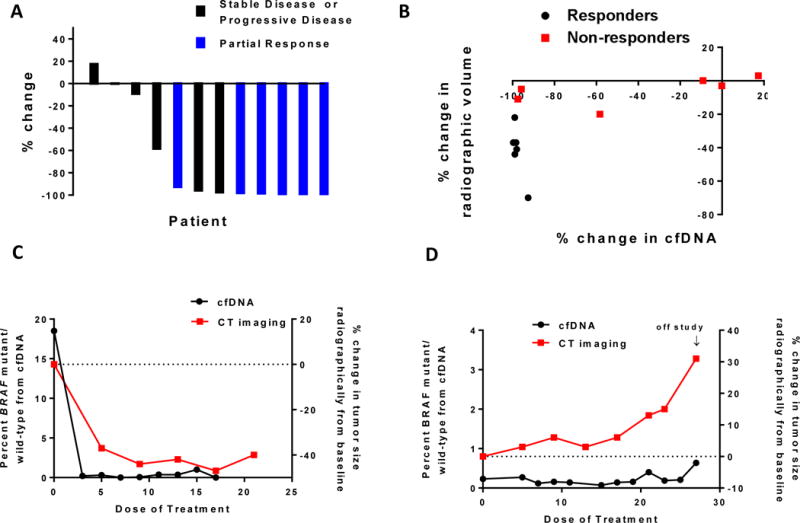
(A) Changes in BRAFV600E cfDNA allele fraction from baseline after one dose of treatment for 12 patients with serial samples available, classified according to radiographic responders (blue) or non-responders (black). (B) Percentage change in BRAFV600E cfDNA fraction versus percent change in radiographic volume of target lesions at the time of first restaging (after 4 doses of treatment), according to radiographic responders (black) or non-responders (red). Representative trends in cfDNA fraction (black) and radiographic changes (red) for patients with radiographic response (C) and progression (D).
Declines in the BRAFV600E cfDNA allele fraction within two weeks of treatment initiation exceeded a 90% reduction from baseline in all six patients with radiographic responses, well before the time of first radiographic staging for response (Figure 2C and Supplementary Figure 2). In general, trends in BRAFV600E cfDNA with time mirrored those of patients with no radiographic response on study (Figure 2D and Supplementary Figure 2).
Ten patients had sufficient plasma at progression for cfDNA analysis to compare mutation and copy number profiles with matched pretreatment samples using the CLIA-certified 68-gene targeted NGS assay. This cfDNA methodology was orthogonally validated with the digital PCR results, with a near-perfect correlation (R2=.99) between detected BRAFV600E cfDNA fractions for samples from a matched patient and specific time point as assessed by both methodologies (Supplementary Figure 3). Amplifications in copy number of KRAS, MET, and EGFR were detected in one patient. Increases in the fraction of PTENI122N, NF1E977K, EGFRL93I, ERBB2R522S, and GNAS1R201C mutant alleles were found. All of these were detected at lower mutant allele frequencies prior to treatment initiation (Figure 3A), and all of these mutations except for the GNASR201C mutation arose were detected when microsatellite instability was present. Three patients acquired MEK1 mutations (2 with MEK1C121S and 1 with MEK1K57T) not present at the start of treatment, and one patient each acquired novel ARAFS490T and GNAS1R201C mutations (Figure 3A).
Figure 3.
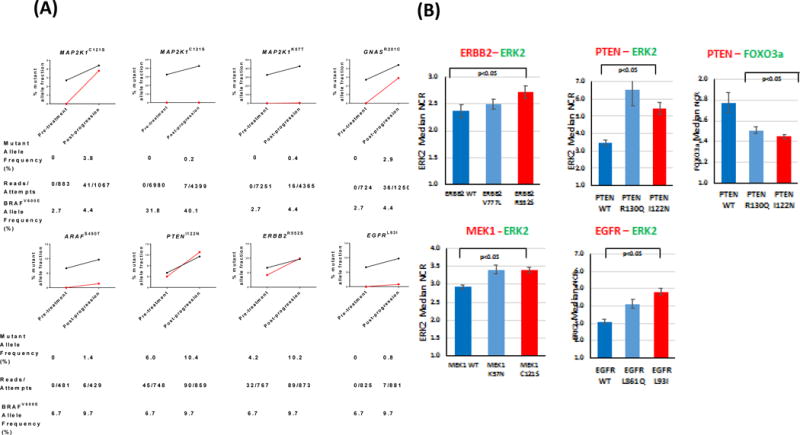
(A) Relative changes in mutant allele fractions for various oncogenes implicated in MAPK signaling (red) pre-treatment and post-progression, with superimposed BRAFV600E cfDNA allele fraction (black). (B) Median nuclear: cytoplasmic ratios for ERK2 and FOXO3a as surrogate measures of MAPK and PI3K/Akt signaling for variants detected in plasma samples from patients at progression. Decreasing nuclear:cytoplasmic ratio for ERK2 and FOXO3a represents decreasing MAPK and increasing PI3K/Akt signaling, respectively.
To characterize the functional significance of the mutations identified from cfDNA profiling, we measured their oncogenic activity via microscopic quantification of nuclear transcription factor localization.
Of the mutations that increased in fraction following treatment, the ERBB2R552S variant/mutation was found to be active through the MAPK/ERK pathway (Figure 3B). Similarly, the PTENI122N mutation elicited significant activation of the MAPK/ERK pathway as well as the PI3K/AKT pathway, performing similarly to the known PTENR130Q mutation(19). The MEK1C121S and MEK1K57T alterations were found to elicit significant oncogenic activation of the MAPK/ERK pathway. Similarly, the EGFRL93I variant/mutation was profiled and found to elicit high oncogenic activation of MAPK/ERK pathway when compared to a known exon 21 driver mutation (EGFRL861Q).
Xenograft Studies
We examined two xenograft models of BRAFV600E mCRC – one derived from a patient at our institution (B1003) and the other from a vemurafenib-resistant cell line (RKO) – for sensitivity to the various agents used in this clinical trial. As seen in Figure 4A, the treatment of the patient-derived xenograft B1003 model with single-agent irinotecan or the combination of vemurafenib plus cetuximab led to a relatively static treatment response substantially different to the untreated control group. However, a regression in mean tumor volumes in the mice treated with the triple combination of irinotecan plus vemurafenib and cetuximab was seen at the end of the experiment when compared to those treated with irinotecan alone (P=.002) or the vemurafenib and cetuximab doublet (P<.0001). Similar findings were noted with the cell-line derived xenograft model of BRAFV600E mCRC, whereby the triple combination of irinotecan, cetuximab, and vemurafenib was associated with a reduction in mean tumor volume after 21 days not observed with the other treatments (Figure 4B). Waterfall plots (Figures 4C and 4D) for each the individual mice treated on this study demonstrates that all mice administered the triple combination therapy experienced reduction in their tumor volumes, whereas the majority of mice in both the single-agent irinotecan cohort, as well as the vemurafenib plus cetuximab group, had tumor growth by day 21.
Figure 4.
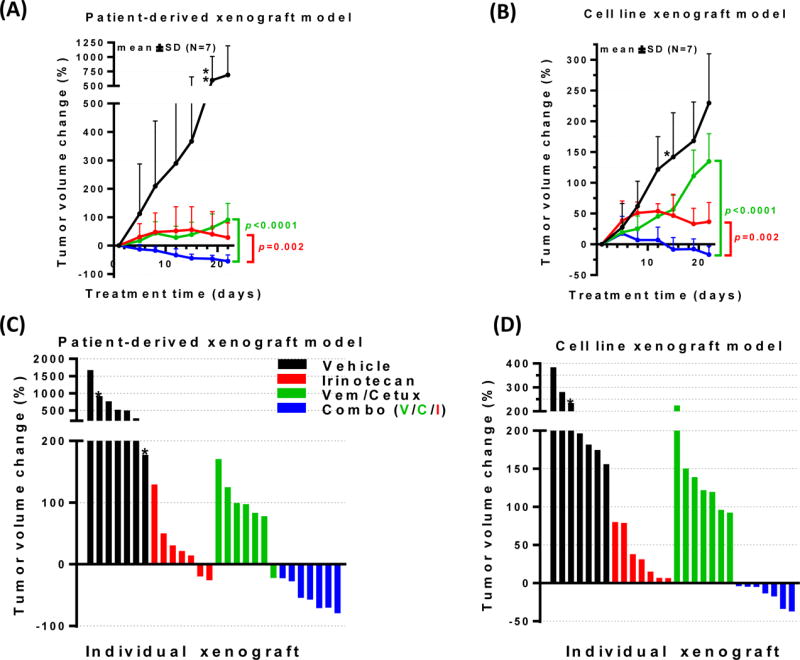
Xenograft studies using two BRAFV600E models of mCRC reveal reductions in mean tumor volumes (A, B) and in individual mice (C, D) treated with the triple combination of irinotecan plus vemurafenib and cetuximab relative to mice treated with irinotecan alone or the vemurafenib/cetuximab doublet.
Cell Proliferation Assays
We also tested two cell lines of MSI-H, BRAFV600E mCRC – RKO and B1003 (derived from the xenograft detailed above) – by treating with vemurafenib and cetuximab in combination with one of three additional third agents - SN-38 (active metabolite of irinotecan; Figures 5A and B), BYL719 (PI3K inhibitor; Figures 5C and 5D), or trametinib (Figures 5E and 5F). Based on the median effective doses (ED50), the addition of SN-38 to vemurafenib plus cetuximab resulted in a moderately synergistic cytotoxic effect, with ED50 values of .66 and .84 for B1003 and RKO, respectively. Synergy was also seen upon the addition of BYL719 (ED50 of .48 and .77 for B1003 and RKO, respectively) or trametinib (ED50 0.06 and 0.18, respectively) to vemurafenib and cetuximab.
Figure 5.
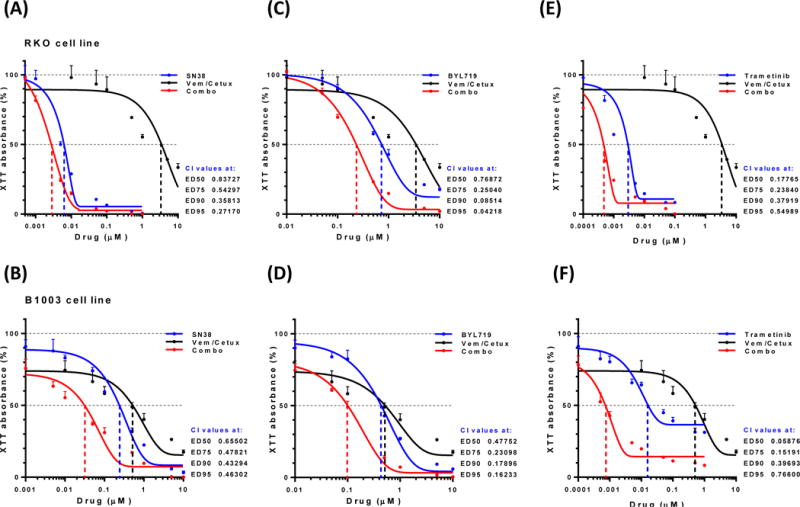
In vitro analyses of two BRAFV600E cell lines - RKO and B1003, respectively - demonstrated synergism (combination index [CI] < 1) when SN-38 (metabolite of irinotecan; A and B), BYL719 (PI3K inhibitor; C and D), and trametinib (MEK inhibitor; E and F) were all added to vemurafenib and cetuximab.
Discussion
Our current trial is the first in which irinotecan has been added to vemurafenib and cetuximab for patients with BRAF-mutated metastatic colorectal cancer. This triple combination had an acceptable toxicity profile. Three patients in our study experienced dose-limiting toxic effects, but only one, a patient with dose-limiting diarrhea, withdrew consent as a result. The other two patients, one with arthralgia who experienced resolution of symptoms with a reduction in the vemurafenib dose and one with diarrhea who experienced improvement in symptoms after reduction in the irinotecan dose, were able to remain on study and achieved partial responses.
The rates of grade 3 diarrhea (26%) and neutropenia (21%) across all cycles were similar to those in a prior second-line colorectal cancer study of cetuximab and irinotecan (28% and 32%, respectively). Notably, even with irinotecan treatment, only one case of grade 1 transaminitis was observed, and this was not associated with a rise in the alkaline phosphatase level. In contrast, in a previous study of vemurafenib and panitumumab in patients with BRAFV600E metastatic colorectal cancer, one patient had grade 4 transaminitis related to vemurafenib, and four had grade 3 elevations in alkaline phosphatase level (33% incidence of grade 3–4 hepatotoxicity)(15). Our findings showed fewer unacceptably severe toxic effects on the liver with this three-drug approach.
Durable disease control was noted in patients with pretreated BRAF-mutated metastatic colorectal cancer treated with this triple combination. Antitumor activity was observed at all dose levels of vemurafenib, and the median progression-free survival was 7.7 months. The clinical responses observed here suggest reason for optimism regarding improvement of outcomes for patients with BRAFV600E-mutated colorectal cancer. In multiple studies of irinotecan with cetuximab in patients with metastatic colorectal cancer who experienced disease progression with prior systemic chemotherapy(17, 20–22), median progression-free survival times of 3–4 months were estimated, although outcomes were not characterized by BRAF mutation status. Two other groups have published results of BRAF inhibitor combined with anti-EGFR therapy for patients with BRAF-mutant metastatic colorectal cancer. One cohort of 27 patients treated with vemurafenib and cetuximab noted a single partial response (4%) with a median progression-free survival of 3.7 months(16). In a study of 15 patients treated with vemurafenib and panitumumab(15), 2 of 12 evaluable patients (17%) had partial responses, and the median progression-free survival was only 3.2 months. Unlike our study, patients were excluded if they had received prior anti-EGFR therapy. In our study, seven patients had experienced disease progression on prior cetuximab therapy, and of these, one had a partial response and five had stable disease with vemurafenib, irinotecan, and cetuximab. We also observed stable disease in 9 evaluable patients (53%), which may provide additional insight into the longer median PFS in this cohort relative to prior reports. Our findings suggest that patients with BRAFV600E mutations may derive some clinical benefit from this triple combination despite disease progression on prior anti-EGFR therapy.
With the mixed pattern of effect from treatment observed here, next-generation sequencing was performed on pretreatment samples to identify other mutations present which could be associated with a favorable response to treatment. Prior preclinical work has implicated alterations in the PI3K/Akt pathway as a mechanism for innate resistance to BRAF inhibition for BRAFV600E mCRC(23). However, in this cohort, only one of 17 patients had a PIK3CA mutation, which limited further analysis of any possible association here. Similarly, only three patients harbored MSI-H tumors, and no association was seen with response.
Patients with radiographic responses to vemurafenib with irinotecan and cetuximab demonstrated declines in the percentage of BRAFV600E cfDNA before their first restaging scan, and the magnitudes of these changes were maintained at the time of first restaging. The six patients with stable disease with serial blood available for analysis had lower percent reductions in BRAFV600E cfDNA at the time of first restaging than their responder counterparts, and these trends appeared to last through the time of first restaging. Of interest, two patients with stable disease on study experienced near-resolution of mutant BRAF cfDNA both within two weeks of treatment initiation and also at the time of their first radiographic reassessment. Although the dramatic depth of response in cfDNA for these two patients was not associated with a radiographic partial response, both demonstrated some reduction in volume of their target lesions from baseline. For these two patients then, there appears to be no evidence of an early response followed by rapid adaptive resistance. The pattern of change in cfDNA frequency appears more heterogeneous in those patients with stable/progressive disease relative to the responders on treatment, all of whom demonstrated early, near-complete disappearance of mutant BRAFV600E cfDNA. However, the number of patients analyzed was small, and larger cohorts of patients are needed to investigate these findings further. Since the appearance of the BRAF mutation appears early in the pathogenesis of these tumors, these findings lend further support to the notion that identification of early truncal mutations may serve as reliable biomarkers which can be monitored as personalized, surrogate biomarkers for response through blood-based sequencing methods.
The cfDNA NGS analysis also provided important insights into possible mechanisms of resistance to the combination of irinotecan with vemurafenib and cetuximab. Three patients developed detectable MEK1 mutations at the time of radiographic progression. MEK1C121S has been reported in BRAFV600E-mutated metastatic melanoma with acquired resistance to BRAF inhibition by vemurafenib(24) and, in in vitro models of BRAFV600E melanoma, confers resistance to treatment with the MEK inhibitor selumetinib(24) but not to the combination of dual BRAF and MEK inhibition(25). MEK1C121S has not previously been reported in BRAFV600E-mutated metastatic colorectal cancer following treatment with combined BRAF and EGFR blockade. GNASR201C mutations were also noted in post-progression cfDNA at a higher allelic frequency relative to its paired pretreatment specimen. This mutation has been implicated in the promotion of continued cyclic AMP activity(26) and propagation of downstream signaling of both Wnt/beta-catenin and MAPK pathways(27), both important in CRC pathogenesis. This mutation has not previously been reported as a possible mechanism of acquired resistance to BRAF +/− EGFR targeted therapies in BRAFV600E mCRC, but given its known association with MAPK signaling in colorectal cancer, it is feasible that tumor cells could employ this oncogene as another mechanism to bypass the anti-tumor activity of these targeted drugs. Our findings are also consistent with findings in a prior series of three patients with BRAF-mutated metastatic colorectal cancer treated with a BRAF inhibitor plus a MEK inhibitor and/or anti-EGFR antibody, who were found to have focal amplifications of KRAS and BRAF and acquired mutations in ARAF and MEK1 in post-progression tumor biopsy specimens(28).
Multiple nucleotide variants were detected at the onset of tumor progression that were either amplified relative to baseline or undetected at baseline. Many of these (e.g., ERBB2R522S, ARAFS490T, EGFRL93I, PTENI122N) were associated with the presence of microsatellite instability. Given that higher mutation burdens are found in MSI-H mCRC(29), it was important to understand whether these mutations were deleterious or rather passenger mutations amid an otherwise hypermutated tumor. A functional assay of MAPK activity demonstrated a higher presence of nuclear ERK2 for MEK1C121S, MEK1K57T, ERBB2R522S, PTENI222N and EGFRL93I mutations compared to their wild-type counterparts. Nuclear accumulation of ERK2 is associated with increased activity of MAPK signaling, suggesting that these resistant tumors reactivated MAPK signaling despite the pressures of agents blocking BRAF and EGFR upstream. Other truncal mutations like TP53 were detected in baseline samples (Supplementary Figure 1) and changed in mutation allelic frequency in parallel with the dynamic changes noted in BRAFV600E. Given that many of these mutations predicted to be damaging from genes important in MAPK signaling were undetectable in baseline specimens yet appeared at progression, we surmise that they are not the result of mere tumor growth but rather the consequence of clonal selection.
Prior work in vemurafenib-sensitive xenograft models of mCRC has demonstrated KRAS and NRAS mutations can be detected at low allelic frequencies and can clonally expand upon acquired resistance to vemurafenib(12). In addition, mutations in these oncogenes have also been found in low levels of patients with mCRC that are naïve to exposure with BRAF inhibitors, and aberrations in additional oncogenes like MEK1, EGFR, and MET have all been implicated as mechanisms of acquired resistance to targeted therapies to BRAF inhibitors in melanoma and anti-EGFR therapies in mCRC. We found no evidence of acquired RAS mutations in post-progression samples of our patients, and we found no ectodermal mutations in EGFR hotspots associated with resistance to cetuximab. Amplifications in copy number of EGFR, MET, and KRAS were found together in one patient at progression. While some genetic alterations reported in our work here are consistent with other reports of mechanisms of resistance to targeted therapies against BRAF and EGFR, others are novel. Collectively, our findings suggest that colorectal tumors with a BRAFV600E mutation may rely upon continued propagation of MAPK signaling through a variety of mechanisms at the time of resistance to BRAF and EGFR blockade. We acknowledge the limitation that our analyses here are limited to genomic profiling and did not incorporate gene expression profiling to explore non-genomic mechanisms of resistance like dynamic changes in mRNA expression patterns under epigenetic regulation, which have been implicated to acquired resistance to BRAF inhibitors in melanoma(30). As BRAFV600E mCRC tumors are associated with CpG island hypermethylation(29), it is indeed plausible that epigenetic control influences response to targeted therapies to BRAF and/or EGFR in this subset of patients, even though we were unable to assess this in our present study.
The rationale for this triple combination is also supported by our preclinical evidence showing that the addition of irinotecan to vemurafenib and cetuximab reduces tumor size in xenograft models of BRAFV600E mCRC. The failure of vemurafenib and cetuximab alone to produce sustained reductions in tumor size in our xenograft models mirrors the low response rates noted with the previously described trials targeting BRAF and EGFR in this population. Further support for the cytotoxic benefit of adding irinotecan to the combination of therapies against BRAF and EGFR comes from preclinical xenograft studies of BRAFV600E-mutated metastatic colorectal cancer showing improved response rates and prolonged survival with the addition of irinotecan to vemurafenib and cetuximab(18). B1003 and RKO are both MSI-H models of BRAFV600E mCRC, and susceptibility to irinotecan has been reported in microsatellite unstable CRC cell line models previously relative to microsatellite-stable counterparts(31). Despite a potentially preferential bias towards susceptibility to irinotecan in MSI-H mCRC models, BRAFV600E mutations are nonetheless associated with microsatellite instability. Even though no association between response and MSI-H status was observed here, these findings thereby provide further rationale towards investigating the use of incorporation of irinotecan into a targeted therapy approach for larger series of patients with BRAFV600E mCRC.
We recognize that our findings are limited by the small sample size and by the fact that this study was conducted at a single institution. An expanded cohort at the recommended phase II dose for patients with BRAFV600E mCRC who have progressed on prior anti-EGFR therapy remains ongoing, and those findings will be reported separately. The SWOG 1406 study is an ongoing randomized phase II clinical trial with a completed enrollment of 78 patients with refractory BRAF-mutated metastatic colorectal cancer who will receive irinotecan and cetuximab with or without vemurafenib, and results are awaited. We examined multiple BRAFV600E mCRC cell line assays and found synergism with the addition of all three agents (SN-38, an active metabolite of irinotecan; BYL719, a PI3K inhibitor; and trametinib, a MEK inhibitor) to the BRAF plus EGFR inhibitor doublet. Collectively, this suggests multiple potential strategies that may improve on the clinical activity seen with BRAF plus EGFR inhibition. The findings from this clinical study suggest that this triplet regimen of vemurafenib with cetuximab and irinotecan has a satisfactory toxicity profile and may be effective for patients with BRAFV600E-mutated metastatic colorectal tumors, who are desperately in need of novel therapies.
Methods
Trial Design
This 3+3 phase I dose escalation trial was open to patients with advanced cancer, including metastatic colorectal cancer, with BRAFV600E mutation and KRAS wild-type at codons 12 and 13 as confirmed with tissue biopsy in a CLIA)-certified laboratory. For most patients, screening for BRAF and KRAS mutations was performed using next-generation sequencing at a minimum 250X, with additional profiling for genes including NRAS, PIK3CA, AKT, and TP53. Prior exposure to irinotecan and to anti-EGFR therapies was allowed, as was prior treatment with a BRAF inhibitor or MEK inhibitor. Additional eligibility criteria were Eastern Cooperative Oncology Group performance status of 0 to 2, normal organ and bone marrow function, and life expectancy greater than 3 months.
All patients received irinotecan 180 mg/m2 and cetuximab 500 mg/m2 intravenously on day 1 in 14-day cycles. These doses of irinotecan and cetuximab had previously been FDA approved and remained fixed unless toxic effects were clearly not attributable to vemurafenib, in which case reduction of the irinotecan or cetuximab dose was permitted. The vemurafenib dose was escalated over the course of the study according to a 3+3 design with the following predetermined dose levels: 480 mg twice daily (dose level 1), 720 mg twice daily (dose level 2), and 960 mg twice daily (dose level 3). Patients were evaluated clinically every 2 weeks, and response to treatment was assessed radiographically every 8 weeks. Patients remained on study until disease progression according to the Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1, unacceptable toxic effect, death, or withdrawal of informed consent. The National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0, were used to assess for dose-limiting toxic effects during the first 28 days and for new and ongoing adverse events until 30 days after the last dose of study treatment (additional details appear in the protocol in the Appendix).
The primary objectives of this study were to define the maximum tolerated dose of vemurafenib with irinotecan and cetuximab and to evaluate the safety profile of this combination. Secondary objectives included characterization of the mechanisms of acquired resistance, utility of measuring circulating cfDNA for tracking tumor response, and clinical response rate.
Oversight
This single-center trial was approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center and conducted in accordance with the Declaration of Helsinki. Every patient provided written informed consent before treatment initiation.
Statistical Analysis
Safety and efficacy data are reported for all patients who received at least one dose of vemurafenib with irinotecan and cetuximab. The data cutoff date was May 5, 2015. Median survival was estimated according to Kaplan-Meier analysis. Odds ratios were used to assess for an association between the presence/absence of a radiographic response and various clinical and pathologic features. This trial was registered in clinicaltrials.gov (identifier NCT01787500).
Translational Studies
Patients were asked for consent to participate in an optional laboratory component to measure mutant BRAF alleles as a proportion of total BRAF alleles in circulating cfDNA over time. Serial plasma samples were collected at screening, at each treatment date, and at progression. cfDNA was extracted using a Circulating Nucleic Acid kit (Qiagen, Valencia, CA). The presence of BRAFV600E mutant cfDNA in serially collected patient samples was assessed with the ddPCR BRAF screening kit (Bio-Rad Laboratories, Hercules, CA). The fractional concentration or mutant allele frequency for a given mutation was calculated as the fraction of circulating tumor DNA harboring that mutation in a background of wild-type cfDNA fragments. For the ten patients with serial plasma samples on which BRAFV600E mutant allele fraction was quantified with ddPCR, percentage changes from baseline levels were calculated prior to the second dose of therapy, and mean magnitudes of change were calculated and compared between the cohort patients with radiographic responses according to RECIST 1.1 criteria and those without partial responses (i.e., stable disease or progressive disease) using an independent t-test.
To compare pretreatment and post-progression mutational profiles, genomic alterations in cfDNA from the corresponding plasma samples were analyzed by NGS of a targeted 68-gene panel (Supplementary Figure 4) utilizing an Illumina Hi-Seq 2500 platform at a CLIA-certified laboratory(32) (Guardant Health, Redwood City, CA).
Xenograft Studies
Under an IACUC-approved laboratory protocol, a xenograft model of BRAFV600E mCRC was established from a biopsy of a patient treated at our institution under an IRB-approved protocol. Tumor was implanted into the subcutaneous flanks of 28 female, nude mice. Once tumor volumes reached a mean volume of 250 mm3, mice were randomized into four equal arms to receive a vehicle, irinotecan (TEVA Pharmaceuticals), vemurafenib (Plexxikon) plus cetuximab (BMS Lilly), or the triple combination of irinotecan plus vemurafenib and cetuximab. Vemurafenib was provided continuously as a chow at 417 mg/kg. Irinotecan (40 mg/kg) was administered intraperitoneally every 4 days, and cetuximab (16 mg/kg) was provided intraperitoneally twice weekly. Tumor volumes were measured twice weekly for a total of 21 days, at which point mice were sacrificed. A one-way ANOVA model was used to assess for differences between the four groups and to derive the pairwise comparison results.
Cell Proliferation Assays
Cell lines of BRAFV600E mCRC including RKO (obtained 2/2010 and resuscitated with no intermediary passages following receipt from frozen specimen) and B1003 (used as a primary cell culture from a xenograft tumor in 6/2016) were used to measure cellular proliferation with an XTT assay (Trevigen). RKO was authenticated by Short Tandem Repeat analysis for specific loci including CSF1PO, D13S317, D16S539, D5S818, D75820, TH01, and vWA from a stock solution received directly from ATCC,. B1003 was established as a primary cell culture directly from the xenograft tumor, and was therefore not authenticated. Cells were plated in 96-well plates at a density of 2,500/well for RKO cell line or 5,000 cells/well for B1003 cell lines in a volume of 100 μL. Twenty-four hours after cell plating, fresh growth media was exchanged containing single drugs or drug combinations in a total volume of 100 μl. The cetuximab concentration was 100 μg/mL. All treatments were performed in triplicates, and the average values were used in the analysis. Cells were assayed for proliferation 3 days after treatment. The graphs were prepared in GraphPad Prism software (Version 6.07) and represents nonlinear regressions (mean ± standard deviation). Combinational Indexes (CI) were calculated using CompuSyn software (Version 1.0).
Functional Studies
The functional significance of mutations in 4 genes (EGFRL93I, ERBB2R552S, MEK1C122S, PTENI122N) was analyzed using the oncogenic activity system at a CLIA-certified laboratory (NovellusDx, Jerusalem, Israel). Mutations were generated on a wild-type expression vector backbone. Then, the mutations and a specific signaling pathway reporter were transfected into a live-cell assay. The signaling pathway reporter here was a florescent-tagged signaling protein which translocates from the cytoplasm to the nucleus upon pathway activation. The live-cell assay was then scanned by a fluorescent microscope to detect reporter localization. The oncogenic activity of each pair of mutation-reporter was separately analyzed, and the NCR was determined. The NCR for each condition, with the standard error and calculated p-values, was calculated from three repeated experiments, and mutations were compared to the matched wild-type gene using a student’s t-test.
Supplementary Material
Statement of Significance.
Vemurafenib, in combination with irinotecan and cetuximab, was well tolerated in patients with refractory, BRAF-mutated mCRC, and both survival outcomes and response rates exceeded prior reports for vemurafenib and for irinotecan plus cetuximab in BRAFV600E mCRC. In vivo models demonstrated regressions with the triplet, in contrast to vemurafenib and cetuximab alone. Circulating cell-free DNA predicted radiographic response and identified mutations reactivating MAPK pathway upon progression.
Acknowledgments
We thank the Barbara Rattay Foundation for instrumental support to this research and to all patients who participated in the study.
DSH reports research support from Genentech. MJO declares research support from Roche and Bristol Meyer Squibb. VS declares research support from Roche and Genentech. AT declares grant and research support from Onyx, EMD Serono, Baxter, and Foundation Medicine. NS is an employee of Genentech and owns stock in Roche. RL is an employee at Guardant Health. FMB serves on the advisory board and is a paid consultant for Genentech and also received grant and research support from Genentech. SK is a consultant for Amgen, Bayer, Genentech, Array, MolecularMatch, and Taiho.
Grant Support: Research was funded by the NCI R01CA187238 (SK, DSH), R01CA172670 (SK), and R01CA184843 (SK).
Abbreviations
- cfDNA
circulating cell-free DNA
- CI
combination index
- CLIA
Clinical Laboratory Improvement Amendment
- ED50
median effective dose
- MAPK
mitogen activated protein kinase
- mCRC
metastatic colorectal cancer
- MSI-H
microsatellite-high
- NCR
nuclear: cytoplasmic ratio
- NGS
next-generation sequencing
- OR
odds ratio
Footnotes
Conflicts of Interests: All other authors declare no competing interests.
Contributions
DSH, VKM, BEO, and SK designed the study. DSH and SK supervised the conduct of the study. DSH, FJ, SF, MJO, SPP, VS, BK, AT, DF, IS, FMB, and SK recruited patients onto the study and provided relevant data. DSH, VKM, AVS, FJ, JB, HH, JA, and GT collected and assembled the data. VKM did the statistical analysis. DSH, VKM, AVS, FJ, HH, JA, GT, NS, RL, and SK analyzed and interpreted the data. DSH, VKM, and SK wrote the original report, and all authors revised the report and approved the final version.
References
- 1.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26(35):5705–12. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. The Lancet Oncology. 2010;11(8):753–62. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 4.Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117(20):4623–32. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29(15):2011–9. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- 6.Tol J, Nagtegaal ID, Punt CJ. BRAF mutation in metastatic colorectal cancer. The New England journal of medicine. 2009;361(1):98–9. doi: 10.1056/NEJMc0904160. [DOI] [PubMed] [Google Scholar]
- 7.Morris V, Overman MJ, Jiang ZQ, Garrett C, Agarwal S, Eng C, et al. Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clinical colorectal cancer. 2014;13(3):164–71. doi: 10.1016/j.clcc.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England journal of medicine. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim KB, Cabanillas ME, Lazar AJ, Williams MD, Sanders DL, Ilagan JL, et al. Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAF(V600E) mutation. Thyroid: official journal of the American Thyroid Association. 2013;23(10):1277–83. doi: 10.1089/thy.2013.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munoz J, Schlette E, Kurzrock R. Rapid response to vemurafenib in a heavily pretreated patient with hairy cell leukemia and a BRAF mutation. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31(20):e351–2. doi: 10.1200/JCO.2012.45.7739. [DOI] [PubMed] [Google Scholar]
- 12.Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J Clin Oncol. 2015 doi: 10.1200/JCO.2015.63.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–3. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 14.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer discovery. 2012;2(3):227–35. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yaeger R, Cercek A, O’Reilly EM, Reidy DL, Kemeny N, Wolinsky T, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21(6):1313–20. doi: 10.1158/1078-0432.CCR-14-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. The New England journal of medicine. 2015;373(8):726–36. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. The New England journal of medicine. 2004;351(4):337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 18.Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer research. 2012;72(3):779–89. doi: 10.1158/0008-5472.CAN-11-2941. [DOI] [PubMed] [Google Scholar]
- 19.Han SY, Kato H, Kato S, Suzuki T, Shibata H, Ishii S, et al. Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer research. 2000;60(12):3147–51. [PubMed] [Google Scholar]
- 20.Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W, Abubakr YA, Lutz MP, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26(14):2311–9. doi: 10.1200/JCO.2007.13.1193. [DOI] [PubMed] [Google Scholar]
- 21.Pfeiffer P, Nielsen D, Bjerregaard J, Qvortrup C, Yilmaz M, Jensen B. Biweekly cetuximab and irinotecan as third-line therapy in patients with advanced colorectal cancer after failure to irinotecan, oxaliplatin and 5-fluorouracil. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2008;19(6):1141–5. doi: 10.1093/annonc/mdn020. [DOI] [PubMed] [Google Scholar]
- 22.Martin-Martorell P, Rosello S, Rodriguez-Braun E, Chirivella I, Bosch A, Cervantes A. Biweekly cetuximab and irinotecan in advanced colorectal cancer patients progressing after at least one previous line of chemotherapy: results of a phase II single institution trial. British journal of cancer. 2008;99(3):455–8. doi: 10.1038/sj.bjc.6604530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R, Jr, Dayyani F, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(3):657–67. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29(22):3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nature communications. 2014;5:5694. doi: 10.1038/ncomms6694. [DOI] [PubMed] [Google Scholar]
- 26.Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340(6236):692–6. doi: 10.1038/340692a0. [DOI] [PubMed] [Google Scholar]
- 27.Wilson CH, McIntyre RE, Arends MJ, Adams DJ. The activating mutation R201C in GNAS promotes intestinal tumourigenesis in Apc(Min/+) mice through activation of Wnt and ERK1/2 MAPK pathways. Oncogene. 2010;29(32):4567–75. doi: 10.1038/onc.2010.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahronian LG, Sennott EM, Van Allen EM, Wagle N, Kwak EL, Faris JE, et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer discovery. 2015;5(4):358–67. doi: 10.1158/2159-8290.CD-14-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell. 2015;162(6):1271–85. doi: 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vilar E, Scaltriti M, Balmana J, Saura C, Guzman M, Arribas J, et al. Microsatellite instability due to hMLH1 deficiency is associated with increased cytotoxicity to irinotecan in human colorectal cancer cell lines. British journal of cancer. 2008;99(10):1607–12. doi: 10.1038/sj.bjc.6604691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lanman RB, Mortimer SA, Zill OA, Sebisanovic D, Lopez R, Blau S, et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PloS one. 2015;10(10):e0140712. doi: 10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.