

Abstract
Human babesiosis caused by Babesia microti is an emerging tick-borne zoonosis of increasing importance due to rising incidence and expanding geographic range1. Infection with this organism, an intraerythrocytic parasite of the phylum Apicomplexa, causes a febrile syndrome similar to malaria2. Relapsing disease is common among immunocompromised and asplenic individuals3,4, and drug resistance has recently been reported5. To investigate the origin and genetic diversity of this parasite, we sequenced the complete genomes of 42 B. microti samples from around the world, including deep coverage of clinical infections at endemic sites in the continental United States. Samples from the continental US segregate into a Northeast lineage and a Midwest lineage, with subsequent divergence of subpopulations along geographic lines. We identify parasite variants that associate with relapsing disease, including amino acid substitutions in the atovaquone-binding regions of cytochrome b (cytb) and the azithromycin-binding region of ribosomal protein subunit L4 (rpl4). Our results shed light on the origin, diversity, and evolution of B. microti, suggest possible mechanisms for clinical relapse, and create the foundation for further research on this emerging pathogen.
Human babesiosis is a tick-borne zoonosis caused by piroplasms of the genus Babesia (Apicomplexa: Piroplasmida)1. In the US, the vast majority of cases are due to B. microti, a parasite transmitted by Ixodes scapularis ticks and enzootic in the white-footed mouse, Peromyscus leucopus, and other small mammals6. Human infection occurs predominantly through the bite of an infected tick but can also occur via transfusion of infected blood products7 or trans-placental passage8. Disease is characterized by fever and hemolysis, which, as in malaria, is thought to be a driving pathogenic process9. Infections range from asymptomatic to fulminant and can be complicated by respiratory failure, disseminated intravascular coagulation, or organ failure10. In historical series, death occurs in 3-5% of hospitalized patients11,12 and up to 20% of immunocompromised individuals3. In this latter population, prolonged parasitemia and relapsing disease are common3, and resistance to first-line atovaquone/azithromycin therapy has recently been described5.
Human infection with B. microti is reported in Europe13, Asia14, and Australia15, but most cases occur in the US, where there are foci in the Northeast (NE) and upper Midwest (MW)1. Since the first report of B. microti in the US on Nantucket in 196916, the geographic range17,18 and incidence18,19 have been increasing, and the CDC now classifies human babesiosis as an emerging and nationally notifiable disease.
In order to gain insight into the evolution, geographic expansion, and drug resistance of B. microti, we sequenced the complete genomes of 42 samples, including 32 human cases, two samples from ticks, and four from rodents (Figure 1a–b, Supplemental Table 1). We sequenced the 6.4Mb genome to a mean depth of 153-fold on Illumina HiSeq instruments using a paired-end protocol. Among the 39 B. microti sensu stricto samples (BMSS – see Supplement for a glossary of geographic terms), we identified a total of 114,336 SNPs, approximately 1 per 56 bases (Methods).
Figure 1. Phylogeny of Global B. microti.
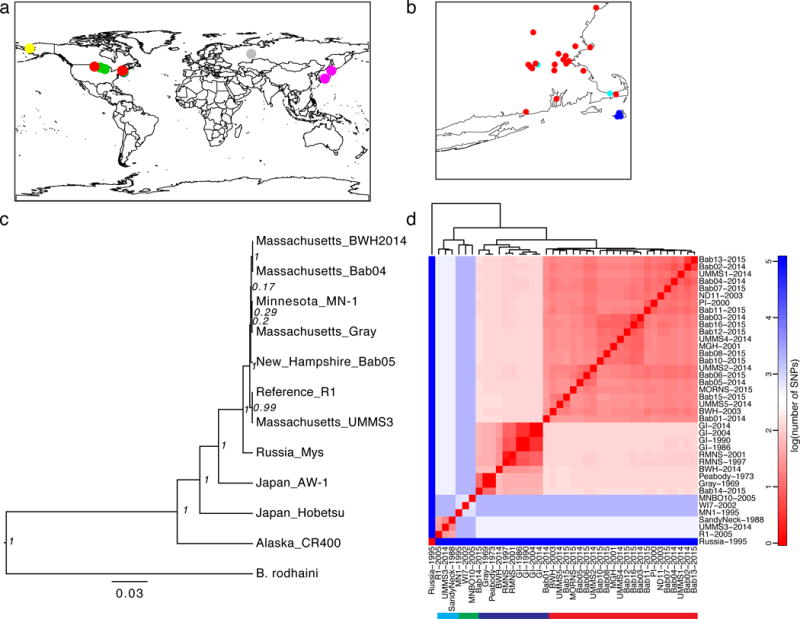
A) Sites of sample origin (Supplemental Table 1), colored by lineage (red: MNE; blue: NAN; green: MW; cyan: REF; or individually for global samples, gray: Russia; pink: Japan; yellow: Alaska). B) Inset of northeastern United States. C) Maximum credibility tree of concatenated COX1-CYTB-COX3 mitochondrial protein sequences with node posterior support. D) Heatmap of the number of SNPs across nuclear chromosomes, mitochondrial, and apicoplast sequences separating BMSS samples on a logarithmic scale (base 10); solid bar underneath denotes lineages, with colors as in A).
We first examined the origin of B. microti. We established a rooted phylogeny for our cohort using concatenated COX1-CYTB-COX3 mitochondrial protein sequences with B. rodhaini as an outgroup (Figure 1c). This analysis confirmed the extensive worldwide diversity and polyphyletic nature of B. microti in the US, with the Alaskan sample separated from the continental US (CUS) lineages with Russian and Japanese samples intervening. Within BMSS, the Russian sample is distinct from, and basal to, samples from CUS.
We next used the genomic data to establish the phylogeny of CUS strains with the Russian sample as the outgroup. This revealed a structured CUS epidemic with deep divergence between NE and MW samples (Figure 1d, Supplemental Figure 3). In the NE group, there were three sub-populations (Figure 1d), labeled here as NAN (Nantucket), MNE (mainland New England) and REF (R1 reference group). The MNE lineage in our dataset typed with the Connecticut/Rhode Island lineage reported previously20. A single sample from North Dakota grouped with the MNE strains. As the travel history was not available for this patient, we cannot establish whether this was an imported or autochthonous case. We identified one case of probable locally imported babesiosis from Nantucket to South Dennis, MA (see Supplement).
NE populations had minimal nucleotide diversity (π = 3.1×10−6 – 6.1×10−6) and strong population differentiation (FST 0.94 – 0.96; Table 1; Supplemental Figure 4a–b), indicating small, geographically isolated, and inbred populations. Most of the variation separating the MNE lineage from the NAN lineage was found in two segments of chromosome 2, segments in which both lineages also share an unusually large amount of variation with MW strains (see Supplement). We found evidence of recombination among CUS as a set, but not within individual NE lineages (Supplemental Figure 5). The MNE lineage demonstrated evidence of recent population expansion, with strongly negative values throughout the genome for Tajima’s D and other statistics of neutral evolution (Table 1 and Fig. 2b), consistent with epidemiological data18. We did not detect a significant relationship between D and π (Supplemental Figure 4c–d). Longitudinal sampling of MNE and NAN samples demonstrated evidence of an empirical molecular clock (Supplemental Figure 6a–b). Using BEAST21, we estimated the time to most recent common ancestry (TMRCA) for CUS lineages (Figure 3). MNE, REF, NAN all emerged within the last 600 years, while TMRCA for CUS was much earlier, between 1,350 and 14,700 years (95% highest posterior density (HPD), median 5,043 years), under the best-supported UCED model (see Supplement).
Table 1. Population Genetic Summary Statistics.
π, pairwise nucleotide diversity; Fst, Wright’s fixation index (calculated from π, and compared to all others); N = the number of samples in each population.
| Population | π | FST | N | Tajima’s D | Fu and Li’s F | Fu and Li’s D |
|---|---|---|---|---|---|---|
| MNE | 2.57E-06 | 0.96 | 23 | −2.2 | −3.92 | −3.73 |
| NAN | 6.04E-06 | 0.96 | 5 | 0.44 | 0.39 | 0.28 |
| REF | 3.03E-06 | 0.95 | 3 | NA | 1.44 | −1.44 |
| MW | 1.10E-04 | 0.96 | 3 | NA | 0.48 | 0.48 |
Figure 2. Genome-Wide Population Genetic Summary Statistics.
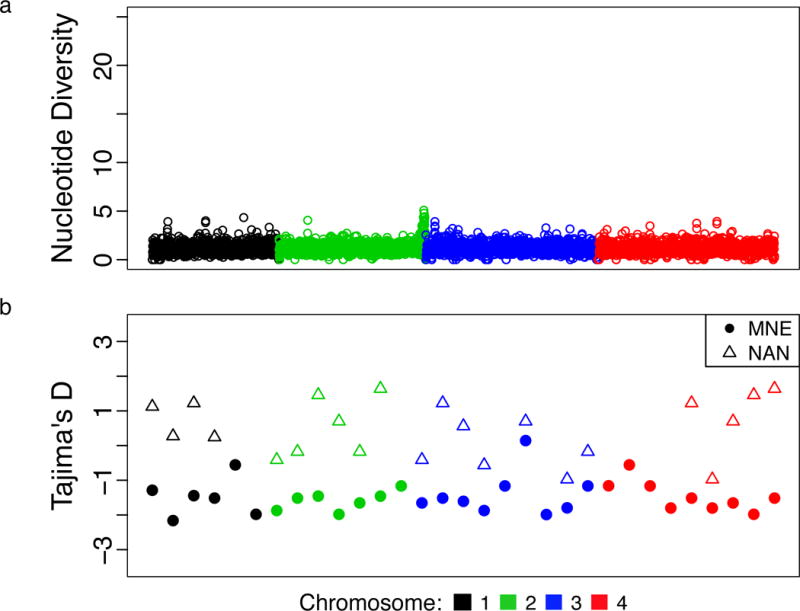
A) Sliding window analysis of pairwise nucleotide diversity (π) in 1 kilobase (Kb) for the set of 36 BMSS samples. B) Tajima’s D statistic in 200Kb windows for MNE and NAN (the lineages for which the number of isolates was sufficient to test neutrality).
Figure 3. Time to Most Recent Common Ancestry for Continental US Samples.
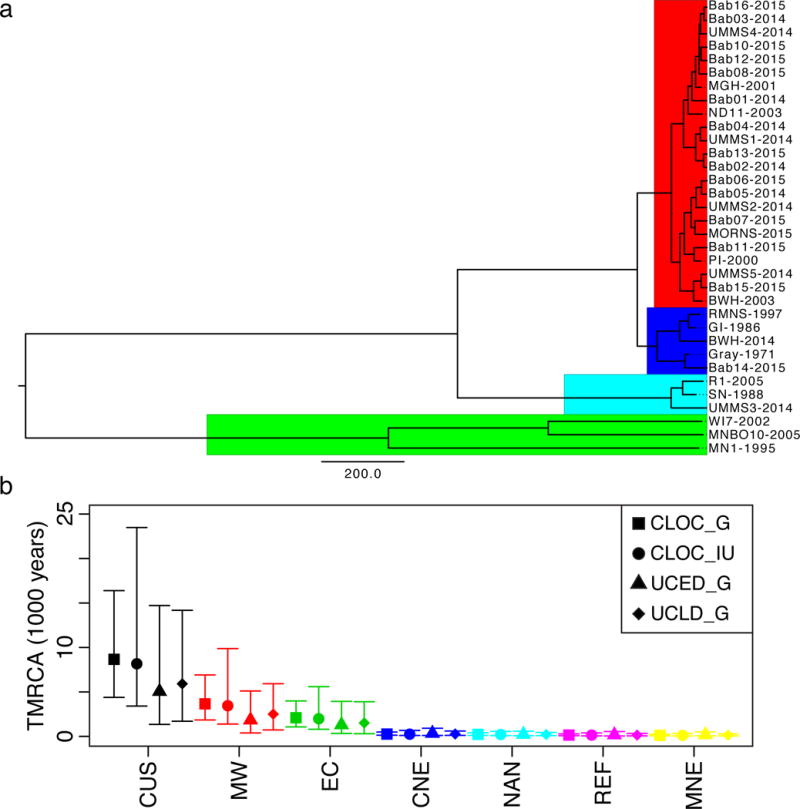
A) Maximum credibility tree colored by lineage (Red: MNE; Blue: NAN; Cyan: REF; Green: MW). Scale bar is years (under the UCED_G model). B) Median and 95% HPD intervals for TMRCA for CUS samples (black) and subpopulations (MW – red, EC – green, CNE – blue, NAN – cyan, Ref – purple, MNE – yellow) for a strict clock with infinite uniform (CLOC_IU, circles) or gamma prior (CLOC_G, squares) and a relaxed clock with gamma prior (uncorrelated exponential – UCED_G, triangles; uncorrelated lognormal – UCLD_G, diamonds).
We next examined evidence for genome evolution. The density of variants was uniform throughout the genome (Fig. 2a), except in subtelomeric and select intrachromosomal regions, where numerous substitutions were detected (Supplemental Figure 7a, Supplemental Table 3). In this respect, the structure of B. microti genomic diversity resembles that of many other parasites and bacteria (Supplemental Note). We calculated dN/dS ratios for all protein coding genes between Russian and US B. microti (Supplemental Figure 8, Supplemental Table 4). Most proteins demonstrated strong evidence of purifying selection (mean = 0.21 +/− 0.41), particularly those encoded by the mitochondrial genome (adjusted P = 8.8×10−6, 1.6×10−3, 1.2×10−2 for cox1, cytochrome b (cytb), cox3 respectively, Methods). We found significant positive selection in only a single gene, BBM_I00435 (adjusted P = 0.036), of unknown function (Supplemental Table 4). Among four additional hits with adjusted P values < 0.1, two, BBM_I03535 and BBM_I00004, were BMN genes, a sero-reactive gene family located at chromosome ends and hypothesized to play a role in immune evasion22. As a family, BMN genes showed evidence of positive selection (P = 1.9 × 10−6; Wilcoxon Rank Sum Test) and an elevated substitution rate (Supplemental Figure 8e–f).
Our cohort included five cases of relapsing babesiosis (Bab05, Bab14, MGH20014, BWH2003, and MORNS2015), studied out of concern that they harbored a resistant phenotype after failing drug treatment. To search for genes that may be involved in relapse, we first inspected the non-synonymous variants that separated each relapsing case from its nearest, non-relapsing neighbor (Supplemental Table 7), a small set due to the limited genetic diversity within a given lineage. All five cases contained non-synonymous variants in cytochrome B, and three out of five contained variants in rpl4, exceeding the number expected due to chance (adjusted P = 3.9 ×10−13 for cytb, 1.3×10−4 for rpl4; Figure 4a, Methods). Zero variants were observed in non-relapsing cases from lineage-matched (MNE and NAN) controls (adjusted P = 0.01 for cytb, Fisher’s exact test; adjusted P = 1 for rpl4; unadjusted P = 5.6 × 10−3, the second-strongest signal of association after cytb, Supplemental Figure 9).
Figure 4.
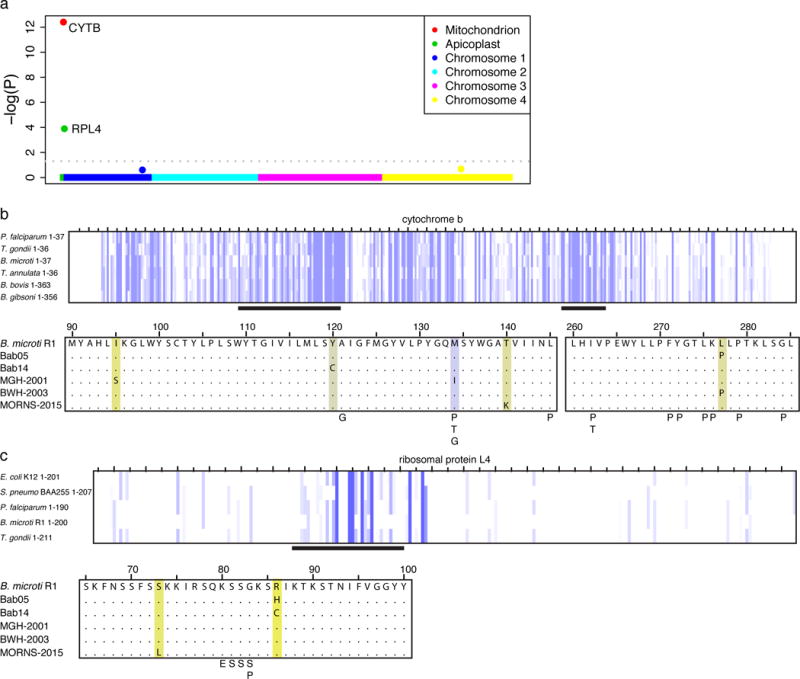
A) Genome-wide P-values for increased rate of non-synonymous variants among relapsing cases. B) CYTB mutations identified in relapsing cases of human B. microti babesiosis are shown in a multiple sequence alignment with the B. microti reference. Highlighting ranges from purple to yellow, indicating greater to less conservation based on amino acid identity and physico-chemical properties. Mutations associated with atovaquone resistance in other Apicomplexa (Plasmodium spp26 - P, Toxoplasma gondii29 - T, and Babesia gibsoni25 – G, see supplement) are also indicated. C) Variants in RPL4 associated with azithromycin resistance in B. microti are shown as in B; also shown are variants associated with azithromycin resistance in P. falciparum – P26, S. pneumoniae30 – S, and E. coli - E28).
Cytochrome b is the known target of atovaquone in other Apicomplexa (Figure 4b)23. CYTB is a highly conserved mitochondrial protein under strong purifying selection (adjusted P = 1.6×10−3). The identified amino acid changes occur in the atovaquone-binding regions of cytochrome B and resemble atovaquone resistance mutations observed in other parasites (Figure 4b, Supplemental Figure 10). The M134I substitution observed in MGH2001 has been studied in the P. falciparum CYTB ortholog and results in a 25-fold decrease in susceptibility to atovaquone in P. falciparum24 (Supplemental Table 8). This variant has also been associated with relapsing canine babesiosis due to B. gibsoni25. The L277P variant was present in two relapsing cases (Figure 4b and Supplemental Table 7), despite its absence in phylogenetically intermediate samples, suggesting adaptive evolution. Together, these findings suggest that mutations in cytb may promote relapse by decreasing atovaquone binding.
We next considered mutations in rpl4, a subunit of the 50S ribosome encoded in the apicoplast genome. Two relapsing cases contained substitutions in the same codon, arginine 86 (R86H in Bab05 and R86C in Bab14), which is highly conserved across multiple bacteria and parasites (Figure 4c). One of these cases, Bab05, presented with a severe relapse while on azithromycin monotherapy (Supplemental note, Supplemental Figure 9c), indicating high-level clinical azithromycin resistance. The mutated arginine is three residues C-terminal to G83, the codon that mediates azithromycin resistance in P. falciparum (Figure 4c)26 and adjacent to the azithromycin binding pocket of the 50S ribosomal pore, the site of azithromycin resistance in multiple species of bacteria (Supplemental Figure 11, Supplemental Table 8)27,28. A third relapsing case (MORNS-2015) was found to have an S73L substitution, and during manual reexamination of the locus, a fourth relapsing case (BWH2003) was also found to contain a C103Y variant in 28/74 reads. These results suggest that mutations in rpl4 may promote relapse by diminishing azithromycin susceptibility.
In summary, we have characterized the emergence of B. microti in the US and identified variants in cytb and rpl4 that associate with relapsing babesiosis. CUS strains show strong geographic structure, possess minimal diversity as a group, and differ greatly from their nearest neighbor, a Russian strain. North American B. microti is polyphyletic, suggesting at least two introductions into the subcontinent. The rarity of interbreeding and timing of divergence are consistent with proposed models of allopatric speciation due to glaciation6. The genes under positive selection between US and Russian B. microti are candidates for investigating how this parasite adapts to new environments or acquires virulence. Despite considerable evolutionary distance, B. microti appears to share principles of genomic organization and mechanisms of drug resistance with other parasitic protozoa. These commonalities may prove useful in understanding mechanisms of virulence and in developing new therapies against this class of pathogens.
Methods
Short Read Alignment and Variant Detection
We aligned short reads to the Babesia microti R1 reference genome using bwa mem31. The original R1 reference contained chromosome 3 as a supercontig of chromosomes 3 and 4. We obtained the revised versions of chromosome 3 (GI: 908660426) and 4 (GI:908661396) from Genbank, which we incorporated into a new reference genome, along with the annotated apicoplast sequence (GI:658131431) and isoform I of the mitochondrion (GI:908661431). Variants were called using the UnifiedGenotyper tool from the Genome Analysis ToolKit (GATK)32 with best practices and with the ploidy set equal to 1 and a minimum quality score of 50 for single nucleotide polymorphisms (SNPs). To establish a set of high-confidence variants, we filtered the identified variants to require each variant to be supported by > 70% of the reads aligning at that position using a custom python script. We considered variants as present or absent. This assumes clonality or near-clonality of infection. We tested this assumption by examining the distribution of the proportion of reads supporting the alternate allele (Supplemental Figure 1g–h), which was close to or equal to 1 in nearly all cases. The number of variants in each gene was counted using R and functions from the Biostrings package33,34. P-values for substation rate were calculated using a one-sample, two-tailed Z test of location for each gene. Short reads for all samples sequenced in this study are freely available in the European Nucleotide Archive under accession code PRJEB13263.
Population Genetic Analysis
We used R to calculate population genetic summary statistics. We performed principal component analysis using the MDSscale() function. We used the PopGenome package35 to calculate nucleotide diversity, Wright’s fixation index, neutrality statistics, and to compute population genetic summary statistics in sliding windows. Geographic data were plotted in R using the package rworldmap36. Serial replicates of strains propagated in rodent models (later timepoints of GI and RMNS) were excluded from population genomic studies, calculation of population summary statistics, and BEAST analysis (as described below), as were identical strains felt to represent possible stock contamination (Gray and Peabody) (Table 1). dN/dS ratios were calculated using the kaks() function in SeqinR37 P-values for dN/dS ratios were calculated for each gene from the standard normal with test statistic Z = (dN – dS)/sqrt(var(dS) + var(dN)) and a two-tailed test.
Phylogenetic analysis
For mitochondrial protein sequence analysis, we downloaded the Genbank sequences of B. rodhaini (AB624357.1) and extracted the mitochondrial sequences from the mitochondrial proteins of B. microti sequenced in this analysis. For global B. microti isolates, we obtained this sequence from translated mitochondrial fragments (as identified by Promer, as below) using Artemis38. For continental US isolates, given minimal sequence variability, we called variants as above and then, using Artemis, extracted mitochondrial gene sequence based on the R1 annotation coordinates. Concatenated COX1, CYTB, and COX3 protein sequences were aligned using MUSCLE39 and then analyzed in BEAST v1.8.121. Alignments were visualized in JalView40, which was also used to color residues by physico-chemical properties. For whole genome phylogenetic inference, after calling variants as described above, we created an alignment using the FastaAlternateReferenceMaker from the GATK. Sites at which we lacked coverage to make a definitive call in all samples were considered invariant and defaulted to the reference. For all samples, greater than 99% of the genome was covered by 2 or more reads (supplemental figure 2b). We excluded serial replicates of strains cultivated in rodent models (i.e. GI and RMNS). Root-to-tip analysis was performed in Path-O-Gen (http://tree.bio.ed.ac.uk/software/pathogen/) using the best-fit root on a maximum-likelihood tree inferred in RAxML version 841, with sample collection dates for the tip dates. Given unusual ancestry of portions chromosomes 2 (see below), analysis in Path-O-Gen and BEAST was performed on alignments with chromosome 2 excluded. For tip dates, we used the date of genomic DNA isolation, except for the Gray and Peabody strains, for which we used date of collection (1969 and 1973) as these were obtained from ATCC and minimally propagated in our hands. These genomes were identical and possibly representation contamination of one stock with another. We treated them as a single strain with a date of 1971 and uncertainty of +/- 2 years in the BEAST and Path-O-Gen analysis. The reference isolate was assigned a tip date of 2005 +/- 5 years as the date of collection was not available in reference42. Regression models were fit in R. The geographic location of samples, when available, was assigned from the location of collection (ticks and historical laboratory isolates), or town of residence for patients who enrolled in the consented protocol (see ‘Collection of Clinical Samples’). For discarded clinical specimens, the location of hospital of collection is labeled in Figure 1b.
Testing for variants associated with relapse
In order to construct Supplemental Table 7, the nearest neighbor of each relapsing case was first determined by minimum p-distance. The number of non-synonymous variants was identified by calculating string distance of the translated genomic sequence to that of the nearest neighbor using Biostrings33 in Bioconductor34. To test whether genes demonstrated excess substitution, we calculated a background rate of non-synonymous substitution for non-synonymous sites among nearest neighbors by counting the number of pairwise non-synonymous variants between nearest neighbors for relapsing as well as non-relapsing cases within MNE and Nantucket lineages. In the set of pairwise comparisons, for all samples i != j, if a given sample j was the nearest neighbor for more one sample i, only the first comparison was included in order to avoid redundant comparisons (results without this correction were essentially unchanged: using all pairwise comparisons, adjusted P = 2.04×10−13 for cytb and 9.3 ×10−5 for rpl4). The mean number of non-synonymous variants was then divided by the total number of non-synonymous sites in the B. microti genome, counted using the method of Nei and Gojobori43. We then tested whether the rate for each gene was equal to this background rate under a Poisson model. Apicoplast proteins appeared to accumulate non-synonymous variants at an increased rate (Supplemental Figure S8, p = 9.8 × 10−10, Wilcoxon rank-sum test), with a mean of 2.0-fold the number of non-synonymous variants as compared to nuclear genes (Supplemental Figure S8). We therefore adjusted the background rate by a factor of 2.0 for apicoplast genes in these tests. Mitochondrial variants accumulated variation at a reduced rate (mean 0.66 fold), but this was not significant (p = 0.44, Wilcoxon rank-sum test), and we therefore did not adjust the background rate for mitochondrial comparisons. UMMS1-5, SN-1988, and PI-2000 had minimal coverage of apicoplast sequences and were excluded from this analysis. We also applied Fisher’s exact test to the number of relapsing and lineage-matched non-relapsing cases with non-synonymous mutations identified. For all approaches, P-values were adjusted for multiple comparisons using the method of Benjamini and Hochberg44.
Structural Modeling of Mutations
The S. cerevisiae cytochrome bc1 complex (PDB: 4PD4)45 and T. thermophilus ribosomal protein L446 (PDB: 4V7Y, bundle 2) crystal structure coordinates were acquired from the Protein Data Bank (http://www.rcsb.org) and viewed using Mac Pymol (Schrodinger LLC). Residues were mutated as indicated in the figure legends using the Pymol Mutagenesis wizard and selecting for rotamers with the highest likelihood of occurrence based on backbone-restrained rotamer probabilities.
Whole Genome Assembly
We used ABySS47 with k set to 60 to perform de novo assemblies of B. microti sensu lato isolates from Alaska and Japan. We aligned the resulting contigs to the Babesia microti reference genome using Promer as implemented in MUMmer3.2348. Draft assemblies are available in supplementary files.
Code Availability
Computer code used to generate results is included as a supplemental file.
Collection of Clinical Samples
We identified patients through the Massachusetts General Hospital (MGH) microbiology laboratory and at University of Massachusetts Medical School (UMMS). At MGH and Brigham and Women’s Hospital (BWH), we obtained informed consent according to Partners IRB protocol 2014P000948 and collected a venous blood sample of 5-10mL in a heparin tube at the time of enrollment, storing it at 4°C until for less than 3 weeks. Patients with a positive Babesia smear or B. microti PCR test were eligible for the study. Exclusion criteria included pregnancy, age under 18, and specific vulnerable populations (medical students and prisoners). We extracted DNA from infected red blood cells after cellulose filtration as described below. In some cases, in which patients were hospitalized briefly or we were unable to approach for participation in this study, we used parasite-only material as IRB-exempted de-identified discarded specimens, but only in cases where we were unable to contact the patient. At UMMS, IRB-exempted de-identified discarded peripheral blood samples were obtained from the hospital clinical laboratory. Historical laboratory-adapted strains and tick samples were obtained from Tufts School of Veterinary medicine. Serial isolates of the GI strain were grown continuously with recurrent tick and rodent passage from 1986 to 2014 (supplemental table 1); The RMNS strain was maintained in a similar manner but frozen intermittently for brief periods between 1997 and 2001. Historical samples were obtained from The Mayo Clinic as discarded, de-identified specimens.
Isolation of B. microti-infected RBC
For patient samples obtained at MGH that underwent cellulose enrichment, we filtered 1-5mL of venous blood through a filter consisting of a 5 cm cellulose (Sigma P/N# C6288-100G) column in a 10mL syringe. The filtrate contained infected and uninfected red blood cells, whereas human leukocytes were retained in the column. Giemsa stain of the flow-through confirmed leukocyte depletion. Historical strains of B. microti were propagated in adult Golden 40-50 gram (3-4 week) Syrian hamsters (Mesocricetus auratus) from Charles River Laboratories. For these strains and some patient samples collected as described above, we directly sequenced DNA extracted from whole blood without leukocyte depletion. All animal work was done under approved protocols from the Laboratory of Animal Medicine Services at Tufts School of Veterinary Medicine. Patient samples had parasitemia between 0.1 – 20%. For these, cellulose filtration or hybrid selection enriched the ratio of parasite to host nucleic acid, yielding an average enrichment of 4.9-fold (Supplemental Fig. 1a).
Genomic DNA Preparation
We used Qiagen DNEasy kit (Qiagen P/N#69504) to extract genomic DNA. We added a volume of 200 microliters of whole blood to each reaction per manufacturer instructions and pooled DNA from 2-4 extractions to generate enough material for sequencing.
Sequencing
We performed DNA sequencing at the Broad Institute and UMMS on an Illumina HiSeq instruments.
Sequencing
For libraries sequenced at the Broad Institute, we sheared 100 nanograms – 1 micrograms purified genomic DNA on a Covaris S220 instrument to a mean fragment size of 400 base pairs. The total shearing time was 1 minute 12 seconds made up of 6 cycles of 9 seconds each. Peak power was 140.0, Duty Factor 10.0, Cycle/Burst 200. Samples were held at ~6 degrees C. We then performed library construction on an IntegenX Apollo 324 robot according to the PrepX 320 base pair library preparation. During library construction, we added 6bp NEXTFlex adapters (BioO Scientific P/N#514102) so that up to 12 samples could be sequenced per lane. For quality control, quantification, and sizing, we ran completed library material on an Agilent Bioanalyzer with a High Sensitivity DNA kit (Agilent P/N#5067-4627). Libraries were pooled at a minimum concentration of 2nM and sequenced on HiSeq instruments. When library concentration was inadequate for sequencing, we amplified finished libraries with 6-8 cycles of PCR using an NEB polymerase (NEBNext High-Fidelity 2x PCR Master Mix P/N#M0541S and NEXTFlex DNA-PCR Primer Mix.)
For libraries sequenced at UMMS, we sheared 100 nanograms – 1 micrograms purified genomic DNA on a Covaris S2 instrument to a mean fragment size of 300 base pairs. We then performed library construction according to standard TruSeq protocols, with the substitution of indexed NEXTFlex adapters.
Hybrid Selection
1. Agilent SureSelect
We developed a custom Agilent SureSelect developer hybrid selection library containing sequencings for known I. scapularis pathogens, including Babesia microti, Borrelia burgdorferi, Borrelia miyamotoi, Anaplasma phagocytophilum, and Powassan Virus. The nucleotide sequences provided to Agilent for fabrication of the SureSelect library are included as a supplemental file. We performed hybrid selection according to manufacturer protocol with 6-8 cycles of amplification prior to hybrid selection and 10-12 cycles of amplification afterward, according to the protocol above. Libraries were prepared on the Apollo as above.
2. Whole genome bait (WBG)
2 milliliters of whole blood was collected over sodium heparin from B. microti-infected rag-deficient C57BL/6J laboratory mice housed at Tufts Medical Center. At UMMS, blood was then subjected to centrifugation with Lympholyte-M (Cedarlane). Genomic DNA was extracted from erythrocyte-enriched fractions using the QIAmp DNA Mini Kit (Qiagen). 100 nanograms of this DNA were amplified using the Genomiphi V3 kit (GE). 5 micrograms of whole genome-amplified product were used as input to prepare DNA templates for biotinylated RNA baits according to a protocol modified from Melnikov et al.49 Input DNA was sheared for 280 s on a Covaris S2 instrument set to duty factor 10%, intensity 5, 200 cycles per burst to obtain a fragment size distribution with a mode of ~150 base pairs. End repair, 3' adenylation, and adapter ligation were performed according to the Illumina genomic DNA sample preparation kit protocol, with the exceptions of using adapter oligonucleotides (IDT) specified by Melnikov et al.49 rather than Illumina kit adapters and cleaning reactions after each step using 1.8X volume AMPure XP beads (Beckman Coulter). Cleaned ligation products were amplified by 8-14 cycles PCR in Q5 HF buffer (NEB) using forward and reverse PCR primers (IDT) from Melnikov et al.49 Initial denaturation was 30 seconds at 98°C, and each cycle was 10 seconds at 98°C, 30 seconds at 55°C, and 30 seconds at 72°C. PCR products were size-selected on a 1.5% agarose gel followed by MinElute gel extraction (Qiagen). Gel-purified PCR products were re-amplified as above using the T7 promoter-containing forward primer (IDT) from Melnikov et al.49 300 nanograms of AMPure XP-cleaned PCR product was used as template to prepare biotinylated RNA baits in a 25 microliters MEGAscript T7 transcription (Ambion) containing 7.5 mM ATP, 7.5 mM CTP, 7.5 mM GTP, 5.625 mM UTP, and 1.875 mM Biotin-16-UTP (Roche). After 18 hours at 37°C, Turbo DNase (Ambion) was added to the reaction to remove DNA template. Unincorporated nucleotides were removed by cleanup with 1.8X volume reaction RNAclean AMPure XP beads (Beckman Coulter). Cleaned baits were stored in the presence of 1 U/microliter SUPERase-In RNase inhibitor (Ambion) at -80°C. Yield was typically 10-20 micrograms of biotinylated RNA as determined by RNA 6000 Nano Bioanalyzer assay (Agilent).
Hybrid selection was conducted as previously described by Gnirke et al.50 For each whole genome fragment library prepared with indexed Nextflex paired-end adapter sequences (BioO Scientific) from a B. microti-infected diagnostic sample, 250 microliters “pond” library was hybridized with 500 nanograms of whole genome bait at 65°C for 72 hours in a 13 microliter reaction volume, with all components besides baits scaled down proportionately from Gnirke et al.50 Captured DNA was pulled down using 250 nanograms (25 microliters) MyOne Streptavidin T1 Dynabeads (Invitrogen). Beads were then washed at room temperature for 15 minutes in 250 microliters 0.5 ml 1 × SSC/0.1% SDS, followed by three 10 minute washes at 65°C with 250 microliters 0.1 × SSC/0.1% SDS pre-warmed to 65°C. Beads were incubated with 25 microliters 0.1M NaOH for 10 minutes at room temperature to elute selected DNA. Eluate was subsequently transferred to a tube containing 35 microliters 1M Tris-HCl, pH 7.5 and desalted and concentrated by cleanup with AMPure XP beads. Hybrid-selected library was amplified for 10-14 cycles in Q5 HF buffer using Nextflex genomic library primers. Initial denaturation was 30 s at 98°C, followed by cycles of 10 seconds at 98°C, 30 seconds at 65°C, and 30 seconds at 72°C. AMPure XP-cleaned PCR product was quantified using qPCR in Power SYBR Green master mix (Life) and NEXTFlex primers. Four indexed, hybrid-selected libraries were sequenced with one lane of Illumina HiSeq 100-bp paired-end reads at UMMS.
Quantitative PCR
Prior to sequencing, parasite DNA enrichment was assessed using a qPCR primer set designed to amplify the B. microti-specific amplicon within the gene BBM_III07585 in the B. microti reference genome. Each Power SYBR Green qPCR reaction contained 5 nanograms pre- or post-selection library as quantified by Quant-iT PicoGreen dsDNA assay kit (Life). Oligonucleotide sequences were from Melnikov et al.49 B. microti qPCR primer Amplicon Chr 3 (GenBank FO082874.1), gene BBM_III07585. Forward primer: 5′-GGTTCCCTAATGGAGACCCTACTA-3′ Reverse primer: 5′-GGCGCCCTTCTTTAAATCCA-3′
Supplementary Material
Acknowledgments
We wish to acknowledge Ryan Tewhey, Anne Piantadosi, and James Maguire for helpful feedback and advice, and Jon Robbins, Joel Katz, Jeff Gelfand, and Tad Wieczorek for helpful discussions and assistance with sample collection. We also wish to thank members of the parasitology and hematology labs at Massachusetts General Hospital and Brigham and Women’s Hospital for assistance with case identification. PCS and this work are supported by the Broad Institute SPARC program and the Bill and Melinda Gates Foundation and the Howard Hughes Medical Institute. This work was supported in part by an Infectious Disease Society of America Medical Scholars award, a Harvard/MIT Division of Health Sciences and Technology Research Assistantship to JEL, and NIH MSTP grants T32GM007753 to JEL and AL. SRT and HKG are supported by NIH U01AI109656 and R41AI078631 and by grants from the Evelyn Lilly Lutz Foundation, Dorothy Harrison Egan Foundation, and the Bill and Melinda Gates Foundation. EV was supported by a grant from the National Research Fund for Tick-Borne Diseases.
Footnotes
Accession codes: Sequence reads are available at the European Nucleotide Archive under accession code PRJEB13263.
Contributions: Performed experiments, analyzed data, wrote the paper: JEL. Performed experiments, analyzed data: ADT, HG. Performed experiments: LMF. Analyzed data: KGA. Analyzed data, wrote the paper: SFS, AL, SRT, ER, JAB, PCS. Contributed reagents/materials, SB, GM, LS, DM. Contributed materials/reagants, wrote the paper: EV, BP.
References
- 1.Vannier E, Krause PJ. Human Babesiosis. N Engl J Med. 2012;366:2397–2407. doi: 10.1056/NEJMra1202018. [DOI] [PubMed] [Google Scholar]
- 2.Ruebush TK, Spielman A. Human Babesiosis in the United States. Ann Intern Med. 1978;88:263–263. doi: 10.7326/0003-4819-88-2-263_1. [DOI] [PubMed] [Google Scholar]
- 3.Krause PJ, et al. Persistent and relapsing babesiosis in immunocompromised patients. Clin Infect Dis. 2008;46:370–376. doi: 10.1086/525852. [DOI] [PubMed] [Google Scholar]
- 4.Vyas JM, Telford SR, Robbins GK. Treatment of refractory Babesia microti infection with atovaquone-proguanil in an HIV-infected patient: case report. Clin Infect Dis. 2007;45:1588–1590. doi: 10.1086/523731. [DOI] [PubMed] [Google Scholar]
- 5.Wormser GP, et al. Emergence of resistance to azithromycin-atovaquone in immunocompromised patients with Babesia microti infection. Clin Infect Dis. 2010;50:381–386. doi: 10.1086/649859. [DOI] [PubMed] [Google Scholar]
- 6.Telford SR., III Babesial infections in humans and wildlife. Parasitic protozoa. 1993;5:1–47. [Google Scholar]
- 7.Herwaldt BL, et al. Transfusion-Associated Babesiosis in the United States: A Description of Cases. Ann Intern Med. 2011;155:509–519. doi: 10.7326/0003-4819-155-8-201110180-00362. [DOI] [PubMed] [Google Scholar]
- 8.Yager PH, Luginbuhl LM, Dekker JP. Case 6-2014. N Engl J Med. 2014;370:753–762. doi: 10.1056/NEJMcpc1208155. [DOI] [PubMed] [Google Scholar]
- 9.Clark IA, Jacobson LS. Do babesiosis and malaria share a common disease process. Annals of tropical medicine and …. 1998 doi: 10.1080/00034989859456. [DOI] [PubMed] [Google Scholar]
- 10.Hatcher JC, Greenberg PD, Antique J, Jimenez-Lucho VE. Severe babesiosis in Long Island: review of 34 cases and their complications. Clin Infect Dis. 2001;32:1117–1125. doi: 10.1086/319742. [DOI] [PubMed] [Google Scholar]
- 11.Meldrum SC, Birkhead GS, White DJ, Benach JL, Morse DL. Human Babesiosis in New York State: An Epidemiological Description of 136 Cases. Clin Infect Dis. 1992;15:1019–1023. doi: 10.1093/clind/15.6.1019. [DOI] [PubMed] [Google Scholar]
- 12.Menis M, et al. Babesiosis Occurrence among the Elderly in the United States, as Recorded in Large Medicare Databases during 2006–2013. PLoS ONE. 2015;10:e0140332. doi: 10.1371/journal.pone.0140332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hildebrandt A, et al. First confirmed autochthonous case of human Babesia microti infection in Europe. Eur J Clin Microbiol Infect Dis. 2007;26:595–601. doi: 10.1007/s10096-007-0333-1. [DOI] [PubMed] [Google Scholar]
- 14.Wei Q, et al. Human babesiosis in Japan: isolation of Babesia microti-like parasites from an asymptomatic transfusion donor and from a rodent from an area where babesiosis is endemic. J Clin Microbiol. 2001;39:2178–2183. doi: 10.1128/JCM.39.6.2178-2183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Senanayake SN, et al. First report of human babesiosis in Australia. Med J Aust. 2012;196:350–352. doi: 10.5694/mja11.11378. [DOI] [PubMed] [Google Scholar]
- 16.Western KA, Benson GD, Gleason NN, Healy GR, Schultz MG. Babesiosis in a Massachusetts Resident. N Engl J Med. 1970;283:854–856. doi: 10.1056/NEJM197010152831607. [DOI] [PubMed] [Google Scholar]
- 17.Centers for Disease Control and Prevention (CDC) Babesiosis surveillance - 18 States, 2011. MMWR Morb Mortal Wkly Rep. 2012;61:505–509. [PubMed] [Google Scholar]
- 18.Krause PJ, et al. Increasing health burden of human babesiosis in endemic sites. American Journal of Tropical Medicine and Hygiene. 2003;68:431–436. [PubMed] [Google Scholar]
- 19.Stafford KC, III, et al. Expansion of Zoonotic Babesiosis and Reported Human Cases, Connecticut, 2001–2010. J Med Entomol. 2014;51:245–252. doi: 10.1603/me13154. [DOI] [PubMed] [Google Scholar]
- 20.Goethert HK, Telford SR. Not ‘out of Nantucket’: Babesia microti in southern New England comprises at least two major populations. Parasites Vectors. 2014;7:546. doi: 10.1186/s13071-014-0546-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed Phylogenetics and Dating with Confidence. Plos Biol. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Homer MJ, et al. A Polymorphic Multigene Family Encoding an Immunodominant Protein from Babesia microti. J Clin Microbiol. 2000;38:362–368. doi: 10.1128/jcm.38.1.362-368.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Srivastava IK, Morrisey JM, Darrouzet E, Daldal F, Vaidya AB. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol Microbiol. 1999;33:704–711. doi: 10.1046/j.1365-2958.1999.01515.x. [DOI] [PubMed] [Google Scholar]
- 24.Korsinczky M, et al. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob Agents Chemother. 2000;44:2100–2108. doi: 10.1128/aac.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsuu A, Miyamoto K, Ikadai H, Okano S, Higuchi S. Short report: cloning of the Babesia gibsoni cytochrome B gene and isolation of three single nucleotide polymorphisms from parasites present after atovaquone treatment. American Journal of Tropical Medicine and Hygiene. 2006;74:593–597. [PubMed] [Google Scholar]
- 26.Sidhu ABS, et al. In vitro efficacy, resistance selection, and structural modeling studies implicate the malarial parasite apicoplast as the target of azithromycin. Journal of Biological Chemistry. 2007;282:2494–2504. doi: 10.1074/jbc.M608615200. [DOI] [PubMed] [Google Scholar]
- 27.Malbruny B, et al. Resistance to macrolides in clinical isolates of Streptococcus pyogenes due to ribosomal mutations. Journal of Antimicrobial Chemotherapy. 2002;49:935–939. doi: 10.1093/jac/dkf038. [DOI] [PubMed] [Google Scholar]
- 28.Chittum HS, Champney WS. Ribosomal protein gene sequence changes in erythromycin-resistant mutants of Escherichia coli. J Bacteriol. 1994;176:6192–6198. doi: 10.1128/jb.176.20.6192-6198.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McFadden DC, Tomavo S, Berry EA, Boothroyd JC. Characterization of cytochrome b from Toxoplasma gondii and Qo domain mutations as a mechanism of atovaquone-resistance. Mol Biochem Parasitol. 2000;108:1–12. doi: 10.1016/s0166-6851(00)00184-5. [DOI] [PubMed] [Google Scholar]
- 30.Pihlajamäki M, et al. Ribosomal mutations in Streptococcus pneumoniae clinical isolates. Antimicrob Agents Chemother. 2002;46:654–658. doi: 10.1128/AAC.46.3.654-658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pages H, Aboyoun P, Gentleman R, DebRoy S. Bioconductor - Biostrings; Memory efficient string containers, string matching algorithms, and other utilities, for fast manipulation of large biological sequences or sets of sequences. bioconductor.fhcrc.org. at < http://bioconductor.fhcrc.org/packages/release/bioc/html/Biostrings.html>.
- 34.Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:1. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfeifer B, Wittelsbürger U, Onsins SER, Lercher MJ. PopGenome: An efficient swiss army knife for population genomic analyses in R. Molecular Biology and Evolution. 2014;31:msu136–1936. doi: 10.1093/molbev/msu136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.South A. rworldmap: a new R package for mapping global data. The R Journal. 2011;3:35–43. [Google Scholar]
- 37.Charif D, Lobry JR. Structural Approaches to Sequence Evolution. Springer Berlin Heidelberg; 2007. pp. 207–232. [DOI] [Google Scholar]
- 38.Rutherford K, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 39.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cornillot E, et al. Sequencing of the smallest Apicomplexan genome from the human pathogen Babesia microti. Nucleic Acids Research. 2012;40:9102–9114. doi: 10.1093/nar/gks700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Molecular Biology and Evolution. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 44.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 1995;57:289–300. [Google Scholar]
- 45.Birth D, Kao WC, Hunte C. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nature Communications. 2014;5 doi: 10.1038/ncomms5029. [DOI] [PubMed] [Google Scholar]
- 46.Bulkley D, Innis CA, Blaha G, Steitz TA. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proceedings of the National Academy of Sciences. 2010;107:17158–17163. doi: 10.1073/pnas.1008685107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simpson JT, et al. ABySS: a parallel assembler for short read sequence data. Genome Research. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delcher AL, et al. Alignment of whole genomes. Nucleic Acids Research. 1999;27:2369–2376. doi: 10.1093/nar/27.11.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melnikov A, et al. Hybrid selection for sequencing pathogen genomes from clinical samples. Genome Biol. 2011;12:R73. doi: 10.1186/gb-2011-12-8-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gnirke A, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.