

Abstract
Granulins (GRNs) are a family of small (∼6 kDa) proteins generated by the proteolytic processing of their precursor, progranulin (PGRN), in many cell types. Both PGRN and GRNs are implicated in a plethora of biological functions, often in opposing roles to each other. Lately, GRNs have generated significant attention due to their implicated roles in neurodegenerative disorders. Despite their physiological and pathological significance, the structure‐function relationships of GRNs are poorly defined. GRNs contain 12 conserved cysteines forming six intramolecular disulfide bonds, making them rather exceptional, even among a few proteins with high disulfide bond density. Solution NMR investigations in the past have revealed a unique structure containing putative interdigitated disulfide bonds for several GRNs, but GRN‐3 was unsolvable due to its heterogeneity and disorder. In our previous report, we showed that abrogation of disulfide bonds in GRN‐3 renders the protein completely disordered (Ghag et al., Prot Eng Des Sel 2016). In this study, we report the cellular expression and biophysical analysis of fully oxidized, native GRN‐3. Our results indicate that both E. coli and human embryonic kidney (HEK) cells do not exclusively make GRN‐3 with homogenous disulfide bonds, likely due to the high cysteine density within the protein. Biophysical analysis suggests that GRN‐3 structure is dominated by irregular loops held together only by disulfide bonds, which induced remarkable thermal stability to the protein despite the lack of regular secondary structure. This unusual handshake between disulfide bonds and disorder within GRN‐3 could suggest a unique adaptation of intrinsically disordered proteins towards structural stability.
Keywords: granulin, progranulin, intrinsically disordered proteins, cysteine‐rich proteins
Introduction
Granulin‐3 (GRN‐3) is one of seven granulins generated by the proteolytic cleavage of its precursor, progranulin (PGRN), also called granulin‐epithelin precursor (GEP).1, 2, 3 PGRN is expressed in many cell types, but its expression is highly upregulated in microglia during inflammation resulting from an injury or an insult.4, 5, 6, 7, 8 Both PGRNs and GRNs have been implicated in a myriad of functions, including embryonic development, wound healing, tissue repair, and immune response modulation.1, 2, 9 In addition, PGRN has also been implicated in several pathological processes, such as tumor progression10 and development of neurodegenerative diseases, including frontotemporal dementia (FTD), Alzheimer disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS).11, 12, 13 GRN‐3 is also known to trigger the production and release of interleukin‐8 (IL8) in epithelial cells, suggesting a pro‐inflammatory role.14 Despite their multifunctional roles, many aspects related to the structure‐function relationship of GRNs remain unknown.
GRNs (1 – 7) are small ∼6 kDa proteins, and all but GRN‐1, are characterized by the presence of twelve cysteines that form six disulfide bonds.3, 15 GRN‐1 contains ten conserved cysteines that form five disulfide bonds. GRNs are among the family of proteins that possess high degree of disulfide bonds, with cysteines accounting for 20% of the total amino acid content. Kringle domains (which are triple‐looped, disulfide cross‐linked domains of ∼80 residues in length that participate in protein‐protein interactions16) present in plasminogen17 and the epithelial growth factor (EGF)‐like domain (which has a sequence of 30 to 40 amino‐acid residues) present in multiple copies in EGF and transforming growth factor‐α (TGF‐α) are the closest related family with three disulfide bonds.18, 19 The positions of the cysteines are highly conserved in all GRNs, with four adjacently placed pairs towards the center of the protein flanked by two single cysteines at both the N‐ and C‐termini.1 The role of these conserved cysteines in both structure and function remains ambiguous. In our previous study, we showed that the abrogation of the disulfide bonds in GRN‐3 (fully reduced form; abbreviated as rGRN‐3) renders the protein disordered,20 indicating that disulfide bonds are important for the overall structure of the protein. Interestingly, rGRN‐3 also activated NF‐ κB in SH‐SY5Y human neuroblastoma cells in a dose‐dependent manner, implicating that GRN‐3 may be functional, at least partly, even in a state without disulfide bonds characterized by a complete loss of structure.20
Intrigued by the behavior of rGRN‐3 and motivated by the desire to understand the structure‐function relationship of this protein, we investigated the cellular expression and the structure of oxidized, native GRN‐3. An earlier attempt to characterize the GRN‐3 structure was unsuccessful, but NMR structures of GRNs 4, 5, and 2 have been solved.21 In this previous report, the authors indicated that the structure of GRN‐3 could not be resolved due to the high degree of structural heterogeneity and disorder within the protein.21 The NMR structure of GRN‐4, however, showed a relatively ordered N‐terminal domain containing two antiparallel β‐strands, with a disordered C‐terminal region.21 The authors observed a structure dominated by loops for GRNs 2 and 5 as well.21
Here, we report several important findings on GRN‐3 structure. First, we show that only 5% of GRN‐3 expressed in E. coli is a monomeric protein (henceforth referred to as GRN‐3), with all 12 cysteines forming intramolecular disulfide bonds. GRN‐3 expressed in human embryonic kidney (HEK) cells (henceforth referred to as hGRN‐3) is not exclusively made as an intramolecular disulfide‐bonded monomers, although the disulfide bond homogeneity was much greater than that of the protein expressed in E. coli. Furthermore, both E. coli and HEK cells form heterogeneous isoforms containing multimeric GRN‐3 ensembles (henceforth referred to as mGRN‐3) consisting of scrambled disulfide bonds to varying degrees. Second, our computational and biophysical analyses show that disulfide bonds predominantly dictate the structure of GRN‐3. We reason that the role of disulfide bonds is to impart structural stability to an otherwise disordered protein, and yet retain parts of unstructured/flexible regions, which may, in part, account for the multifunctional roles observed for human GRNs. This unusual handshake between disulfide bonds and disorder could also be an additional adaptation feature of the structurally and functionally versatile intrinsically disordered proteins (IDPs).
Results
Recombinant GRN‐3 in E. coli is expressed predominantly as disulfide bond scrambled multimers
GRN‐3 was expressed recombinantly in E. coli SHuffleTM cells (NEB, Ipswich, MA) as a thioredoxin fusion protein (GRN‐3‐trxA), as previously reported.20 This fusion construct and cells were chosen because of two distinct advantages. First, SHuffleTM cells are engineered to constitutively express protein disulfide bond isomerase (DsbC) in the cytoplasm, which facilitates the formation of disulfide bonds and reduces scrambling.22 Second, the cells carry knockout mutations of the thioredoxin and glutathione reductase genes (ΔtrxB, Δgor), which help maintaining an oxidative environment in the cytoplasm conducive for disulfide bond formation. Additionally, the thioredoxin tag is known to assist in disulfide bond formation.23 Furthermore, to facilitate spectroscopic characterization of GRN‐3, a conservative Y28W mutation was introduced in the sequence [Fig. 1(A);*]. Tryptophan has a higher extinction coefficient and high quantum yield when compared to tyrosine, making it a better fluorophore. Since some of the spectroscopic measurements were carried out at low micromolar concentrations, it was experimentally necessary to have a stronger fluorophore. Furthermore, since both aromatic amino acids W and Y belong to the group of order promoting amino acids, and since all GRNs, except GRN‐3 and GRN‐1, have a conserved W at this position, replacing Y with W is not expected to significantly affect the protein structure or function. After nickel affinity purification and thioredoxin cleavage by thrombin (details in Materials and Methods), GRN‐3 was fractionated on a C18 reverse‐phase HPLC column. The protein was found in multiple fractions between fractions 47 to 68, indicating the presence of many isoforms [Fig. 1(B), dashed box and Fig. 1(C)]. The cleaved thioredoxin (trxA) eluted at later fractions ∼90.
Figure 1.
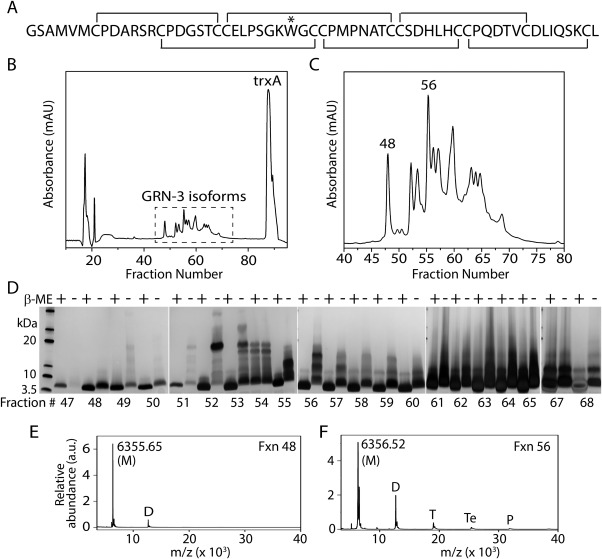
Characterization of GRN‐3. (Α) Primary amino acid sequence of GRN‐3 with putative disulfide bonding pattern. “*” represents the Y28W mutation. (B) HPLC fractionation of GRN‐3 after thrombin cleavage. The peaks corresponding to the different disulfide bond isoforms of GRN‐3 are marked with a dashed box. (C) Zoomed image of the different disulfide bond GRN‐3 isoforms showing distinctly resolved isoforms. Two of the fractions characterized in this work are marked. (D) SDS‐PAGE analysis of the HPLC fractions 47 to 65, 67, and 68 under reducing (βME +) and non‐reducing (βME ‐) conditions. The numbers indicated below the gels are the corresponding fraction numbers. Of all the fractions, fraction 48 shows a single band under reducing and non‐reducing conditions corresponding to the GRN‐3 monomer (Mw = 6.3 kDa). Fraction 56 shows a single band corresponding to monomeric GRN‐3 in reducing conditions, and bands corresponding to monomer, dimer, and trimer under non‐reducing conditions. (E, and F) MALDI‐ToF spectra of fractions 48 showing a predominant signal corresponding to GRN‐3 monomer (M) and a relatively minor peak corresponding to the dimer (D) and fraction 56 showing signals corresponding to the monomer (M), dimer (D), trimer (T), tetramer (Te), and Pentamer (P).
To investigate the molecular mass and disulfide bond integrity of the recombinant protein in fractions 47–68, SDS‐PAGE analysis was conducted under reducing and non‐reducing conditions. As expected, a single band at 6.3 kDa corresponding to the average mass of a monomeric GRN‐3 was observed under reducing conditions in all fractions GRN‐3 [Fig. 1(D), lanes β‐ME +]. However, under non‐reducing conditions, in addition to the monomeric band, bands corresponding to dimeric (∼12.7 kDa) and trimeric (∼19.1 kDa) GRN‐3 were observed for all the fractions except fraction 48 [Fig. 1(D), lanes β‐ME ‐]. This indicates that fractions 49–68 contain multimeric isoforms of GRN‐3 with intermolecular disulfide bonds. Fraction 48 is the only one that showed monomeric protein containing intramolecular disulfide bonds. Although a faint band corresponding to a dimer was observed in fraction 48 under non‐reducing conditions, the percentage of dimer was negligible compared to the other fractions. Indeed, the MALDI‐ToF mass spectrometric analysis of fraction 48 showed a predominant signal at m/z value of 6355.65 Da (M), which corresponds to the molecular weight of monomeric GRN‐3 along with a minor signal (less than 10%) corresponding to dimeric GRN‐3 (D), indicating the presence of small amounts of dimeric GRN‐3 [Fig. 1(E)]. In contrast, the mass spectrum of fraction 56 showed signals corresponding to a monomer (m/z 6356.52 Da; M) along with those corresponding to a dimer (D), trimer (T), tetramer (Te), and a pentamer (P) [Fig. 1(F)]. Similar multimeric peaks were observed in all the fractions between 49 and 68 [Supporting Information Fig. S1(A)].
To further confirm the integrity of disulfide bonds, Ellman's and alkylation assays were performed. Analysis of the freshly purified GRN‐3 and mGRN‐3 samples by Ellman's assay indicated that less than 5% of the cysteines were present in the reduced form (this corresponds to ∼1 cysteine per protein molecule). The iodoacetamide alkylation assay confirmed this observation and showed that only 1 – 2 cysteines per protein molecule were present as free sulfhydryls [Supporting Information Fig. S2(B)]. Fraction 56 was chosen as mGRN‐3 for further analyses as it showed highest concentration of GRN‐3 multimers among all other fractions. From the SDS‐PAGE and MALDI‐ToF analyses it is apparent that intramolecularly disulfide bonded, monomeric GRN‐3 elutes exclusively at fraction 48. Only 5% of GRN‐3 is in this monomeric form, based on quantitation of fraction 48 compared to total GRN‐3 levels in fractions 47 to 68, suggesting that despite the presence of thioredoxin, DsbC, and oxidized environment, E. coli does not generate high‐levels of correct disulfide bonded GRN‐3 perhaps due to the high cysteine content as well as the placement of multiple cysteines in adjacent positions in GRN‐3 [Fig. 1(A)].
GRN‐3 expressed in human embryonic kidney (HEK) cells also form multimers with intermolecular disulfide bonds
It could be argued that the inability of E. coli to generate GRN‐3 with proper disulfide bonds is due to the less efficient post‐translational processing machinery, compared to that of the eukaryotic cells. Therefore, to test this hypothesis, hGRN‐3 was expressed and purified from human embryonic kidney (HEK) cells. Purified hGRN‐3 protein was then subjected to the SDS‐PAGE and MALDI‐ToF analyses similar to those of GRN‐3 expressed in E. coli. SDS‐PAGE, under reducing conditions, showed a distinct band at ∼ 12 kDa corresponding to monomeric hGRN‐3 (the expression construct contains a twin‐Strep and FLAG tag) along with a smear between 15 and 20 kDa [Fig. 2(A), lane 1]. The smear likely reflects glycosylation of hGRN‐3. In the absence of boiling of the sample prior to the electrophoresis, the sample did not show a large change in banding pattern, suggesting a portion of the observed bands arise due to covalent interactions [Fig. 2(A), lane 3]. hGRN‐3 sample electrophoresed under the non‐reducing conditions both with and without heat treatment showed a faint band corresponding to monomeric (single arrow), predominant band corresponding to dimeric (double arrow), and another faint band corresponding to trimeric (triple arrow) hGRN‐3, which was absent in the reduced samples [Fig. 2(A), lanes 2 and 4]. This is indicative of the presence of intermolecular disulfide bonded multimers. Similarly, the MALDI‐ToF spectrum of hGRN‐3 under reducing conditions showed the presence of a predominant monomeric peak [Fig. 2(B), single arrow] along with a relatively minor dimeric peak [Fig. 2(B), double arrow]. In contrast, under non‐reducing conditions, distinct signals corresponding to dimers [Fig. 2(B), double arrow] and trimers [Fig. 2(B), triple arrow] in addition to the monomeric signal [Fig. 2(B), single arrow] were observed. The possibility of multimers arising from non‐covalent interactions is excluded by the presence of identical banding pattern in heat‐treated and untreated samples. These data demonstrate that a portion of hGRN‐3 expressed in mammalian cells contains disulfide‐bonded multimers. In fact, a previous report indicates that full‐length PGRN, when expressed in HEK cells, also shows disulfide bonded multimers both in the cell lysate and in the media (secreted).24 Taken together, it is clear that irrespective of the cell type, the post‐translational machinery involved in disulfide bond formation seems to be inefficient in generating homogenous disulfide bonds in GRN‐3 when overexpressed at high levels, although E. coli is far less efficient than HEK cells. Due to the limitation in the amounts of protein produced in HEK cells for biophysical analyses, E. coli expressed protein was used for further analyses in this study.
Figure 2.
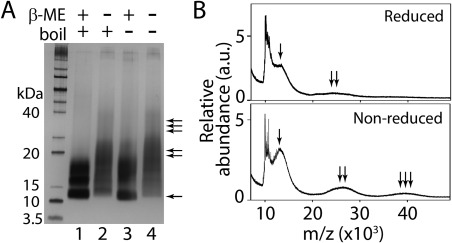
Characterization of hGRN‐3 expressed and purified from human embryonic kidney (HEK) cells. (Α) SDS‐PAGE of hGRN‐3 under reduced, non‐reduced, and boiled (lanes 1 and 2, respectively), and reduced, non‐reduced and unboiled (lanes 3 and 4, respectively). The presence of monomer, dimers, and trimers in lanes 2 and 4 are marked with single, double, and triple arrows, respectively. (Β) MALDI‐ToF spectra of hGRN‐3 showing signals corresponding to monomeric hGRN‐3 (single arrow), dimeric hGRN‐3 (double arrows), and trimeric hGRN‐3 (triple arrows) under reduced and non‐reduced conditions. The results are indicative of three consistent repeats.
Intrinsic disorder in PGRN and GRNs: Evidence for varying disorder propensities among GRNs
To understand how disorder propensity is encoded within the human PGRN sequence and to see if individual GRNs have some unique disorder characteristics, the PGRN precursor protein and individual GRNs were subjected to the computational analyses. The presence of intrinsic disorder identifiable by computer algorithms in these proteins is not a trivial question, since PGRN/GRNs are characterized by extremely high cysteine residue content. This is significant since cysteines are considered as the strongest order‐promoting residues according to the established classification of amino acids in relation to their potential effects on the intrinsic disorder propensity of a protein (based on their depletion or enrichment in IDPs/IDPRs, amino acids are grouped into the order‐promoting (C, W, I, Y, F, L, H, V, and N) and disorder‐promoting residues (R, T, S, K, Q, E, and P).25, 26, 27, 28, 29, 30 We had previously shown that abrogation of disulfide bonds in GRN‐3 (rGRN‐3) results in the protein becoming an IDP.20 Therefore, based on the fact that 14.8% residues of PGRN are cysteines (and the corresponding values are even higher for individual GRNs that contain from 17.9% to 22.2% cysteine residues) it was expected that PGRN/GRNs would be predicted to be ordered. However, only one of the three computational tools used in this study (PONDR® VLXT) showed that human PGRN (UniProt ID: P28799) is expected to contain significant levels of order, whereas according to the other two predictors (PONDR® VL3 and PONDR® VSL2) this protein was shown to be predominantly disordered [Fig. 3(A)]. Curiously, the PONDR® VL3‐ and PONDR® VSL2‐based disorder profiles of this protein were characterized by a peculiar pattern containing several “dips”, whose positions only weakly correlated with the positions of individual GRNs within the PGRN sequence. However, although the results of PONDR® VLXT analysis indicated that human PGRN is expected to be ordered, the disorder profile generated by this computational tool clearly showed that inter‐GRN regions were expected to be either disordered (i.e., had disorder scores above the 0.5 threshold) or possess at least some conformational flexibility (i.e., having disorder scores noticeably deviating from zero). In fact, Figure 3(A) shows that, according to the PONDR® VLXT, individual GRNs are always surrounded by flexible/disordered regions. This is an important observation clearly related to the biogenesis of individual GRNs, which are generated by the proteolytic processing of the PGRN precursor protein. Since proteolytic digestion is orders of magnitude faster in unstructured compared to structured protein regions,31, 32, 33, 34, 35, 36 it is extremely important for the protein cleavage process that the sites of cleavage are located in regions that lack structure or possess high structural flexibility.
Figure 3.
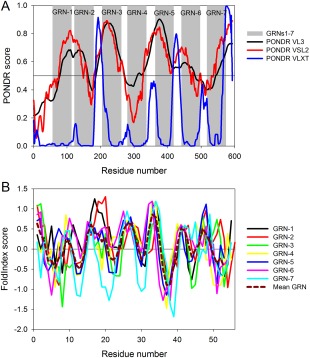
Intrinsic disorder propensity of human PGRN and individual GRNs. (A) Evaluation of intrinsic disorder propensity in human PGRN (UniProt ID: P28799) by PONDR® VL3 (black curve), PONDR® VSL2 (red curve), and PONDR® VLXT algorithms (green curve). In this analysis, regions with scores above 0.5 are considered intrinsically disordered. Position of individual GRNs are shown by gray shaded areas. (B) Intrinsic disorder propensities of individual human GRNs by the FoldIndex algorithm. Data for the individual GRNs are presented by differently colored solid curves. In this analysis, positive values represent regions/residues likely to be folded, and negative values represent those likely to be intrinsically disordered. Dashed dark red curve describes the behavior of mean Grn. The corresponding data were obtained by simple averaging of disorder profiles of individual GRNs.
Curiously, analysis of the Figure 3(A) (see disorder profiles generated by PONDR® VL3 and PONDR® VSL2) indicate that the disorder propensities of individual GRNs are rather different. Furthermore, based on their intrinsic disorder predispositions, GRNs form a peculiar high‐low pattern within the PGRN sequence, where apparently the more disordered GRN‐1, GRN‐3, GRN‐5, and GRN‐7 are interspersed by the more ordered GRN‐2, GRN‐4, and GRN‐6. The conclusion on the inequality of intrinsic disorder predispositions of individual GRNs is further illustrated by Figure 3(B), which represents the results of the evaluation of intrinsic disorder propensities of individual GRNs by a graphic web server FoldIndex37 that implements an algorithm based on the average residue hydrophobicity and net charge of the sequence30 and calculates a separate score for each individual residue.37 Figure 3(B) shows that such charge‐hydropathy‐based disorder profiles are different for individual GRNs, and Table 1 provides an overview of all these observations by summarizing some specific disorder‐based characteristics of individual GRNs. It is likely that the differences in intrinsic disorder propensities between individual GRNs are related to the differences in their biological functions. All these data taken together indicate that GRN‐3 experimentally analyzed in this study is expected to show the highest degree of disorderness compared to the other human GRNs.
Table 1.
Analysis of the Intrinsic Disorder Predisposition of Individual Human GRNs
| Name (length) | FoldIndexa | Mean PONDR disorder scoreb | |||
|---|---|---|---|---|---|
| NIDPR | Longest IDPR | NIDRes | % disorder | ||
| GRN‐1 (57) | 3 | 5 | 20 | 35.1 | 0.683 (0.593) |
| GRN‐2 (58) | 4 | 4 | 21 | 36.2 | 0.570 (0.564) |
| GRN‐3 (57) | 5 | 10 | 28 | 49.1 | 0.843 (0.778) |
| GRN‐4 (57) | 2 | 6 | 22 | 38.6 | 0.482 (0.416) |
| GRN‐5 (55) | 5 | 5 | 24 | 43.6 | 0.844 (0.733) |
| GRN‐6 (56) | 3 | 5 | 18 | 32.1 | 0.566 (0.538) |
| GRN‐7 (57) | 7 | 7 | 32 | 56.1 | 0.609 (0.560) |
| Mean GRN (55) | 4 | 4 | 24 | 43.6 | |
Table represents some of the disorder‐based characteristics of query proteins: “NIDPR” represents the number of intrinsically disordered protein regions (IDPRs) longer than 3 residues; “Longest IDPR” shows the length of the longest disordered region; “NIDRes” represents the number of predicted disordered residues; “% disorder” represents content of all disordered residues.
This column represents mean disorder score values generated by PONDR® VL3 and PONDR® for various human GRNs analyzed individually. Values in brackets show the region‐averaged disorder scores of GRNs within the PGRN sequence.
GRN‐3 is more ordered than mGRN‐3 and rGRN‐3
Based on our computational predictions, GRN‐3 has a higher degree of disorder among all the human GRNs. To further assess the structural effect of disulfide bonds, we investigated the structure of GRN‐3 compared to mGRN‐3 and rGRN‐3 by NMR, far‐UV circular dichroism (CD) spectroscopy, and molecular modeling. 1H‐15N heteronuclear multiple quantum coherence (HMQC) spectrum of GRN‐3 showed well‐resolved resonances and a good spectral dispersion between 6.9 and 9.7 ppm for 1H, and between 104 and 128 ppm for 15N dimensions in the amide region [Fig. 4(A), GRN‐3]. The resonances for all the backbone amides and side chain amide nitrogens, except for the six proline residues, could be accounted for. The broad signals observed at 7.4 and 7.8 ppm on the 1H and at 104 and 109 ppm on the 15N dimensions are likely to be those of arginine and lysine residues.38, 39, 40 The negative peaks at 7.0 and 129 ppm reflect potential contaminants, and likely not part of the protein. The spectrum for mGRN‐3 ensembles, on the other hand, was not as well dispersed as that of GRN‐3, although it indicated well‐resolved resonances for a majority of residues [Fig. 4(A), mGRN‐3]. Furthermore, significant line broadening was observed for several resonances. This indicated that mGRN‐3 possessed a structure that was only partly folded with a large part resembling a molten globule with regions of collapsed hydrophobic core. Abrogation of all disulfide bonds rendered the protein (rGRN‐3) completely disordered as evidenced by the narrow spectral dispersion on the 1H dimension between 8 and 8.9 along with a relatively well dispersed 15N dimension between 125 and 116 ppm, which is an indicator of disorder [Fig. 4(A), rGRN‐3].41 Furthermore, significant overlap and line broadening among the resonances were also observed, confirming that rGRN‐3 was completely disordered. Together, the NMR results show that disulfide bonds play a major role in imparting structure to GRN‐3. To confirm these observations, the samples were also analyzed by far‐UV CD. Surprisingly, GRN‐3 showed a random coil structure as indicated by a spectrum with a strong minimum at 198 and a weak maximum at 225 nm, commonly observed for disordered proteins [Fig. 4(B), solid line]. A nearly identical spectrum was also observed for rGRN‐3 [Fig. 4(B), short dash] as previously reported.20 mGRN‐3 ensembles, on the other hand, displayed a spectrum with a minimum at 201 nm and a less intense maximum at ∼230 nm corresponding to a poly proline type‐II (PPII) helix structure [Fig. 4(B), dash].42, 43 It is well established that many IDPs display PPII conformation44, 45, 46, 47 mainly due to the similarity in arrangement between amide dipoles within PPII and random coil geometries.48
Figure 4.
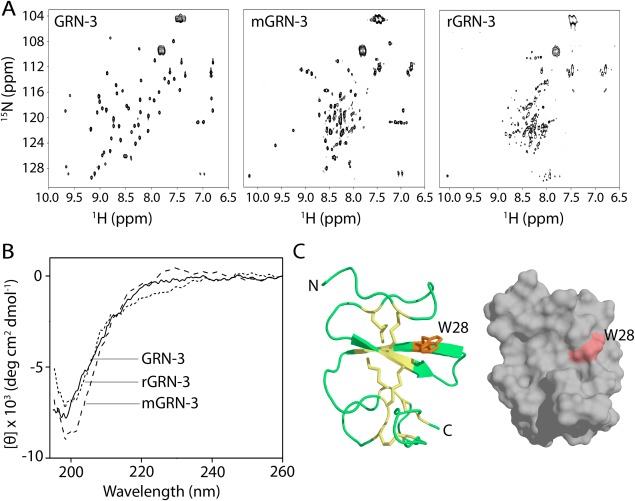
Structural characterization of GRN‐3, mGRN‐3, and rGRN‐3. (Α) 1H‐15N HMQC NMR spectra GRN‐3, mGRN‐3, and rGRN‐3. (B) Far‐UV circular dichroism (CD) spectra of GRN‐3 (solid line), mGRN‐3 (dashed line), and rGRN‐3 (short dashed line). Both GRN‐3 and rGRN‐3 show minima at 198 nm typical of random coil conformation whereas mGRN‐3 shows a minimum at 202 nm and a maximum at ∼230 nm corresponding to a PP‐II helix. (C) Structural model (ribbon and space‐filled representations) of GRN‐3 obtained using I‐TASSER with disulfide bonds represented as sticks (yellow) and W28 (orange) exposed on the surface.
To further probe the potential contradictory results from NMR and CD, the structure prediction tool, I‐TASSER (Iterative Threading ASSEmby Refinement) was used to model GRN‐3.49, 50 The software predicted four possible structures for GRN‐3, and the one with the highest confidence score (c‐score of 0.51) showed that the 82% of the structure is made of irregular loops [Fig. 4(C)]. All the other structures generated by the model also showed structures that are abundant in disordered loops (Supporting Information Fig. S2). The only secondary structure observed (β‐strands) accounted for 18% of the structure, making the predicted structure of GRN‐3 predominantly disordered. Importantly, this structure showed a great similarity to the GRN‐4 structure (this aspect is further elaborated in the Discussion section). Therefore, the potentially conflicting observations from NMR and CD data for GRN‐3 can be explained based on the observation that disulfide bonds, which, if present in a ladder‐like arrangement [Fig. 4(C)] as also observed for GRN‐4,21 hold the loops such that the protein backbone show minimal conformational freedom. This may show a distinct magnetic environment responsible for a good spectral dispersion in NMR, yet the backbone amide dipoles are not in a typical β‐sheet environment to display π→* transition at 216 nm in CD. These findings seem to indicate that although disulfide bonds are important to induce structural order in GRN‐3, they do not seem to induce well‐defined secondary structures but rather disordered loops with less conformational freedom.
mGRN‐3 ensembles have more solvent‐exposed hydrophobic surfaces as compared to GRN‐3
To further evaluate the structural differences between GRN‐3 and mGRN‐3 ensembles, the solvent‐exposed hydrophobic surfaces were qualitatively determined using the fluorescent dye, 8‐anilino‐1‐naphthalene sulfonic acid (ANS). ANS is an extrinsic dye that exhibits weak fluorescence in polar environments but shows strong fluorescence with a distinct blue shift in the emission wavelength upon binding to exposed hydrophobic pockets in the protein.51 ANS binding has been widely used to study protein folding/unfolding and structure of molten globule states and other partially folded intermediates.52 Upon incubating 25 μM GRN‐3 or mGRN‐3 with 500 μM ANS, the fluorescent intensity with mGRN‐3 was significantly higher than that with GRN‐3 [Fig. 5(A)], indicative of mGRN‐3 containing more solvent‐exposed hydrophobic surfaces than GRN‐3. This evaluation is in agreement with the 1H‐15N HMQC NMR data, which shows mGRN‐3 ensembles to be more disordered and collapsed than GRN‐3. In addition, the solvent accessibility of the single tryptophan residue (W28) in GRN‐3 was examined using Stern‐Volmer dynamic quenching analysis, where changes in the fluorescence intensity of the W28 residue were monitored as a function of the increasing concentration of an external quencher (acrylamide). A relatively large incline of the Stern‐Volmer plot was observed for the GRN‐3 [Fig. 5(B), •], which suggested that tryptophan undergoes dynamic quenching indicative of the probe being solvent exposed,53 which is in agreement with our predictive model of GRN‐3 [Fig. 4(C)]. In contrast, the slope of the corresponding plot for mGRN‐3 was noticeably smaller, indicating more efficient protection of tryptophan from solvent in these conformational ensembles [Fig. 5(B), ○]. Thus, W28 seems to be more solvent accessible in GRN‐3 than in mGRN‐3, and it is likely that W28 is buried within the hydrophobic core of a collapsed mGRN‐3 structure.
Figure 5.
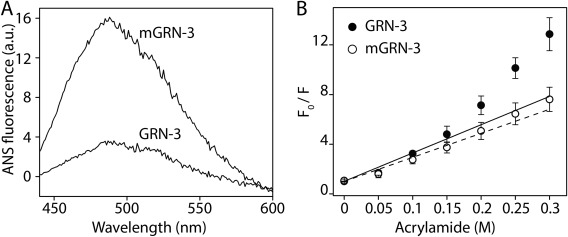
Structural differences between GRN‐3 and mGRN‐3. (Α) Differential interaction of 8‐anilino‐1‐naphthalene sulfonic acid (ANS) with GRN‐3 and mGRN‐3 at 25 μM, indicating differences in the exposed hydrophobic surfaces. (Β) Stern‐Volmer plot of GRN‐3 (•) and mGRN‐3 (○) using acrylamide as the quenching agent.
Disulfide bonds contribute overwhelmingly to the overall stability of GRN‐3
To obtain specific insights into the role of disulfide bonds in the structure and stability of GRN‐3, the protein was subjected to detergent‐induced thermal denaturation. It is known that high concentrations of SDS (> 1%) induce rod‐like or necklace‐like structures to polypeptides that exhibit an α‐helical structure.54, 55, 56 It has also been shown that SDS‐induced thermal denaturation of proteins represents a convenient way to monitor protein stability by CD. Furthermore, in these experiments, proteins that lack ordered secondary structure typically gain an α‐helical structure.57 Therefore, GRN‐3, mGRN‐3, and rGRN‐3 samples were treated with 1% SDS (35 mM), and the conversion of the coil‐like to helical structure was monitored as a function of temperature by CD spectroscopy. As expected, disordered rGRN‐3 instantly gained significant α‐helical structure in presence of SDS prior to the heat treatment at 10°C [Fig. 6(A), rGRN‐3, dotted line] and remained α‐helical over the temperature range up to 90°C [Fig. 6(A), dashed line and Fig. 6(B), Δ]. GRN‐3, on the other hand, showed no appreciable changes in the random coil structure at 10°C, and it remained disordered up to 90°C [Fig. 6(A), GRN‐3, dotted and dashed lines, respectively and Fig. 6(B), ▪], with only a small deviation from the spectrum characteristic of random coil towards a spectrum reflecting presence of partial α‐helical structure with a minimum at 207 nm and an increased negative ellipticity at 220 nm [Fig. 6(A), GRN‐3, dashed line, and Fig. 6(B), ▪]. A similar response was observed for mGRN‐3, with the protein ensembles remaining disordered throughout the entire temperature range [Fig. 6(A), mGRN‐3 and Fig. 6(B), ○]. Furthermore, hGRN‐3 expressed in HEK cells also showed remarkable resistance to the SDS thermal denaturation and its melting curves resembled those of mGRN‐3 derived from E. coli (Supporting Information Fig. S3), suggesting the presence of scrambled disulfide bonds in the protein. It is clear that both GRN‐3 and mGRN‐3 resist SDS‐induced structural alteration and keep their original conformation even at elevated temperatures, thereby exhibiting robust thermal stability. This can possibly be attributed to the presence of six intramolecular disulfide bonds in GRN‐3, which preclude the formation of ‘necklace’ or ‘rod‐like’ helical structures known to appear when SDS interacts with polypeptides.54, 55, 56, 58 If disulfide bonds are the main reason for the structural stability, abrogation of these should make the protein amenable to SDS‐induced thermal denaturation, which is precisely what is observed with rGRN‐3. This contention can be further reinforced by the charge‐hydropathy plots30 for GRN‐3 along with those for the all Cys to Ala (alaGRN‐3) and all Cys to Ser (serGRN‐3) mutations of GRN‐3 (Supporting Information Fig. S5). These data indicate that serGRN‐3, which models the fully reduced form of GRN‐3, renders the protein to move towards disorder as compared to the oxidized form. This supports the idea that the disulfide bonds are solely responsible for the structure and stability of GRN‐3.
Figure 6.
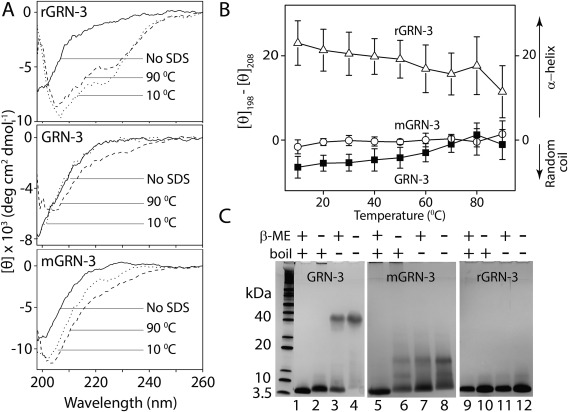
Structural stability of GRN‐3, mGRN‐3, and rGRN‐3. (Α) Far‐UV CD spectra without SDS (solid line), and in presence of 1% SDS at 10°C (dotted line), and 90°C (dashed line). (Β) Molar ellipticity difference between 198 nm (random coil) and 208 nm (α‐helix) for GRN‐3 (▪), mGRN‐3 (○), and rGRN‐3 (Δ) plotted as a function of increasing temperature in presence of 1% SDS. (C) SDS‐PAGE under reduced and non‐reduced conditions with heat treatment (lanes 1, 2, 5, 6, 9, and 10, respectively) and reduced and non‐reduced conditions without the heat treatment (lanes 3, 4, 7, 8, 11, and 12, respectively).
Next, in light of the high stability of GRN‐3 and mGRN‐3 to SDS denaturation, we wanted to see whether this stability is reflected in electrophoretic mobility. As observed earlier [Fig. 1(C), fraction 48], under both reducing and non‐reducing conditions with heat treatment (boiling the sample with 1% SDS for five minutes), GRN‐3 migrates as a single 6.3 kDa band corresponding to the molecular mass of the monomeric GRN‐3 [Fig. 6(D), lanes 1 and 2]. However, under both reducing and non‐reducing conditions, but without heat treatment, a GRN‐3 band is observed between 40 and 50 kDa [Fig. 6(D), lanes 3 and 4]. Furthermore, in the presence of the reducing agent (βME), an additional monomeric band was observed which was absent in the βME‐untreated sample, reinforcing the inference that disulfide bonds are solely responsible for the stability and abnormal electrophoretic mobility of GRN‐3. This unusual electrophoretic mobility was not observed for mGRN‐3 or rGRN‐3 [Fig. 6(D), lanes 5 to 12] even though both mGRN‐3 and rGRN‐3 are more disordered than GRN‐3 for reasons that remain unknown.
Discussion
The results presented in this study bring forth many intriguing aspects of the GRN‐3 structure, which could also be relevant to the disulfide bonded IDPs at large. First, both E. coli and HEK cells are inefficient in exclusively generating monomers with intramolecular disulfide bonds. In E. coli, only 5% of the total GRN‐3 protein is expressed as monomeric species, while the rest are different disulfide bond scrambled isoforms, despite the presence of thioredoxin tag and the use of engineered bacterial cells with an oxidizing cytoplasmic environment. Expression of GRN‐3 in mammalian HEK293T cells to overcome the possible lack of appropriate post‐translational machinery in E. coli did not generate GRN‐3 monomers exclusively, although the extent of disulfide scrambled multimers in this case was lower compared to E. coli. It should be noted that in Metazoa, GRNs are always expressed in the larger context of the full‐length PGRN precursor protein, which contains linker regions that may play a role in proper folding. Another plausible reason for the ineffective disulfide bond formation could be the high concentration of cysteines within the protein (20%), which could lead to incorrect cysteine linkages, especially in the context of overexpression to generate recombinant GRN‐3. In addition, the location of cysteines within the protein sequence [four pairs present adjacent to one another; Fig. 1(A)] could exacerbate the errors in catalysis conducted by protein disulfide isomerases (PDIs). For the same reasons, it is also uncertain whether scrambling of disulfide bonds occurs within the expressed monomeric GRN‐3. Furthermore, the complications of forming homogenous disulfide bonds due to high cysteine content could be far greater for the precursor, PGRN, which has 90 cysteines forming 45 disulfide bonds (7.5 repeats of GRN modules). Indeed, the presence of multiple disulfide‐bonded isoforms of PGRN have also been observed previously in the protein expressed in HEK cells.24 Furthermore, the observation that proteolysis of PGRN often generates products much larger than individual GRN units indicates the possibility of the presence of scrambled disulfide bonds within PGRN.59 Considering that GRNs are not directly expressed in cells but are generated by the proteolytic processing of PGRN, the presence of multiple disulfide bond scrambled isoforms of GRNs under physiological conditions seems to be realistic. Whether or exactly how the problem of disulfide scrambling is overcome in PGRN in mammalian cells is not clear as of yet, but one possibility is that the linker regions between the GRN motifs in PGRN could facilitate the binding and specificity of PDIs that may generate correct disulfide bonds.
A second intriguing aspect of GRN‐3 is that the structure and stability of this protein seems to be dictated solely by disulfide bonds, since the abrogation of these bonds renders the protein completely disordered.20 Our NMR data based on HMQC spectra also show rGRN‐3 to be intrinsically disordered. In addition, our data suggest that mGRN‐3 with scrambled disulfide bond, is disordered, although it shows a greater degree of order when compared to rGRN‐3. However, the HMQC spectrum of monomeric GRN‐3 shows a remarkable spectral dispersion suggesting a great deal of structural order within the protein and directly implicates the disulfide bonds to be the main driving force for its structural integrity. The predicted molecular model of GRN‐3 supports these conclusions and indicates that the disulfide bonds anchor the overall structural fold of the protein. Based on our model, the conserved cysteines in monomeric GRN‐3 [Fig. 4(C)] form a ladder‐like disulfide bond arrangement as observed for GRN‐4 (Fig. 7), which forms the fulcrum of the structure stabilizing the loops surrounding them. Such a structure has minimal secondary structure content (only short segments of β‐strands are observed) and the conformational freedom of the loops seems to be constrained by the central shaft made of disulfide bonds [Fig. 4(C)]. This model agrees with the reported GRN‐4 structure (PDB ID: 2jye) with an RMSD of 1.02 Å (Fig. 7).
Figure 7.
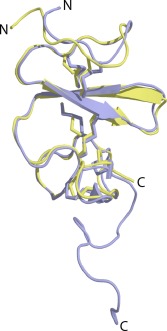
Alignment of GRN‐3 structure (green) predicted using I‐TASSER with that of NMR structure of GRN‐4 (blue) using PyMol. The rms deviation is 1.02 Å.
Structures similar to GRNs are abundant in nature, among which the EGF and kringle domain families bear the closest resemblance to this fold (Supporting Information Fig. S4). Known structures of both EGF and kringle domains contain irregular loops without any well‐defined secondary structure that are stabilized only by three disulfide bonds made from six conserved cysteines. Proteins containing the EGF fold include the epidermal growth factor (EGF), and the transforming growth factor α (TGF‐α).18, 19 Proteins with kringle fold include hepatocyte growth factor60, 61 and several proteases involved in blood clotting such as prothrombin, and plasminogen,62 apolipoprotein,63 and tissue plasminogen activator.64 Murine EGF exhibits a striking resemblance to the unique stacked β‐sheet domain observed for the N‐terminal domain of GRN‐4.65 It is noteworthy that the human EGF shows well‐dispersed resonances in HSQC spectrum,66 while displaying a spectrum corresponding to a PP‐II‐helix in CD [Supporting Information Fig. S4(A)], similar to what is observed for GRN‐3 reported in our work. Furthermore, the predicted structure of GRN‐3 and the solution NMR structure of human EGF (pdb ID: 2kv4)66 are characterized by similar folds [Supporting Information Fig. S4(B)]. The kringle domains of several blotting clotting factors and the EGF‐like domains of EGF and TGF‐α exhibit striking resemblance to the unique stacked β‐sheet domain observed for the N‐terminal domain of GRNs 2, 4 and 5,2, 18 although the former contains only half the number of disulfide bonds as those found in GRNs.
Perhaps the most intriguing question that arises from these studies is why do disulfide bonds induce structural order to what is clearly a disordered protein? Furthermore, why such a structural ordering does not seem to generate secondary structures within the protein? It is clear from our data that the presence of disulfide bonds induces remarkable thermal stability in GRN‐3, and hence one could speculate that these disulfide bonds provide stability to a conformationally flexible protein and yet maintain plasticity. By holding the loops in place, the disulfide bonds are able to accomplish precisely that [Fig. 4(C)]. For the same reasons, one could also hypothesize that the ineffective post‐translational processing of GRN‐3 may not be entirely detrimental for its physiological functions as the plasticity in loop conformations could facilitate interaction with other proteins in both monomeric and multimeric forms. Such pluripotent structures of IDPs with multiple functionality are now well‐established.41, 67, 68 Furthermore, we speculate that the cysteines in GRNs may also be involved in attenuating oxidative stress by redox mediated mechanism, which could justify the observation of multiple isoforms of the protein. However, this needs to be tested and verified. Nevertheless, this report brings forth some interesting aspects of the role of disulfide bonds in the structure and function of GRNs, the most striking of which is the delicate handshake between disulfides and disorder towards maintaining protein stability and, perhaps, functional plasticity.
Materials and Methods
Cloning of human GRN‐3 in E. coli
The GRN‐3 protein was expressed as a trxA fusion using the pET32b plasmid (Novagen) with a conservative Y24W mutation to facilitate biophysical investigations of GRN‐3 as described in previous study.20 Tryptophan (W) is conserved at this position in all the granulins except GRN‐3 and GRN‐1, and hence is not expected to affect its function greatly. The plasmid containing the GRN‐3 construct was expressed in the E. coli SHuffleTM cells (New England Biolabs, MA). The transformed cells with pET32b:GRN‐3‐trxA plasmid were grown at 30°C in 2 L baffled culture shake flasks containing 1 L LB medium supplemented with 100 μg mL−1 of ampicillin. Expression of the protein was induced by adding isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM when the optical density of the culture media was between 0.5 and 0.7 AU, as measured by UV‐absorbance at 600 nm. After 6 hours of induction at 30°C, cells were harvested by centrifuging (14000g, 4°C) and used immediately or stored at −20°C until further use.
Purification of recombinant GRN‐3
Cell pellets from 4 L of culture were thawed at room temperature and resuspended in 120 mL of equilibration buffer (50 mM Tris, pH 6.5, 300 mM NaCl, and 10 mM imidazole) containing 0.5 mM phenylmethylsulfonyl fluoride (PMSF). The cell suspension was sonicated for 4 minutes (40 second burst with 1 minute rest) on ice and centrifuged at 9200x g to remove debris. The supernatant was incubated with Ni2+‐NTA beads pre‐equilibrated with five bed volumes of the equilibration buffer and stirred gradually at 4°C for 1 hour. The supernatant was then allowed to flow through the beads using a peristaltic pump and collected as a flow‐through fraction. The beads were then washed with 200 ml of wash buffer (50 mM Tris, pH 6.5, 300 mM NaCl) containing 120 mM imidazole. The GRN‐3‐trxA fusion protein was eluted using 50 mL of 400 mM imidazole in 50 mM Tris, pH 6.5, and 300 mM NaCl. The eluate was then dialyzed at 4°C against a buffer of 2 mM Tris, pH 6.5, and 5 mM NaCl and using a 10 kDa MWCO dialysis membrane (Spectrum Labs). The dialysate was then concentrated 10 times using vacuum evaporation such that the final buffer concentration was 20 mM Tris, pH 6.5, and 50 mM NaCl. Concentration of the fusion protein was measured using UV‐Vis spectroscopy with a molar extinction coefficient of 20355 M−1cm−1 at 280 nm.69 On average, 10–12 mg of fusion protein was obtained per liter of cells. The fusion protein was then digested by addition of restriction grade thrombin (Novagen) 1U per 2 mg of the protein to cleave both trxA and the His‐tag. The reaction was incubated in a 37°C water bath for 22–24 hours. GRN‐3 protein was then fractionated using a semi‐preparative Jupiter® 5 μm 10x250 mm C18 reverse phase HPLC column (Phenomenex, CA), using a gradient elution of 80–60% acetonitrile containing 0.1% TFA. The concentration of the protein was estimated spectrophotometrically using the calculated molar extinction coefficient of 6250 M−1cm−1 at 280 nm.69 Freshly purified GRN‐3 was used for every experiment.
SOFAST heteronuclear multiple‐quantum coherence (HMQC)
The NMR spectra for GRN‐3, mGRN‐3, or rGRN‐3 was acquired on the Bruker Advance – III‐HD 850 MHz NMR spectrometer equipped with a Bruker TCI cryoprobe at the high field NMR facility of University of Alabama, Birmingham. SOFAST HMQC was used as it allows reduction of the recycle delays (T scan) to ≤ 100 ms while maintaining high sensitivity, thereby decreasing the overall experimental time.70 HMQC spectra was obtained for 40 μM (152 μg) of GRN‐3 or mGRN‐3 and 10 μM (38 μg) of rGRN‐3 resuspended in 20 mM Tris, pH 6.5 with 10% D2O inside a Shigemi NMR tube. The spectra were collected with 2048 data points in F2 (1H) with 128 scans coadded for each of the 160 F1 (15N) increments. A 1J(NH) of 90 Hz was used with a 100 ms relaxation delay. The delay interval from F1 to F2 was set at 0.32 ms. The HMQC spectra were acquired at 25°C and were processed using Bruker TopSpin 3.5 analysis software with standard methods with phase corrections in both dimensions.
Evaluation of intrinsic disorder propensity of human PGRN and individual GRNs
Intrinsic disorder predisposition of human PGRN (UniProt ID: P28799) was evaluated by three members of the PONDR family (PONDR® VLXT,25 PONDR® VL3,71 and PONDR® VSL272), whereas peculiarities of disorder propensity in individual GRNs were studied using the FoldIndex graphic web server.37 PONDR® VLXT is one of the first‐generation disorder predictors. Although its accuracy is not too high, this predictor is known to have high sensitivity to local sequence peculiarities and can be used for identifying disorder‐based interaction sites.25, 27, 73, 74 In fact, because it was trained on a limited set of then available proteins with experimentally validated intrinsically disordered status, PONDR® VLXT may underestimate the occurrence of long disordered regions in proteins. PONDR® VL3 was specifically designed to accurately predict long disordered regions.71 PONDR® VSL2B is one of the more accurate stand‐alone disorder predictors.72, 75, 76 FoldIndex is a method developed from the charge‐hydrophobicity plots30 by rearranging the terms in the basic equation and by adding the technique of sliding windows.37 Here, by applying a sliding window centered at a specific residue (the length of window can be selected by the user), the position of this segment on charge‐hydrophobicity plot can be calculated, and the distance of this position away from the boundary line is taken as an indication whether the central residue is disordered or not.37
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website.
Supporting Information
Acknowledgments
The authors also wish to thank the MS INBRE for use of its core facilities at USM, and Prof. Timothy Logan (Florida State University) for his help on NMR data analysis. The authors are thankful to Alexey Uversky for careful reading and editing of this manuscript.
References
- 1. Bateman A, Belcourt D, Bennett H, Lazure C, Solomon S (1990) Granulins, a novel class of peptide from leukocytes. Biochem Biophys Res Commun 173:1161–1168. [DOI] [PubMed] [Google Scholar]
- 2. Bateman A, Bennett H (1998) Granulins: the structure and function of an emerging family of growth factors. J Endocrin 158:145–151. [DOI] [PubMed] [Google Scholar]
- 3. Bhandari V, Palfree R, Bateman A (1992) Isolation and sequence of the granulin precursor cDNA from human bone marrow reveals tandem cysteine‐rich granulin domains. Proc Natl Acad Sci USA 89:1715–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Naphade SB, Kigerl KA, Jakeman LB, Kostyk SK, Popovich PG, Kuret J (2010) Progranulin expression is upregulated after spinal contusion in mice. Acta Neuropath 119:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moisse K, Volkening K, Leystra‐Lantz C, Welch I, Hill T, Strong MJ (2009) Divergent patterns of cytosolic TDP‐43 and neuronal progranulin expression following axotomy: implications for TDP‐43 in the physiological response to neuronal injury. Brain Res 1249:202–211. [DOI] [PubMed] [Google Scholar]
- 6. Tanaka Y, Matsuwaki T, Yamanouchi K, Nishihara M (2013) Exacerbated inflammatory responses related to activated microglia after traumatic brain injury in progranulin‐deficient mice. Neuroscience 231:49–60. [DOI] [PubMed] [Google Scholar]
- 7. Revuelta GJ, Rosso A, Lippa CF (2008) Association between progranulin and β‐amyloid in dementia With Lewy bodies. Am. J Alzheimer's Dis Other Dementias 23:488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pereson S, Wils H, Kleinberger G, McGowan E, Vandewoestyne M, Van Broeck B, Joris G, Cuijt I, Deforce D, Hutton M (2009) Progranulin expression correlates with dense‐core amyloid plaque burden in Alzheimer disease mouse models. J Pathol 219:173–181. [DOI] [PubMed] [Google Scholar]
- 9. Zanocco‐Marani T, Bateman A, Romano G, Valentinis B, He Z‐H, Baserga R (1999) Biological activities and signaling pathways of the granulin/epithelin precursor. Cancer Res 59:5331–5340. [PubMed] [Google Scholar]
- 10. Toh H, Chitramuthu BP, Bennett HP, Bateman A (2011) Structure, function, and mechanism of progranulin; the brain and beyond. J Mol Neurosci 45:538–548. [DOI] [PubMed] [Google Scholar]
- 11. Baker M, Mackenzie IR, Pickering‐Brown SM al. e (2006) Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919. [DOI] [PubMed] [Google Scholar]
- 12. Brouwers N, Nuytemans K, van der Zee J, et al. (2007) ALzheimer and parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol 64:1436–1446. [DOI] [PubMed] [Google Scholar]
- 13. Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar‐Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin‐positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924. [DOI] [PubMed] [Google Scholar]
- 14. Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument‐Bromage H, Tempst P, Wright CD, Ding A (2002) Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell 111:867–878. [DOI] [PubMed] [Google Scholar]
- 15. Plowman GD, Green JM, Neubauer MG, Buckley SD, McDonald VL, Todaro GJ, Shoyab M (1992) The epithelin precursor encodes two proteins with opposing activities on epithelial cell growth. J Biol Chem 267:13073–13078. [PubMed] [Google Scholar]
- 16. Goretzki L, Lombardo CR, Stallcup WB (2000) Binding of the NG2 proteoglycan to kringle domains modulates the functional properties of angiostatin and plasmin(ogen). J Biol Chem 275:28625–28633. [DOI] [PubMed] [Google Scholar]
- 17. Stec B, Yamano A, Whitlow M, Teeter MM (1997) Structure of human plasminogen kringle 4 at 1.68 Å and 277 K. A possible structural role of disordered residues. Acta Cryst D53:169–178. [DOI] [PubMed] [Google Scholar]
- 18. Montelione GT, Winkler ME, Burton LE, Rinderknecht E, Sporn MB, Wagner G (1989) Sequence‐specific 1H‐NMR assignments and identification of two small antiparallel beta‐sheets in the solution structure of recombinant human transforming growth factor alpha. Proc Natl Acad Sci USA 86:1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kohda D, Inagaki F (1991) 3D Structures of EGF and TGF‐alpha determined by NMR. Anal Sci 7:853–856. [Google Scholar]
- 20. Ghag G, Wolf LM, Reed RG, Van Der Munnik NP, Mundoma C, Moss MA, Rangachari V (2016) Fully reduced granulin‐B is intrinsically disordered and displays concentration‐dependent dynamics. Prot Eng Des Sel. 29:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tolkatchev D, Malik S, Vinogradova A, Wang P, Chen Z, Xu P, Bennett HP, Bateman A, Ni F (2008) Structure dissection of human progranulin identifies well‐folded granulin/epithelin modules with unique functional activities. Protein Sci 17:711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bessette PH, Åslund F, Beckwith J, Georgiou G (1999) Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc Natl Acad Sci USA 96:13703–13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stewart EJ, Åslund F, Beckwith J (1998) Disulfide bond formation in the Escherichia coli cytoplasm: an in vivo role reversal for the thioredoxins. EMBO J 17:5543–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang J, Van Damme P, Cruchaga C, Gitcho MA, Vidal JM, Seijo‐Martínez M, Wang L, Wu JY, Robberecht W, Goate A (2010) Pathogenic cysteine mutations affect progranulin function and production of mature granulins. J Neurochem 112:1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z (2001) Intrinsically disordered protein. J Mol Graph Model 19:26–59. [DOI] [PubMed] [Google Scholar]
- 26. Williams RM, Obradovi Z, Mathura V, Braun W, Garner EC, Young J, Takayama S, Brown CJ, Dunker AK (2001) The protein non‐folding problem: amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput 89–100. [DOI] [PubMed] [Google Scholar]
- 27. Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK (2001) Sequence complexity of disordered protein. Proteins 42:38–48. [DOI] [PubMed] [Google Scholar]
- 28. Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK (2007) Intrinsic disorder and functional proteomics. Biophys J 92:1439–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vacic V, Uversky VN, Dunker AK, Lonardi S (2007) Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinform 8:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Uversky VN, Gillespie JR, Fink AL (2000) Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41:415–427. [DOI] [PubMed] [Google Scholar]
- 31. Fontana A, Fassina G, Vita C, Dalzoppo D, Zamai M, Zambonin M (1986) Correlation between sites of limited proteolysis and segmental mobility in thermolysin. Biochemistry 25:1847–1851. [DOI] [PubMed] [Google Scholar]
- 32. Polverino dLP, De Filippis V, Di Bello M, Zambonin M, Fontana A (1995) Probing the molten globule state of alpha‐lactalbumin by limited proteolysis. Biochemistry 34:12596–12604. [DOI] [PubMed] [Google Scholar]
- 33. Fontana A, de Laureto PP, De Filippis V, Scaramella E, Zambonin M (1997) Probing the partly folded states of proteins by limited proteolysis. Fold Des 2:R17–R26. [DOI] [PubMed] [Google Scholar]
- 34. Iakoucheva LM, Kimzey AL, Masselon CD, Bruce JE, Garner EC, Brown CJ, Dunker AK, Smith RD, Ackerman EJ (2001) Identification of intrinsic order and disorder in the DNA repair protein XPA. Protein Sci 10:560–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fontana A, de Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M (2004) Probing protein structure by limited proteolysis. Acta Biochim Polon 51:299–322. [PubMed] [Google Scholar]
- 36. de Laureto PP, Tosatto L, Frare E, Marin O, Uversky VN, Fontana A (2006) Conformational properties of the SDS‐bound state of α‐synuclein probed by limited proteolysis: unexpected rigidity of the acidic C‐terminal tail. Biochemistry 45:11523–11531. [DOI] [PubMed] [Google Scholar]
- 37. Prilusky J, Felder CE, Zeev‐Ben‐Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL (2005) FoldIndex©: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 21:3435–3438. [DOI] [PubMed] [Google Scholar]
- 38. André I, Linse S, Mulder FAA (2007) Residue‐specific pKa determination of lysine and arginine side chains by indirect 15N and 13C NMR spectroscopy: Application to apo calmodulin. J Am Chem Soc 129:15805–15813. [DOI] [PubMed] [Google Scholar]
- 39. Poon DKY, Schubert M, Au J, Okon M, Withers SG, McIntosh LP (2006) Unambiguous determination of the ionization state of a glycoside hydrolase active site lysine by 1H−15N heteronuclear correlation spectroscopy. J Am Chem Soc 128:15388–15389. [DOI] [PubMed] [Google Scholar]
- 40. Henry GD, Sykes BD (1995) Determination of the rotational dynamics and pH dependence of the hydrogen exchange rates of the arginine guanidino group using NMR spectroscopy. J Biomol NMR 6:59–66. [DOI] [PubMed] [Google Scholar]
- 41. Tompa P, Fersht A. (2009) Molecular Functions of Disordered Proteins, In Structure and Function of Intrinsically Disordered Proteins. pp 163–188, hapman and Hall/CRC, Boca Raton, FL. [Google Scholar]
- 42. Woody RW (1992) Circular dichroism and conformation of unordered polypeptides. Adv Biophys Chem 2:37–79. [Google Scholar]
- 43. Bovey F, Hood F (1967) Circular dichroism spectrum of poly‐L‐proline. Biopolymers 5:325–326. [DOI] [PubMed] [Google Scholar]
- 44. Adzhubei AA, Sternberg MJE, Makarov AA (2013) Polyproline‐II helix in proteins: Structure and function. J Mol Biol 425:2100–2132. [DOI] [PubMed] [Google Scholar]
- 45. Creamer TP, Campbell MN (2002) Determinants of the polyproline II helix from modeling studies. Adv Prot Chem 62:263–282. [DOI] [PubMed] [Google Scholar]
- 46. Hu K‐N, Havlin RH, Yau W‐M, Tycko R (2009) Quantitative determination of site‐specific conformational distributions in an unfolded protein by solid‐state nuclear magnetic resonance. J Mol Biol 392:1055–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Syme CD, Blanch EW, Holt C, Jakes R, Goedert M, Hecht L, Barron LD (2002) A Raman optical activity study of rheomorphism in caseins, synucleins and tau. Eur J Biochem 269:148–156. [DOI] [PubMed] [Google Scholar]
- 48. Woody RW (2009) Circular dichroism spectrum of peptides in the poly (Pro) II conformation. J Am Chem Soc 131:8234–8245. [DOI] [PubMed] [Google Scholar]
- 49. Roy A, Kucukural A, Zhang Y (2010) I‐TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang Y (2008) I‐TASSER server for protein 3D structure prediction. BMC Bioinform 9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stryer L (1965) The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. J Mol Biol 13:482–495. [DOI] [PubMed] [Google Scholar]
- 52. Semisotnov G, Rodionova N, Razgulyaev O, Uversky V, Gripas A, Gilmanshin R (1991) Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 31:119–128. [DOI] [PubMed] [Google Scholar]
- 53. Lakowicz JR. (1983). Protein Fluorescence, In Principles of Fluorescence Spectroscopy, pp 341–381, Springer US, Boston, MA. [Google Scholar]
- 54. Reynolds JA, Tanford C (1970) The gross conformation of protein‐sodium dodecyl sulfate complexes. J Biol Chem 245:5161–5165. [PubMed] [Google Scholar]
- 55. Mattice WL, Riser JM, Clark DS (1976) Conformational properties of the complexes formed by proteins and sodium dodecyl sulfate. Biochemistry 15:4264–4272. [DOI] [PubMed] [Google Scholar]
- 56. Montserret R, McLeish MJ, Böckmann A, Geourjon C, Penin F (2000) Involvement of electrostatic interactions in the mechanism of peptide folding induced by sodium dodecyl sulfate binding. Biochemistry 39:8362–8373. [DOI] [PubMed] [Google Scholar]
- 57. Moore BD, Rangachari V, Tay WM, Milkovic NM, Rosenberry TL (2009) Biophysical analyses of synthetic amyloid‐β (1− 42) aggregates before and after covalent cross‐linking. Implications for deducing the structure of endogenous amyloid‐β oligomers. Biochemistry 48:11796–11806. [DOI] [PubMed] [Google Scholar]
- 58. Otzen DE, Oliveberg M (2002) Burst‐phase expansion of native protein prior to global unfolding in SDS. J Mol Biol 315:1231–1240. [DOI] [PubMed] [Google Scholar]
- 59. Salazar DA, Butler VJ, Argouarch AR, Hsu T‐Y, Mason A, Nakamura A, McCurdy H, Cox D, Ng R, Pan G (2015) The progranulin cleavage products, granulins, exacerbate TDP‐43 toxicity and increase TDP‐43 levels. J Neurosci 35:9315–9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Matsumoto K, Takehara T, Inoue H, Hagiya M, Shimizu S, Nakamura T (1991) Deletion of kringle domains or the N‐terminal hairpin structure in hepatocyte growth factor results in marked decreases in related biological activities. Biochem Biophys Res Commun 181:691–699. [DOI] [PubMed] [Google Scholar]
- 61. Nakamura T, Nishizawa T, Hagiya M, Seki T, Shimonishi M, Sugimura A, Tashiro K, Shimizu S (1989) Molecular cloning and expression of human hepatocyte growth factor. Nature 342:440–443. [DOI] [PubMed] [Google Scholar]
- 62. Magnusson S, Petersen TE, Sottrup‐Jensen L, Claeys H (1975) Complete primary structure of prothrombin: Isolation, structure and reactivity of ten carboxylated glutamic acid residues and regulation of prothrombin activation by thrombin. Proteases Biol Control 2:123–149. [Google Scholar]
- 63. McLean JW, Tomlinson JE, Kuang W‐J, Eaton DL, Chen EY, Fless GM, Scanu AM, Lawn RM (1987) cDNA sequence of human apolipoprotein (a) is homologous to plasminogen. Nature 330:132–137. [DOI] [PubMed] [Google Scholar]
- 64. De Vos AM, Ultsch MH, Kelley RF, Padmanabhan K, Tulinsky A, Westbrook ML, Kossiakoff AA (1992) Crystal structure of the kringle 2 domain of tissue plasminogen activator at 2.4‐. ANG. resolution. Biochemistry 31:270–279. [DOI] [PubMed] [Google Scholar]
- 65. Hrabal R, Chen Z, James S, Bennett H, Ni F (1996) The hairpin stack fold, a novel protein architecture for a new family of protein growth factors. Nat Struct Biol 3:747–752. [DOI] [PubMed] [Google Scholar]
- 66. Huang H‐W, Mohan SK, Yu C (2010) The NMR solution structure of human epidermal growth factor (hEGF) at physiological pH and its interactions with suramin. Biochem Biophys Res Commun 402:705–710. [DOI] [PubMed] [Google Scholar]
- 67. Tompa P (2002) Intrinsically unstructured proteins. Trends Biochem Sci 27:527–533. [DOI] [PubMed] [Google Scholar]
- 68. Tompa P, Szász C, Buday L (2005) Structural disorder throws new light on moonlighting. Trends Biochem Sci 30:484–489. [DOI] [PubMed] [Google Scholar]
- 69. Gasteiger E, Hoogland C, Gattiker A, Duvaud Se, Wilkins M, Appel R, Bairoch A (2005) Protein Identification and Analysis Tools on the ExPASy Server In: Walker J, Ed. (2005) The proteomics protocols handbook. Humana Press, pp. 571–607. Humana Press, Totowa, NJ. [Google Scholar]
- 70. Schanda P, Brutscher B (2005) Very fast two‐dimensional NMR spectroscopy for real‐time investigation of dynamic events in proteins on the time scale of seconds. J Am Chem Soc 127:8014–8015. [DOI] [PubMed] [Google Scholar]
- 71. Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z (2006) Length‐dependent prediction of protein intrinsic disorder. BMC Bioinform 7:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, Obradovic Z (2005) Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol 3:35–60. [DOI] [PubMed] [Google Scholar]
- 73. Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK (2005) Coupled folding and binding with α‐helix‐forming molecular recognition elements. Biochemistry 44:12454–12470. [DOI] [PubMed] [Google Scholar]
- 74. Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, Dunker AK (2007) Mining α‐helix‐forming molecular recognition features with cross species sequence alignments. Biochemistry 46:13468–13477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Peng Z‐L, Kurgan L (2012) Comprehensive comparative assessment of in‐silico predictors of disordered regions. Curr Prot Peptide Sci 13:6–18. [DOI] [PubMed] [Google Scholar]
- 76. Fan X, Kurgan L (2014) Accurate prediction of disorder in protein chains with a comprehensive and empirically designed consensus. J Biomol Struct Dynam 32:448–464. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website.
Supporting Information