

Abstract
Background
STIM/ORAI-mediated store-operated Ca2+ entry (SOCE) mediates a myriad of Ca2+-dependent cellular activities in mammals. Genetic defects in STIM1/ORAI1 lead to devastating severe combined immunodeficiency; whereas gain-of-function mutations in STIM1/ORAI1 are intimately associated with tubular aggregate myopathy. At molecular level, a decrease in the Ca2+ concentrations within the lumen of endoplasmic reticulum (ER) initiates multimerization of the STIM1 luminal domain to switch on the STIM1 cytoplasmic domain to engage and gate ORAI channels, thereby leading to the ultimate Ca2+ influx from the extracellular space into the cytosol. Despite tremendous progress made in dissecting functional STIM1-ORAI1 coupling, the activation mechanism of SOCE remains to be fully characterized.
Objective and Methods
Building upon a robust fluorescence resonance energy transfer assay designed to monitor STIM1 intramolecular autoinhibition, we aimed to systematically dissect the molecular determinants required for the activation and oligomerization of STIM1.
Results
Here we showed that truncation of the STIM1 luminal domain predisposes STIM1 to adopt a more active conformation. Replacement of the single transmembrane (TM) domain of STIM1 by a more rigid dimerized TM domain of glycophorin A abolished STIM1 activation. But this adverse effect could be partially reversed by disrupting the TM dimerization interface. Moreover, our study revealed regions that are important for the optimal assembly of heterooligomers composed of full-length STIM1 with its minimal STIM1-ORAI activating region, SOAR.
Conclusions
Our study clarifies the roles of major STIM1 functional domains in maintaining a quiescent configuration of STIM1 to prevent preactivation of SOCE.
Keywords: Calcium signaling, ORAI1, STIM1, protein-protein interaction, store-operated calcium entry, calcium release-activated calcium channel, FRET, imaging
Introduction
Store-operated Ca2+ entry (SOCE) constitutes one of the most important routes of Ca2+ flux from the extracellular space into the cytosol. Ca2+ release-activated Ca2+ (CRAC) channel composed of STIM1 and ORAI1 is a prototypical example of SOCE, in which the Ca2+ store depletion in the endoplasmic reticulum (ER) elicits a cascade of conformational changes within STIM1 that culminates in the engaging and opening of ORAI channels to conduct Ca2+ permeation across the plasma membrane [1-8]. SOCE is known to be present in most cell types including non-excitable cells (e.g., endothelial cells and cells of the immune system) and excitable cells such as skeletal muscle and neurons [1, 2, 9-12]. Recent studies on naturally occurring loss- or gain-of-function mutations in either ORAI1 or STIM1 have highlighted the crucial roles of STIM1-ORAI1 signaling in mediating a number of physiological functions, including, but not limited to immune response and muscle function [2, 10, 13-18]. The loss-of-function mutations in ORAI1 (e.g., R91W, and A103E/L194P) and STIM1 (e.g., R429C and P165Q) impair or abolish CRAC channel function to cause CRAC channelopathy, as clinically characterized by severe combined immunodeficiency (SCID), autoimmunity, muscular hypotonia, and ectodermal dysplasia [14-16, 18, 19]. By contrast, the gain-of-function somatic mutations in ORAI1 (e.g., G98S, L138F and P245L) or STIM1 (e.g., R304W) result in constitutive or augmented activation of CRAC channels [18, 20-22]. Patients with such gain-of-function mutations show nonsyndromic tubular aggregate myopathy (TAM) and Stormorken syndromes [18, 20-22]. The myopathy caused by either loss- or gain of-function mutations in ORAI1 and STIM1 highlights the importance of Ca2+ homeostasis maintained by CRAC channels in skeletal muscle function [18, 19, 21]. The cellular dysfunction and clinical diseases caused by mutations in ORAI1 and STIM1 provide critical information regarding the role of CRAC channels in human physiology and disease. A thorough understanding of CRAC channel activation and regulatory mechanisms will likely provide further insights into the development of novel therapeutic strategies to treat human diseases associated with aberrant CRAC channel function.
The molecular basis of functional STIM1-ORAI1 coupling has been studied in great detail over the past decade through collective efforts from multiple groups [1-3, 23-25]. STIM1 is a type I single-pass transmembrane protein that acts as an ER-resident Ca2+ sensor and is generally regarded to exist as a dimer at rest. The luminal EF-SAM (EF, EF-hand; SAM, sterile alpha motif) domain binds Ca2+ with a dissociation constant of approximately 200 μM in vitro [26, 27], a level that is comparable to the magnitude of free Ca2+ concentration in ER lumen [28, 29]. Following extracellular stimulation or pharmacological perturbations that lead to the depletion of ER Ca2+ store depletion, Ca2+ dissociates from the STIM1 luminal EF-hand to trigger conformational changes in the EF-hands and the SAM domain, thus destabilizing the luminal EF-SAM domain to drive its dimerization/oligomerization [26, 28]. This substantial structural changes bring about the close apposition of the two ends of the STIM1 transmembrane domain (TM), probably via the rearrangement of the inter-helical angle or spacing of the adjacent TM segments of STIM1 dimers [30], to transmit luminal signals toward the cytoplasmic domain of STIM1 (STIM1ct). Close apposition of a dimeric STIM1ct at the N-termini disrupts the intramolecular autoinhibition mediated by putative coiled-coil interplays [31, 32] between the juxtamembrane coiled coil 1 (CC1) region and a minimal STIM1-ORAI activating region known as SOAR (aa 344-442) or CAD (aa 342-448) [33, 34]. To aid the visualization of the intramolecular trapping of STIM1, we have recently developed a real-time fluorescence assay to monitor the CC1-SOAR interaction and map the CC1-SOAR contact interface (L258-L261/V416-V419) in physiologically-relevant conditions [30]. Once the inhibitory clamp of CC1-SOAR is disrupted, STIM1ct switches from a ‘folded-back’ conformation to a more extended conformation to fully expose SOAR/CAD to gate ORAI1 Ca2+ channels [30, 31, 35]. Physical interactions of SOAR/CAD with both termini of ORAI1 lead to the rearrangement of the ORAI pore and selectively permit extracellular Ca2+ flux into the cytosol [1-3, 23, 36-40]. Despite all the progress in the mechanistic dissection of SOCE activation, two outstanding questions remain unclarified: how does each functional domain contribute to the initial activation of a dimeric STIM1? What are the molecular determinants driving the intermolecular oligomerization of STIM1 following its initial activation?
In the current study, building upon a Ca2+ influx assay together with a FRET or colocalization assay designed to monitor STIM1 intramolecular interaction mediated by CC1 and SOAR/CAD, we dissected the role of STIM1 luminal and TM domains in signal transduction across the ER membrane. In parallel, by mixing full-length STIM1 with a series of truncated STIM1ct fragments, we scrutinized the molecular determinants that regulate the inter-molecular interaction of STIM1. Our findings yield further insights into the molecular mechanism driving STIM1 activation during SOCE.
Results
STIM1 Luminal Domain is Required to Keep the Cytoplasmic Domain of STIM1 Inactive at Rest
At rest, the isolated STIM1 luminal EF-SAM domain exists as a Ca2+-bound monomer; meanwhile, the cytoplasmic domain of STIM1 (STIM1ct) adopts an inactive folded-back configuration through autoinhibitory interactions between its CC1 and SOAR/CAD (Fig. 1A-B) [1, 30, 32]. To test if the luminal domain is required to keep STIM1 quiescent at rest (without store depletion), we generated a construct devoid of the EF-SAM domain (STIM1209-685; ΔN in Fig. 1B). Transient expression of this truncated construct in HEK293 cells stably expressing ORAI1 (thereafter designated as “HEK293-ORAI1”) induced robust constitutively Ca2+ influx, with the maximal signals reaching nearly half of Ca2+ responses detected in HEK293-ORAI1 cells expressing SOAR/CAD (red and blue; Fig. 1C-D). As a negative control, the heterologous expression of the full-length STIM1 (aa 1-685) in HEK293-ORAI1 cells did not elicit significant constitutive Ca2+ influx and failed to respond to the changes of Ca2+ concentrations in the external medium (black; Fig. 1C-D). Consistent with the Ca2+ influx results, GFP-STIM1209-685 expressed in HeLa cells was preactivated, as reflected by spontaneous puncta formation and colocalization with ORAI1 without store depletion (Fig. 1E). Further truncation of the transmembrane domain (TM) resulted in a cytosolic protein, STIM1233-685 (STIM1ct), which exhibited marginal constitutive Ca2+ influx (green; Fig. 1C-D). These results clearly demonstrated that the luminal region of STIM1 works in connection with its TM domain to keep STIM1 inactive at rest.
Fig. 1. STIM1 luminal domain is required to maintain STIM1 in an inactive state at rest.
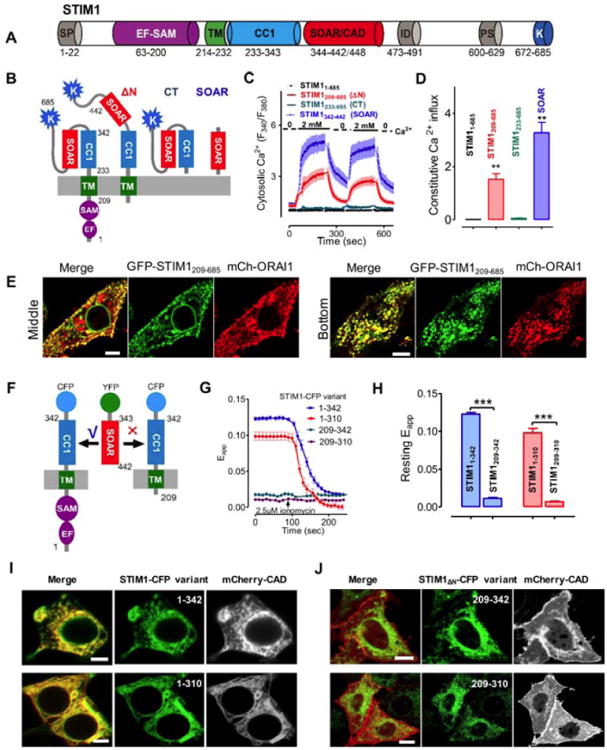
(A). Domain organization of the full-length human STIM1. EF, Ca2+-binding EF-hand motif; SAM, sterile alpha motif; TM, transmembrane domain; CC1, putative coiled-coil 1 region; SOAR/CAD, minimal ORAI-activating domain; ID, inhibitory domain; PS, proline/serine-rich region; K, polybasic domain.
(B-D) Ca2+ influx in HEK293-ORAI1 stable cells transiently expressing STIM1 or its truncated fragments monitored by Fura-2 fluorescence ratio. Constitutive Ca2+ entry was assessed by switching the external media between 0 and 2 mM Ca2+. (B) Schematic of STIM1 variants used in the assay. ΔN, STIM1209-685; CT, STIM1233-685 and SOAR, STIM1343-442. (C) Representative traces of cytosolic Ca2+ response from three independent experiments are shown here. Cells were kept in nominally Ca2+ free solution, 2 mM Ca2+ were present in the external medium only at times indicated by solid bars. (D) Statistics of the mean constitutive Ca2+ influx (n = 3, 20-50 cells each time).
(E) Confocal images (left, middle section; right, footprint layer) of HeLa cells co-expressing GFP-STIM1209-685 and mCherry- ORAI1. Scale bar, 5 μm.
(F-H) FRET measurements on HEK293 cells co-expressing YFP-SOAR with each of the indicated STIM1-CFP variants. (F) Schematic of the donor-acceptor pair used in the FRET assay. YFP-SOAR bound to STIM11-342-CFP (or STIM11-310-CFP), but failed to associate with the variants devoid of the N-terminus (aa 1-209) (G) Representative traces of FRET signals during the course of ionomycin (2.5 μM)-induced store depletion. (H) Bar graphs showing the statistical results of the resting FRET signals. (I-J) Confocal images of HeLa cells co-expressing mCherry-CAD with the indicated STIM1-CFP (I) or STIM1Δn-CFP variants (J). Scale bar, 5 μm.
All data were presented as mean ± s.e.m. **P 0.01 and ***P 0.001 (paired Student's t-test).
To evaluate how STIM1 luminal domain affects the conformation of its cytoplasmic domain, we resorted to a FRET assay recently developed by us to visualize the autoinhibitory CC1-SOAR interactions at real-time (Fig. 1F) [30]. Under resting conditions without store depletion, strong FRET signals were detected in HEK293 cells coexpressing YFP-SOAR with STIM11-310 or STIM11-342 (Fig. 1G-H). Confocal imaging further revealed that mCherry-CAD displayed an ER-like distribution pattern in HeLa cells and colocalized with ER-resident STIM11-310 or STIM11-342 (Fig. 1I). Following rapid store depletion induced by ionomycin, we observed a substantial decrease in the FRET signals between YPF-SOAR and STIM11-310 or STIM11-342. These findings imply that SOAR/CAD is tightly docked to the cytoplasmic side of ER via its interaction with CC1 when the Ca2+ store is full. By contrast, the deletion of luminal domain (aa 1-208) within the corresponding STIM1 proteins abolished the docking of mCherry-CAD toward ER membrane (Fig. 1J). The majority of mCherry-CAD were either dispersed into the cytosol or translocated to the plasma membrane, presumably by binding to endogenous ORAI proteins. As a result, the resting FRET signals in those cells remained extremely low and showed no response to store depletion (blue and purple; Fig. 1G-H). Collectively, results from both imaging and Ca2+ influx assays were converged to suggest that the deletion of the luminal domain disrupts the resting CC1-SOAR autoinhibitory interactions to cause STIM1 activation.
Structural Flexibility of the Transmembrane Domain of STIM1 is Required for Efficient Lumen-to-Cytosol Signal Transmission
The full-length STIM1 remains largely inactive without store depletion and the removal of its luminal domain efficiently overcame auto-inhibition to activate STIM1 (Fig. 1). The deletion of both the luminal and TM domains, nonetheless, makes STIM1 returning to a less activated state [31, 34]. Thus, in the absence of conformational restraints imposed by the luminal domain, the TM domain itself seems to be able to trap STIM1ct in a more activated conformational state. We, therefore, reasoned that some structural flexibility of two adjacent TM helices within a STIM1 homodimer might be needed to switch on STIM1. To test this idea, we generated a chimeric STIM1 by replacing the STIM1-TM (aa 214-230) with a model transmembrane segment (aa 73-94) derived from glycophorin A (GpA). GpA is a primary sialoglycoprotein of human erythrocyte membranes which forms a noncovalent dimer via sequence-specific (79GXXXGXXG86) interactions between two TM helices (Fig. 2A) [41-45]. The stability of the dimeric GpA-TM can be disrupted by introducing mutations in the helix–helix contact interface (e.g., G83I) [41-43, 46]. We found that both the chimeric GpA-STIM1s and their mutant GpAG83I-STIM1, similar to wild-type STIM1, displayed typical ER-like tubular distribution patterns in the HeLa cells (Fig. 2C-D), confirming that the introduction of GpA-TM into STIM1 did not alter its subcellular localization. Ca2+ imaging results in HEK293-ORAI1 cells expressing GpA-STIM1 or GpAG83I-STIM1 which further showed that the replacement of TM did not spontaneously activate the chimeric STIM1 or alter Ca2+ release from ER store (the first peak; Fig. 2B). Notably, upon store depletion, the cells expressing GpA-STIM1 failed to produce the second Ca2+ response peak (representative of SOCE) in a typical “Ca2+ add-back” experiment (blue; Fig. 2B), suggesting that GpA-TM abolished the activation of chimeric STIM1. Interestingly, the dimer-disruptive chimera GpAG83I-STIM1 partly (∼ 30%, compared to WT STIM1) recovered the second phase of Ca2+ influx (red; Fig. 2B). Confocal imaging results also showed that GFP-GpA-STIM1 failed to form puncta or colocalize with mCherry-ORAI after store depletion (Fig. 2E). GFP-GpAG83I-STIM1, on the other hand, readily formed puncta to colocalize with mCherry-ORAI1 following TG-induced store depletion (Fig. 2F). GpA-TM has been thoroughly characterized as a dimer even in aqueous detergent micelles or in the presence of SDS, which suggests that the GpA-TM dimer is very stable and rigid [42, 43]. We speculate that the Ca2+ depletion-induced conformational changes in the EF-SAM domain might not be sufficient to trigger the repacking of GpA-TM helices and thus fail to transmit ER luminal signal to the cytosol. By contrast, the G83I mutation disrupts the tight helix-helix packing to enable a more flexible rearrangement of the contact interface, thereby enabling at least partial signal transduction across the ER membrane.
Fig. 2. Replacement of STIM1-TM by GpA-TM impairs the lumen-to-cytosol signal transmission.
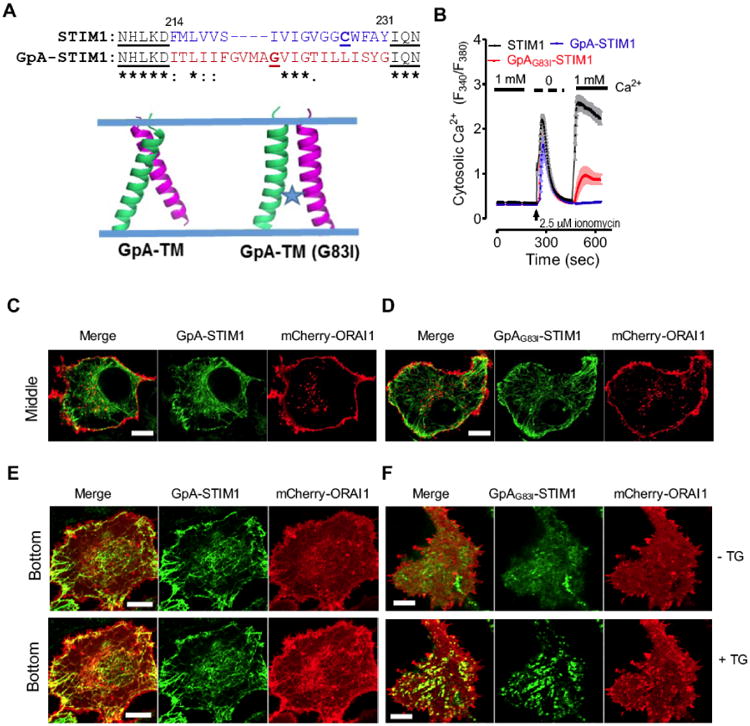
(A) A dimeric TM domain from glycophorin A (GpA; aa 73-94) was used to replace STIM1-TM (aa 214-230). Top, sequence alignment of STIM1-TM with GpA-TM; Bottom, modeled structures of a dimeric GpA-TM dimer. G83I is a well-known disruptive mutant of GpA that destabilizes its dimerization.
(B) Ca2+ response curves in HEK293-ORAI1-CFP stable cells transiently transfected with wild-type STIM1, chimeric GpA-STIM1 or the mutant GpAG83I-STIM1 monitored by ratiometric Fura-2 fluorescence (F340 nm/F380 nm). Store depletion was induced by 2.5 μM ionomycin. Shown were representative traces from three independent experiments (n = 3, 20∼30 cells per measurement). The solid bar above the curves indicates the presence of 1 mM Ca2+ in the external medium.
(C-D) Confocal images of the middle planes of HeLa cells co-expressing mCherry-ORAI1 and GFP-GpA-STIM1 (C) or the mutant GFP-GpAG83I-STIM1 (D). Both GpA-STIM1 and its mutant GpAG83I-STIM1 showed good ER distribution at rest. Scale bar, 5 μm.
(E-F) Confocal images of footprints of HeLa cells co-expressing mCherry-ORAI1 and GFP-GpA-STIM1 (E) or the mutant GFP-GpAG83I-STIM1 (F). 1 μM thapsigargin (TG) was added to trigger store depletion. Scale bar, 5 μm.
Overexpressed SOAR/CAD Forms Heteromer with STIM1 and Inhibits STIM1 Activation
Our prior study showed that SOAR/CAD is capable of forming a stable heteromeric complex with STIM11-442, regardless of the Ca2+ filling status in the ER [30]. Here we further confirmed the interactions of YFP-SOAR/mCherry-CAD with two more STIM1-CFP variants (aa 1-448, 1-491; Fig. S1), as well as the full-length STIM1 (aa 1-685). As shown in Fig. 3A-C, the cells co-expressing mCherry-CAD and STIM1-YFP showed clear colocalization nearby the ER network. After TG-induced store depletion, both STIM1 fragments and SOAR retained their ER-like distribution. The same scenario was visualized between YFP-SOAR and other STIM1 variants (Fig. S1). Clearly, SOAR/CAD tightly docks to STIM1 or its truncated variants independent of store depletion, suggesting the formation of very stable heteromeric complexes between SOAR/CAD and ER-resident STIM1 variants.
Fig. 3. SOAR/CAD forms heterodimer with full length STIM1 to modulate STIM1 activation.
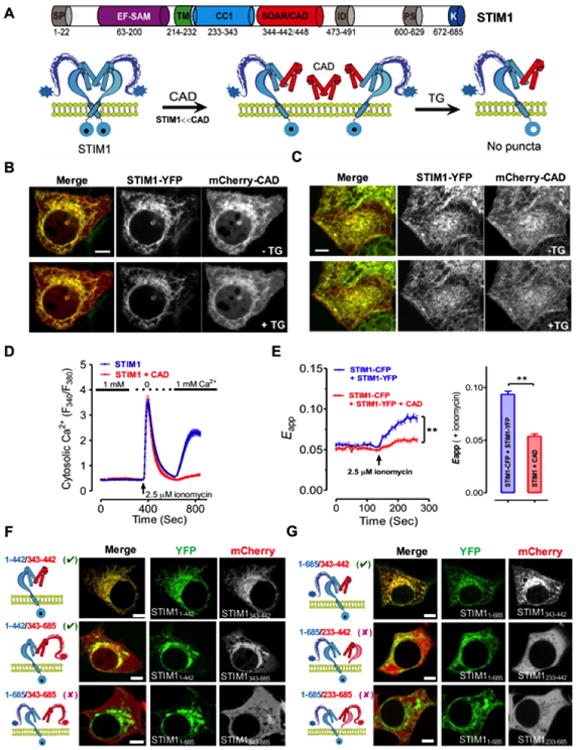
(A) A proposed model for the formation of possible CAD/STIM1 heteromeric complexes before and after TG-induced store depletion. When CAD is more abundant than STIM1, CAD may compete with STIM1 homodimer to form CAD:STIM1 heterodimer, but this may prevent the activation of a monomeric STIM1 in the heterodimer due to lack of signal transduction (requiring concerted efforts of TMs and CC1s from another copy of STIM1) across the ER membrane after store depletion. Therefore, puncta formation and Ca2+ response will be counter-intuitively suppressed.
(B-C) Confocal images of the middle planes (B) and footprints (C) of HeLa cells co-expressing STIM1-YFP and excessive amounts of mCherry-CAD constructs before (upper) and after (lower) store depletion induced by 1 μM TG. Scale bar, 5 μm.
(D) Ca2+ responses in HEK293 cells with overexpressed STIM1 alone (blue) or STIM1-YFP-T2A-mCherry-CAD (red) monitored by Fura-2 fluorescence ratio. The solid bar above the curves indicates 1 mM Ca2+ in the external medium.
(E) FRET signals in HEK293 cells co-expressing STIM1-CFP and STIM1-YFP, in the presence (red) or absence (blue) of excessive amounts of mCherry-CAD. Store depletion was induced by 2.5 μM ionomycin. The FRET signals after store depletion were plotted as bar graph. **P 0.01, paired Student's t-test.
(F) The combinations of possible STIM1 heterodimers tested in the colocalization assay (left) and confocal images of HeLa cells co-expressing STIM11-442-YFP or STIM1-YFP (middle) with the indicated mCherry-tagged STIM1 fragments (STIM1343-442 orSTIM1343-685; right). Scale bar, 5 μm.
(G) Constructs used for the colocalization assay (left) and confocal images of HeLa cells co-expressing STIM1-YFP (middle with the indicated STIM1 fragments: mCherry-STIM1343-442, mCherry-STIM1233-442, or mCherry-STIM1233-685 (right). Scale bar, 5 μm.
We then examined how the formation of such heteromeric complex impinged on Ca2+ influx and STIM1 activation (Fig. 3A-E). SOAR/CAD is well known to cause constitutive activation of endogenous ORAI channels when expressed alone in HEK293, HeLa or T cells [33, 34]. Counterintuitively, when overexpressing STIM1 with an equal amount CAD in HEK293 cells using the 2A self-cleaving peptide-based multicistronic vector [47], mCherry-CAD surprisingly suppressed ionomycin-induced SOCE (Fig. 3D), indicating that STIM1's ability to activate ORAI channel is compromised by CAD. To further examine how SOAR/CAD affects STIM1 oligomerization, we employed a FRET assay by fusing donor (CFP) or acceptor (YFP) fluorophores in the C-terminus of STIM1 to examine whether mCherry-CAD overexpression disturbed STIM1-STIM1 homomerization at the cytoplasmic domain. Following store depletion, the FRET signals of both the STIM1-CFP/STIM1-YFP pairs increased by ∼50% due to the formation of STIM1 oligomers. Overexpression of mCherry-CAD greatly diminished the ionomycininduced increases in FRET signals between C-terminally tagged STIM1-YFP and STIM1-CFP (Fig. 3E). This finding indicates that, after store depletion, the cytosolic oligomerization of STIM1 is significantly impaired by mCherry-CAD.
To further determine which regions might perturb the formation of STIM1-SOAR heteromers, we coexpressed STIM1-YFP or STIM11-442-YFP with mCherry-tagged STIM1ct (aa 233-685) or its truncated variants (aa 233-448 or 343-685) in HeLa cells, and examined YFP/mCherry colocalization by confocal microscopy (Fig. 3F&G). Unlike SOAR or CAD that was predominantly docked toward the ER in HeLa cells expressing STIM11-442-YFP (Figs. 3F and S1), only about half of the expressed STIM1343-685 was associated with STIM11-442, with the rest distributed in the cytosol or adjacent to plasma membrane via interaction with endogenous ORAI (Fig. 3F). We explained that C-terminal regions (aa 443-685) downstream from SOAR/CAD (aa 344-442) might partially cover critical residues required to mediate the STIM1-CAD heteromerization. Indeed, the inclusion of C-terminal regions in both molecules (STIM11-685 + STIM1343-685) completely abolished the heteromeric interaction (Fig. 3F). Similarly, we further found that full-length STIM1 failed to interact with STIM1233-442 or STIM1233-685, both of which contains the CC1 region upstream of the SOAR/CAD domain (Fig. 3G). Taken together, regions adjacent to CAD (STIM1233-343 and STIM1448-685) may introduce steric hindrance to interfere with the heteromeric interaction between STIM1 and SOAR/CAD.
Discussion
It has been generally accepted that STIM1 exists as dimer and adopts an inactive conformation when the ER Ca2+ store is full [1-3, 23, 36]. Truncation of the luminal domain of STIM1 (STIM1209-685), as shown by this study and an earlier study [48], leads to constitutive Ca2+ influx and spontaneous puncta formation. The present study provides further evidence to support the notion that STIM1 without its luminal domain adopts an activated conformation by overcoming its intramolecular autoinhibition (Fig. 1). On the contrary, with further truncation of TM domain in STIM1209-685 (STIM1233-685 or STIM1ct), the resultant cytosolic protein becomes largely inactive. Therefore, at least two “brakes” are in place to lock resting STIM1 in an inactive configuration: one is the luminal EF-SAM domain that not only senses ER luminal Ca2+ change [26, 28, 29] but also may impose conformational constraints on the downstream TM domain; and the other is an intramolecular clamp mediated by CC1-SOAR interactions in the cytoplasm [1, 30, 32].
The removal of luminal domain might render more structural flexibility of STIM1-TM to cause the rearrangement of transmembrane segments, thereby perturbing the juxtapositioning of two adjacent TM helices within one STIM1 homodimer. A tight and rigid packing of TM helices, even in a dimeric form (e.g., GpA-TM), abolishes the signal transmission from ER lumen toward the cytosol (Fig. 2). Our findings strongly suggest that the flexible positioning, rather than the oligomeric status of TM helices, plays a more important role in regulating STIM1-mediated signal transduction across the ER membrane. This view is further corroborated by results from our previous mutagenesis studies on STIM1-TM, in which STIM1-TM underwent a local conformational change rather than a “monomer-to-dimer” transition after store depletion [30]. Specifically, we identified two “gain-of-function” mutations (I220W and C227W) in STIM1-TM that exhibit local structural rearrangements (interhelical angle changes) to prompt conformational changes in the cytosolic CC1 region and ultimately abolish the autoinhibitory CC1-SOAR interaction [30]. Collectively, results from studies on both the “gain-of-function” mutants (I220W and C227W) [30] and the “loss-of-function” GpA-STIM1 chimera (Fig. 2) reinforce the conclusion that STIM1-TM plays an indispensable role in transmitting signals, rather than simply connecting the luminal domain with the cytoplasmic domain.
Following the initial activation of STIM1, molecular determinants driving further oligomerization of STIM1 remain unclarified. The most popular model argues that STIM1 oligomers may form through interdimeric interactions among STIM1 homodimers, with the exposed SOAR/CAD domain acting as a “crosslinker” [1]. This view is supported by indirect evidence garnered from FRET assays and changes in STIM1 diffusion rate after store depletion [49, 50]. In the current study, we found that overexpression of mCherry-CAD was capable of forming a stable heteromeric complex with full-length STIM1 even before store depletion. Most notably, overexpressed mCherry-CAD can perturb the FRET signals between the STIM1-CFP and STIM1-YFP homodimer (Fig. 3E), strongly supporting the capability of CAD to mediate intermolecular interactions in the cytosolic region (Fig. 3). Since both the upstream domain (CC1) and the downstream domain (443-685) of SOAR/CAD inhibit the heteromeric interaction between STIM1 and SOAR/CAD, we explained that SOAR/CAD has to be exposed in order to mediate intermolecular associations among STIM1 homodimers.
Conclusion
To conclude, we have shown that the luminal domain of STIM1 acts as a “brake” to prevent the activation of STIM1 and that the transmembrane domain plays a crucial role in STIM1-mediated inside-out signaling. Furthermore, SOAR/CAD domain, once exposed following the store depletion, can mediate further oligomerization of STIM1 to drive the full activation of STIM1. These findings not only afford novel insights into the SOCE activation mechanism, but also provide guidance for future optimization of STIM1-based optogenetic tools to control Ca2+ signaling [51-54]. Two outstanding questions remain to be addressed in the future: what is the exact STIM1:ORAI1 stoichiometry at ER-PM junctional sites? How does STIM1 binding to ORAI1 lead to the permeation of Ca2+ ions through the ORAI1 channel at the plasma membrane?
Methods
Constructs
Full-length cDNA of human STIM1 was subcloned into the pCMV6-XL5 vector (Origene) [55] with the insertion of EYFP between two additional NarI sites introduced immediately after residue N39. To generate full length STIM1-CFP/YFP and truncated STIM1-CFP/YFP variants, human STIM1 and its fragments were amplified by standard PCR and inserted into pECFP/YFP-N1 between XhoI and BamHI restriction sties. YFP/mcherry-STIM1 variants were made by inserting the corresponding STIM1 fragments into pEYFP-C1 or pmCherry-C1 between the XhoI and EcoRI sites. To generate the STIM1-GpA chimera, the transmembrane domain of STIM1 was substituted by GpA-TM domain in pCMV6-XL5-YFP-STIM1. The mutant construct G83I was subsequently made by using the QuikChange Lightning site-directed mutagenesis kit (Agilent). mCherry-CAD [34] and YFP-SOAR [33] were generated as previously described. To generate multicistronic vector STIM1-YFP-T2A-mCherry-CAD, multiple DNA fragments STIM1-YFP, T2A, mCherry-CAD and vector were amplified by standard PCR and then assembled by NEBuilder HiFi DNA Assembly Master Mix.
Real-time Intracellular Ca2+ Measurements
Intracellular Ca2+ levels were measured with Fura-2 AM by following our previous procedures [39, 40, 56, 57]. In brief, HEK293-ORAI1-CFP or STIM1-CFP stable cells cultured on cover slips were kept in a dye loading solution (107 mM NaCl, 7.2 mM KCl, 1.2 mM MgCl2, 1 mM CaCl2, 11.5 mM glucose, 20 mM HEPES-NaOH (pH 7.2) with 2 μM Fura-2 AM for 30 min. The cells were then kept in Fura-2 AM free solution for another 30 min. For HEK293-ORAI1 cells transfected with constitutively active STIM1 fragments, a dye loading solution containing 300 μM Ca2+ or nominally Ca2+ free solution was used to keep the cells healthy. Fura-2 signals were recorded using a ZEISS oberserver-A1 microscope equipped with a Lambda DG4 light source (Sutter Instruments), Brightline filter sets (part number: FURA2-C-000, Semrock Inc.), a 40× oil objective (NA = 1.30), and an iXon3 EMCCD camera (Oxford Instruments), and the MetaFluor software (Molecular Devices). Emission fluorescence at 505 nm generated by 340 nm excitation light (F340) and 380 nm light (F380) was collected every two seconds, and intracellular Ca2+ levels are shown as F340/F380 ratio. All experiments were carried out at room temperature. Traces shown are representative of at least three independent repeats with each including 30–60 single cells.
Confocal Microscopy
HEK293 and HeLa cells purchased from ATCC were used for fluorescence imaging. All the cells were grown in Dulbecco's modified Eagle's medium (DMEM, Sigma) supplemented with 10 mM HEPES and 10% heat-inactivated fetal bovine serum, unless otherwise noted. Transfections were performed using Lipofectamine 3000 (Life Technologies) following the manufacturer's instructions. For ER Ca2+ store depletion, the DMEM medium was firstly substituted by pre-warmed Ca2+-free Hank's Balanced Salt Solution (HBSS) before imaging. 1 μM thapsigargin or 2.5 μM ionomycin was used to induce store depletion. Live cell imaging was performed at room temperature with 60× oil lens on an inverted Nikon Eclipse Ti-E microscope customized with A1R-A1 confocal and motorized total internal reflection fluorescence (TIRF) modules by using argon-ion (405 nm and 488 nm) and helium-neon (543 nm) or diode (561 nm) as laser sources. Image analysis was performed using the NIS-Elements software (Nikon) or ImageJ (NIH).
FRET Measurements
FRET measurements were recorded using a ZEISS oberserver-A1 microscope equipped with a Lambda DG4 light source (Sutter Instruments), an Optosplit II Image Splitter (Cairn Research Limited), a 40× oil objective (NA = 1.30), and an ANDOR iXon3 EMCCD camera (Oxford Instruments), and the MetaFluor software (Molecular Devices). CFP (428.9±5.5Ex/465±32Em), YFP (502.6±11.2Ex/549±21Em), FRETraw (428.9±5.5Ex/549±21Em) filters were used to capture images (FCFP, FYFP and Fraw, respectively) every 10 seconds at room temperature. Three-channel corrected FRET was calculated as previously described [56-58). FRET signal was calculated using the following formula: FRETc = Fraw-Fd/Dd*FCFP-Fa/Da*FYFP where FRETc represents the corrected total amount of energy transfer, Fd/Dd represents measured bleed-through of CFP into the FRET filter (0.826), and Fa/Da represents measured bleed-through of YFP through the FRET filter (0.048). To reduce variations caused by differences in expression levels, FRETc values were normalized against donor fluorescence (FCFP) to generate N-FRET (normalized FRET) signal. To eliminate instrument-dependent factors, apparent FRET efficiency, Eapp, was calculated using the following equation: Eapp= N-FRET/(N-FRET+G) [58], where G (4.59) is the system-dependent factor. It is obtained with partial YFP photo-bleaching method: G = (FRETc-FRETcpost)/(FCFPpost –FCFP), where FRETcpost and FCFPpost correspond to FRETc and FCFP values after partial photo-bleach of YFP [58]. The intensity of the light used to bleach YFP was carefully chosen so that it will not bleach CFP at the same time. All fluorescence images were collected and briefly processed with MetaFluor software, and then the resulting data were further analyzed with Matlab R2012b software and plotted with Prism5 software. Representative traces of at least three independent experiments performed on 15-30 cells are shown as mean ± s.e.m.
Statistical Analyses
Unless otherwise noted, quantitative data are expressed as the mean and standard deviation of the mean (s.e.m.). Statistical significance was determined with two-tailed Student's t-test. *P<0.05; **P<0.01; ***P<0.001.
Supplementary Material
Acknowledgments
This work was supported by grants from the Welch Foundation (BE-1913 to YZ), the National Natural Science foundation of China (NSFC-31471279 to YW), the Recruitment Program for Young Professionals of China (to YW), the Program for New Century Excellent Talents in University (NCET-13-0061 to YW), the American Cancer Society (RSG-16-215-01-TBE to YZ) and the National Institutes of Health (R01GM112003 to YZ).
Footnotes
Authors' Contributions: YZ, YW and GM conceived the ideas and directed the work. GM, YZ, YW and SZ designed the study. GM, YK, LH and YZ designed and generated all the plasmid constructs. GM, SZ, LZ, YZ and YW performed all the fluorescence imaging and other cell-based experiments. GM, YZ, YW and SZ analyzed data, with input from the other authors. YZ, GM and YW wrote the manuscript.
Conflict Of Interest: The authors state that they have no competing interests.
Supplementary Material: Supplementary material is available on the publisher's website along with the published article.
Disclaimer: The above article has been published in Epub (ahead of print) on the basis of the materials provided by the author. The Editorial Department reserves the right to make minor modifications for further improvement of the manuscript.
References
- 1.Prakriya M, Lewis RS. Store-Operated Calcium Channels. Physiol Rev. 2015;95(4):1383–436. doi: 10.1152/physrev.00020.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012;13(9):549–65. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vig M, Peinelt C, Beck A, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312(5777):1220–3. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feske S, Gwack Y, Prakriya M, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441(7090):179–85. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 6.Zhang SL, Yu Y, Roos J, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437(7060):902–5. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roos J, DiGregorio PJ, Yeromin AV, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169(3):435–45. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liou J, Kim ML, Heo WD, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15(13):1235–41. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moccia F, Zuccolo E, Soda T, et al. Stim and Orai proteins in neuronal Ca(2+) signaling and excitability. Front Cell Neurosci. 2015;9:153. doi: 10.3389/fncel.2015.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trebak M, Zhang W, Ruhle B, et al. What role for store- operated Ca(2)(+) entry in muscle? Microcirculation. 2013;20(4):330–6. doi: 10.1111/micc.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiviluoto S, Decuypere JP, De Smedt H, Missiaen L, Parys JB, Bultynck G. STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet Muscle. 2011;1(1):16. doi: 10.1186/2044-5040-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stiber J, Hawkins A, Zhang ZS, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10(6):688–97. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W, Halligan KE, Zhang X, et al. Orai1-mediated I (CRAC) is essential for neointima formation after vascular injury. Circ Res. 2011;109(5):534–42. doi: 10.1161/CIRCRESAHA.111.246777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Picard C, McCarl CA, Papolos A, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360(19):1971–80. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev. 2009;231(1):189–209. doi: 10.1111/j.1600-065X.2009.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gwack Y, Srikanth S, Oh-Hora M, et al. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol. 2008;28(17):5209–22. doi: 10.1128/MCB.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baba Y, Nishida K, Fujii Y, Hirano T, Hikida M, Kurosaki T. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat Immunol. 2008;9(1):81–8. doi: 10.1038/ni1546. [DOI] [PubMed] [Google Scholar]
- 18.Lacruz RS, Feske S. Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci. 2015;1356:45–79. doi: 10.1111/nyas.12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCarl CA, Picard C, Khalil S, et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol. 2009;124(6):1311–1318.e7. doi: 10.1016/j.jaci.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Endo Y, Noguchi S, Hara Y, et al. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca(2)(+) channels. Hum Mol Genet. 2015;24(3):637–48. doi: 10.1093/hmg/ddu477. [DOI] [PubMed] [Google Scholar]
- 21.Nesin V, Wiley G, Kousi M, et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc Natl Acad Sci U S A. 2014;111(11):4197–202. doi: 10.1073/pnas.1312520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morin G, Bruechle NO, Singh AR, et al. Gain-of-Function Mutation in STIM1 (P.R304W) Is Associated with Stormorken Syndrome. Hum Mutat. 2014;35(10):1221–32. doi: 10.1002/humu.22621. [DOI] [PubMed] [Google Scholar]
- 23.Gudlur A, Zhou Y, Hogan PG. STIM-ORAI interactions that control the CRAC channel. Curr Top Membr. 2013;71:33–58. doi: 10.1016/B978-0-12-407870-3.00002-0. [DOI] [PubMed] [Google Scholar]
- 24.Deng X, Wang Y, Zhou Y, Soboloff J, Gill DL. STIM and Orai: dynamic intermembrane coupling to control cellular calcium signals. J Biol Chem. 2009;284(34):22501–5. doi: 10.1074/jbc.R109.018655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frischauf I, Fahrner M, Jardin I, Romanin C. The STIM1: Orai Interaction. Adv Exp Med Biol. 2016;898:25–46. doi: 10.1007/978-3-319-26974-0_2. [DOI] [PubMed] [Google Scholar]
- 26.Stathopulos PB, Zheng L, Li GY, Plevin MJ, Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 2008;135(1):110–22. doi: 10.1016/j.cell.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J Biol Chem. 2006;281(47):35855–62. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- 28.Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454(7203):538–42. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131(7):1327–39. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma G, Wei M, He L, et al. Inside-out Ca(2+) signalling prompted by STIM1 conformational switch. Nat Commun. 2015;6:7826. doi: 10.1038/ncomms8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Y, Srinivasan P, Razavi S, et al. Initial activation of STIM1, the regulator of store-operated calcium entry. Nat Struct Mol Biol. 2013;20(8):973–81. doi: 10.1038/nsmb.2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fahrner M, Muik M, Schindl R, et al. A coiled-coil clamp controls both conformation and clustering of stromal interaction molecule 1 (STIM1) J Biol Chem. 2014;289(48):33231–44. doi: 10.1074/jbc.M114.610022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11(3):337–43. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park CY, Hoover PJ, Mullins FM, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136(5):876–90. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muik M, Fahrner M, Schindl R, et al. STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 2011;30(9):1678–89. doi: 10.1038/emboj.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Derler I, Jardin I, Romanin C. Molecular mechanisms of STIM/Orai communication. Am J Physiol Cell Physiol. 2016;310(8):C643–62. doi: 10.1152/ajpcell.00007.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hou X, Pedi L, Diver MM, Long SB. Crystal structure of the calcium release-activated calcium channel Orai. Science. 2012;338(6112):1308–13. doi: 10.1126/science.1228757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443(7108):230–3. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 39.Zhou Y, Ramachandran S, Oh-Hora M, Rao A, Hogan PG. Pore architecture of the ORAI1 store-operated calcium channel. Proc Natl Acad Sci U S A. 2010;107(11):4896–901. doi: 10.1073/pnas.1001169107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Y, Meraner P, Kwon HT, et al. STIM1 gates the store- operated calcium channel ORAI1 in vitro. Nat Struct Mol Biol. 2010;17(1):112–6. doi: 10.1038/nsmb.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bormann BJ, Knowles WJ, Marchesi VT. Synthetic peptides mimic the assembly of transmembrane glycoproteins. J Biol Chem. 1989;264(7):4033–7. [PubMed] [Google Scholar]
- 42.Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry. 1992;31(51):12719–25. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 43.MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: structure and implications. Science. 1997;276(5309):131–3. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 44.Mineev KS, Bocharov EV, Volynsky PE, et al. Dimeric structure of the transmembrane domain of glycophorin a in lipidic and detergent environments. Acta Naturae. 2011;3(2):90–8. [PMC free article] [PubMed] [Google Scholar]
- 45.Trenker R, Call ME, Call MJ. Crystal Structure of the Glycophorin A Transmembrane Dimer in Lipidic Cubic Phase. J Am Chem Soc. 2015;137(50):15676–9. doi: 10.1021/jacs.5b11354. [DOI] [PubMed] [Google Scholar]
- 46.Russ WP, Engelman DM. TOXCAT: a measure of transmembrane helix association in a biological membrane. Proc Natl Acad Sci U S A. 1999;96(3):863–8. doi: 10.1073/pnas.96.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Radcliffe PA, Mitrophanous KA. Multiple gene products from a single vector: ‘self-cleaving’ 2A peptides. Gene Therapy. 2004;11:1673–4. [Google Scholar]
- 48.Li Z, Lu J, Xu P, Xie X, Chen L, Xu T. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem. 2007;282(40):29448–56. doi: 10.1074/jbc.M703573200. [DOI] [PubMed] [Google Scholar]
- 49.Covington ED, Wu MM, Lewis RS. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol Biol Cell. 2010;21(11):1897–907. doi: 10.1091/mbc.E10-02-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci U S A. 2007;104(22):9301–6. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma G, Wen S, He L, Huang Y, Wang Y, Zhou Y. Optogenetic toolkit for precise control of calcium signaling. Cell Calcium. 2017 doi: 10.1016/j.ceca.2017.01.004. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Huang L, Li Z, Ma G, Zhou Y, Han G. Illuminating Cell Signaling with Near-Infrared Light-Responsive Nanomaterials. ACS Nano. 2016;10(4):3881–5. doi: 10.1021/acsnano.6b02284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan P, He L, Han G, Zhou Y. Optogenetic Immunomodulation: Shedding Light on Antitumor Immunity. Trends Biotechnol. 2017;35(3):215–26. doi: 10.1016/j.tibtech.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He L, Zhang Y, Ma G, et al. Near-infrared photoactivatable control of Ca(2+) signaling and optogenetic immunomodulation. Elife. 2015;4 doi: 10.7554/eLife.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Z, Liu L, Deng Y, et al. Graded activation of CRAC channel by binding of different numbers of STIM1 to Orai1 subunits. Cell Res. 2011;21(2):305–15. doi: 10.1038/cr.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Deng X, Zhou Y, et al. STIM protein coupling in the activation of Orai channels. Proc Natl Acad Sci U S A. 2009;106(18):7391–6. doi: 10.1073/pnas.0900293106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Wang Y, Zhou Y, et al. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat Commun. 2014;5:3183. doi: 10.1038/ncomms4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zal T, Gascoigne NR. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys J. 2004;86(6):3923–39. doi: 10.1529/biophysj.103.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.