

Abstract
Purpose
Age-related macular degeneration (AMD) is the leading cause of permanent vision loss among the elderly in many industrialized countries, and the complement system plays an important role in the pathogenesis of AMD. Inhibition of complement factor B, a key regulator of the alternative pathway, is implicated as a potential therapeutic intervention for AMD. Here we investigated the effect of liver factor B reduction on systemic and ocular factor B levels.
Methods
Second-generation antisense oligonucleotides (ASOs) targeting mouse and monkey factor B mRNA were administered by subcutaneous injection to healthy mice or monkeys, and the level of factor B mRNA was assessed in the liver and the eye. In addition, the factor B protein level was determined in plasma and whole eyes from the treated animals.
Results
Mice and monkeys treated with factor B ASOs demonstrated a robust reduction in liver factor B mRNA levels with no change in ocular factor B mRNA levels. Plasma factor B protein levels were significantly reduced in mice and monkeys treated with factor B ASOs, leading to a dramatic reduction in ocular factor B protein, below the assay detection levels.
Conclusions
The results add to the increasing evidence that the liver is the main source of plasma and ocular factor B protein, and demonstrate that reduction of liver factor B mRNA by an ASO results in a significant reduction in plasma and ocular factor B protein levels. The results suggest that inhibition of liver factor B mRNA by factor B ASOs would reduce systemic alternative complement pathway activation and has potential to be used as a novel therapy for AMD.
Introduction
Age-related macular degeneration (AMD) is a progressive disease of the macula and is the leading cause of central vision impairment in persons over the age of 50 years in developed countries [1]. In the early and intermediate stages of AMD, the disease is characterized by the deposition of drusen, protein, and lipid-rich extracellular deposits between the RPE cells and Bruch’s membrane [2,3]. As part of the natural course of the disease, there is development of atrophic areas, which enlarge continuously and correspond with an absolute scotoma and geographic atrophy [4]. The late stage of dry AMD, known as geographic atrophy (GA), may be associated with RPE cell death, overlying photoreceptor loss, and underlying choriocapillaris atrophy [5-8].
The complement system, an important component of innate immunity, is the most widely accepted pathogenic pathway of the immune system implicated in AMD. Recent genetic and pathophysiological studies have provided evidence that the progression of AMD may be due in part to an inflammatory state sustained by aberrant activity of the complement alternative pathway [9-17].
The initial step in the alternative pathway is proteolysis of complement C3, which plays a central role in the activation of all complement pathways [18,19]. While the scaffold protein, complement component 3 (C3), and serine proteases factor B and factor D are required and sufficient for formation of the alternative pathway convertase, several complement proteins, including complement factor H (CFH), act as negative regulators of this enzyme complex [20]. Common genetic variants in complement genes component 3 (OMIM 120700), CFH (OMIM 134370), and factor B are associated with AMD risk and progression [9,12,13,21-25]. Specific polymorphisms of CFH, which is the negative regulator of alternative pathway (AP), confer increased risk for AMD [15,26]. In contrast, specific polymorphisms of CFB (OMIM 138470), the positive regulator of the alternative complement pathway, confer protection of AMD [23,26-28]. Elevated plasma levels of some key proteins of the alternative pathway, including factor D, factor B, and activation fragments C3a, factor Ba, and factor Bb, have been reported in patients with AMD [13,29,30]. Furthermore, a study, using well-characterized human donor eyes, showed that AMD disease severity and complement genotypes are associated with complement activation in the eye. Moreover, C3, factor B, and factor D were found in vitreous and the Bruch’s membrane choroid interface of the macula [20].
Factor B, a 93-kDa single-chain glycoprotein, is required for the initiation and propagation of alternative pathway activation. When factor B associates with a cleavage product of complement protein C3 (C3b) it is cleaved by factor D to release an N-terminal fragment (Ba) and the bound carboxyl-terminal serine proteinase (Bb). Because C3 is a component of the C3bBb enzyme, as well as its substrate, this pathway serves as a positive amplification loop for both pathways of complement activation. Factor B is synthesized in the liver and at low levels in several extrahepatic sites [31-34]. The liver is implicated as the primary source of plasma complement factor B [35-37]. Ocular factor B is located predominately in the choroidal capillaries and Bruch’s membrane region and not evident in the neural retina [16,19,20]. Thus, choroidal factor B appears to be derived from systemic sources.
In this study, we used second-generation antisense oligonucleotides (ASOs) to target and reduce the expression of factor B mRNA and factor B protein to determine whether the liver is the source of ocular factor B. This antisense approach used an RNase H1 mechanism to degrade factor B mRNA species. The results show that systemic administration of factor B ASOs results in a significant reduction in liver mRNA and plasma factor B levels, leading to a robust reduction in ocular factor B protein in mice and non-human primates. These results provide further evidence that the liver is the main source of ocular factor B protein.
Methods
ASO synthesis and chemistry
The ASOs used were 20 nucleotides long and chemically modified with phosphorothioate in the backbone, five residues at each terminus, and a central deoxynucleotide region of ten residues (5–10–5 gapmer). 2′-O-methoxyethyl modified antisense phosphorothioate oligonucleotides (2′-MOE ASOs) were synthesized at Ionis Pharmaceuticals, Inc. (Carlsbad, CA) as described previously [38]. Oligonucleotides were synthesized using an Applied Biosystems 380B automated DNA synthesizer (PerkinElmer Life and Analytical Sciences–Applied Biosystems, Foster City, CA) and purified as previously described [39]. The ASO sequences were as follows: murine factor B ASO, ION-516323, 5′-TCC ACC CAT GTT GTG CAA GC-3′; control ASO ION-141923, 5′-CCT TCC CTG AAG GTT CCT CC-3′; monkey ASO, ION-588548, 5′-TTA ATT CAA TCC CAC GCC CC-3′. The ASOs were dissolved in PBS (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4, pH7.4; Ca-Mg-; Invitrogen, Thermo Fisher Scientific, Carlsbad, CA) for the in vivo experiments.
Culture of hepatocytes and treatment with ASOs
Mouse or monkey primary hepatocytes were prepared using the standard collagenase procedure described previously [40]. Electroporation of ASOs was performed using the HT-200 BTX Electroporator with the ElectroSquare Porator (ECM 830) voltage source in 96-well electroporation plates (BTX, 2 mm; Harvard Apparatus, Holliston, MA). Cells were harvested 16 h after electroporation. Cells were electroporated in the presence of factor B ASOs at the indicated concentrations and plated. Sixteen hours after transfection, total cellular RNA was isolated, and the amount of factor B mRNA was quantified using a quantitative real-time PCR (qRT-PCR) assay (TaqMan, Applied Biosystems, Thermo Fisher Scientific).
Quantitative real-time PCR
The cultured cells were lysed, and total RNA was extracted with a QIAGEN (Valencia, CA) RNeasy column. Animal tissues were homogenized in a guanidine isothiocyanate solution (Invitrogen) supplemented with 8% 2-mercaptoethanol (Sigma-Aldrich, St Louis, MO). Total RNA was prepared using the RNeasy mini kit instructions (QIAGEN). The qRT-PCR analyses were performed using an ABI Prism 7700 sequence detector (Applied Biosystems). PCR results were normalized to total RNA measure with Quant-iT RiboGreen RNA Reagent (Molecular Probes, Eugene, OR). The sequences of primers and probes used were as follows: for mouse factor B: forward: 5′-GGG CAA ACA GCA ATT TGT GA-3′, reverse: 5′-TGG CTA CCC ACC TTC CTT GT-3′, probe: 5′-Fam-CTG GAT ACT GTC CCA ATC CCG GTA TTC C-Tamra-3′; monkey factor B: forward: 5′-CGA AGA AGC TCA GTG AAA TCA A-3′, reverse: 5′-TGC CTG GAG GGC CCT CTT-3′, probe: 5′-Fam- AGA CCA CAA GTT GAA GTC-Tamra-3′; mouse cyclophilin A: forward: 5′-TCG CCG CTT GCT GCA-3′, reverse: 5′-ATC GGC CGT GAT GTC GA-3′, probe: 5′-CCA TGG TCA ACC CCA CCG TG TTC-Tamra-3′; monkey cyclophilin A: forward: 5′-CGA CGG CGA GCC TTT G-3′, reverse: 5′-TCT GCT GTC TTT GGA ACC TTG TC-3′, probe: 5′-CGC GTC TCC TTC GAG CTG TTT GC-Tamra-3′. PCR conditions, holding stage: 15 min at 50 °C, 5 min at 95 °C. Cycling stage: 15 msec at 95 °C, 1 min at 60 °C for 40 cycles. PCR results were normalized to total RNA measure with Quant-iT RiboGreen RNA Reagent (Molecular Probes).
Animal studies
Mouse studies: Male wild-type C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were housed four animals per cage at 22–25 °C with a 12 h: 12 h light-dark cycle and free access to food and water. The ASO drugs were prepared in PBS and were administered by subcutaneous injection. Dosages are reported as the weekly dose for all the animal studies, and the mice were dosed subcutaneously once per week. On the day of necropsy, the mice were euthanized with isoflurane inhalation. Plasma was collected by cardiac puncture and frozen at −70 °C or less. Liver and whole eyes were collected when the mice were euthanized approximately 72 h after dosing was completed.
Monkey study: Male cynomolgus monkeys received loading doses of 0 (vehicle control, n = 6) or 40 mg/kg of ION-588548 (n = 4), every 2 days during the first week of the study (on days 1, 3, 5, and 7) followed by once a week administration thereafter, commencing on day 14 (40 mg/kg/wk). Doses were administered by subcutaneous injection for 12 consecutive weeks and calculated based on the most recently recorded bodyweight. Plasma for factor B analysis was collected at days −6, 1, 30, 58, 72, and 86. At the terminal necropsy, liver samples and whole eyes were collected for factor B mRNA and protein analysis. On the day of the necropsy, the monkeys were sedated, weighed, and anesthetized followed by exsanguination and necropsy. The study was conducted at Korea Institute of Toxicology (KIT).
Study approval
All the animal studies were conducted under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of KIT and Ionis Pharmaceuticals. The monkey study was conducted at an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited facility that has an Animal Welfare Assurance issued by the Office of Laboratory Animal Welfare (OLAW), is registered with the United States Department of Agriculture (USDA), and has an Institutional Animal Care and Use Committee (IACUC) that is responsible for compliance with applicable laws and regulations regarding the use of laboratory animals. The study protocol was approved by the IACUC at the testing facility before dose administration.
Factor B western blot
Blood was collected under anesthesia via cardiac puncture into sample tubes coated with the anticoagulant EDTA. Intracardiac blood collection was performed once at the end of the study, before the necropsy. Blood was centrifuged at 4,000 ×g for 15 min, and platelet-poor plasma was collected and stored at –80 °C before western blotting. One microliter of plasma samples from all groups was analyzed with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (PAGE) followed by immunoblotting with antibodies to factor B. The antifactor B antibody was obtained from Sigma (Cat #HPA001817), was produced in rabbits, and was specific to human, mouse, and rat full-length factor B protein.
In the perfusion experiments, 9-week-old, wild-type C57BL/6J mice were subjected to trans-cardial perfusion with Dulbecco’s PBS (137.9 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4, pH7.3; DPBS, no calcium, no magnesium; Invitrogen, Thermo Fisher Scientific, Cat.: 14190144) to remove the blood from the tissues. Briefly, a catheter infusion set was connected to a 20 ml syringe, and the needle was inserted in the left ventricle. The right atria was cut, and 15–20 ml of DPBS+10 U/ml heparin (Sigma, Cat: H3393) was slowly pumped through the heart. A steady flow of about 10 ml/min was applied. Following perfusion with DPBS+heparin, an additional 5 ml of DPBS was pumped through the heart to wash out any residual heparin. The eye and liver samples were collected and snap frozen in liquid nitrogen.
One eye from each animal was used for protein extraction and western blot analysis. The eye was homogenized in buffer containing protease inhibitors and incubated for 30 min on ice. The eye homogenate was then centrifuged for 10 min at 17,949 ×g. The resulting supernatant was used for western blot analysis. The plasma and the protein extracts from the eye and liver were analyzed with sodium dodecyl sulfate PAGE followed by immunoblotting with an anti-human polyclonal antibody against factor B (Sigma HPA001817, 1:1,000). To ensure equal loading on the gel, an antibody anti-apolipoprotein E (ApoE; Abcam, Cambridge, MA, 1:5,000) was used for the plasma samples. Anti-alpha tubulin antibody (Abcam, ab7291, 1:10,000) was used for the eye and liver samples. The quantification of the western blot images was performed using Image J software. Recombinant factor B (Abcam, ab151392) was included as a control.
Plasma factor B analysis
Plasma factor B levels were analyzed at the Complement Laboratory at National Jewish Health. Factor B levels were measured with radial immunodiffusion (RID; CL.0012), using an anti-human Factor B antibody preparation incorporated into an agarose gel to form immunoprecipitation in a ring around the application well. The area of the precipitin ring (minus the area of the well) is proportional to the amount of antigen (factor B) that is present in the test specimen. The reference range for cynomolgus monkeys is 127.6–278.5 µg/ml.
Statistical analysis
Values presented represent the mean ± standard error of the mean (SEM). Statistical differences between groups were determined using one-way ANOVA with Tukey honest significant difference (HSD) multiple comparisons. A p value of less than 0.05 was considered statistically significant.
Results
Identification of mouse factor B ASO (ION-516323) and monkey factor B ASO (ION-588548)
ASOs are short synthetic strings of nucleotides designed to prevent the expression of a targeted protein by selectively binding to the RNA that encodes the targeted protein with high affinity and thus preventing translation [41]. The factor B ASO hybridizes to its complementary mRNA target through well-characterized Watson–Crick base pairing and triggers mRNA cleavage by RNase H1, leading to the inhibition of target RNA translation and subsequent reduction in target protein levels [39].
To identify active ASOs that specifically target mouse or monkey factor B mRNA for in vivo studies, we designed and screened in vitro more than 150 ASOs for each species. The efficacy of the ASOs was determined in mouse or monkey primary hepatocytes as previously described [42]. All the ASOs tested were 20 base chimeric 2’-MOE phosphorothioate oligonucleotides incorporating a 5–10–5 design, that is, five 2′-O-methoxyethyl nucleotides at the 5′ end, ten deoxynucleotides in the center, five 2′-O-methoxyethyl nucleotides at the 3′ end, and phosphorothioate backbone substitution throughout [39]. An example of the range of activities of several of the ASOs tested is presented in Appendix 1. Active ASOs were further evaluated in dose–response studies. The IC50 achieved for the ASOs selected for in vivo studies were 0.5 μM for ION-516323 (mouse) and 0.7 μM for ION-588548 (monkey).
Administration of factor B ASO in wild-type mice effectively reduces liver factor B mRNA and plasma factor B protein levels
To establish the relationship between liver factor B mRNA reduction by the factor B ASO and plasma factor B levels, the factor B ASO were administered to wild-type C57BL/6J mice, four mice for each treatment group, by subcutaneous injections at doses of 12, 25, 50, 75, or 100 mg/kg/wk, for 6 weeks (Figure 1A). Liver factor B mRNA levels were reduced in a dose-dependent manner, reaching a maximum reduction of 95±0.1% of saline (Figure 1B). The calculated ED50 of the murine factor B ASO (ION-516323) was 18 mg/kg/wk (Figure 1C). Liver factor B mRNA levels in the control ASO-treated (50 mg/kg/week) mice were not different from those in the saline-treated control group. ASO drugs are distributed broadly to many organs with the exception of the central nervous system (CNS) and the eyes, as ASO drugs are unable to pass the blood–brain or blood–retina barrier [43]. Therefore, as expected, the ocular mRNA levels of factor B were not changed with systemic administration of the factor B ASO (Figure 1D). Although the factor B mRNA levels were measurable in the whole eye preparations, the ocular level of factor B mRNA was less than 1% (0.86+0.03%) of the liver levels.
Figure 1.

Factor B ASO effectively reduces hepatic factor B mRNA and plasma factor B of normal mice. C57BL/6J mice were treated for 6 weeks with the indicated weekly doses of factor B antisense oligonucleotide (ASO) via subcutaneous injection (n = 4 per group). A: Study design. B: Dose-dependent reduction in hepatic factor B mRNA after subcutaneous administration of factor B ASO. C: ED50 of hepatic factor B mRNA reduction by mouse factor B ASO. D: Ocular factor B mRNA level. E: Dose-dependent reduction in plasma factor B protein levels after the administration of the factor B ASO. F: Factor B protein level was measured with western blot with anti-factor B antibody. Quantification of the western blot using Image J. Factor B band intensity was normalized to the level of plasma immunoglobulin G (IgG). Results represent mean ± standard error of the mean (SEM). *p<0.05; one-way ANOVA with Tukey honest significant difference (HSD) multiple comparisons. G: Correlation between liver factor B mRNA level and plasma factor B protein level of mice after treatment with the factor B ASO. The Pearson correlation was calculated using Prism software, and the calculated R squared was 0.655.
We next analyzed the effect of the administration of the factor B ASO on plasma factor B levels with western blot. The analysis showed that administration of the factor B ASO resulted in a dose-dependent reduction in the plasma factor B levels (Figures 1E,F). The reduction in the factor B mRNA level correlated with the reduction in factor B plasma protein (Figure 1G).
Administration of factor B ASO effectively reduces ocular factor B protein levels in mice and in cynomolgus monkeys
To evaluate the effect of systemically administered factor B ASOs on ocular factor B levels, species-specific factor B ASOs were administered to mice or cynomolgus monkeys by subcutaneous injection for 6 or 13 weeks, respectively. In both species, following the subcutaneous administration of the ASO, the ASO binds to plasma proteins and transfer rapidly from blood to tissues with a distribution half-life of 1–2 h. Once the ASO is in tissues, clearance is dramatically slower than that observed in plasma, with tissue half-life reported to range from about 1 to 4 weeks [44]. The long half-lives of ASOs allow for infrequent (once weekly or less) dosing, coupled with a robust safety profile, including chronic toxicology [45].
Wild-type C57BL/6J mice (n = 4 per treatment group) received 25, 50, 75, and 100 mg/kg/wk the factor B ASO by subcutaneous injections for 6 weeks. A control mismatch ASO at 100 mg/kg/wk was used as negative control. As observed in the previous experiment, the administration of the factor B ASO resulted in a significant and dose-dependent reduction in the factor B mRNA levels in the liver. The maximum reduction achieved was 79±10% with factor B ASO ION-516323 (Appendix 2). Plasma factor B protein levels were analyzed with a western blot using 1 µl of plasma. As shown in Figure 2A, the levels of factor B protein were reduced in a dose-dependent manner. Moreover, systemic administration of the factor B ASO resulted in marked reductions in ocular factor B protein levels in mice and in the 50 and 100 mg/kg/week treatment groups. Total whole eye protein extract was purified from all mice from the various treatment groups. Ocular factor B protein levels were determined with western blot (Figure 2B). The robust reduction in ocular protein corresponded with the reductions of factor B in the plasma.
Figure 2.

Factor B ASO effectively reduces plasma and ocular factor B protein levels. C57BL/6J mice were treated for 6 weeks with the indicated weekly doses of the factor B antisense oligonucleotide (ASO) via subcutaneous injection (n = 4 per group). A: Dose-dependent reduction in plasma factor B protein levels after administration of the factor B ASO. One microliter of plasma from all individual mice from the various treatment groups was analyzed with western blot. B: Dose-dependent reduction in ocular factor B protein with the administration of the factor B ASO. One whole eye from each mouse was used for total protein isolation. The factor B protein level was measured with western blot with the anti-factor B antibody.
To demonstrate that the major source of ocular factor B protein is the circulating factor B, we removed all blood from wild-type mice by trans-cardial perfusion with PBS. Subsequently, we evaluated the level of ocular factor B protein level with western blot. The results show that the ocular factor B levels are significantly reduced following perfusion (Figure 3A,B). However, the factor B levels in the perfused livers were comparable to the levels in the unperfused tissues (Figure 3C,D). In addition, we compared factor B expression in the murine liver and eye. Using qRT-PCR and western blot analysis, we determined at the mRNA and protein levels that ocular factor B expression is about ten- to 100-fold less than the hepatic factor B expression (data not shown).
Figure 3.

Reduction in ocular factor B protein following trans-cardial perfusion of wild-type mice. C57BL/6J mice were subjected to trans-cardial perfusion with PBS. Unperfused mice served as control (n = 3 per group). A: The factor B protein levels were measured in the ocular extracts with western blot analysis. Total protein extracts were obtained from one whole eye. Different amounts of recombinant human factor B protein (rhFB) were loaded on the gel to estimate the ocular factor B levels. Immunoblotting for tubulin was used to ensure equal protein loading on the gel. B: Quantification of panel A. C: Analysis of liver factor B protein levels with western blot. Immunoblotting for tubulin was used to ensure equal protein loading on the gel. D: Quantification of panel C.
The in vivo efficacy of the monkey-specific factor B ASO, ION-588548 was also evaluated with subcutaneous administration to cynomolgus monkeys. Two- to four-year-old male monkeys were dosed with 40 mg/kg/wk of ION-588548 (n = 4) or saline (n = 6) for 12 weeks. Plasma, eye, and liver samples were collected after the animals were euthanized and analyzed for factor B mRNA and protein levels. As expected, a significant reduction in the hepatic complement factor B mRNA levels was observed following the administration of the factor B ASO (−66±7% compared to saline control). Similar to the observations in mice, the ocular factor B mRNA levels in animals treated with the factor B ASO were not significantly different from those in animals treated with saline (−19±8% of saline control). Moreover, the monkeys treated with the factor B ASO demonstrated a time-dependent reduction in plasma factor B levels, achieving an 80±6% reduction in plasma factor B at day 72 (Figure 4). Furthermore, the level of factor B protein in the monkey eye, as determined with western blot, was dramatically reduced after the systemic factor B ASO treatment, reaching undetectable levels (Figure 5).
Figure 4.
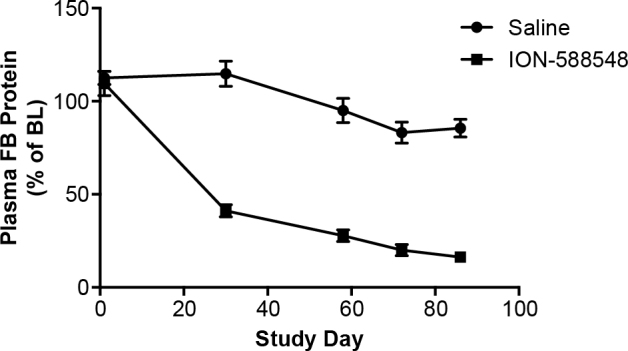
Reduction in plasma factor B protein levels of cynomolgus monkeys treated with the factor B ASO for 13 weeks. Cynomolgus monkeys were dosed with saline (n = 6) or ION-588548 (n = 4) at 40 mg/kg/wk for 13 weeks. Plasma samples were obtained at days −6, 1, 30, 58, 72, and 86 of the study and analyzed for the level of factor B protein. Data presented as % of baseline (day −6) for the individual monkeys and mean ± standard error of the mean (SEM) for each time point.
Figure 5.
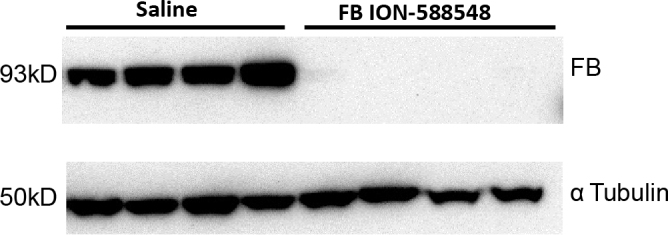
Reduction in ocular factor B protein of monkeys treated with the factor B ASO for 13 weeks. Cynomolgus monkeys were dosed with saline (n = 4) or ION-588548 (n = 4) at 40 mg/kg/wk for 13 weeks. One whole eye from each monkey was used for total protein isolation. The factor B protein levels were measured with western blot.
Discussion
AMD is strongly associated with the over-activation of complement pathways [16]. Supporting evidence comes from the detection of various complement proteins in the drusen of patients with AMD [9] and the close association between AMD and the alternative pathway. Furthermore, the association of genetic variants in complement genes, including the human factor B gene, is a risk factor for all forms of AMD [5,9,22,24]. In addition, it has been shown that polymorphisms of genes encoding the alternative pathway proteins CFH, factor B, and C3, as well as the classical pathway protein C2, are also associated with AMD [23,46]. Studies in animal models of choroidal neovascularization (CNV), a major pathologic association with wet AMD, also supports a role for complements in AMD. Furthermore, a targeted inhibitor specific for the AP of complement significantly reduces CNV and the physiologic consequences of CNV on retinal function [11,47].
Additional evidence for a role of the AP pathway in AMD exists. For example, plasma concentrations of AP activation products were found to be significantly elevated in patients with AMD compared to controls [13,20,29]. In addition, various independent studies have shown that the concentrations of several complement proteins, such as C3, C4, factor B, and factor D, as well as the activation products Ba, C3a, C3d, C5a, and sC5b-9, are greater in the blood of patients with AMD compared to age-matched controls [48]. These studies suggest that the ongoing systemic activation of the alternative complement pathway plays an important role in AMD pathogenesis and adds to the increasing evidence that AMD is a systemic disease with local disease manifestation in the aging macula [49].
However, it is clear that local activation of the complement pathway also plays a significant role in AMD disease pathogenesis [16,20]. Supporting evidence of local complement activation comes from the detection of various complement proteins in the drusen of patients with AMD [9] and the fact that increased activation of the alternative complement pathway in vitreous is controlled by disease stage and genetic variation in the complement pathway [20]. Extensive work by multiple laboratories has demonstrated that some complement components are expressed in the RPE and choroidal layers of the eye. Interestingly, CFH is expressed at a high level in RPE layers [9,10]. In contrast, other complement components, such as C3, C5, factor B, and factor D, are expressed predominantly in the choroidal layers rather than the RPE or the retina [16]. A study in mice expressing factor B uniquely in the RPE cells demonstrated that local factor B expression is sufficient to drive pathology in the eye. However, the same group showed that in the absence of locally produced complement factor B (CFB-KO), systemic factor B derived from wild-type serum injected via tail vein can drive pathology [50]. In vitro studies have demonstrated that cultured RPE cells constitutively express higher levels of complement regulators, including factor B and C3, and under inflammatory conditions, activated macrophages could promote the alternative pathway of complement activation in the retina via induction of RPE cell complement factor B and C3 expression. Macrophages and RPE cells may play an important regulatory role in complement activation at the retina–choroidal interface under pathophysiologic conditions, such as age-related macular degeneration [51]. The importance of the dysregulation of complements in the choroid has been well documented [17,52,53]. Specifically, deposition of membrane attack complex (MAC) in the choriocapillaris begins at early ages, continues throughout aging, and is greater in patients with AMD [53]. It has been hypothesized that the MAC-induced disruption of choriocapillaris is a primary event in the etiogenesis of AMD [17].
The liver has been implicated as the primary source of plasma complement factor B, and a systemic complement factor B inhibitor could potentially provide benefit to patients with AMD by slowing disease progression [35-37]. In the human eye, the choroidal expression of factor B is substantially less than that observed in the liver [16], which we confirmed in the present animal studies. In this study, we further investigated the source of plasma and ocular factor B protein in mice and monkeys. Using second-generation ASOs, we demonstrated that reductions in liver factor B mRNA lead to robust reductions in plasma and ocular factor B protein levels. However, although marked reductions in liver factor B mRNA were demonstrated, no reductions in ocular factor B mRNA levels could be detected. These results add to the increasing evidence that the liver is the main source for circulating and ocular factor B. These studies are also consistent with the premise that the ongoing systemic activation of the alternative complement pathway plays an important role in AMD pathogenesis and adds to the increasing evidence that AMD is a systemic disease with local disease manifestation at the aging macula [49].
In view of the potential pivotal role of the complement system in the development of AMD, several trials have been initiated to investigate the effect of complement inhibitors on this disease. Recent clinical trials have used humanized monoclonal antibodies (mAb) against C3, C5, and factor D [54]. Preliminary findings suggest that blocking the alternative pathway may offer a good approach to treat AMD. Lampalizumab (Genentech, Inc., South San Francisco, CA) is the antigen-binding fragment (Fab) of a humanized monoclonal antibody (mAb) that inhibits complement factor D, which is the rate-limiting enzyme in the activation and amplification of the alternative complement pathway [55]. The MAHALO study, a phase 2 clinical trial, demonstrated that intravitreal injected lampalizumab inhibited disease progression in patients with geographic atrophy [54]. Here we propose that inhibition of the alternative pathway by systemic administration of factor B ASO drugs could benefit AMD. In contrast to the therapeutic potential of inhibiting the complement pathways, deficiencies within the complement system can lead to inappropriate inflammation, impair host defense, and increase the risk of infection. There have been only a few recorded cases of factor B deficiency, and the individuals reported presented with meningococcemia but no history of any autoimmune disorder [56,57]. Antisense technology is a highly effective approach for reducing the target expression in liver. However, treatment with ASOs usually achieves a significant knockdown (not knockout) of the target protein, leaving some residual activity of the target [58]. In vitro studies have evaluated the ability of various concentrations of wild-type and polymorphs of factor B protein to maintain serum hemolytic activity demonstrated that more than 20% of normal serum hemolytic activity could be maintained with less than 10% of normal factor B levels [26]. Therefore, since factor B hemolytic activity is achieved with low concentrations of plasma factor B, the risk of infection should be minimal with systemic inhibition using factor B ASOs.
In summary, we propose an alternative strategy for AMD therapeutics that involves inhibition of the alternative pathway by systemic administration of factor B ASOs. We suggest that the reduction in systemic complement activation would lead to reduced complement activation in the eye, which will potentially slow AMD disease progression and provide benefit to patients with AMD.
Acknowledgments
The Authors are thankful to Tracy Reigle (Ionis Pharmaceuticals) for assistance with graphics and figures preparation.
Appendix 1. FB ASOs screen in primary hepatocytes
FB ASOs were electroporated to mouse primary hepatocytes. Cells were harvested 16 h after electroporation total cellular RNA was isolated, and the amount of FB mRNA was quantified using a quantitative qRT-PCR) assay. Values are presented as % of untreated control (UTC). The arrow is pointing to ION-516323 ASO that was selected for the in vivo studies. To access the data, click or select the words “Appendix 1”.
Appendix 2. Dose dependent reduction in hepatic FB mRNA after SC administration of FB ASOs.
C57BL/6J mice were treated for 6 weeks with the indicated weekly doses of FB ASO via subcutaneous injection (n=4 per group). Results represent mean ± SEM. To access the data, click or select the words “Appendix 2”.
References
- 1.Ratnapriya R, Chew EY. Age-related macular degeneration-clinical review and genetics update. Clin Genet. 2013;84:160–6. doi: 10.1111/cge.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bird AC, Bressler NM, Bressler SB, Chisholm IH, Coscas G, Davis MD, de Jong PT, Klaver CC, Klein BE, Klein R. An international classification and grading system for age-related maculopathy and age-related macular degeneration. The International ARM Epidemiological Study Group. Surv Ophthalmol. 1995;39:367–74. doi: 10.1016/s0039-6257(05)80092-x. [DOI] [PubMed] [Google Scholar]
- 3.van Lookeren Campagne M, LeCouter J, Yaspan BL, Ye W. Mechanisms of age-related macular degeneration and therapeutic opportunities. J Pathol. 2014;232:151–64. doi: 10.1002/path.4266. [DOI] [PubMed] [Google Scholar]
- 4.Lindblad AS, Lloyd PC, Clemons TE, Gensler GR, Ferris FL, 3rd, Klein ML, Armstrong JR. Age-Related Eye Disease Study Research G. Change in area of geographic atrophy in the Age-Related Eye Disease Study: AREDS report number 26. Arch Ophthalmol. 2009;127:1168–74. doi: 10.1001/archophthalmol.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein R, Klein BE, Knudtson MD, Meuer SM, Swift M, Gangnon RE. Fifteen-year cumulative incidence of age-related macular degeneration: the Beaver Dam Eye Study. Ophthalmology. 2007;114:253–62. doi: 10.1016/j.ophtha.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 6.Sunness JS, Gonzalez-Baron J, Applegate CA, Bressler NM, Tian Y, Hawkins B, Barron Y, Bergman A. Enlargement of atrophy and visual acuity loss in the geographic atrophy form of age-related macular degeneration. Ophthalmology. 1999;106:1768–79. doi: 10.1016/S0161-6420(99)90340-8. [DOI] [PubMed] [Google Scholar]
- 7.Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol Aspects Med. 2012;33:295–317. doi: 10.1016/j.mam.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao S, Wang JC, Gao J, Wong M, To E, White VA, Cui JZ, Matsubara JA. CFH Y402H polymorphism and the complement activation product C5a: effects on NF-kappaB activation and inflammasome gene regulation. Br J Ophthalmol. 2016;100:713–8. doi: 10.1136/bjophthalmol-2015-307213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–32. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 10.Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–31. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- 11.Bora NS, Kaliappan S, Jha P, Xu Q, Sohn JH, Dhaulakhandi DB, Kaplan HJ, Bora PS. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: role of factor B and factor H. J Immunol. 2006;177:1872–8. doi: 10.4049/jimmunol.177.3.1872. [DOI] [PubMed] [Google Scholar]
- 12.Francis PJ, Hamon SC, Ott J, Weleber RG, Klein ML. Polymorphisms in C2, CFB and C3 are associated with progression to advanced age related macular degeneration associated with visual loss. J Med Genet. 2009;46:300–7. doi: 10.1136/jmg.2008.062737. [DOI] [PubMed] [Google Scholar]
- 13.Reynolds R, Hartnett ME, Atkinson JP, Giclas PC, Rosner B, Seddon JM. Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Invest Ophthalmol Vis Sci. 2009;50:5818–27. doi: 10.1167/iovs.09-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kijlstra A, La Heij E, Hendrikse F. Immunological factors in the pathogenesis and treatment of age-related macular degeneration. Ocul Immunol Inflamm. 2005;13:3–11. doi: 10.1080/09273940590909185. [DOI] [PubMed] [Google Scholar]
- 15.Donoso LA, Vrabec T, Kuivaniemi H. The role of complement Factor H in age-related macular degeneration: a review. Surv Ophthalmol. 2010;55:227–46. doi: 10.1016/j.survophthal.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Rickman CB, Bok D, Hageman GS, Johnson LV. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010;29:95–112. doi: 10.1016/j.preteyeres.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitmore SS, Sohn EH, Chirco KR, Drack AV, Stone EM, Tucker BA, Mullins RF. Complement activation and choriocapillaris loss in early AMD: implications for pathophysiology and therapy. Prog Retin Eye Res. 2015;45:1–29. doi: 10.1016/j.preteyeres.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller-Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem. 1988;57:321–47. doi: 10.1146/annurev.bi.57.070188.001541. [DOI] [PubMed] [Google Scholar]
- 19.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–97. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loyet KM, Deforge LE, Katschke KJ, Jr, Diehl L, Graham RR, Pao L, Sturgeon L, Lewin-Koh SC, Hollyfield JG, van Lookeren Campagne M. Activation of the alternative complement pathway in vitreous is controlled by genetics in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2012;53:6628–37. doi: 10.1167/iovs.12-9587. [DOI] [PubMed] [Google Scholar]
- 21.Francis PJ, Schultz DW, Hamon S, Ott J, Weleber RG, Klein ML. Haplotypes in the complement factor H (CFH) gene: associations with drusen and advanced age-related macular degeneration. PLoS One. 2007;2:e1197. doi: 10.1371/journal.pone.0001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 23.Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Group AMDGCS, Hageman GS, Dean M, Allikmets R. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–62. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 25.Maller J, George S, Purcell S, Fagerness J, Altshuler D, Daly MJ, Seddon JM. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38:1055–9. doi: 10.1038/ng1873. [DOI] [PubMed] [Google Scholar]
- 26.Heurich M, Martinez-Barricarte R, Francis NJ, Roberts DL, Rodriguez de Cordoba S, Morgan BP, Harris CL. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci USA. 2011;108:8761–6. doi: 10.1073/pnas.1019338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montes T, Tortajada A, Morgan BP, Rodriguez de Cordoba S, Harris CL. Functional basis of protection against age-related macular degeneration conferred by a common polymorphism in complement factor B. Proc Natl Acad Sci USA. 2009;106:4366–71. doi: 10.1073/pnas.0812584106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mantel I, Ambresin A, Moetteli L, Droz I, Roduit R, Munier FL, Schorderet DF. Complement factor B polymorphism and the phenotype of early age-related macular degeneration. Ophthalmic Genet. 2014;35:12–7. doi: 10.3109/13816810.2013.766217. [DOI] [PubMed] [Google Scholar]
- 29.Scholl HP, Charbel Issa P, Walier M, Janzer S, Pollok-Kopp B, Borncke F, Fritsche LG, Chong NV, Fimmers R, Wienker T, Holz FG, Weber BH, Oppermann M. Systemic complement activation in age-related macular degeneration. PLoS One. 2008;3:e2593. doi: 10.1371/journal.pone.0002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hecker LA, Edwards AO, Ryu E, Tosakulwong N, Baratz KH, Brown WL, Charbel Issa P, Scholl HP, Pollok-Kopp B, Schmid-Kubista KE, Bailey KR, Oppermann M. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum Mol Genet. 2010;19:209–15. doi: 10.1093/hmg/ddp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsumoto M, Fukuda W, Circolo A, Goellner J, Strauss-Schoenberger J, Wang X, Fujita S, Hidvegi T, Chaplin DD, Colten HR. Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc Natl Acad Sci USA. 1997;94:8720–5. doi: 10.1073/pnas.94.16.8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whaley K. Biosynthesis of the complement components and the regulatory proteins of the alternative complement pathway by human peripheral blood monocytes. J Exp Med. 1980;151:501–16. doi: 10.1084/jem.151.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strunk RC, Eidlen DM, Mason RJ. Pulmonary alveolar type II epithelial cells synthesize and secrete proteins of the classical and alternative complement pathways. J Clin Invest. 1988;81:1419–26. doi: 10.1172/JCI113472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ripoche J, Mitchell JA, Erdei A, Madin C, Moffatt B, Mokoena T, Gordon S, Sim RB. Interferon gamma induces synthesis of complement alternative pathway proteins by human endothelial cells in culture. J Exp Med. 1988;168:1917–22. doi: 10.1084/jem.168.5.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan BP, Gasque P. Extrahepatic complement biosynthesis: where, when and why? Clin Exp Immunol. 1997;107:1–7. doi: 10.1046/j.1365-2249.1997.d01-890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koskimies S, Lokki ML, Hockerstedt K. Changes in plasma complement C4 and factor B allotypes after liver transplantation. Complement Inflamm. 1991;8:257–60. doi: 10.1159/000463194. [DOI] [PubMed] [Google Scholar]
- 37.Marsh JE, Zhou W, Sacks SH. Local tissue complement synthesis--fine tuning a blunt instrument. Arch Immunol Ther Exp (Warsz) 2001;49(Suppl 1):S41–6. [PubMed] [Google Scholar]
- 38.Baker BF, Lot SS, Condon TP, Cheng-Flournoy S, Lesnik EA, Sasmor HM, Bennett CF. 2′-O-(2-Methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligonucleotides selectively increase the ICAM-1 mRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells. J Biol Chem. 1997;272:11994–2000. doi: 10.1074/jbc.272.18.11994. [DOI] [PubMed] [Google Scholar]
- 39.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–93. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 40.Quistorff B, Dich J, Grunnet N. Preparation of isolated rat liver hepatocytes. Methods Mol Biol. 1990;5:151–60. doi: 10.1385/0-89603-150-0:151. [DOI] [PubMed] [Google Scholar]
- 41.Geary RS, Yu RZ, Watanabe T, Henry SP, Hardee GE, Chappell A, Matson J, Sasmor H, Cummins L, Levin AA. Pharmacokinetics of a tumor necrosis factor-alpha phosphorothioate 2′-O-(2-methoxyethyl) modified antisense oligonucleotide: comparison across species. Drug Metab Dispos. 2003;31:1419–28. doi: 10.1124/dmd.31.11.1419. [DOI] [PubMed] [Google Scholar]
- 42.Grossman TR, Hettrick LA, Johnson RB, Hung G, Peralta R, Watt A, Henry SP, Adamson P, Monia BP, McCaleb ML. Inhibition of the alternative complement pathway by antisense oligonucleotides targeting complement factor B improves lupus nephritis in mice. Immunobiology. 2016;221:701–8. doi: 10.1016/j.imbio.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Banks WA. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009;9(Suppl 1):S3. doi: 10.1186/1471-2377-9-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2009;5:381–91. doi: 10.1517/17425250902877680. [DOI] [PubMed] [Google Scholar]
- 45.Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46–51. doi: 10.1016/j.addr.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 46.Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, Armbrecht AM, Dhillon B, Deary IJ, Redmond E, Bird AC, Moore AT. Genetic Factors in AMDSG. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–61. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 47.Rohrer B, Long Q, Coughlin B, Wilson RB, Huang Y, Qiao F, Tang PH, Kunchithapautham K, Gilkeson GS, Tomlinson S. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:3056–64. doi: 10.1167/iovs.08-2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kijlstra A, Berendschot TT. Age-Related Macular Degeneration: A Complementopathy? Ophthalmic Res. 2015;54:64–73. doi: 10.1159/000432401. [DOI] [PubMed] [Google Scholar]
- 49.Smailhodzic D, Klaver CC, Klevering BJ, Boon CJ, Groenewoud JM, Kirchhof B, Daha MR, den Hollander AI, Hoyng CB. Risk alleles in CFH and ARMS2 are independently associated with systemic complement activation in age-related macular degeneration. Ophthalmology. 2012;119:339–46. doi: 10.1016/j.ophtha.2011.07.056. [DOI] [PubMed] [Google Scholar]
- 50.Schnabolk G, Coughlin B, Joseph K, Kunchithapautham K, Bandyopadhyay M, O’Quinn EC, Nowling T, Rohrer B. Local production of the alternative pathway component factor B is sufficient to promote laser-induced choroidal neovascularization. Invest Ophthalmol Vis Sci. 2015;56:1850–63. doi: 10.1167/iovs.14-15910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo C, Zhao J, Madden A, Chen M, Xu H. Complement expression in retinal pigment epithelial cells is modulated by activated macrophages. Exp Eye Res. 2013;112:93–101. doi: 10.1016/j.exer.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 52.Chirco KR, Tucker BA, Stone EM, Mullins RF. Selective accumulation of the complement membrane attack complex in aging choriocapillaris. Exp Eye Res. 2015 doi: 10.1016/j.exer.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mullins RF, Dewald AD, Streb LM, Wang K, Kuehn MH, Stone EM. Elevated membrane attack complex in human choroid with high risk complement factor H genotypes. Exp Eye Res. 2011;93:565–7. doi: 10.1016/j.exer.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yaspan BL, Williams DF, Holz FG, Regillo CD, Li Z, Dressen A, van Lookeren Campagne M, Le KN, Graham RR, Beres T, Bhangale TR, Honigberg LA, Smith A, Henry EC, Ho C, Strauss EC, MAHALO Study Investigators Targeting factor D of the alternative complement pathway reduces geographic atrophy progression secondary to age-related macular degeneration. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aaf1443. [DOI] [PubMed] [Google Scholar]
- 55.Katschke KJ, Jr, Wu P, Ganesan R, Kelley RF, Mathieu MA, Hass PE, Murray J, Kirchhofer D, Wiesmann C, van Lookeren Campagne M. Inhibiting alternative pathway complement activation by targeting the factor D exosite. J Biol Chem. 2012;287:12886–92. doi: 10.1074/jbc.M112.345082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slade C, Bosco J, Unglik G, Bleasel K, Nagel M, Winship I. Deficiency in complement factor B. N Engl J Med. 2013;369:1667–9. doi: 10.1056/NEJMc1306326. [DOI] [PubMed] [Google Scholar]
- 57.Botto M, Kirschfink M, Macor P, Pickering MC, Wurzner R, Tedesco F. Complement in human diseases: Lessons from complement deficiencies. Mol Immunol. 2009;46:2774–83. doi: 10.1016/j.molimm.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 58.Antisense Drug Technology STC. Principles, Strategies, and Applications. 2008 ed. Boca Raton, FL: CRC Press; 2008. p. 273-303. [Google Scholar]