

ABSTRACT
The signaling molecule cyclic diguanylate (c-di-GMP) mediates physiological adaptation to extracellular stimuli in a wide range of bacteria. The complex metabolic pathways governing c-di-GMP synthesis and degradation are highly regulated, but the specific cues that impact c-di-GMP signaling are largely unknown. In the intestinal pathogen Clostridium difficile, c-di-GMP inhibits flagellar motility and toxin production and promotes pilus-dependent biofilm formation, but no specific biological functions have been ascribed to any of the individual c-di-GMP synthases or phosphodiesterases (PDEs). Here, we report the functional and biochemical characterization of a c-di-GMP PDE, PdcA, 1 of 37 confirmed or putative c-di-GMP metabolism proteins in C. difficile 630. Our studies reveal that pdcA transcription is controlled by the nutrient-regulated transcriptional regulator CodY and accordingly increases during stationary phase. In addition, PdcA PDE activity is allosterically regulated by GTP, further linking c-di-GMP levels to nutrient availability. Mutation of pdcA increased biofilm formation and reduced toxin biosynthesis without affecting swimming motility or global intracellular c-di-GMP. Analysis of the transcriptional response to pdcA mutation indicates that PdcA-dependent phenotypes manifest during stationary phase, consistent with regulation by CodY. These results demonstrate that inactivation of this single PDE gene is sufficient to impact multiple c-di-GMP-dependent phenotypes, including the production of major virulence factors, and suggest a link between c-di-GMP signaling and nutrient availability.
KEYWORDS: CodY, biofilm, c-di-GMP, cyclic diguanylate, flagella, flagellar motility, nutrient, toxin
INTRODUCTION
The anaerobic, Gram-positive bacterium Clostridium difficile is the most common cause of nosocomial infections in the developed world, resulting in symptoms ranging from mild diarrhea to potentially lethal pseudomembranous colitis (1). C. difficile exposure rarely results in symptomatic infections in healthy individuals, as a healthy microbiota creates an environment that hinders C. difficile growth (2, 3). The main risk factor for an initial C. difficile infection is disruption of the gut microbiota by antibiotic treatment, which creates a transient “nutrient bloom” due to drug-induced disruption of normal bacterial metabolism (4–7).
The C. difficile life cycle has two distinct phases with different roles in disease: metabolically active vegetative cells and dormant spores. C. difficile spores are resistant to oxygen, heat, and many commercial disinfectants, making their eradication from contaminated environments difficult (8–10). In the anaerobic mammalian intestine, spores germinate into vegetative cells, a process triggered by chemical signals including certain amino acids and bile acids (11, 12). Vegetative C. difficile cells secrete protein cytotoxins that drive the development of disease symptoms (13, 14). These cytotoxins, TcdA and TcdB, are internalized by host epithelial cells, where they glucosylate and inactivate the Rho and Rac GTPases that regulate the host actin cytoskeleton (15–18). Intoxication leads to actin depolymerization, with cytopathic effects including cell rounding and disruption of cell-cell tight junctions, which effectively permeabilizes the intestinal epithelium (13, 14, 19). C. difficile toxins are capable of inducing apoptosis and necrosis in intoxicated cells (20, 21). The effects of these toxins result in diarrhea and trigger a massive inflammatory response in the host (14, 22, 23).
During infection, C. difficile cells can either remain vegetative or differentiate into spores, which are shed in stool and allow transmission of C. difficile to new hosts (24). The factors that influence the balance between vegetative growth and sporulation in vivo are poorly understood. There is growing evidence that nutrient uptake from the environment inhibits the onset of sporulation, as spore formation is negatively regulated by nutrient transporters (25, 26). In vegetative C. difficile, nutrient availability also modulates the activity of CodY, a transcriptional regulator that is conserved among low-GC Gram-positive bacteria. When CodY is activated by binding of branched-chain amino acids (BCAA), it binds to target DNA sequences and primarily represses gene transcription (27). In some organisms, including C. difficile, CodY is also activated by GTP (28–30). When BCAA and GTP are abundant, such as in exponential phase, CodY represses the transcription of stationary-phase metabolic genes such as amino acid synthetases and peptide transporters (31, 32). Intracellular stores of BCAA and GTP become limited during stationary phase (28, 33, 34), relieving CodY repression of these genes. In a number of Gram-positive pathogens, including C. difficile, Staphylococcus aureus, and Listeria monocytogenes, CodY represses the transcription of virulence factor genes during exponential growth (30, 35–38). Microarray analysis and affinity purification of CodY binding targets have revealed that CodY regulates the transcription of hundreds of C. difficile genes, including some with predicted roles in signaling by the second messenger cyclic diguanylate (c-di-GMP) (27).
In a multitude of Gram-negative bacteria, c-di-GMP regulates transitions between motile, planktonic lifestyles and sessile modes of growth, often involving biofilm formation (39). In several Gram-negative pathogens, c-di-GMP also influences virulence (40–45). The regulatory roles of c-di-GMP in Gram-positive bacteria are less well characterized but appear to be largely conserved with Gram-negative species, with elevated c-di-GMP inhibiting swimming motility and promoting nonmotile lifestyles (46).
c-di-GMP is synthesized from GTP by diguanylate cyclase (DGC) enzymes containing a characteristic GGDEF domain (47) and degraded by two families of phosphodiesterase (PDE) enzymes identified by conserved EAL or HD-GYP domains (48, 49). Bacterial genomes commonly contain multiple genes encoding these domains, which are often coupled to sensory or regulatory domains (50, 51). The C. difficile 630 genome encodes 37 DGCs and PDEs (52). The c-di-GMP-metabolizing activity of 28 of these enzymes has been confirmed by heterologous expression in Vibrio cholerae and/or Bacillus subtilis (53, 54). Elevated cytoplasmic levels of c-di-GMP repress C. difficile flagellar biosynthesis and toxin production and enhance type IV pilus biogenesis and biofilm formation (55–60). c-di-GMP also appears to regulate the production of clostridial proteins that interact with the mammalian extracellular matrix, suggesting a role for c-di-GMP in the regulation of adhesion to host cells (56, 61–63). While c-di-GMP regulates multiple processes with a known or potential role in virulence, specific functions have not yet been ascribed to any of the individual C. difficile DGCs or PDEs.
In this study, we examined the transcriptional and posttranslational regulation of the C. difficile c-di-GMP PDE PdcA and determined its role in the regulation of c-di-GMP-dependent processes. We show that pdcA is a stationary-phase gene regulated by CodY and that PdcA enzymatic activity is regulated by GTP via its GGDEF domain, indicating that nutrient availability impacts c-di-GMP signaling in C. difficile. Notably, mutation of pdcA impacts multiple c-di-GMP-regulated processes, including biofilm formation and toxin production. Thus, this study identified a single c-di-GMP PDE capable of broadly affecting C. difficile c-di-GMP signaling during stationary phase.
RESULTS
The PAS and GGDEF domains affect PdcA function in vitro and in vivo.
We previously reported that full-length PdcA and its EAL domain alone exhibit c-di-GMP PDE activity in vitro but that overproduction of the PdcA EAL domain alone had no detectable effect on the cytoplasmic c-di-GMP concentration or flagellar motility of C. difficile (55). Heterologous overproduction of full-length PdcA in V. cholerae or B. subtilis increases motility and decreases V. cholerae biofilm formation, consistent with decreased c-di-GMP (53, 54), leading us to hypothesize that the other domains of PdcA are required for activity of the EAL domain in vivo. Full-length PdcA is a PAS-GGDEF-EAL fusion protein (Fig. 1A). The PdcA EAL domain contains a valine substitution for the alanine in the canonical EAL motif (i.e., EVL). While the glutamic acid in the EAL sequence is required for PDE activity, the alanine residue has been shown previously to be dispensable (42, 55). The PAS domain is part of a family of regulatory domains that can bind diverse small-molecule ligands and often regulates protein activity through protein-protein interactions (64). The PdcA GGDEF domain contains a degenerate active site (DGDEM) and has been demonstrated to be catalytically inactive (55). In some homologous proteins with tandem GGDEF-EAL domains, catalytically inactive GGDEF domains are capable of binding GTP, which enhances the PDE activity of the associated EAL domains (65–68). Crystal structures of catalytically active and inactive DGCs suggest that the small side chain of the glycine at position 2 creates a binding pocket for the guanine base of GTP, while the aspartic acid at position 4 coordinates a magnesium ion that interacts with the phosphates (69–71). These residues are conserved in PdcA, suggesting a regulatory role for its GGDEF domain.
FIG 1.

PdcA enzymatic activity is controlled by regulatory domains in vitro and in vivo. (A) Schematic of the PdcA derivatives used in these experiments with the corresponding symbols and colors used in panels B to D. The EAL domain of PdcA contains a substitution of valine for alanine in the EAL motif, noted here as EVL. (B) Percentages of initial c-di-GMP substrate hydrolyzed by full-length PdcA, PdcA(GA), PdcAΔPAS, and PdcA-EAL, determined at 1-min intervals after initiation of the reaction upon addition of the substrate. Open circles indicate values for buffer-only negative controls. The data shown are mean values and standard deviations from three [WT and PdcA(GA)] or two (PdcAΔPAS and PdcA-EAL) separate protein purifications. (C) PDE activity, expressed as a percentage of the starting c-di-GMP hydrolyzed at 1 min with increasing concentrations of GTP. The data shown are mean values and standard deviations. The activity of full-length PdcA (blue) and PdcAΔPAS (purple) at a given GTP concentration was compared to the activity of the same protein in the absence of GTP by one-way analysis of variance (*, P < 0.05; **, P < 0.01; ***, P < 0.001; asterisks are color coded according to comparison). The activities of PdcA(GA) and PdcA-EAL in the presence of GTP were compared to their activities in the absence of GTP by one-way analysis of variance and were not statistically significantly different at any of the GTP concentration tested (not shown). (D) C. difficile strains with (left to right) the vector (v), pPdcA, pPdcA(GA), pPdcAΔPAS, or pPdcA-EAL were assayed for motility in BHIS-Tm with 0.3% agar supplemented with 0 or 5 μg/ml nisin. Motility diameters were measured after 72 h at 37°C. Data were analyzed by two-way analysis of variance and Bonferroni's multiple-comparison test comparing values to the average vector control value (**, P < 0.01).
To examine the potential regulatory effects of the PAS and GGDEF domains on PdcA enzymatic activity, we analyzed the full-length protein and three derivatives: full-length PdcA in which the putative DGDEM cyclase active-site residues are mutated to alanines [PdcA(GA)], a truncated protein with no N-terminal PAS domain (PdcA-ΔPAS), and the isolated PDE domain (PdcA-EAL) (Fig. 1A). The enzymatic activities of these recombinant proteins, along with wild-type (WT) PdcA, were assayed in vitro with radioactive c-di-GMP as a substrate. All of these proteins hydrolyzed c-di-GMP to various degrees (Fig. 1B). To determine whether GTP binding to the GGDEF domain stimulates hydrolytic activity by the EAL domain, we assayed c-di-GMP cleavage in the presence of increasing amounts of GTP. WT PdcA activity was increased 50% by the addition of one molar equivalent of GTP and increased in a dose-dependent manner at higher GTP concentrations (Fig. 1C). PdcA-ΔPAS, which has an intact DGDEM sequence, was less responsive to low levels of GTP than the WT but also displayed a dose-dependent GTP-stimulated increase in PDE activity (Fig. 1C). The enzymatic activities of PdcA(GA) and PdcA-EAL, both of which lack the DGDEM putative GTP-binding motif, were unaffected by GTP (Fig. 1C). The results are consistent with allosteric activation of PDE activity of the EAL domain as a result of GTP binding to the DGDEM sequence in the GGDEF domain, as has been observed for several other tandem GGDEF-EAL proteins with degenerate GGDEF domains (65–68). The PdcA-ΔPAS protein was slightly less active than the full-length protein in vitro (Fig. 1B), suggesting an as-yet-undefined regulatory function for the N-terminal PAS domain.
We next evaluated the roles of the PdcA regulatory domains in vivo by comparing the effects of overproducing WT PdcA or the PdcA(GA), PdcA-ΔPAS, or PdcA-EAL derivative in C. difficile. The genes for these PdcA variants were expressed in C. difficile with a plasmid-borne expression system in which sublethal concentrations of the antimicrobial peptide nisin induce gene expression (55). We did not assay biofilm formation in these strains, as the C. difficile 630 strain does not form very dense biofilms (60, 72), so the anticipated decreases upon PDE overproduction would likely be subtle. We have previously reported that overproduction of PdcA-EAL does not impact flagellar motility within 96 h (55). Here, C. difficile overproducing WT full-length PdcA exhibited a significant, nisin-dependent increase in motility compared to the vector control strain by 72 h, consistent with reduced c-di-GMP upon WT PdcA overproduction (Fig. 1D). Overproduction of PdcA(GA), PdcA-ΔPAS, or PdcA-EAL did not significantly affect motility, indicating that the PAS and GGDEF regulatory domains are necessary for maximal PdcA enzymatic activity in vivo.
pdcA transcription is regulated by CodY.
Previous studies have demonstrated that PdcA, 1 of 19 EAL domain proteins encoded in the C. difficile genome, is a functional c-di-GMP PDE (53–55). However, the contribution of PdcA to c-di-GMP signaling in C. difficile is unknown. Previous transcriptional profiling studies suggested that pdcA expression is greater during stationary phase than during exponential growth and is reduced in C. difficile lacking SigH, a sigma factor involved in the onset of stationary phase and the initiation of spore formation (73). In addition, a study aimed at identifying DNA sequences bound by the transcription factor CodY in C. difficile identified a sequence overlapping the 5′ end of the pdcA coding sequence (CD630_15150) (27, 30). To determine whether pdcA transcription shows CodY-dependent growth phase regulation, we performed quantitative reverse transcription-PCR (qRT-PCR) to evaluate transcript abundance in C. difficile mRNA harvested during exponential-phase and early stationary-phase growth. Two stationary-phase genes previously shown to be regulated by CodY, ilvC and tcdR, served as positive controls (30). In WT cells (JIR8084, a derivative of C. difficile 630), the pdcA transcript was three times as abundant in stationary phase as in log phase (Fig. 2). Similar results were obtained for ilvC and tcdR, which were 12- and 20-fold more abundant during stationary phase, respectively. In a codY mutant, transcript levels of pdcA, ilvC, and tcdR were higher than in the parent strain (2-, 12-, and 20-fold higher, respectively) during exponential phase, indicating derepression of these genes in the absence of CodY (Fig. 2). Stationary phase onset in the codY null strain caused no significant further increase in pdcA or ilvC transcripts, and the tcdR transcript showed a modest 3-fold increase (Fig. 2). Thus, expression of all three genes during exponential growth was repressed in WT C. difficile and not in the codY null strain, indicating that pdcA is a bona fide CodY-regulated gene expressed during stationary phase (27).
FIG 2.
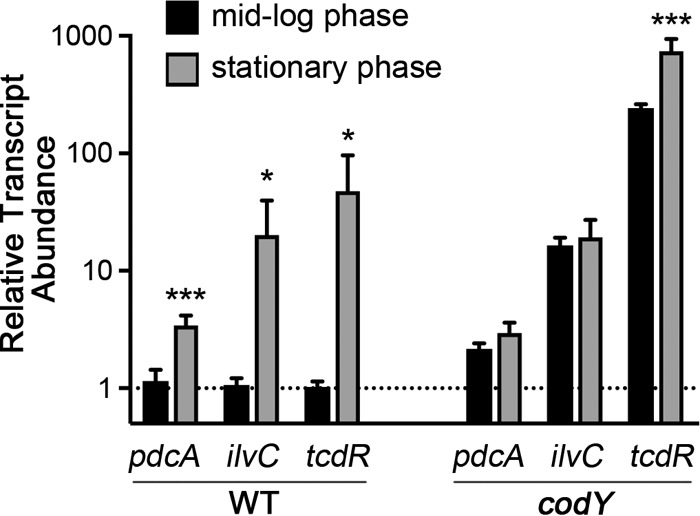
CodY regulates pdcA transcription. Transcript levels of the genes indicated in WT JIR8094 (erythromycin-sensitive derivative of C. difficile strain 630) and an isogenic codY-null strain in exponential phase (black) and stationary phase (gray) were measured by qRT-PCR. The data were analyzed by the ΔΔCT method as described in Materials and Methods, here with exponential-phase WT cells as the reference condition. The mean values and standard deviations from three biological replicates are shown. The data were analyzed by unpaired t test comparing the exponential- and stationary-phase transcript levels of each gene (*, P < 0.05; ***, P < 0.001).
Though Dineen et al. identified a DNA region encompassing the pdcA translational start site by affinity purification with CodY, the putative pdcA promoter region lacked a recognizable CodY consensus binding sequence by the specified criteria (27, 30). We used electrophoretic mobility shift assays (EMSAs) to determine whether CodY binds pdcA promoter DNA. We chose a DNA region containing the 194-bp sequence pulled down by CodY, which includes the first 146 bp of the pdcA coding sequence (27), plus an additional 261 bp of the upstream sequence. GTP and BCAA leucine, isoleucine, and valine, which stimulate CodY binding to target DNA in C. difficile, were included in the binding reaction mixtures, and the amino acids were added to the electrophoresis buffer (30). CodY had no effect on the migration of the 133-bp nonspecific, negative control DNA fragment of V. cholerae gbpA included in each binding reaction mixture. As shown previously, CodY bound to and shifted the migration of the ilvC promoter region, which served as a positive control (Fig. 3A) (27). Similarly, CodY bound to the pdcA promoter fragment (Fig. 3B). These results suggest that CodY negatively regulates pdcA transcription by binding directly to the pdcA promoter.
FIG 3.

CodY binds directly to the pdcA promoter region. Purified six-histidine-tagged C. difficile CodY was tested for the ability to bind the pdcA promoter region with EMSAs. Serial 2-fold dilutions of CodY were incubated with the ilvC promoter region previously shown to be directly bound by CodY (A) or the pdcA promoter region (B). As a negative control, a 133-bp V. cholerae DNA fragment was included in each binding reaction mixture (asterisks). The concentrations of CodY used were (left to right) 0, 7.2, 3.6, 1.8, 0.9, 0.45, 0.23, 0.12, and 0.06 μM. In each reaction mixture and in the electrophoresis buffer, isoleucine, leucine, and valine were added to promote CodY binding. A representative of three independent experiments is shown. The values to the left are molecular sizes in kilodaltons.
Mutation of pdcA specifically affects stationary-phase phenomena.
Despite using multiple targeting sequences, we were unsuccessful in generating a pdcA mutant via insertional inactivation by a targeted intron (74). As an alternative, we replaced the pdcA open reading frame (ORF) with the catP gene by allelic exchange. We cloned the 2,000-bp sequences upstream and downstream of pdcA into pMC234 flanking the catP gene (see Fig. S1 in the supplemental material). The construct was introduced into C. difficile 630 by conjugation. After the identification of a thiamphenicol-resistant recombinant strain in which the plasmid had recombined into the chromosome at the pdcA locus, it was passaged without selection to allow plasmid loss. The resulting thiamphenicol-sensitive isolates were screened by PCR to identify an isolate in which pdcA had been replaced on the chromosome with catP by double homologous recombination (Fig. S1) (75).
The resulting pdcA::catP mutant was assessed for altered c-di-GMP and related phenotypes, including flagellar motility, biofilm formation, and toxin production (55, 57). The cytoplasmic c-di-GMP concentrations of the WT and pdcA::catP deletion C. difficile strains grown to early stationary phase were assayed by ultraperformance liquid chromatography coupled to mass spectrometry (UPLC-MS) and found to be indistinguishable (Fig. 4A), indicating that the absence of PdcA does not impact global c-di-GMP levels under these growth conditions. C. difficile motility through brain heart infusion medium with 0.5% yeast extract (BHIS) and 0.3% agar, which is inhibited by high levels of c-di-GMP, was also not altered in the pdcA::catP mutant (Fig. 4B). In contrast, biofilm formation, which is positively regulated by c-di-GMP (56, 60), was modestly but significantly higher in the pdcA::catP mutant strain than in the WT (Fig. 4C). The pdcA::catP mutant expressing pdcA from a plasmid (pRT1214) under the control of its native promoter produced biofilm comparably to the WT bearing the control vector (pRT1099) (Fig. 4D and E). Expression of an allele of pdcA encoding an enzymatically inactive PdcA protein in the pdcA::catP mutant (pdcA E479A allele, pRT1662) did not restore biofilm formation to WT levels (Fig. 4D and E), indicating that PdcA controls biofilm production via c-di-GMP hydrolysis. Thus, although the pdcA mutation does not affect global c-di-GMP levels during early stationary phase and does not alter motility through 0.3% agar, increased c-di-GMP as a result of the pdcA mutation resulted in increased biofilm formation.
FIG 4.

pdcA affects biofilm formation but not swimming motility or global c-di-GMP levels of C. difficile. (A) c-di-GMP in the cytoplasm of the 630 WT and pdcA::catP mutant C. difficile strains was quantified by UPLC-MS and normalized to the total cellular protein level. The mean values and standard deviations of six biologically independent samples are shown. (B) Swimming motility through BHIS–0.3% agar was measured at 24, 48, and 72 h. The mean values and standard deviations of six biologically independent samples are shown. (C) Biofilm formation assayed by crystal violet staining after 24 h of growth. The mean values and standard deviations of five biologically independent samples are shown. **, P < 0.01 by unpaired t test. (D, E) Complementation analysis of biofilm formation after 24 h of growth. pRT1099 is the vector control, pRT1214 encodes PdcA, and pRT1662 encodes a catalytically inactive form of PdcA. (D) Representative image of crystal violet-stained biofilms. (E) Quantification of biofilm biomass by crystal violet staining. The mean values and standard deviations of three biologically independent samples are shown. **, P < 0.05 by one-way analysis of variance and Dunnett's multiple-comparison test.
pdcA affects toxin production and cytopathicity.
Production of the C. difficile toxins TcdA and TcdB is a stationary-phase phenomenon governed by multiple regulatory factors, including c-di-GMP and CodY (76). Production of TcdR, a sigma factor that positively regulates tcdA and tcdB expression, is repressed by CodY when GTP and BCAA are abundant in the cytoplasm (27). Expression of tcdR is positively regulated by the sigma factor SigD (57, 58). Expression of sigD is repressed by elevated c-di-GMP, making c-di-GMP a negative regulator of toxin gene expression (55, 57, 58). To determine whether PdcA activity during stationary phase impacts toxin production, TcdA protein produced by the 630 WT and pdcA::catP mutant strains, bearing the vector or complementation plasmids indicated, was detected by Western blot analysis. The pdcA::catP mutant strain with the vector (pRT1099) produced less TcdA than the WT with the vector (Fig. 5A). Expression of pdcA under the control of its native promoter (pRT1214) partially restored toxin production in the pdcA::catP mutant strain (Fig. 5A). In contrast, expression of the pdcA E479A allele (pRT1662) encoding catalytically inactive PdcA did not restore WT levels of toxin production (Fig. 5A). These results support a role for PdcA in the regulation of TcdA production, specifically, the ability to hydrolyze c-di-GMP.
FIG 5.

pdcA influences C. difficile toxin production and cytopathicity. (A) TcdA production by 630/pRT1099 (vector control), pdcA::catP/pRT1099 (vector control), pdcA::catP/pRT1214 (complementation with pdcA WT allele), and pdcA::catP/pRT1662 (complementation with pdcA E479A mutant allele). Strains were grown for 16 h in TY medium, and lysates were probed for TcdA by Western blot analysis. Samples were normalized by adjustment to the OD of the culture. The data are expressed as a percentage of the 630/pRT1099 value in each respective experiment. The mean values and standard errors from four independent assays are shown. Data were analyzed by one-way analysis of variance and Tukey's multiple-comparison test (**, P < 0.01; ****, P < 0.0001 compared to 630/pRT1099; #, P < 0.01 for the comparisons indicated). A representative Western blot analysis for TcdA is shown at the top. (B) Relative viability of MDCK-LA cells after incubation with supernatants from 630 WT and pdcA::catP mutant strain stationary-phase cultures. Data shown are mean values and standard deviations of three biological replicates and were analyzed by two-way analysis of variance and Dunnett's posttest (n.t., not treated; n.s. not significant; *, P < 0.05; ***, P < 0.001). (C) Cytopathicity of 630 WT and pdcA::catP mutant strain supernatants against MDCK-LA cells after 24 h of incubation. Scale bars, 10 μm. (D) Effects of the 630 WT and pdcA::catP mutant strains on the actin cytoskeleton (red). TIRF microscopy shows stress fibers at the basolateral cell surface. Epifluorescence shows punctate microvilli at the apical surface. Scale bars, 10 μm.
To confirm that diminished TcdA production in the pdcA::catP mutant strain corresponds to reduced cytopathicity against mammalian cells, we incubated MDCK-LA epithelial cell monolayers overnight with filtered supernatants from stationary-phase cultures of the WT and pdcA::catP mutant strains. C. difficile supernatants, as well as the purified TcdA and TcdB toxins, disrupt the actin cytoskeleton and cause MDCK cell rounding and death (77). We found that a 1:40 dilution of WT supernatant disrupted cell-cell junctions and caused the epithelium to dissociate from the substrate (Fig. 5C). A 1:40 dilution of pdcA::catP supernatant resulted in some individual cell detachment but left the monolayer largely intact (Fig. 5C). A 1:80 dilution of WT supernatant caused partial cell rounding and dissociation, leaving isolated patches of monolayer intact (Fig. 5C). A 1:80 dilution of pdcA::catP supernatant had almost no visible effect on monolayer integrity (Fig. 5C). These observations are supported by quantitative evaluation of cell viability after incubation with C. difficile supernatants. ATP levels, indicating cell viability, were significantly lower in MDCK-LA cells treated with supernatant from the WT strain than in cells treated with supernatant from the pdcA::catP mutant strain, indicating reduced cytotoxicity of the pdcA::catP mutant (Fig. 5B).
To determine whether this reduced cytopathicity was due to reduced toxin production, as opposed to another effect of the pdcA::catP mutation, we assessed the integrity of the actin cytoskeleton, which is fluorescently labeled in MDCK-LA cells (78). Healthy epithelial cells contain actin stress fibers at the basolateral surface and actin-based microvillus protrusions at the apical surface (Fig. 5D, mock treated) (78). Incubation with WT C. difficile lysate resulted in actin depolymerization and perturbed both of these structures (Fig. 5D). Incubation with the same dilution of pdcA::catP mutant supernatant left these subcellular actin structures largely intact (Fig. 5D). These data suggest that while the pdcA::catP mutation causes a moderate reduction in toxin synthesis, it is a functionally significant difference and is sufficient to render the pdcA mutant strain less cytopathic and cytotoxic to mammalian cells.
Growth phase regulation of pdcA expression limits the cellular behaviors affected by PdcA.
c-di-GMP inhibits swimming motility and toxin biosynthesis by repressing the expression of flagellum and toxin genes in C. difficile (57, 58), and c-di-GMP promotes biofilm formation (56, 60). However, the pdcA mutation only had a detectable effect on biofilm formation and toxin production. These results were surprising, because regulation of tcdA and tcdB by c-di-GMP occurs via SigD, which is encoded in the flgB operon, whose expression is directly inhibited by c-di-GMP (57, 58). We speculated that the directed effect of PdcA on a subset of c-di-GMP-regulated processes could be due to restriction of pdcA expression to specific conditions and/or to posttranslational control of PdcA function. Given that pdcA is a stationary-phase gene (Fig. 2) and the observed biofilm and toxin phenotypes involve stationary-phase bacteria, we hypothesized that temporal regulation of pdcA transcription limits PdcA activity to stationary phase. If this is so, we predict that while pdcA expression during the motility assay may not be high enough for the pdcA mutation to have a measurable effect on bacterial swimming, the pdcA::catP mutation will still affect the expression of both flagellum and toxin genes under stationary-phase conditions. To test this, we compared the expression levels of toxin and flagellar genes in the 630 WT and pdcA::catP mutant strains during exponential growth and during stationary phase. We found that transcript levels of tcdA, the early flagellar gene flgB, and the flagellar filament gene fliC were unaffected by the pdcA::catP mutation during exponential phase (Fig. 6A). During stationary phase, when pdcA is expressed in WT cells, transcript levels of tcdA and flgB were significantly lower in the pdcA::catP mutant strain than in the parental strain (Fig. 6B). The 2-fold reduction observed in toxin gene expression was consistent with the 2-fold reduction in TcdA protein production due to the pdcA::catP mutation (Fig. 5A), supporting the conclusion that pdcA influences toxin production by regulating gene expression. The fliC transcript was also somewhat reduced in the pdcA::catP mutant strain during stationary phase, though the differences were not statistically significant (P = 0.059) (Fig. 6B). Expression of the WT pdcA gene in the pdcA::catP mutant partially restored tcdA and fliC expression, though the differences were not statistically significant (Fig. S3). The expression of the pdcA E479A mutant allele had a more modest or no effect (Fig. S3). The challenges of complementing the changes in gene expression in the pdcA::catP mutant are likely due to the small effects of the pdcA mutation on transcript levels. Thus, the pdcA mutation affects the regulation of flagellar genes in addition to the toxin genes under nutrient-limited conditions in which pdcA is expressed, suggesting that genes expressed during stationary phase have limited effects on motility through soft agar.
FIG 6.
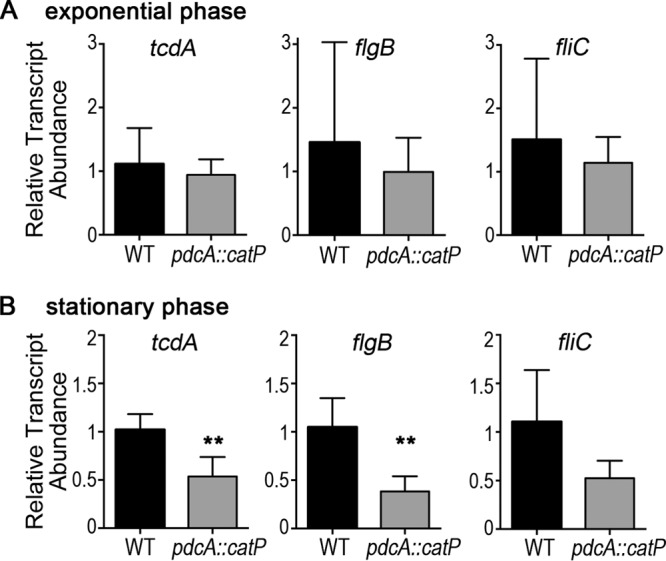
Transcriptional regulation in the pdcA::catP mutant. (A) Relative transcript levels of tcdA, flgB, and fliC in WT (black) and pdcA::catP mutant (gray) cells during exponential growth. (B) Relative transcript levels during stationary phase. The mean values and standard deviations of five biological replicates are shown. The data were analyzed by unpaired t test (**, P < 0.05).
DISCUSSION
In this work, we expanded our prior understanding of c-di-GMP regulation in C. difficile by characterizing the regulation and function of PdcA, 1 of 19 known or putative c-di-GMP PDEs in C. difficile strain 630. We had previously reported that overexpression of the PdcA-EAL domain had no discernible effect on C. difficile physiology or behavior and speculated that cytoplasmic c-di-GMP levels were low enough under the conditions studied to preclude a significant reduction upon ectopic PDE overproduction (55). Here, we show that the activity of the PdcA regulatory domains and growth phase regulation of pdcA expression determine PdcA-dependent phenotypes.
Disruption of pdcA significantly impacts biofilm formation and toxin biosynthesis. Disruption of pdcA does not affect flagellar motility through soft agar, although the pdcA mutation does affect flagellar gene expression during stationary phase. We speculate that this distinction is due to the conditions of the motility assay. As flagellar motility assays involve bacterial cells continuously expanding into conditions of low cell density and fresh, nutrient-rich medium, the effects of stationary-phase genes may not be readily apparent. In contrast, C. difficile toxins are produced in stationary-phase cultures in vitro (27) and biofilms are likely to contain stationary-phase bacteria, making it possible to detect the effects of a pdcA mutation. Thus, CodY inhibition of pdcA transcription during exponential growth results in temporal regulation that directs PdcA activity toward the c-di-GMP-regulated processes that occur during stationary phase.
In vitro assays of PDE activity of PdcA and mutant derivatives indicate that the PAS domain is required for full activity and suggest that GTP binding to the GGDEF domain stimulates the enzymatic activity of the EAL domain. These results are supported by in vivo analyses. While ectopic expression of WT, full-length pdcA significantly increased swimming motility, expression of the mutant derivatives did not, although they were catalytically active. Together, these data indicate that PdcA activity is regulated at the protein level. The ability of degenerate GGDEF domains to regulate a tandem EAL domain in response to GTP has been demonstrated (68, 79), and it remains possible that the PdcA PAS domain provides another as-yet-uncharacterized means of posttranslational regulation. Of the 19 C. difficile EAL family c-di-GMP PDEs, 18 contain degenerate GGDEF domains (80), suggesting that c-di-GMP turnover in C. difficile may be generally linked to GTP availability (65–67). As c-di-GMP synthesis utilizes GTP as a substrate, linking c-di-GMP hydrolysis to GTP availability may limit c-di-GMP turnover under nutrient-limiting growth conditions when intracellular GTP availability is limited (33, 34, 65, 81–83).
A DNA region encompassing the 5′ end of the pdcA ORF and putative promoter was identified as a target of CodY binding (27). That study did not identify a consensus CodY binding site in the pdcA promoter region. However, we demonstrate that CodY binds directly to the pdcA promoter region in vitro. We note that Dineen et al. allowed up to three mismatches from the CodY consensus, whereas the putative CodY binding site upstream of the pdcA coding sequence contains four bases that deviate from the consensus (Fig. S2). Lower concentrations of CodY bound to and shifted the DNA fragment containing the ilvC promoter, suggesting higher affinity of CodY for the ilvC promoter than for the pdcA promoter, consistent with the greater transcriptional repression by CodY observed for ilvC.
Regulation of pdcA transcription by CodY further links PdcA activity to cytoplasmic GTP. Interestingly, CodY and PdcA appear to have dramatically different affinities for GTP in vitro. Purified C. difficile CodY is activated in vitro only by very large (10,000-fold) molar excesses of GTP (30, 84), while purified PdcA is activated by an equimolar amount of GTP, suggesting that PdcA activity may be stimulated by GTP concentrations too low to activate CodY. This suggests that fluctuations in cytoplasmic GTP levels can impact the transcription of the pdcA gene, as well as modulate the activity of previously expressed PdcA protein (Fig. 7). At limiting GTP concentrations, the enzymatic activity of PdcA is likely to be low, but deactivation of CodY would allow pdcA transcription to increase the overall level of PdcA in the cell cytoplasm. At the other extreme, very high GTP levels would inhibit pdcA transcription via CodY, but the activity of existing PdcA molecules would be maximal. In both B. subtilis and S. aureus, CodY functions as a “dimmer” rather than a binary on-off switch, repressing certain genes differentially in response to moderate or severe levels of nutrient limitation (85, 86). It is likely that in C. difficile many circumstances in vivo could feature intermediate cytoplasmic GTP levels low enough to allow some pdcA expression but high enough to permit stimulation of PdcA enzymatic activity (Fig. 7). As intracellular GTP concentrations in bacteria are highly dynamic and responsive to the addition of nutrients to the growth medium, with transient 70 to 80% decreases in the cytoplasmic GTP concentration and concomitant rises in ppGpp occurring upon stationary phase onset or during starvation (32–34, 81–84, 87), the net PdcA activity in the cell may be highly responsive to intracellular GTP availability and extracellular nutrient availability.
FIG 7.
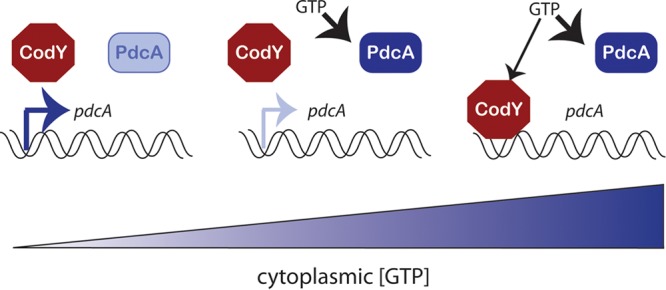
GTP levels inversely regulate the production and activity of PdcA enzymes. At very low cytoplasmic GTP levels, the activity level of existing PdcA proteins will be low, but CodY will be deactivated to permit additional pdcA transcription. At intermediate concentrations, both pdcA transcription and PdcA activity will be responsive to GTP fluctuations. At high GTP levels, pdcA transcription will be inhibited but existing PdcA will be very active.
c-di-GMP signaling is affected by nutrient limitation in other bacterial species. For example, multiple Pseudomonas species have been shown to increase the expression of PDE genes in response to nutrient starvation, although these genes have been implicated in biofilm dispersal rather than biofilm formation or motility (88, 89). Similarly, a bifunctional DGC-PDE protein affects the long-term survival of Mycobacterium smegmatis under nutrient-limiting conditions (90). Nutrient limitation is a clinically relevant stress for intestinal pathogens: the healthy mammalian large intestine is not rich in usable nutrient sources for C. difficile, as mono- and disaccharides and amino acids are largely absorbed by the host in the small intestine (91, 92). The recent discovery that a “nutrient bloom” in response to antibiotic disruption of the gut microbiome increases susceptibility to C. difficile infection suggests that nutrient availability is a major barrier to C. difficile survival in a healthy host (93). It is worth noting that pdcA (CD630_15150) expression is also increased by heat shock, another clinically relevant stress (94), raising the possibility that this PDE controls a more general stress response in C. difficile.
This is the first report linking any c-di-GMP-regulated phenotypes in C. difficile to the activity of a specific c-di-GMP metabolism protein, underscoring the importance of analyzing individual PDE and DGC enzymes. Future studies to more precisely identify the extracellular nutrient signals that affect PdcA activity will help define the environmental conditions that influence c-di-GMP signaling in C. difficile. Further work to determine the chemical signals that modulate intracellular c-di-GMP in C. difficile is vital, given the role of this second messenger in the control of processes central to pathogenesis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table S1. C. difficile 630 and derivative strains were routinely grown in BHIS, supplemented as specified, in an anaerobic chamber with an atmosphere of 85% N2, 5% CO2, and 10% H2. Medium for growth of strains containing pMC123 derivative plasmids was supplemented with 10 μg/ml thiamphenicol (BHIS-Tm). Medium for growth of strains containing the integrated pSD21 codY insertional mutation was supplemented with 2 μg/ml erythromycin (BHIS-Erm). Medium for growth of strains containing pRT1099, pRT1214, or pRT1662 was supplemented with 500 μg/ml spectinomycin unless indicated otherwise. Escherichia coli strains were propagated in Luria-Bertani (LB) medium or on LB agar plates containing 50 μg/ml ampicillin, 10 μg/ml chloramphenicol, and/or 50 μg/ml spectinomycin, as needed, at 37°C under aerobic conditions.
Plasmid and strain construction.
All restriction enzymes and DNA polymerase (Phusion) were purchased from New England BioLabs. The sequences of the oligonucleotides used in this study are listed in Table S2.
The creation of pMMBneo::pdcA and pMMBneo::pdcA-EAL has previously been described (55). Purified pMMBneo::pdcA (55) was used as the PCR template to generate E. coli expression constructs. A mutant allele of pdcA (CD630_1515) in which the GGDEF motif, sequence DGDEM, is mutated to alanines was generated by splicing by overhang extension (SOE) PCR. Primers pdcAF and pdcAgaR were used to amplify the upstream fragment, and primers pdcAgaF and pdcAR were used to amplify the downstream fragment, and they were subsequently spliced together. A PdcA truncation lacking the N-terminal PAS domain (residues 1 to 251) was amplified from pMMBneo::pdcA with primers pdcAdpF and pdcAR. The resulting fragments were digested with KpnI and PstI and ligated to similarly digested pMMBneo (95). Kanamycin-resistant transformants of E. coli DH5α were screened for the desired plasmids by PCR with gene-specific primers and plasmid-specific primers 67EHF and 67EHR. The plasmids were introduced by electroporation into E. coli BL21 for gene expression and protein purification.
For gene expression in C. difficile, pPdcA (55) was the PCR template for generating a GGDEF point mutant and a ΔPAS truncation with the primers listed above. The resulting fragments were digested with KpnI and PstI and ligated to similarly digested pMC-Pcpr. Chloramphenicol-resistant transformants of E. coli DH5α were screened for the desired plasmids by PCR with primers pUCmscF and m13R. These plasmids were transformed by electroporation into strain HB101(pRK24) to allow conjugation with C. difficile. The HB101(pRK24) plasmid donor strains were mated with C. difficile strain 630 as previously described (55). Transconjugants were selected on BHIS-Tm supplemented with 100 μg/ml kanamycin and screened by PCR with pUCmscF and m13R to confirm the presence of a plasmid with an insert of the correct size and with primers tcdBF and tcdBR to confirm the isolation of C. difficile. At least two isolates were obtained for each conjugation. The generation of pPdcA and pPdcA-EAL and their introduction into C. difficile were previously described (55).
The pdcA::catP mutant was created by double homologous recombination essentially as previously described, with some modifications (75). Allelic-exchange vector pMC234 was created by the stepwise addition of two DNA fragments to pUC19 (96). A 390-bp oriT fragment was PCR amplified from pJIR1456 (97) with primers oMC15 and oMC16, digested with EcoO1091/AatII, and ligated into pUC19 to give plasmid pMC95 (98). A 1,043-bp fragment containing the C. perfringens catP gene was then amplified from pJIR1456 (97) with primers oMC2 and oMC143, digested with BamHI, and ligated into pMC95 to yield plasmid pMC234. The 2,000-bp regions upstream and downstream of the pdcA ORF were amplified with primers pdcAf1 and pdcAr1 or pdcAf2 and pdcAr2, respectively. The 5′ flanking region was digested with EcoRI and KpnI, and the 3′ flanking region was digested with SalI and PstI. Fragments were sequentially ligated into pMC234, flanking the catP gene, to create pMC234::pdcA::catP, which was transformed by electroporation into HB101(pRK24) for conjugation with C. difficile 630. Isolates with a single-crossover integration of pMC234::pdcA::catP were selected on BHIS-Tm plates supplemented with 100 μg/ml kanamycin to select against HB101(pRK24). Thiamphenicol-resistant single-crossover isolates were confirmed by PCR with primers within the sequences flanking the pdcA ORF, which yield a 2.1-kb product when amplifying pdcA from the chromosome and a 1.1-kb product when amplifying catP from the integrated plasmid. Primers PdcAF and PdcAR, which amplify the pdcA ORF but not the flanking sequences, were used to specifically detect pdcA, and oMC143 and oMC2 were used to specifically detect catP. Additional screening was performed with the flanking primer pair pdcAbamHI and pdcApstI or pdcAbamHI and PdcAR0, which will amplify the WT pdcA ORF on the chromosome but yield no product from the integrated vector. Isolates were passaged once in BHIS-Tm broth and subsequently in BHIS with no antibiotic. Each passage was plated on BHIS agar, and individual colonies were screened by PCR with multiple pairs of primers. After eight passages, we obtained a strain in which the pdcA gene (2,094 bp) was replaced with the catP gene (1,043 bp). The loss of integrated pMC234::pdcA::catP was confirming by PCR screening for the bla gene on the vector with primers blaF and blaR.
To create vectors encoding spectinomycin resistance, the Enterococcus faecalis aad9 gene was amplified from pBTS (gift from A. R. Richardson) (99) with primers aad9F_EcoRV and aad9R_EcoRV and digested with EcoRV. pMC123 was digested with SapI and PciI to remove the catP cassette and then treated with Klenow fragment to blunt the ends. The aad9 gene was ligated into pMC123 lacking catP, generating pRT1099. pdcA and the 521 bp upstream of the translational start site were amplified from C. difficile genomic DNA with primers pdcApromF and pdcApromR and ligated into pRT1099 at the SphI and BamHI restriction sites to create pRT1214. A pdcA E479A mutant allele with an alanine substitution of glutamic acid in the EAL motif was generated by SOE PCR with primers pdcAeaF and pdcAeaR. Primers pdcApromF and pdcAeaR were used to amplify the upstream fragment of C. difficile 630 genomic DNA, and primers pdcAeaF and pdcApromR were used to amplify the downstream fragment. The fragments were spliced together and cloned into pRT1099 at the SphI and BamHI restriction sites, yielding pRT1662. The plasmids were confirmed by PCR and sequencing of the cloned fragment and then introduced into C. difficile by conjugation via HB101(pRK24).
RNA isolation and qRT-PCR.
RNA was isolated as described previously from C. difficile cultures grown in BHIS with the appropriate antibiotics to exponential or early stationary phase as previously indicated (55). Exponential-phase samples were collected at an optical density at 600 nm (OD600) of 0.4 to 0.6, and stationary-phase samples were collected 1 to 2 h after logarithmic growth ended. After DNase treatment (Ambion), cDNA samples were prepared from 500 ng of RNA with random hexamers and the Tetro cDNA synthesis kit (Bioline). Real-time PCR was done with 4 ng of template cDNA with the SensiMix SYBR and fluorescein kit (Bioline). Primers (Table S2) were designed with the PrimerQuest tool from IDT DNA Technologies, and forward and reverse primers were named in accordance with the pattern gene-qF and gene-qR, respectively. At least three independent samples were analyzed. The rpoC transcript was used as the reference gene (55). Controls with no reverse transcriptase were included for all templates and all primer sets. The data were analyzed by the 2−ΔΔCT method, with normalization to rpoC and the stated reference condition or strain (55).
Protein purification.
BL21 expression strains were grown at 37°C to an OD600 of 0.1 to 0.15, at which point the cultures were shifted to 25°C and expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 16 h. Cells were collected by centrifugation and lysed by sonication in His6 buffer (10 mM Tris-HCl [pH 7.8], 300 mM NaCl, 50 mM NaH2PO4, 10% glycerol) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and 1 mg/ml lysozyme (55). Lysates were clarified by centrifugation at 12,000 × g for 30 min at 4°C, and the soluble fractions were incubated with HisPur Ni-nitrilotriacetic acid (NTA) resin (Thermo Scientific) for 2 h at 4°C. Samples were centrifuged at 700 × g for 2 min at 4°C, and then the resin was suspended in His6 buffer, rocked for 15 min at 4°C, and centrifuged at 700 × g for 2 min at 4°C. This washing protocol was repeated twice with His6 buffer containing 100 mM imidazole. Proteins were eluted in His6 buffer with 250 mM imidazole. Purified proteins were confirmed by SDS-PAGE and staining with Coomassie brilliant blue (55). Protein concentrations were measured by UV spectroscopy by using ε280 values calculated from ExPASY ProtParam (100). Purified proteins were stored at 4°C and used for enzymatic assays within 5 days of purification.
Histidine-tagged CodY was purified from E. coli BL21 bearing pEAV1 (30). The strain was grown at 37°C to an OD600 of 0.4 to 0.5, and then IPTG was added to a final concentration of 1 mM. After an additional 5 h of growth at 37°C, cells were collected by centrifugation. The pellet was suspended in BugBuster Lysis Solution (EMD Millipore), and cells were lysed in accordance with the manufacturer's protocol. The soluble fraction was collected by centrifugation and incubated with HisPur Ni-NTA resin for 2 h at 4°C. The resin was washed with CodY buffer (20 mM Tris [pH 8], 125 mM KCl, 50 mM NaH2PO4, 10% glycerol, 1 mM PMSF) containing 50 mM imidazole. CodY was eluted with CodY buffer containing 200 mM imidazole. Amicon Ultrafree columns with a 3,000-Da molecular mass cutoff (EMD Millipore) were used for buffer exchange and protein concentration into CodY buffer. Protein concentration was determined with the Pierce BCE Protein Assay kit (Thermo Scientific). Glycerol was added to a final concentration of 20%, and the protein was stored at −20°C.
In vitro enzymatic assays.
Purified PdcA, PdcA(GA), PdcA-ΔPAS, and PdcA-EAL were assayed for PDE activity as previously described (55). The radiolabeled c-di-GMP substrate was synthesized by using the characterized C. difficile DGC DccA and purified as described previously (101, 102). The reaction was initiated by the addition of radiolabeled substrate, and 1-μl aliquots were taken for analysis at 6, 30, 60, and 120 s. Reaction mixtures lacking enzyme were used as negative controls. Where specified, 0, 1, 20, or 100 μM GTP was added to the reaction mixtures. Aliquots were analyzed by thin-layer chromatography as described previously (102). All reaction mixtures were prepared in triplicate. Enzymatic activity was recorded as the percentage of 32P-labeled c-di-GMP cleaved over time.
EMSAs.
Binding of CodY to the pdcA promoter, as well as an ilvC promoter positive control, was assessed by EMSA essentially as described previously (30). A region encompassing the first 145 nucleotides of the pdcA coding sequence and 261 upstream bases was amplified from C. difficile 630 chromosomal DNA by PCR with pdcAemsaF and pdcAemsaR. The 423-bp ilvC promoter region was amplified with OSD107 and OSD108 (27). As a negative control, a 133-bp fragment of the gbpA gene was amplified from V. cholerae C6706 chromosomal DNA with primers gbpAqF and gbpAqR (103). Binding reactions between CodY and putative target DNA took place in a mixture of 20 mM Tris (pH 8); 50 mM sodium glutamate; 10 mM MgCl2; 5 mM EDTA; 0.1% Tween 20; 5% glycerol; 2 mM GTP; and 100 mM each leucine, isoleucine, and valine (30). Binding reaction mixtures contained serially diluted CodY, 50 ng of V. cholerae gbpA DNA, and 70 ng of either ilvC promoter DNA or pdcA promoter DNA and were incubated at room temperature for 30 min. Samples were separated by electrophoresis in 10% Mini-PROTEAN TGX precast gels (Bio-Rad) in a running buffer consisting of 25 mM Tris base, 192 mM glycine, and 10 mM isoleucine-leucine-valine. Gels were stained for 1 h with GelRed (Biotium) and imaged under UV light.
c-di-GMP quantification by UPLC-MS.
Nucleotides were extracted from C. difficile and quantified as previously described (60). Briefly, individual colonies of the 630 WT and pdcA::catP mutant strains were grown anaerobically at 37°C for 7 h. These starter cultures were diluted 1:100 in 50 ml of BHIS medium and grown anaerobically at 37°C for 10 h to an OD600 of 1.6 to 1.8. Nucleotides were isolated from the supernatant through methanol-acetonitrile extraction as previously described (60). The total protein content of the insoluble fraction of each sample was quantified by the colorimetric BCA assay kit (Thermo Scientific). Nucleotide samples were dried overnight with a vacuum concentrator at room temperature and suspended in 100 μl of distilled H2O/200 μl of extracted nucleotide. Samples were stored at −80°C until use. Aliquots (10 μl) were injected into a TSQ Quantum Ultra triple-quadrupole mass analyzer (Thermo Scientific, Waltham, MA). The analytes were separated on a Waters HSS T3 UPLC column (2.1 by 100 mm, 1.8 μm) by gradient elution from 99.9% solvent A (10 mM ammonium formate in water) to 40% solvent B (10 mM ammonium formate in methanol) for 2 min, followed by column flushing at 90% solvent B for 2.5 min and column re-equilibration for 3.5 min. The column effluent was diverted to waste for the first 2 min. The mass spectrometer parameters were as follows: positive-ion electrospray mode; spray voltage, 3.5 kV; vaporizer temperature, 250°C; sheath gas, 35 arbitrary units; auxiliary gas, 30 arbitrary units; capillary temperature, 285°C; collision gas pressure, 1.5 mtorr (0.19 Pa). c-di-GMP was detected by monitoring precursor ion-to-fragment ion transitions (m/z 691 to >152 [collision energy, 35] and m/z 691 to >540 [collision energy, 21]) and quantified by using a calibration curve of known concentrations of pure c-di-GMP (Biolog Life Science Institute, Bremen, Germany) ranging from 0.1 to 1,000 nM. Cytoplasmic c-di-GMP concentrations were normalized to the total protein content of the samples. Four independent samples were analyzed for each strain.
Motility assays.
Motility experiments were performed as previously described (55). Briefly, individual colonies were inoculated with sterile toothpicks into 0.3% agar plates containing BHIS or BHIS-Tm with 0 or 5 μg/ml nisin. The plates were incubated at 37°C in an anaerobic chamber for 72 h. The diameters of motility growth were measured every 24 h for three independent experiments.
Detection of TcdA by Western blot analysis.
Overnight cultures were diluted 1:100 in tryptone-yeast (TY) medium with 250 μg/ml spectinomycin (104). After 16 h of growth under anaerobic conditions at 37°C, bacteria were collected from 3 ml of culture by centrifugation and suspended in SDS-PAGE loading buffer. Samples were normalized to the ODs of the cultures for loading onto 4 to 15% TGX polyacrylamide gradient gels (Bio-Rad). After electrophoresis, the samples were transferred to nitrocellulose membranes. Blots were probed with a mouse anti-TcdA antibody (Novus Biologicals) and an IR800-conjugated goat anti-mouse secondary antibody (Thermo Fisher) and visualized on an Odyssey Imager (Li-COR Biotechnology). The anti-TcdA signal was quantified with Image Studio (Li-COR Biotechnology). Four independent experiments were done, and the data are expressed as percentages of the strain 630/pRT1099 (WT with vector) value in that experiment.
Biofilm assays.
Untreated polystyrene 24-well culture plates (Corning) were kept in an anaerobic chamber for a minimum of 72 h prior to use. Overnight cultures were diluted 1:50 in 1 ml of BHIS supplemented with 1% glucose and 50 mM sodium phosphate buffer (pH 7.5). For experiments with C. difficile bearing pRT1099 and derivatives, 250 μg/ml spectinomycin was added to the medium. After incubation at 37°C for 24 h under anaerobic conditions, the culture supernatant was removed and the adherent biomass was washed once with phosphate-buffered saline (PBS) and stained for 30 min with 0.1% (wt/vol) crystal violet in water. Excess crystal violet was removed, and the wells were washed twice with PBS. The bound crystal violet was solubilized with 95% ethanol. The absorbance at 570 nm was measured with a Synergy HT microplate reader (BioTek). Experiments were done at least three times.
Analysis of cytopathic effects on target cells.
The WT and pdcA::catP mutant C. difficile strains were grown in BHIS medium supplemented with 50 mM sodium phosphate buffer (pH 7.5) for 24 h. Cultures were centrifuged for 5 min at 2,000 × g, and supernatants were filtered through 0.45-μm sterile nylon filters (Fisher). MDCK cells stably expressing LifeAct-tagRFP-T (ibidi USA) (78) were generated as described in reference 105. Briefly, type II MDCK cells (clone T23; Invitrogen) were cotransfected with pLL5/LifeAct-tagRFP-T and pTK-Hyg. The transfected cells were cultured with selection medium containing 250 μg/ml hygromycin B (Roche). Clones were selected by red fluorescent protein (RFP) fluorescence and confirmed by Western blot analysis for tagRFP protein and are termed MDCK-LA cells here. For cytopathicity assays, MDCK-LA cells were seeded at high density and grown for 2 days after confluence was reached in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (F2442; Sigma) in 24-well tissue culture plates (3524; Costar) at 37°C and 5% CO2. Samples were then incubated with the dilutions of bacterial supernatant indicated at 37°C and 5% CO2 for 24 h. For quantitative measurements of cell viability, ATP content was measured with the CellTiter-Glo Luminescent Cell Viability assay (Promega).
MDCK cells were imaged with a Nikon TiE microscope equipped with a Hamamatsu Orca-Flash 4.0 camera. Cells in plates were imaged with Nikon 10×/numerical aperture (NA) 0.25 (Plan, Ph1 DL) and 20×/NA 0.75 (Plan Apo, Ph2 DM) objectives in conjunction with phase-contrast and differential interference contrast optical components, as indicated. For visualization of the actin cytoskeleton, cells were grown in 35-mm glass bottom dishes (P35G-1.5-20-C; MatTek) and visualized with the same microscope with a laser with a 568-nm wavelength for total internal reflection fluorescence (TIRF) and epifluorescence through a 60×/NA 1.45 (Plan Apo TIRF) objective. All imaging data were collected with Nikon Elements. Image analysis was performed with Nikon Elements and ImageJ.
Supplementary Material
ACKNOWLEDGMENTS
We thank Abraham Sonenshein for the JIR8094/pSD21 strain and for pEAV1.
This research was supported by NIH grants U54-AI057157 and R01-AI107029 to R.T., R01-DC003299 to R.E.C., and K01-DK087763 to S.M.M. E.B.P. was supported by T32-DK007737 to the Center for Gastrointestinal Biology and Disease, and D.S.C. was supported by T32-CA009156 to the Lineberger Comprehensive Cancer Center. UPLC-MS was performed at the University of North Carolina Environmental Sciences and Engineering Biomarker Mass Spectrometry Core Facility, which is supported in part by a grant from the National Institute of Environmental Health Sciences (P30ES010126). The contents of this report are solely the responsibility of the authors and do not necessarily represent the official views of the funding bodies.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00347-17.
REFERENCES
- 1.Miller M. 2010. Fidaxomicin (OPT-80) for the treatment of Clostridium difficile infection. Expert Opin Pharmacother 11:1569–1578. doi: 10.1517/14656566.2010.485614. [DOI] [PubMed] [Google Scholar]
- 2.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MR, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, Pamer EG. 2015. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Britton RA, Young VB. 2014. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology 146:1547–1553. doi: 10.1053/j.gastro.2014.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de La Cochetière M-F, Montassier E, Hardouin J-B, Carton T, Vacon F, Durand T, Lalande V, Petit J, Potel G, Beaugerie L. 2010. Human intestinal microbiota gene risk factors for antibiotic-associated diarrhea: perspectives for prevention. Microb Ecol 59:830–837. doi: 10.1007/s00248-010-9637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manges AR, Labbe A, Loo VG, Atherton JK, Behr MA, Masson L, Tellis PA, Brousseau R. 2010. Comparative metagenomic study of alterations to the intestinal microbiota and risk of nosocomial Clostridium difficile-associated disease. J Infect Dis 202:1877–1884. doi: 10.1086/657319. [DOI] [PubMed] [Google Scholar]
- 6.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, Sonnenburg JL. 2013. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theriot CM, Koenigsknecht MJ, Carlson PE Jr, Hatton GE, Nelson AM, Li B, Huffnagle GB, Li J, Young VB. 2014. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sambol SP, Tang JK, Merrigan MM, Johnson S, Gerding DN. 2001. Infection of hamsters with epidemiologically important strains of Clostridium difficile. J Infect Dis 183:1760–1766. doi: 10.1086/320736. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez-Palacios A, Lejeune JT. 2011. Moist-heat resistance, spore aging, and superdormancy in Clostridium difficile. Appl Environ Microbiol 77:3085–3091. doi: 10.1128/AEM.01589-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vohra P, Poxton IR. 2011. Comparison of toxin and spore production in clinically relevant strains of Clostridium difficile. Microbiology 157:1343–1353. doi: 10.1099/mic.0.046243-0. [DOI] [PubMed] [Google Scholar]
- 11.Sorg JA, Sonenshein AL. 2008. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol 190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howerton A, Ramirez N, Abel-Santos E. 2011. Mapping interactions between germinants and Clostridium difficile spores. J Bacteriol 193:274–282. doi: 10.1128/JB.00980-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Bella S, Ascenzi P, Siarakas S, Petrosillo N, di Masi A. 2016. Clostridium difficile toxins A and B: insights into pathogenic properties and extraintestinal effects. Toxins (Basel) 8:E134. doi: 10.3390/toxins8050134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Just I, Fritz G, Aktories K, Giry M, Popoff MR, Boquet P, Hegenbarth S, von Eichel-Streiber C. 1994. Clostridium difficile toxin B acts on the GTP-binding protein Rho. J Biol Chem 269:10706–10712. [PubMed] [Google Scholar]
- 16.Just I, Selzer J, von Eichel-Streiber C, Aktories K. 1995. The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J Clin Invest 95:1026–1031. doi: 10.1172/JCI117747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dillon ST, Rubin EJ, Yakubovich M, Pothoulakis C, LaMont JT, Feig LA, Gilbert RJ. 1995. Involvement of Ras-related Rho proteins in the mechanisms of action of Clostridium difficile toxin A and toxin B. Infect Immun 63:1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burridge K, Wennerberg K. 2004. Rho and Rac take center stage. Cell 116:167–179. doi: 10.1016/S0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 19.Ottlinger ME, Lin S. 1988. Clostridium difficile toxin B induces reorganization of actin, vinculin, and talin in cultured cells. Exp Cell Res 174:215–229. doi: 10.1016/0014-4827(88)90156-5. [DOI] [PubMed] [Google Scholar]
- 20.Ng J, Hirota SA, Gross O, Li Y, Ulke-Lemee A, Potentier MS, Schenck LP, Vilaysane A, Seamone ME, Feng H, Armstrong GD, Tschopp J, Macdonald JA, Muruve DA, Beck PL. 2010. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 139:542–552, 552 e541–543. doi: 10.1053/j.gastro.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Chen S, Sun C, Wang H, Wang J. 2015. The role of Rho GTPases in toxicity of Clostridium difficile toxins. Toxins (Basel) 7:5254–5267. doi: 10.3390/toxins7124874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuipers EJ, Surawicz CM. 2008. Clostridium difficile infection. Lancet 371:1486–1488. doi: 10.1016/S0140-6736(08)60635-2. [DOI] [PubMed] [Google Scholar]
- 23.McCollum DL, Rodriguez JM. 2012. Detection, treatment, and prevention of Clostridium difficile infection. Clin Gastroenterol Hepatol 10:581–592. doi: 10.1016/j.cgh.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 24.Deakin LJ, Clare S, Fagan RP, Dawson LF, Pickard DJ, West MR, Wren BW, Fairweather NF, Dougan G, Lawley TD. 2012. The Clostridium difficile spo0A gene is a persistence and transmission factor. Infect Immun 80:2704–2711. doi: 10.1128/IAI.00147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edwards AN, Nawrocki KL, McBride SM. 2014. Conserved oligopeptide permeases modulate sporulation initiation in Clostridium difficile. Infect Immun 82:4276–4291. doi: 10.1128/IAI.02323-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nawrocki KL, Edwards AN, Daou N, Bouillaut L, McBride SM. 2016. CodY-dependent regulation of sporulation in Clostridium difficile. J Bacteriol 198:2113–2130. doi: 10.1128/jb.00220-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dineen SS, McBride SM, Sonenshein AL. 2010. Integration of metabolism and virulence by Clostridium difficile CodY. J Bacteriol 192:5350–5362. doi: 10.1128/JB.00341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shivers RP, Sonenshein AL. 2004. Activation of the Bacillus subtilis global regulator CodY by direct interaction with branched-chain amino acids. Mol Microbiol 53:599–611. doi: 10.1111/j.1365-2958.2004.04135.x. [DOI] [PubMed] [Google Scholar]
- 29.Handke LD, Shivers RP, Sonenshein AL. 2008. Interaction of Bacillus subtilis CodY with GTP. J Bacteriol 190:798–806. doi: 10.1128/JB.01115-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dineen SS, Villapakkam AC, Nordman JT, Sonenshein AL. 2007. Repression of Clostridium difficile toxin gene expression by CodY. Mol Microbiol 66:206–219. doi: 10.1111/j.1365-2958.2007.05906.x. [DOI] [PubMed] [Google Scholar]
- 31.Sonenshein AL. 2005. CodY, a global regulator of stationary phase and virulence in Gram-positive bacteria. Curr Opin Microbiol 8:203–207. doi: 10.1016/j.mib.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Hendriksen WT, Bootsma HJ, Estevao S, Hoogenboezem T, de Jong A, de Groot R, Kuipers OP, Hermans PW. 2008. CodY of Streptococcus pneumoniae: link between nutritional gene regulation and colonization. J Bacteriol 190:590–601. doi: 10.1128/JB.00917-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopez JM, Marks CL, Freese E. 1979. The decrease of guanine nucleotides initiates sporulation of Bacillus subtilis. Biochim Biophys Acta 587:238–252. doi: 10.1016/0304-4165(79)90357-X. [DOI] [PubMed] [Google Scholar]
- 34.Ratnayake-Lecamwasam M, Serror P, Wong K-W, Sonenshein AL. 2001. Bacillus subtilis CodY represses early-stationary-phase genes by sensing GTP levels. Genes Dev 15:1093–1103. doi: 10.1101/gad.874201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majerczyk CD, Sadykov MR, Luong TT, Lee C, Somerville GA, Sonenshein AL. 2008. Staphylococcus aureus CodY negatively regulates virulence gene expression. J Bacteriol 190:2257–2265. doi: 10.1128/JB.01545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Schaik W, Château A, Dillies M-A, Coppée J-Y, Sonenshein AL, Fouet A. 2009. The global regulator CodY regulates toxin gene expression in Bacillus anthracis and is required for full virulence. Infect Immun 77:4437–4445. doi: 10.1128/IAI.00716-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lobel L, Sigal N, Borovok I, Ruppin E, Herskovits AA. 2012. Integrative genomic analysis identifies isoleucine and CodY as regulators of Listeria monocytogenes virulence. PLoS Genet 8:e1002887. doi: 10.1371/journal.pgen.1002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kreth J, Chen Z, Ferretti J, Malke H. 2011. Counteractive balancing of transcriptome expression involving CodY and CovRS in Streptococcus pyogenes. J Bacteriol 193:4153–4165. doi: 10.1128/JB.00061-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boyd CD, O'Toole GA. 2012. Second messenger regulation of biofilm formation: breakthroughs in understanding c-di-GMP effector systems. Annu Rev Cell Dev Biol 28:439–462. doi: 10.1146/annurev-cellbio-101011-155705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hisert KB, MacCoss M, Shiloh MU, Darwin KH, Singh S, Jones RA, Ehrt S, Zhang Z, Gaffney BL, Gandotra S, Holden DW, Murray D, Nathan C. 2005. A glutamate-alanine-leucine (EAL) domain protein of Salmonella controls bacterial survival in mice, antioxidant defence and killing of macrophages: role of cyclic diGMP. Mol Microbiol 56:1234–1245. doi: 10.1111/j.1365-2958.2005.04632.x. [DOI] [PubMed] [Google Scholar]
- 41.Kulasakara H, Lee V, Brencic A, Liberati N, Urbach J, Miyata S, Lee DG, Neely AN, Hyodo M, Hayakawa Y, Ausubel FM, Lory S. 2006. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc Natl Acad Sci U S A 103:2839–2844. doi: 10.1073/pnas.0511090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tamayo R, Schild S, Pratt JT, Camilli A. 2008. Role of cyclic di-GMP during El Tor biotype Vibrio cholerae infection: characterization of the in vivo-induced cyclic di-GMP phosphodiesterase CdpA. Infect Immun 76:1617–1627. doi: 10.1128/IAI.01337-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tischler AD, Camilli A. 2005. Cyclic diguanylate regulates Vibrio cholerae virulence gene expression. Infect Immun 73:5873–5882. doi: 10.1128/IAI.73.9.5873-5882.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun Y-C, Koumoutsi A, Jarrett C, Lawrence K, Gherardini FC, Darby C, Hinnebusch BJ. 2011. Differential control of Yersinia pestis biofilm formation in vitro and in the flea vector by two c-di-GMP diguanylate cyclases. PLoS One 6:e19267. doi: 10.1371/journal.pone.0019267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He M, Zhang J-J, Ye M, Lou Y, Yang XF. 2014. Cyclic Di-GMP receptor PlzA controls virulence gene expression through RpoS in Borrelia burgdorferi. Infect Immun 82:445–452. doi: 10.1128/IAI.01238-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Purcell EB, Tamayo R. 2016. Cyclic diguanylate signaling in Gram-positive bacteria. FEMS Microbiol Rev 40:753–773. doi: 10.1093/femsre/fuw013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryjenkov DA, Tarutina M, Moskvin OV, Gomelsky M. 2005. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J Bacteriol 187:1792–1798. doi: 10.1128/JB.187.5.1792-1798.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt AJ, Ryjenkov DA, Gomelsky M. 2005. The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J Bacteriol 187:4774–4781. doi: 10.1128/JB.187.14.4774-4781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryan RP, Fouhy Y, Lucey JF, Dow JM. 2006. Cyclic di-GMP signaling in bacteria: recent advances and new puzzles. J Bacteriol 188:8327–8334. doi: 10.1128/JB.01079-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galperin MY, Nikolskaya AN, Koonin EV. 2001. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol Lett 203:11–21. doi: 10.1111/j.1574-6968.2001.tb10814.x. [DOI] [PubMed] [Google Scholar]
- 51.Galperin MY. 2005. The Molecular Biology Database Collection: 2005 update. Nucleic Acids Res 33:D5-24. doi: 10.1093/nar/gki139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeno-Tarraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet 38:779–786. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 53.Bordeleau E, Fortier LC, Malouin F, Burrus V. 2011. c-di-GMP turn-over in Clostridium difficile is controlled by a plethora of diguanylate cyclases and phosphodiesterases. PLoS Genet 7:e1002039. doi: 10.1371/journal.pgen.1002039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao X, Dong X, Subramanian S, Matthews PM, Cooper CA, Kearns DB, Dann CE. 2014. Engineering of Bacillus subtilis strains to allow rapid characterization of heterologous diguanylate cyclases and phosphodiesterases. Appl Environ Microbiol 80:6167–6174. doi: 10.1128/AEM.01638-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Purcell EB, McKee RW, McBride SM, Waters CM, Tamayo R. 2012. Cyclic diguanylate inversely regulates motility and aggregation in Clostridium difficile. J Bacteriol 194:3307–3316. doi: 10.1128/JB.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soutourina OA, Monot M, Boudry P, Saujet L, Pichon C, Sismeiro O, Semenova E, Severinov K, Le Bouguenec C, Coppee JY, Dupuy B, Martin-Verstraete I. 2013. Genome-wide identification of regulatory RNAs in the human pathogen Clostridium difficile. PLoS Genet 9:e1003493. doi: 10.1371/journal.pgen.1003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McKee RW, Mangalea MR, Purcell EB, Borchardt EK, Tamayo R. 2013. The second messenger cyclic di-GMP regulates Clostridium difficile toxin production by controlling expression of sigD. J Bacteriol 195:5174–5185. doi: 10.1128/JB.00501-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.El Meouche I, Peltier J, Monot M, Soutourina O, Pestel-Caron M, Dupuy B, Pons J-L. 2013. Characterization of the SigD regulon of C. difficile and its positive control of toxin production through the regulation of tcdR. PLoS One 8:e83748. doi: 10.1371/journal.pone.0083748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bordeleau E, Purcell EB, Lafontaine DA, Fortier LC, Tamayo R, Burrus V. 2015. Cyclic di-GMP riboswitch-regulated type IV pili contribute to aggregation of Clostridium difficile. J Bacteriol 197:819–832. doi: 10.1128/JB.02340-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Purcell EB, McKee RW, Bordeleau E, Burrus V, Tamayo R. 2015. Regulation of type IV pili contributes to surface behaviors of historical and epidemic strains of Clostridium difficile. J Bacteriol 198:565–577. doi: 10.1128/JB.00816-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peltier J, Shaw HA, Couchman EC, Dawson LF, Yu L, Choudhary JS, Kaever V, Wren BW, Fairweather NF. 2015. Cyclic diGMP regulates production of sortase substrates of Clostridium difficile and their surface exposure through ZmpI protease-mediated cleavage. J Biol Chem 290:24453–24469. doi: 10.1074/jbc.M115.665091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cafardi V, Biagini M, Martinelli M, Leuzzi R, Rubino JT, Cantini F, Norais N, Scarselli M, Serruto D, Unnikrishnan M. 2013. Identification of a novel zinc metalloprotease through a global analysis of Clostridium difficile extracellular proteins. PLoS One 8:e81306. doi: 10.1371/journal.pone.0081306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hensbergen PJ, Klychnikov OI, Bakker D, Dragan I, Kelly ML, Minton NP, Corver J, Kuijper EJ, Drijfhout JW, van Leeuwen HC. 2015. Clostridium difficile secreted Pro-Pro endopeptidase PPEP-1 (ZMP1/CD2830) modulates adhesion through cleavage of the collagen binding protein CD2831. FEBS Lett 589:3952–3958. doi: 10.1016/j.febslet.2015.10.027. [DOI] [PubMed] [Google Scholar]
- 64.Henry JT, Crosson S. 2011. Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu Rev Microbiol 65:261–286. doi: 10.1146/annurev-micro-121809-151631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Christen M, Christen B, Folcher M, Schauerte A, Jenal U. 2005. Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J Biol Chem 280:30829–30837. doi: 10.1074/jbc.M504429200. [DOI] [PubMed] [Google Scholar]
- 66.Kazmierczak BI, Lebron MB, Murray TS. 2006. Analysis of FimX, a phosphodiesterase that governs twitching motility in Pseudomonas aeruginosa. Mol Microbiol 60:1026–1043. doi: 10.1111/j.1365-2958.2006.05156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.An S, Wu Je Zhang L-H. 2010. Modulation of Pseudomonas aeruginosa biofilm dispersal by a cyclic-di-GMP phosphodiesterase with a putative hypoxia-sensing domain. Appl Environ Microbiol 76:8160–8173. doi: 10.1128/AEM.01233-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Enomoto G, Ni Ni W, Narikawa R, Ikeuchi M. 2015. Three cyanobacteriochromes work together to form a light color-sensitive input system for c-di-GMP signaling of cell aggregation. Proc Natl Acad Sci U S A 112:8082–8087. doi: 10.1073/pnas.1504228112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chan C, Paul R, Samoray D, Amiot NC, Giese B, Jenal U, Schirmer T. 2004. Structural basis of activity and allosteric control of diguanylate cyclase. Proc Natl Acad Sci U S A 101:17084–17089. doi: 10.1073/pnas.0406134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wassmann P, Chan C, Paul R, Beck A, Heerklotz H, Jenal U, Schirmer T. 2007. Structure of BeF3-modified response regulator PleD: implications for diguanylate cyclase activation, catalysis, and feedback inhibition. Structure 15:915–927. doi: 10.1016/j.str.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 71.Navarro MV, De N, Bae N, Wang Q, Sondermann H. 2009. Structural analysis of the GGDEF-EAL domain-containing c-di-GMP receptor FimX. Structure 17:1104–1116. doi: 10.1016/j.str.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ðapa T, Leuzzi R, Ng YK, Baban ST, Adamo R, Kuehne SA, Scarselli M, Minton NP, Serruto D, Unnikrishnan M. 2013. Multiple factors modulate biofilm formation by the anaerobic pathogen Clostridium difficile. J Bacteriol 195:545–555. doi: 10.1128/JB.01980-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saujet L, Monot M, Dupuy B, Soutourina O, Martin-Verstraete I. 2011. The key sigma factor of transition phase, SigH, controls sporulation, metabolism, and virulence factor expression in Clostridium difficile. J Bacteriol 193:3186–3196. doi: 10.1128/JB.00272-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. 2007. The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods 70:452–464. doi: 10.1016/j.mimet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 75.Faulds-Pain A, Wren BW. 2013. Improved bacterial mutagenesis by high-frequency allele exchange, demonstrated in Clostridium difficile and Streptococcus suis. Appl Environ Microbiol 79:4768–4771. doi: 10.1128/AEM.01195-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martin-Verstraete I, Peltier J, Dupuy B. 2016. The regulatory networks that control Clostridium difficile toxin synthesis. Toxins (Basel) 8:E153. doi: 10.3390/toxins8050153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miura M, Kato H, Matsushita O. 2011. Identification of a novel virulence factor in Clostridium difficile that modulates toxin sensitivity of cultured epithelial cells. Infect Immun 79:3810–3820. doi: 10.1128/IAI.00051-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. 2008. Lifeact: a versatile marker to visualize F-actin. Nat Methods 5:605. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Christen B, Christen M, Paul R, Schmid F, Folcher M, Jenoe P, Meuwly M, Jenal U. 2006. Allosteric control of cyclic di-GMP signaling. J Biol Chem 281:32015–32024. doi: 10.1074/jbc.M603589200. [DOI] [PubMed] [Google Scholar]
- 80.Schultz J, Milpetz F, Bork P, Ponting CP. 1998. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A 95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chiaverotti TA, Parker G, Gallant J, Agabian N. 1981. Conditions that trigger guanosine tetraphosphate accumulation in Caulobacter crescentus. J Bacteriol 145:1463–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ochi K. 1987. Metabolic initiation of differentiation and secondary metabolism by Streptomyces griseus: significance of the stringent response (ppGpp) and GTP content in relation to A factor. J Bacteriol 169:3608–3616. doi: 10.1128/jb.169.8.3608-3616.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Whitehead KE, Webber GM, England RR. 1998. Accumulation of ppGpp in Streptococcus pyogenes and Streptococcus rattus following amino acid starvation. FEMS Microbiol Lett 159:21–26. doi: 10.1111/j.1574-6968.1998.tb12836.x. [DOI] [PubMed] [Google Scholar]
- 84.Inaoka T, Ochi K. 2002. RelA protein is involved in induction of genetic competence in certain Bacillus subtilis strains by moderating the level of intracellular GTP. J Bacteriol 184:3923–3930. doi: 10.1128/JB.184.14.3923-3930.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Belitsky BR, Brinsmade SR, Sonenshein AL. 2015. Intermediate levels of Bacillus subtilis CodY activity are required for derepression of the branched-chain amino acid permease, BraB. PLoS Genet 11:e1005600. doi: 10.1371/journal.pgen.1005600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Waters NR, Samuels DJ, Behera RK, Livny J, Rhee KY, Sadykov MR, Brinsmade SR. 2016. A spectrum of CodY activities drives metabolic reorganization and virulence gene expression in Staphylococcus aureus. Mol Microbiol 101:495–514. doi: 10.1111/mmi.13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Buckstein MH, He J, Rubin H. 2008. Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J Bacteriol 190:718–726. doi: 10.1128/JB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Newell PD, Monds RD, O'Toole GA. 2009. LapD is a bis-(3′,5′)-cyclic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc Natl Acad Sci U S A 106:3461–3466. doi: 10.1073/pnas.0808933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gjermansen M, Nilsson M, Yang L, Tolker-Nielsen T. 2010. Characterization of starvation-induced dispersion in Pseudomonas putida biofilms: genetic elements and molecular mechanisms. Mol Microbiol 75:815–826. doi: 10.1111/j.1365-2958.2009.06793.x. [DOI] [PubMed] [Google Scholar]
- 90.Bharati BK, Sharma IM, Kasetty S, Kumar M, Mukherjee R, Chatterji D. 2012. A full-length bifunctional protein involved in c-di-GMP turnover is required for long-term survival under nutrient starvation in Mycobacterium smegmatis. Microbiology 158:1415–1427. doi: 10.1099/mic.0.053892-0. [DOI] [PubMed] [Google Scholar]
- 91.Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, Britigan BE, Singh PK. 2011. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334:982–986. doi: 10.1126/science.1211037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hooper LV, Midtvedt T, Gordon JI. 2002. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr 22:283–307. doi: 10.1146/annurev.nutr.22.011602.092259. [DOI] [PubMed] [Google Scholar]
- 93.Ng SC, Lam EF, Lam TT, Chan Y, Law W, Tse PC, Kamm MA, Sung JJ, Chan FK, Wu JC. 2013. Effect of probiotic bacteria on the intestinal microbiota in irritable bowel syndrome. J Gastroenterol Hepatol 28:1624–1631. doi: 10.1111/jgh.12306. [DOI] [PubMed] [Google Scholar]
- 94.Ternan NG, Jain S, Srivastava M, McMullan G. 2012. Comparative transcriptional analysis of clinically relevant heat stress response in Clostridium difficile strain 630. PLoS One 7:e42410. doi: 10.1371/journal.pone.0042410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Osorio CG, Crawford JA, Michalski J, Martinez-Wilson H, Kaper JB, Camilli A. 2005. Second-generation recombination-based in vivo expression technology for large-scale screening for Vibrio cholerae genes induced during infection of the mouse small intestine. Infect Immun 73:972–980. doi: 10.1128/IAI.73.2.972-980.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 97.Lyras D, Rood JI. 1998. Conjugative transfer of RP4-oriT shuttle vectors from Escherichia coli to Clostridium perfringens. Plasmid 39:160–164. doi: 10.1006/plas.1997.1325. [DOI] [PubMed] [Google Scholar]
- 98.McBride SM, Sonenshein AL. 2011. Identification of a genetic locus responsible for antimicrobial peptide resistance in Clostridium difficile. Infect Immun 79:167–176. doi: 10.1128/IAI.00731-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fuller JR, Vitko NP, Perkowski EF, Scott E, Khatri D, Spontak JS, Thurlow LR, Richardson AR. 2011. Identification of a lactate-quinone oxidoreductase in Staphylococcus aureus that is essential for virulence. Front Cell Infect Microbiol 1:19. doi: 10.3389/fcimb.2011.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gasteiger J. 2006. Chemoinformatics: a new field with a long tradition. Anal Bioanal Chem 384:57–64. doi: 10.1007/s00216-005-0065-y. [DOI] [PubMed] [Google Scholar]
- 101.Pratt JT, Tamayo R, Tischler AD, Camilli A. 2007. PilZ domain proteins bind cyclic diguanylate and regulate diverse processes in Vibrio cholerae. J Biol Chem 282:12860–12870. doi: 10.1074/jbc.M611593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tamayo R, Tischler AD, Camilli A. 2005. The EAL domain protein VieA is a cyclic diguanylate phosphodiesterase. J Biol Chem 280:33324–33330. doi: 10.1074/jbc.M506500200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kariisa AT, Grube A, Tamayo R. 2015. Two nucleotide second messengers regulate the production of the Vibrio cholerae colonization factor GbpA. BMC Microbiol 15:166. doi: 10.1186/s12866-015-0506-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Garnier T, Cole ST. 1986. Characterization of a bacteriocinogenic plasmid from Clostridium perfringens and molecular genetic analysis of the bacteriocin-encoding gene. J Bacteriol 168:1189–1196. doi: 10.1128/jb.168.3.1189-1196.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Choi W, Acharya BR, Peyret G, Fardin MA, Mege RM, Ladoux B, Yap AS, Fanning AS, Peifer M. 2016. Remodeling the zonula adherens in response to tension and the role of afadin in this response. J Cell Biol 213:243–260. doi: 10.1083/jcb.201506115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.