

Abstract
Trimethylation of lysine 4 of histone H3 occurs at the 5′ end of active genes and is catalyzed by Set1 in Saccharomyces cerevisiae. Trimethylation requires histone H2B ubiquitylation and the PAF1 complex, which are linked to transcription elongation, but how they activate Set1 is not known. Set1 also bears several conserved domains with uncharacterized contributions to activity. Here, we isolated dominant hyperactive SET1D alleles, which revealed a complex interplay among Set1 regulatory domains. Remarkably, the RNA-recognition motif (RRM) of Set1 is required for H3K4 trimethylation, but not dimethylation. Also, a central autoinhibitory domain was identified that opposes RRM function by inhibiting trimethylation. Furthermore, a G990E replacement in the catalytic domain conferred Set1 hyperactivity and restored trimethylation to a Set1 derivative bearing mutations in the RRM domain. Surprisingly, certain SET1D alleles also partially restored trimethylation to strains lacking histone H2B ubiquitylation or Paf1. Taken together, our data suggest that the catalytic domain of Set1 integrates opposing inputs from the RRM and autoinhibitory domains to link properly H3K4 methylation to the transcript elongation process.
Keywords: chromatin, methylation, RRM, RSC, Set1
Introduction
Histones are modified at specific positions on their conserved amino-terminal ‘tails', and these modifications play central roles in gene regulation (Jenuwein and Allis, 2001; Turner, 2002). For example, acetylation at lysine 14 of histone H3 (H3K14Ac) or methylation of lysine 4 (H3K4Me) is associated with gene activation. In contrast, the lack of acetylation as well as the methylation of lysine 9 of histone H3 (H3K9Me) is correlated with gene repression (Jenuwein and Allis, 2001; Turner, 2002). These two types of modifications are performed by histone acetyltransferases (HATs) and histone methyltransferases (HMTases). These modifying enzymes are members of large multiprotein complexes with other subunits likely serving roles in targeting or regulation. The primary substrates for these enzymes are the amino-terminal tails of the histone proteins. Modified tails function as binding platforms for transcriptional regulatory factors. For example, the histone H3 tail methylated at lysine 9 is bound by the chromodomain, a motif present in HP1 (Paro and Hogness, 1991), a protein often associated with repressed genes. Interestingly, histone methylation may be associated with either repression or activation; H3K9Me is associated with (and helps direct) repression, whereas H3K4Me is primarily associated with activation (Lachner and Jenuwein, 2002). The yeast Saccharomyces cerevisiae lacks H3K9Me, but bears significant H3K4Me. Very recently, the Chd1 protein (a chromatin-remodeling factor with associated HAT activity) has emerged as a strong candidate for binding H3K4Me (Pray-Grant et al, 2005), linking increased H3K4Me to increased remodeling and acetylation.
Several years ago, the Drosophila protein Su(var)3-9 was shown to utilize its SET domain to methylate H3K9 (Rea et al, 2000). This discovery was significant, as many SET domain proteins had been identified as transcriptional regulators, but their mode of action was not understood. In S. cerevisiae, the SET domain Set1 protein was identified as the unique H3K4 methyltransferase, as null mutations in SET1 eliminated all H3K4 methylation (Briggs et al, 2001; Roguev et al, 2001; Krogan et al, 2002). Set1 is a member of a large protein complex called SET1/COMPASS, and most members of the complex are required for H3K4 methylation (Miller et al, 2001; Roguev et al, 2001; Nagy et al, 2002). Null mutations (set1Δ) affect transcription of many genes including INO1 and MET16 (Santos-Rosa et al, 2002, 2003) and genes near telomeres (Nislow et al, 1997; Krogan et al, 2002). Set1 also has roles in regulating silencing in the ribosomal DNA repeat locus (Briggs et al, 2001; Bryk et al, 2002) and is required for efficient DNA repair (Corda et al, 1999).
A central question in chromatin and transcriptional regulation is how histone methylation is regulated. Remarkably, Set1 has the capacity to either mono-, di-, or trimethylate H3K4, and chromatin immunoprecipitation (ChIP) experiments reveal trimethylation restricted primarily to the 5′ end of active genes and their proximal promoter region (Santos-Rosa et al, 2002; Krogan et al, 2003; Ng et al, 2003). H3K4 di- and trimethylation requires members of the PAF1 complex (including Paf1 and Rtf1), which is involved in the transition from initiation to elongation, suggesting that H3K4Me is associated with this process (Krogan et al, 2003; Ng et al, 2003). Set1 activity further requires the ubiquitylation of histone H2B, performed by Rad6 (E2) and Bre1 (E3) (Dover et al, 2002; Sun and Allis, 2002; Wood et al, 2003), and also the function of the proteasome components Rpt6 (Sug1) and Rpt4 (Sug2) (Ezhkova and Tansey, 2004). All of these factors are linked to transcript elongation, further connecting Set1 trimethylation to this process.
Set1 is a large protein with many domains that might help regulate trimethylation: an amino-terminal RNA-recognition motif (RRM) (Nagai et al, 1990), a semiconserved central domain, an N-SET domain, the catalytic SET domain, and the C-terminal post-SET domain (see Figure 1). We find that all five of these domains are conserved in Set1-related proteins from humans (K1AA0339 and K1AA1076), mouse (BC010250 and BC035291), Drosophila (CG40351), and other organisms, although the central region shows considerable variation (data not shown). The SET, N-SET, and post-SET domains are absolutely required for Set1 enzymatic activity, as their omission prevents H3K4Me. However, the roles of the central domain and RRM in regulating trimethylation are not known. Interestingly, these two domains are only present in Set1-related proteins (that methylate H3K4) and not present in SET domain HMTases that are known to methylate H3K9 or other positions. This raises the possibility that the RRM and central region may work together to direct SET domain function to the task of H3K4Me at the 5′ ends of the genes. If so, these domains might work together with factors that control transcript elongation to execute this task. Here, a screen involving chromatin remodeling yielded dominant activating mutations in SET1 (SET1D). Characterization of these SET1D alleles revealed that the RRM and N-SET domains promote trimethylation, whereas the central region restricts trimethylation activity. Our studies further suggest that elongation factors may operate through these domains to affect enzyme activity. Furthermore, we identify a residue in the catalytic pocket that is important for overall activity and for integrating information from the RRM and central region.
Figure 1.
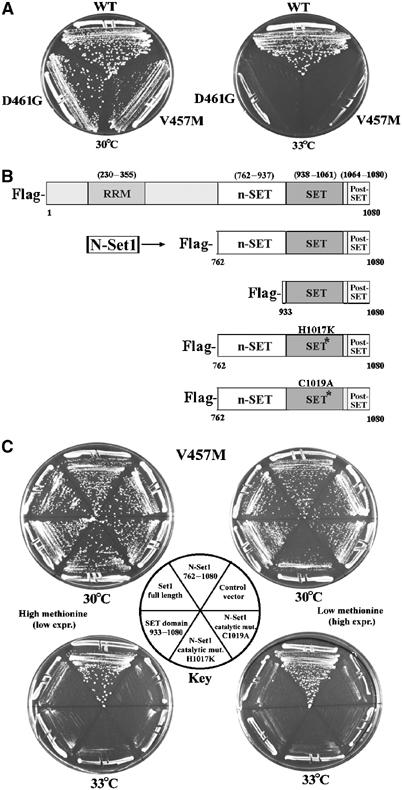
rsc2 alleles are suppressed by the Set1 truncation derivative N-Set1. (A) Growth of strains rsc2-V457M (YBC1111) and rsc2-D461G (YBC1112) on rich medium (YPD). (B) Map of Set1 derivatives: Set11–1080 (p1067), Set1762–1080 (p1102) termed N-Set1, Set1933–1080 (p1066) SET domain, Set1762–1080 H1017K (p1103), Set1762–1080 C1019A (p1104). (C) The N-Set1 derivative of Set1 suppresses rsc2 Ts− alleles. Set1 constructs from (B) were transformed into the rsc2-V457M strain (YBC1111) and grown on media containing (at left) or lacking (at right) methionine at 30 and 33°C.
Results
Isolation of SET1D alleles through the suppression of rsc2 conditional alleles
We isolated SET1D alleles in genetic selections for suppressors of rsc2 alleles. Rsc2 is a member of RSC, an essential and abundant chromatin- remodeling complex. We obtained conditional (temperature-sensitive (Ts−)) rsc2 alleles through a combination of targeted mutagenic PCR and in vivo recombination (see Materials and methods). A complete description of the screen will be published elsewhere, as the focus of this work is on Set1 regulation. Two rsc2 alleles with clear Ts− phenotypes (rsc2-D461G and rsc2-V457M; Figure 1A) were chosen for suppression, which led to the isolation of SET1D alleles.
Suppression of rsc2 yielded the dominant truncation allele N-Set1
Suppressors of rsc2 mutations were isolated using a high-copy plasmid library (yEP24-based) bearing random fragments of yeast genomic DNA. Separate selections were performed using strains bearing rsc2-D461G or rsc2-V457M. Suppressors were selected at 35°C, which prevents growth of these rsc2 alleles in isolation. Remarkably, both screens yielded SET1-containing plasmids as potent suppressors. However, all of these plasmids encoded identical truncated forms of Set1 that lacked the amino-terminal RRM domain and the moderately conserved central region (Figure 1B). This Set1 derivative was likely produced from a weak cryptic promoter present on the backbone of the library plasmid (data not shown). We termed this derivative N-Set1, as it retained the catalytic SET domain and the flanking N-SET and post-SET subdomains, which are required for HMTase activity (Briggs et al, 2001; Bryk et al, 2002).
To better characterize N-Set1, we generated a series of Set1 derivatives bearing the FLAG epitope at their amino-terminus, all driven from the methionine-repressible MET25 promoter. Expression of these derivatives was moderately low in the presence of methionine and moderately high in the absence of methionine (data not shown). Again, N-Set1 suppressed rsc2, whereas full-length Set1 and a catalytically inactive form of N-Set1 (H1017K or C1019A) did not suppress (Figure 1C). This indicated that N-Set1 is a dominant gain-of-function allele (SET1D) whose function requires HMTase activity. N-Set1 is not simply a rogue methyltransferase; N-Set1 interacts with other members of COMPASS (data not shown) and N-Set1 requires the Bre2 or Spp1 components of SET1/COMPASS for rsc2 suppression (Supplementary Figure 1). Taken together, a selection for rsc2 suppressors yielded a dominant truncated form of Set1, N-Set1, that may have acquired new functions and abilities.
Although we investigated several possible functional relationships between RSC and Set1, we have not determined how N-Set1 (or the other SET1D alleles described below) suppresses rsc2. Mutations in rsc2 do not affect H3K4 di- or trimethylation levels in strains bearing wild-type (WT) SET1, suggesting that Rsc2 does not regulate Set1 activity against H3K4. In addition, recombinant Rsc2 does not prefer H3 tail peptides bearing K4 di- or trimethylation, suggesting that Rsc2 does not bind H3K4Me (data not shown). Therefore, the functional link between RSC and Set1 is not known. Regardless, rsc2 suppression provided a bioassay ideal for the isolation of dominant SET1D alleles. As described below, these SET1D alleles are hyperactive and misregulated in otherwise WT (RSC+) strains, showing that these properties are intrinsic to the SET1D allele and independent of rsc2. Below, we isolate nine additional SET1D alleles and use them to reveal domains and residues important for the regulation of H3K4 di- and trimethylation.
Isolation of full-length dominant activating SET1D mutations
To isolate full-length SET1D alleles, we performed saturating random mutagenesis of the full-length SET1 gene and selected for SET1D alleles by rsc2 suppression in a strain bearing WT SET1 (to ensure dominance, see Materials and methods). A search involving an estimated 50 000 amino-acid substitutions yielded nine independent full-length SET1D alleles. As shown below, the identity of the replacements strongly suggests that this screen was saturating. We report SET1D allele characterization in the following order: their identity, their impact on H3K4 methylation in vivo, their H3K4 HMTase activity in vitro, and their ability to bypass normal HMTase regulation.
Identification of SET1D alleles
The nine SET1D alleles were sorted into three classes: (1) those bearing a substitution in the catalytic SET domain, (2) those bearing substitutions in the semiconserved central region, and (3) those bearing substitutions in the flanking N-SET domain. Remarkably, six of the nine SET1D alleles obtained bore a substitution at residue 990 in the catalytic SET domain: G990E (four alleles), G990K (one allele), and G990A (one allele). Each G990E encoding allele was independent based on unique DNA alterations at other positions. However, as one allele (SET1D-G990E) bore only one alteration, dominant behavior can be attributed solely to this replacement. Furthermore, the expressivity of SET1D-G990E was equal to all other alleles bearing G990 and additional replacements (see G990E(+), Figure 2A), further suggesting replacement of G990 as the crucial determinant. The location of this residue in the catalytic pocket and its impact on methylation are discussed in a later section.
Figure 2.
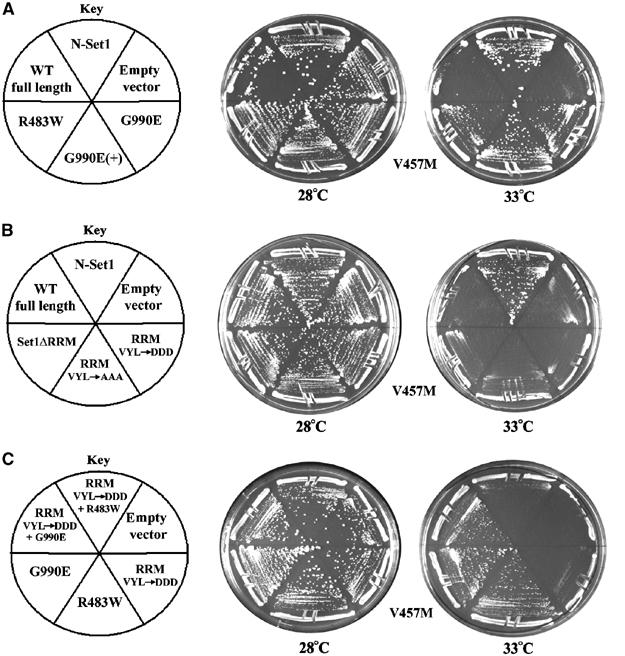
Characterization of SET1D and set1 alleles. (A) Ability of SET1D alleles to suppress rsc2-V457M. Strains bearing rsc2-V457M (YBC1111) were transformed with empty YEp24 vector, N-Set1 (p1102), Set1 (p1067), Set1 R483W, A613T (p1368), Set1G990E (+) S43N, S509F, P975S (p1306), or Set1 G990E (p1305) and grown on media lacking uracil at 28 and 33°C. (B) RRM domain mutants fail to suppress rsc2-V457M. Strain rsc2-V457M (YBC1111) was transformed with empty YEp24 vector, N-Set1 (p1102), Set1 (p1067), Set1ΔRRM (p1377), Set1 VYL295-297AAA, A293T (p1375), or Set1 VYL295-297DDD (p1376) and grown on media lacking uracil at 28 or 33°C. (C) G990E bypasses RRM mutations, enabling suppression of rsc2-V457M. Strain rsc2-V457M (YBC1111) was transformed with empty YEp24 vector, Set1 VYL295-297DDD (p1376), Set1 R483W (p1409), Set1 G990E (p1305), Set1 VYL295-297DDD +R483W (p1431), or Set1 VYL295-297DDD + G990E (p1410) and grown on media lacking uracil at 28 or 33°C.
The second class consists of two independent SET1D alleles that share an R483W replacement (R483W A613T and S104A R483W). R483 is located in the moderately conserved central region, which has not previously been characterized. The R483W replacement was isolated and shown to be sufficient for suppression (Figure 2, and data not shown). This result raises the possibility that the central region negatively regulates Set1 function.
The SET1D-L777F E896K allele bears two substitutions at the two distal ends of the N-SET domain. Our alignments of the N-SET domain show good conservation among N-SET members in the vicinity of L777, and the almost uniform presence of a leucine at this position, whereas conservation of E896 is poor (data not shown). The N-SET domain has been shown to have a positive role in Set1 activity, as its presence is required for SET domain function (Figure 1) (Nislow et al, 1997; Briggs et al, 2001). Whether the N-SET domain has a role in regulating H3K4 trimethylation is currently not known. Although the SET1D-L777F E896K allele might promote the function of the N-SET domain in enzyme activation, its characterization lagged behind the other alleles, and therefore was not included in this work. Therefore, the remainder of this work focuses on an extensive characterization of the first two (more prevalent) classes of SET1D mutations, the function of the RRM domain, and their regulatory relationships.
The central region negatively regulates Set1 activity, whereas the RRM domain positively regulates Set1 activity
To test whether the central region confers negative regulation on Set1 H3K4 activity, we added back this region to N-Set1, creating a Set1 derivative that begins just after the RRM domain, termed Set1ΔRRM. Set1ΔRRM lacked the ability to suppress rsc2 module mutations (Figure 2B), even though it was produced and assembled into SET1/COMPASS complex (Supplementary Figure 2). These results strongly suggest that the central region negatively regulates Set1 activity (autoinhibition) and that the R483W substitution partially relieves this inhibition.
The lack of suppression by Set1ΔRRM suggested that the RRM domain positively regulates Set1. The RRM domain is a common motif found in many RNA-binding proteins and residues utilized for RNA recognition have been identified by mutational studies. A set of 3–4 hydrophobic residues is utilized for interaction with the RNA bases (Kranz and Hall, 1999). Using these studies as a guide, we directed either alanine or aspartic acid substitutions (as a three-residue window) to this location, replacing VYL (residues 295–297) in the Set1 RRM domain. These RRM mutant derivatives were all produced and assembled into SET1/COMPASS complex (Supplementary Figure 2), but were unable to suppress rsc2 mutations (Figure 2B). Furthermore, combining RRM substitutions with the R483W substitution prevented rsc2 suppression (Figure 2C). Taken together, these results are consistent with the Set1 RRM domain positively regulating Set1 activity against H3K4, and reveal a role for putative RNA-interacting residues in Set1 function.
SET1D mutations confer moderate increases in bulk histone H3K4 methylation levels
Our genetic analysis is consistent with the SET1D alleles encoding Set1 derivatives that either increase activity, bypass normal regulation, or both. To test whether these SET1D alleles increase the steady-state levels of bulk histone H3K4 methylation in vivo, we analyzed immunoblots of whole-cell extracts with antibodies that recognize di- or trimethylated H3K4. These antibodies were specific for their epitopes (data not shown, see Materials and methods). N-Set1 and G990E conferred moderate increases in bulk di- and trimethylated H3K4, whereas the R483 alleles conferred only slight enhancement. Interestingly, an allele that combines G990E with R483W has a similar level of H3K4 trimethylation levels, but was a more effective suppressor of rsc2 mutations than were the isolated substitutions (Figure 3, and data not shown). Taken together, these results are consistent with SET1D alleles displaying slight to moderate increases in bulk H3K4 trimethylation in vivo.
Figure 3.
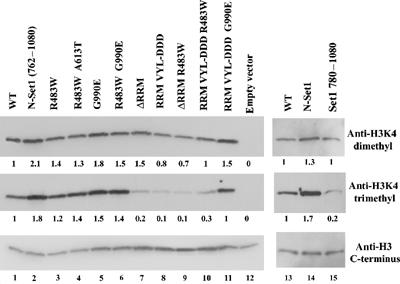
Impact of SET1D and set1 mutations on histone H3K4 methylation levels in vivo. Whole-cell extracts (5 μg) from strain YBC1236 (set1Δ) transformed with plasmids bearing Set1 mutations as indicated (see Figure 2 for plasmid identities) were loaded onto an SDS 10–20% acrylamide gel, transferred to PVDF, and probed with the antibodies indicated. Levels were quantified by normalizing to the WT. We note that bulk H3K4Me levels varied ±20% between individual biological replicates, so many (3–5) replicates were prepared and compared. Trends were clear, and shown is a representative experiment that accurately reflects the trends.
RRM mutants lack appreciable trimethylation
Remarkably, removal of the RRM domain dramatically lowered trimethylation levels while dimethylation levels were largely retained. Likewise, replacements in the RRM domain designed to impair RNA interaction also drastically reduced trimethylation levels (Figure 3) while having only a modest impact on dimethylation. These results identify the RRM as an important positive regulator of trimethylation by Set1.
G990E, but not R483W, suppresses mutations in the RRM domain
These studies identified both positive and negative acting domains and mutations in Set1, which allowed us to explore their interplay in Set1 regulation. To this end, we combined mutations in the RRM domain (VYL-DDD) with the dominant activating SET1-G990E and SET1-R483W mutations and monitored their ability to suppress rsc2 and to methylate H3K4 in vivo. Remarkably, the G990E substitution retained the ability to suppress rsc2 when combined with RRM mutations (Figure 2C) and also restored H3K4 methylation to near WT levels (Figure 3). In contrast, R483W combinations failed to suppress rsc2 (Figure 2C) and only weakly restored H3K4 methylation levels (Figure 3). These results show that the G990E substitution largely bypasses the requirement for the RRM for trimethylation whereas the R483W substitution largely retains reliance on the RRM domain. As removal of the entire autoinhibitory domain (generating N-Set1) fully restores trimethylation, the R483W allele likely represents a weaker allele in which the autoinhibitory domain is only partially functional (see Discussion and Figure 6).
Figure 6.
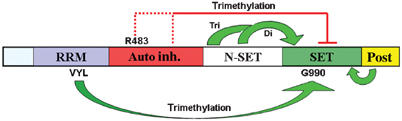
Model depicting the roles of Set1 domains in enzyme regulation. Set1 requires at least two inputs: (1) a positive input from the RRM domain, which relies on hydrophobic residues (VYL) known to interact with RNA in other RRM proteins, and (2) relief of inhibition by a central autoinhibitory region. Here, R483 potentiates this inhibition, but only removal of the entire domain relieves inhibition. Regulation is integrated at the SET domain, where a critical residue (G990) regulates activity and reliance on the RRM domain for trimethylation. Replacements at this position render the enzyme hyperactive against H3K4 and independent of the RRM domain for trimethylation. The N-SET and post-SET domains are required for SET domain activity, with the amino-terminal region of the N-SET domain required for high levels of H3K4 trimethylation.
A role for the N-SET domain in promoting H3K4 trimethylation
The N-Set1 allele obtained through rsc2 suppression (encoding residues 762–1080) is similar to an allele prepared previously by others (encoding 780–1080; Briggs et al, 2001), but was not tested for H3K4 trimethylation. However, our isolation of a SET1D allele bearing a substitution at L777 (and also E896) raised the possibility that this N-terminal region of the N-SET domain (762–779) might influence di- or trimethylation. As we have not separated these mutations, L777 could not be tested in isolation. However, a strain expressing the Set1780–1080 derivative displayed fairly high levels of dimethylation but very low levels of trimethylation (Figure 3, right panel). These results suggest a role for the amino-terminal section of the N-SET domain in promoting trimethylation.
Set1-G990E is a hyperactive histone H3K4 methyltransferase in vitro
Our data are consistent with G990 substitutions conferring hyperactivity and/or misregulation of Set1 activity. Currently, there is no crystal structure of Set1. Therefore, we utilized the crystal structure of the highly related mammalian H3K4 methyltransferase Set7/9 (Xiao et al, 2003), in combination with sequence alignments, to identify the predicted location of G990 in the SET domain of Set1. Interestingly, the residue (G264) in Set7/9 that corresponds to G990 in Set1 resides in the active site within 3.5 Å of the methyl-lysine, and also very near the cofactor product S-adenosylhomocysteine (S-AdHoCys) (Figure 4A), consistent with this substitution altering catalytic activity. Furthermore, alignment of SET domains reveals glycine as the most common residue at this position (data not shown).
Figure 4.
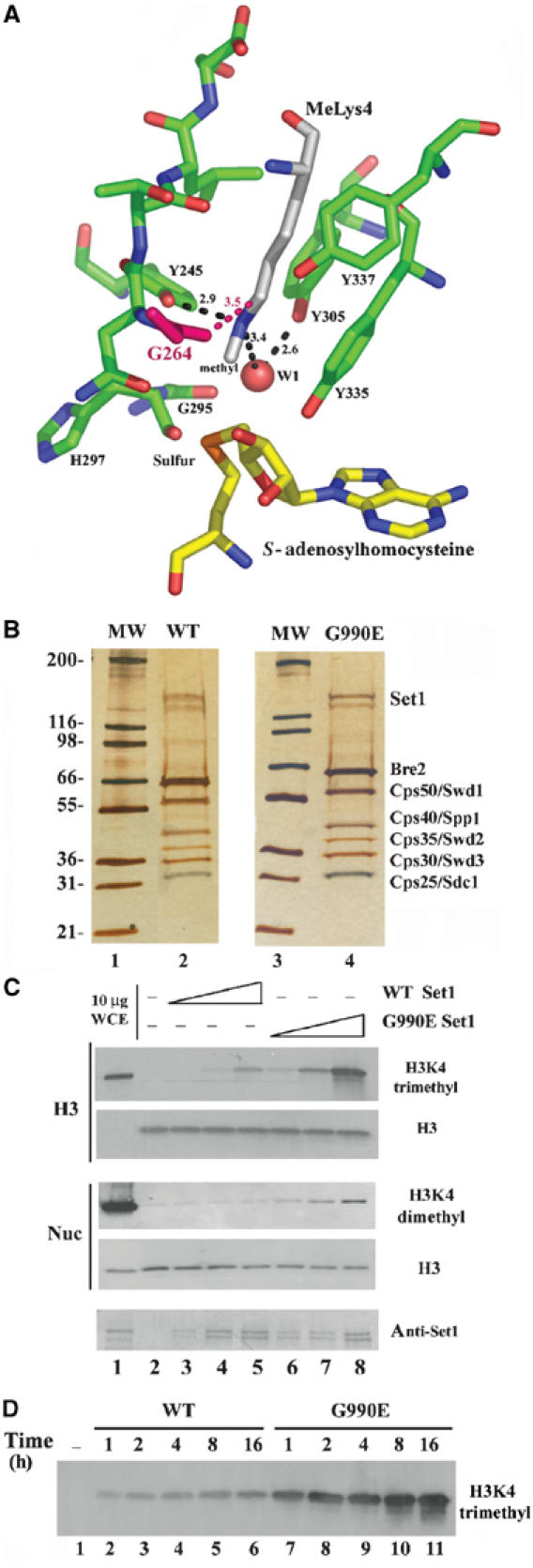
G990E replacement in Set1 creates a hyperactive H3K4 methyltransferase. (A) Structure of the Set7/9 SET domain (Xiao et al, 2003). The G990E dominant mutation in Set1 corresponds to G264 of Set7/9 (pink). (B) Purification of WT and G990E SET1/COMPASS complexes. SET1/COMPASS was purified by TAP purification from strains expressing TAP-tagged BRE2 (YBC1720), and containing plasmids bearing either WT Set1 (p1067) or Set1 G990E (p1305). A 10 μl portion of each purified complex was loaded onto an SDS 4–15% acrylamide gel and stained with silver. (C) HMTase activity of WT and G990E complexes. Purified complexes were incubated with 2.5 μg of recombinant H3, in the presence of S-adenosylmethionine and HMTase buffer for 16 h at 30°C. Whole-cell extract (10 μg) and 10% of each reaction (250 ng H3) were loaded onto an SDS 10–20% acrylamide gel and transferred to PVDF membrane. Immunoblots were incubated with anti-H3K4 trimethyl (upper panel), anti-H3K4 dimethyl (middle panel), anti-Set1, or pan H3 antibody. Note: Lane 1 shows very weak H3 immunoreactivity, as this lane contains whole-cell extract at amounts near the limit of detection for this pan-H3 antibody, whereas other lanes have recombinant H3. Middle panel: Methylation of H3K4 with recombinant unmodified yeast mononucleosomes (Nuc) (172 bp DNA, 300 ng/reaction), probed with anti-H3K4 dimethyl and anti-H3 C-terminus. (D) Time course of HMTase activity. Purified WT and G990E complexes (400 ng) were incubated with 2.5 μg of recombinant H3, in the presence of S-adenosylmethionine and HMTase buffer for 1, 2, 4, 8, or 16 h, and 20% of each reaction was loaded onto an SDS 10–20% acrylamide gel and transferred to PVDF membrane. Immunoblots were probed with anti-H3K4 trimethyl antibodies.
To determine whether the G990E derivative encodes a hyperactive methyltransferase, SET1/COMPASS complexes bearing Set1-G990E or WT Set1 protein were purified to homogeneity in parallel (Figure 4B) and their HMTase activities were compared using recombinant yeast histone H3 or mononucleosomes (Figure 4C). SET1/COMPASS complex bearing Set1-G990E protein is approximately 10-fold more active than WT Set1 on histone H3 when normalized for Set1 protein (Figure 4C). This difference is likely due to increased turnover number and not simply enzyme stability, as the product accumulates in a relatively linear manner, with the G990E enzyme showing proportionately higher levels at all times (Figure 4D). Identical results were obtained from parallel purifications of SET1/COMPASS complex from WT Set1 and a strain bearing an alternative SET1D allele, termed G990E(+), which also bears additional substitutions (S43N, S509F, P975S; data not shown). Remarkably, H3K4 dimethylation of nucleosomal H3 was not apparent with WT SET1/COMPASS, whereas dimethylation with the G990E derivative was clearly detected (Figure 4C). We note that the G990E complex is less active on nucleosomes than on isolated H3, and that our anti-trimethyl antibody has slightly lower avidity than our anti-dimethyl antibody. Therefore, we could not determine whether trimethylation of nucleosomes was occurring with the G990E complex. Taken together, these results demonstrate that G990E substitutions confer a hyperactive SET1/COMPASS complex with the capacity to methylate an unmodified nucleosome at H3K4.
N-Set1 can restore viability to rsc2rad6 combinations
SET1D alleles suppress rsc2 mutations, suggesting that Set1 hyperactivity and/or misregulation suppresses rsc2. If so, then reduced Set1 activity might exacerbate rsc2 mutations. Here, we combined rsc2 with two alleles that by themselves eliminate H3K4Me, set1Δ and rad6Δ. Indeed, rsc2 set1Δ combinations conferred severe sickness, and rsc2 rad6Δ combinations conferred lethality (data not shown). Interestingly, we found that introduction of N-Set1 into rsc2 rad6Δ cells restored viability, raising the possibility that N-Set1 may restore H3K4 methylation to a rad6Δ strain. This prompted us to test whether our other SET1D alleles had acquired the ability to bypass Rad6 and other factors normally required for H3K4Me. In keeping with our focus on Set1 regulation, all of the studies on methylation below were performed in RSC+ strains.
SET1D alleles partially suppress the requirement for Rad6 and Paf1 for H3K4 methylation
In vivo, full H3K4 methylation relies on SET1/COMPASS complex members (such as Set1, Bre2, and Spp1), and proteins required for histone ubiquitylation (Rad6) and transcription elongation (Paf1, Rtf1). To determine whether our SET1D alleles require these factors for H3K4 methylation, the SET1D alleles (or WT SET1) were expressed in strains lacking these factors and the bulk H3K4Me levels were monitored. To more quantitatively assess their impact, our controls included identical levels of protein derived from WT cells (Figure 5, 15 μg, lanes 1) or 10% of those levels (1.5 μg, lanes 2).
Figure 5.
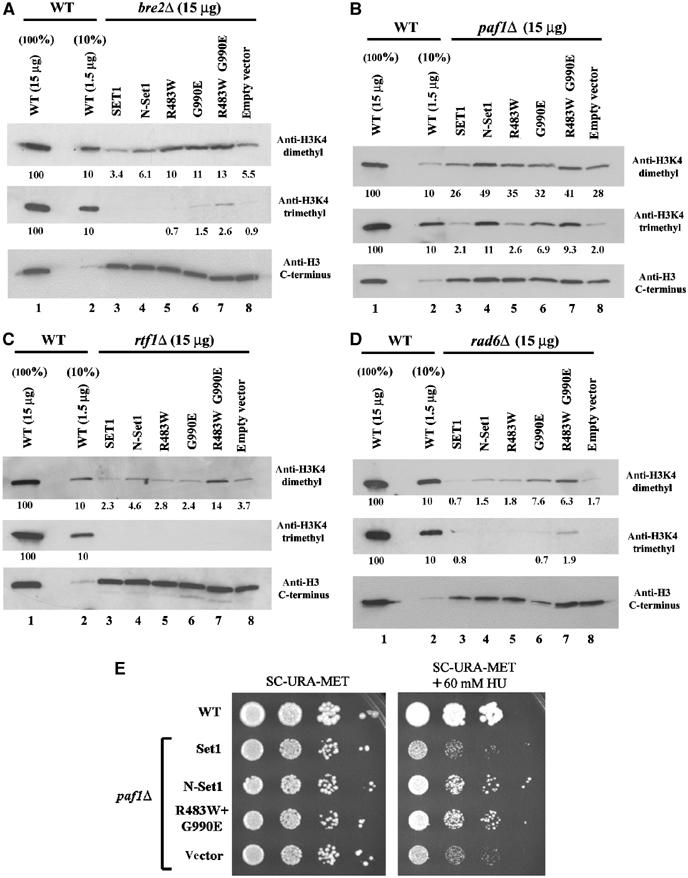
SET1D alleles partially bypass regulatory factors and restore H3K4 methylation. (A) Methylation by SET1D in a bre2Δ strain. Whole-cell extracts from WT (YBC63) and a bre2Δ strain (YBC1366) transformed with Set1 (p1067), N-Set1 (p1102), Set1 R483W (p1409), Set1 G990E (p1305), Set1 R483W G990E (p1638), or empty vector (p525) and grown in synthetic media lacking uracil and methionine were loaded onto SDS 10–20% acrylamide gels, transferred to PVDF, and probed with the antibodies indicated. (B) Methylation by SET1D in a paf1Δ strain. Same as (A) except paf1Δ (GHY878). (C) Methylation by SET1D in an rtf1Δ strain. Same as (A) except rtf1Δ (YBC2259). (D) Methylation by SET1D in a rad6Δ strain. Same as (A) except rad6Δ (YBC1539). (E) SET1D alleles can partially restore growth ability to paf1Δ strains on medium containing hydroxyurea. A paf1Δ strain (GHY880) was transformed with the plasmids indicated and tested for growth ability on selective medium, or selective medium containing 60 mM hydroxyurea (HU), and grown for 6 days.
Strains lacking Bre2 lack trimethylation and bear very low levels of dimethylation. Remarkably, three SET1D alleles restored dimethylation levels to 10–15% of WT (Figure 5A). Additionally, the SET1D-R483W G990E double substitution provided a low but detectable level of trimethylation, raising the possibility that combining SET1D substitutions confers even greater activity/misregulation. Strains lacking Spp1 showed moderately reduced levels of H3K4 dimethylation (∼50% of WT) and significant reductions in trimethylation (∼10% of WT); however, the SET1D alleles did not significantly raise H3K4 methylation (data not shown), suggesting that increased H3K4 methylation by these SET1D alleles requires Spp1. Strains lacking Paf1 lack appreciable trimethylation and bear low levels (∼25% of WT) of dimethylation. Remarkably, the N-Set1 and SET1D-R483W G990E alleles restored dimethylation and trimethylation to much higher levels (Figure 5B). In contrast, the R483W allele was largely ineffective, reminiscent of its inability to suppress RRM mutations (see Discussion). Strains lacking Rtf1 lack H3K4 trimethylation and bear extremely low levels of dimethylation. Here, the SET1D-R483W G990E combination conferred significant H3K4 dimethylation, whereas trimethylation was not appreciably affected (Figure 5C). Strains lacking Rad6 completely lack both histone H2BK123 ubiquitylation and H3K4Me (Sun and Allis, 2002; Wood et al, 2003). Here, most SET1D alleles conferred low to moderate increases in H3K4 dimethylation levels. However, the SET1D-R483W G990E allele conferred significant increases in dimethylation (6% of WT) and a slight restoration of trimethylation levels (Figure 5D). Thus, these SET1D alleles can partially restore di- and trimethylation to strains lacking factors normally required for H3K4Me. However, restoration of activity was most striking in cells lacking Paf1, which is closely linked to transcript elongation; levels of dimethylation are restored to 30–40% of WT levels and levels of trimethylation are restored to 5–15% of WT levels. This restoration has a positive impact on growth ability in vivo. Paf1 has many functions beyond promoting Set1 trimethylation, so a test for suppression by SET1D alleles must involve a phenotype shared by both paf1Δ and set1Δ strains. The clearest phenotype is their inability to grow on medium containing hydroxyurea (an inhibitor of DNA replication), likely due to defects in transcription of genes involved in replication or DNA metabolism. We find that the expression of our SET1D alleles in paf1Δ mutants partially restores growth ability in this condition (Figure 5E). Here, partial restoration was all that was expected, as Paf1 is required for efficient targeting of Set1/COMPASS to the 5′ ends of genes (Krogan et al, 2003; Ng et al, 2003). Therefore, in spite of paf1Δ mutations greatly lowering the targeting and activity of SET1/COMPASS, SET1D alleles can improve paf1Δ growth ability. Combined with the observation that SET1D alleles restore viability to rsc2 rad6 mutants, the function of these SET1D alleles is linked both to Paf1 and Rad6 function.
Discussion
SET domain methyltransferases play central roles in transcriptional regulation, and display exceptionally complex regulation. Set1 is the sole H3K4 HMTase in yeast, and in addition to the catalytic SET domain, Set1 bears multiple conserved domains with the potential for enzyme regulation. Loss-of-function mutations provide information about required domains/residues in enzymes such as Set1, and are easily acquired. However, dominant gain-of-function mutations are much more difficult to acquire, although often prove extremely valuable in understanding enzyme regulation. Here, we isolated dominant SET1D alleles, which were utilized to reveal new domains and properties underlying Set1 regulation.
rsc2 suppression yielded SET1D alleles
Here, a selection for suppressors of rsc2 conditional mutations yielded nine SET1D alleles, which conferred either increased activity against H3K4, partial bypass of normal regulation, or both. How these SET1D alleles suppress rsc2 mutations is not known; rsc2 does not affect H3K4Me levels (in WT SET1 strains) and Rsc2 does not prefer to bind H3K4Me peptides (data not shown). However, one simple model for suppression is that both H3K4Me and Rsc2 (RSC) promote transcription, and that a defect in Rsc2 function can be compensated by increased H3K4Me. A related possibility is that certain genes utilize either RSC or an alternative remodeler that binds H3K4Me, with Chd1 as a strong candidate (Pray-Grant et al, 2005). At these loci, reduced RSC function would require a greater participation from Chd1, which is facilitated through increased H3K4Me by these SET1D alleles. Although this possibility will be interesting to explore, this current work has an entirely different focus: the isolation and characterization of SET1D alleles useful for understanding how methylation by Set1 is regulated.
Set1 regulation is linked to transcription elongation
Set1 regulation is remarkably complex. Set1 activity requires its assembly into a large complex (COMPASS), the presence of histone ubiquitylation at H2BK123, and the presence and activity of several factors important for RNA Pol II CTD phosphorylation and transcript elongation (see Introduction). In addition, both Set1 occupancy and H3K4 trimethylation are localized to the 5′ ends of active genes. In contrast, dimethylation is found on the coding regions of active genes, and also on intergenic segments of active genes and repressed genes that are not packaged into repressive heterochromatin (Bernstein et al, 2002; Santos-Rosa et al, 2002; Krogan et al, 2003; Ng et al, 2003). Currently, how these factors recruit Set1 and activate trimethylation is not well understood. However, it is clear that both CTD phosphorylation (at serine 5) and the PAF1 complex are required for Set1 recruitment to the 5′ end of active genes (Krogan et al, 2003; Ng et al, 2003). Although the roles of individual members of the PAF1 complex in this process are not known, current data are consistent with SET1/COMPASS being recruited to the 5′ end of genes during the transition from initiation to elongation, where the PAF1 complex associates with RNA Pol II phosphorylated at serine 5. Therefore, trimethylation occurs in the region where the nascent RNA is produced. Taken together, several factors involved in the transition from transcript initiation to elongation regulate Set1 recruitment and activity against H3K4.
A role for the RRM domain in promoting trimethylation
A question of particular interest is how the factors described above stimulate trimethylation at the 5′ ends of active genes. Here, our data strongly suggest that RRM domain of Set1 plays a central role in regulating trimethylation. First, its removal essentially eliminates H3K4 trimethylation, with only moderate effects on dimethylation. Furthermore, site-directed mutations designed to impair nucleic acid interaction by the RRM also severely reduce H3K4 trimethylation activity in vivo. These results raise the intriguing possibility that the binding of the nascent mRNA transcript at the 5′ end of genes might help activate H3K4 trimethylation. Here, we found that the addition of purified yeast mRNA to purified SET1/COMPASS did not, by itself, significantly stimulate trimethylation activity in vitro on histone H3 (data not shown). However, as multiple additional factors are required for robust trimethylation in vivo, these same factors may likewise be required in vitro. Thus, a full understanding of Set1 regulation will require reconstitution of the process with all participating factors in vitro.
Alteration at G990 creates a hyperactive enzyme that bypasses RRM mutations for trimethylation
The largest class of SET1D alleles obtained represented three different substitutions at a single position, G990. As the three substituted residues (lysine, alanine, and glutamic acid) are of very different chemical nature, and as no proteins appear to be lost or gained in Set1 G990E purifications, it is likely that a larger side chain itself provides a structural alteration that hyperactivates Set1. Consistent with this interpretation, strains bearing G990E showed moderately higher levels of both H3K4 di- and trimethylation, and purified SET1/COMPASS complex bearing Set1 G990E displayed about 10-fold higher HMTase activity in vitro. In addition, the hyperactive complex is able to methylate H3K4 in otherwise unmodified nucleosomes in vitro, although at reduced levels from free histone H3. Strikingly, G990 substitutions suppress RRM domain mutations, fully restoring trimethylation in vivo. Residue 990 is predicted to reside in the catalytic pocket, within 3.5 Å of the substrate lysine and also near the S-AdHoCys coproduct binding site (Xiao et al, 2003), consistent with the capacity to affect turnover and specificity.
Set1 bears a central domain that inhibits trimethylation
Whereas removal of the RRM domain from Set1 eliminates trimethylation, the additional removal of the central region (creating N-Set1) restores trimethylation. This strongly suggests that the central region is an autoinhibitory domain that negatively regulates trimethylation (Figure 6). SET1D mutations (R483W) were also isolated in this region that weakly restored trimethylation to RRM site-directed mutants, but not to the ΔRRM derivative. We suggest that the R483W mutation weakens autoinhibition, but still leaves the enzyme largely reliant on RRM function for trimethylation. In contrast, removal of the autoinhibitory domain altogether confers trimethylation without RRM stimulation (Figure 6).
The SET domain integrates inputs from multiple domains
The catalytic SET domain and flanking N-SET and post-SET domains are essential for H3K4 methylation, as mutations in (or omission of) these domains eliminate all H3K4 methylation. Our work has focused on identifying and characterizing the domains (and residues within them) that specifically regulate di- and especially trimethylation. Taken together, our data suggest that activation of WT Set1 requires two inputs: positive input from the RRM domain, and relief from the inhibition imposed by the central autoinhibitory region (Figure 6). Here, R483 likely potentiates inhibition by the central region, but only removal of the entire central region renders the enzyme independent of RRM stimulation. Regulation is integrated at the SET domain, where G990 is important for regulating overall activity against H3K4 and maintaining reliance on the RRM domain, as replacements at this residue confer enzyme hyperactivity and independence from the RRM domain for trimethylation. Combining mutations in G990 with R483W provides a highly active and/or misregulated enzyme that can partially bypass a variety of factors normally required for trimethylation. For example, in all cases tested (except for spp1Δ), the SET1DR483W G990E allele conferred significantly higher levels of di- or trimethylation. Our work may also provide some insight into Paf1 function. Here, N-Set1 and G990 replacements significantly suppressed the trimethylation defect in paf1Δ strains, whereas the R483W allele was largely ineffective. Interestingly, SET1D alleles that suppress paf1Δ trimethylation defects likewise suppress RRM mutant trimethylation defects. This result raises the possibility that Paf1 and the RRM domain work together to promote trimethylation. An interesting alternative to a direct stimulation of Set1 by Paf1 is that Set1 (or another member of COMPASS) might recognize the ubiquitin moiety present on histone H2B, which requires Paf1 for its presence.
Taken together, our results suggest that the SET domain integrates opposing inputs from the RRM and autoinhibitory regions to control trimethylation. Our results further suggest a role for the N-SET domain in regulating trimethylation. Interestingly, these three domains (the RRM, N-SET, and central region) are not present in the H3K9 HMTases that regulate repression, suggesting that these domains specialize H3K4 HMTases by adapting the SET domain to regulation by factors present at the 5′ ends of genes, such as the PAF1 complex. Future experiments will aim at revealing how Set1 interacts with factors in the SET1/COMPASS and PAF1 complexes, ubiquitylated H2B, and possibly nascent RNA to influence enzyme activity and specificity.
Materials and methods
Media, genetic methods, and strains
Rich media (YPD), synthetic complete (SC) media, and sporulation media were prepared by standard methods. Standard procedures were used for transformations, sporulation, and tetrad analysis. Full strain genotypes are available in Supplementary Table S1.
SET1 plasmid construction
The SET1 gene was isolated from ATCC cosmid #71214 with ClaI and NruI, and cloned into ClaI/SmaI sites of pRS426 (Sikorski and Hieter, 1989). SET1 derivatives were made by PCR, incorporating a single FLAG tag and an NheI site at the 5′ end, and a HindIII site at the 3′ end. The PCR product was then cloned into SpeI/HindIII sites of p426 Met25 or p425 Met25 (Mumberg et al, 1994). Clones were sequence verified.
Isolation of rsc2 temperature-sensitive mutants and site-directed mutagenesis
To isolate conditional rsc2 alleles, we generated a TRP1-marked plasmid bearing rsc2BAHΔ, in which the essential BAH domain was deleted and replaced with a unique restriction site (SphI). This plasmid was the target for recombination in vivo with mutagenized versions of the BAH region, which were prepared by mutagenic PCR. SphI-cleaved linear plasmid and the mutagenized PCR fragment were cotransformed into a trp1 rsc1Δ rsc2Δ strain bearing RSC2 on a URA3-marked plasmid. Cells that directed the in vivo recombination/repair of the rsc2 domain deletion plasmid were identified by tryptophan prototrophy and screened for conditional phenotypes following the loss of the RSC2∷URA3 plasmid on media containing 5-FOA. A full description of all mutants will be described elsewhere. Site-directed mutations in SET1 were created using the QuikChange method (Stratagene) and were subcloned and sequenced.
Screen for suppressors of rsc2 temperature-sensitive mutants
A multicopy library on the YEp24 vector (gift of Marian Carlson) was transformed into YBC1111 (V457M), and YBC1112 (D461G). Approximately 13 000 transformants for each strain were screened. Transformants that could suppress the temperature sensitivity of rsc2 Ts− mutations on SC medium lacking uracil at 35°C were isolated, retransformed, and plated to SC medium lacking uracil and containing 5-FOA at 35°C. Those plasmids that maintained suppression were sequenced.
Preparation of whole-cell extracts and immunoblots
Cells were grown in YPD or SC media at 30°C to OD600=1–2. Whole-cell extracts were prepared as described previously (Cairns et al, 1999). Extracts were loaded onto SDS–PAGE gels, transferred to PVDF, and probed with the antibodies indicated. Specificity of histone antibodies (Abcam) was confirmed by immunoblotting histone H3, H3K4 dimethyl, and H3K4 trimethyl peptides.
Random mutagenesis of SET1
To isolate full-length suppressors of rsc2 Ts− mutations, 426.Flag.Met25.SET1 was mutagenized with hydroxylamine, and transformed into YBC1111 or YBC1112. Suppressors of rsc2 Ts− mutations on SC medium lacking uracil at 33°C were selected, isolated, and fully sequenced.
Purification of WT and dominant active SET1/COMPASS complex
SET1/COMPASS complex was purified from strain YBC1720 (BRE2.TAP set1Δ∷HIS3MX6 pep4Δ∷kanmx) containing either p426.Met25.SET1 or p426.Met25.SET1.G990E. Cells (12 l) were grown in SC-Met-Ura to OD600=4–5. TAP purification was performed according to Rigaut et al (1999) with minor modifications; details are available on request.
Histone methyltransferase assay
HMTase reactions were carried out in a 25 μl volume at 30°C using purified COMPASS, 2.5 μg recombinant H3 or 600 ng mononucleosome, 100 μM S-adenosylmethionine (ICN), HMTase buffer (Rea et al, 2000), and 1 × protease inhibitor cocktail. Reactions were stopped by adding 10 μl of 4 × SDS sample buffer. Protein was separated on a 10–20% SDS–PAGE gel (Bio-Rad) and immunoblotted. Blots were incubated with the antibodies indicated. For the time course, purified WT and G990E COMPASS complexes (400 ng) were incubated with 2.5 μg of recombinant H3 in the presence of S-adenosylmethionine and HMTase buffer for 1, 2, 4, 8, or 16 h, and then 20% of each reaction was loaded onto an SDS 10–20% acrylamide gel and transferred to PVDF membrane. Immunoblots were probed with anti-H3K4 trimethyl (diluted 1:1000).
Supplementary Material
Supplemental Figure 1
Supplemental Figure 2 Materials and Methods
Supplemental Table
Acknowledgments
We thank Lorraine Pillus for anti-Set1 antibody, Bob Schackmann for oligos, and Andy VanDemark and Heidi Schubert for PYMOL figures. We thank Jacqui Wittmeyer for nucleosomes and histone H3, Grant Hartzog for the paf1Δ null strain, Ali Shilatifard for the Bre2-TAP strain, and David Allis and Vincent Géli for plasmids. We thank Vincent Géli and Ali Shilatifard for sharing unpublished information and valuable discussions. We thank Margaret Kasten, Jacqui Wittmeyer, Don Ayer, and Craig Kaplan for comments on the manuscript. This work was funded by the National Institutes of Health (GM60415 to BRC and CA20414 for core facilities), and by the Howard Hughes Medical Institute. BRC is an assistant investigator with HHMI and an investigator with the Huntsman Cancer Institute.
References
- Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL (2002) Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci USA 99: 8695–8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SY, Winston F, Allis CD (2001) Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev 15: 3286–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryk M, Briggs SD, Strahl BD, Curcio MJ, Allis CD, Winston F (2002) Evidence that Set1, a factor required for methylation of histone H3, regulates rDNA silencing in S. cerevisiae by a Sir2-independent mechanism. Curr Biol 12: 165–170 [DOI] [PubMed] [Google Scholar]
- Cairns BR, Schlichter A, Erdjument-Bromage H, Tempst P, Kornberg RD, Winston F (1999) Two functionally distinct forms of the RSC nucleosome-remodeling complex, containing essential AT hook, BAH, and bromodomains. Mol Cell 4: 715–723 [DOI] [PubMed] [Google Scholar]
- Corda Y, Schramke V, Longhese MP, Smokvina T, Paciotti V, Brevet V, Gilson E, Geli V (1999) Interaction between Set1p and checkpoint protein Mec3p in DNA repair and telomere functions. Nat Genet 21: 204–208 [DOI] [PubMed] [Google Scholar]
- Dover J, Schneider J, Tawiah-Boateng MA, Wood A, Dean K, Johnston M, Shilatifard A (2002) Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J Biol Chem 277: 28368–28371 [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Tansey WP (2004) Proteasomal ATPases link ubiquitylation of histone H2B to methylation of histone H3. Mol Cell 13: 435–442 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Kranz JK, Hall KB (1999) RNA recognition by the human U1A protein is mediated by a network of local cooperative interactions that create the optimal binding surface. J Mol Biol 285: 215–231 [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A (2002) COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 277: 10753–10755 [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, Greenblatt JF, Shilatifard A (2003) The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell 11: 721–729 [DOI] [PubMed] [Google Scholar]
- Lachner M, Jenuwein T (2002) The many faces of histone lysine methylation. Curr Opin Cell Biol 14: 286–298 [DOI] [PubMed] [Google Scholar]
- Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A (2001) COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci USA 98: 12902–12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M (1994) Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res 22: 5767–5768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai K, Oubridge C, Jessen TH, Li J, Evans PR (1990) Crystal structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein A. Nature 348: 515–520 [DOI] [PubMed] [Google Scholar]
- Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML (2002) A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc Natl Acad Sci USA 99: 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K (2003) Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell 11: 709–719 [DOI] [PubMed] [Google Scholar]
- Nislow C, Ray E, Pillus L (1997) SET1, a yeast member of the trithorax family, functions in transcriptional silencing and diverse cellular processes. Mol Biol Cell 8: 2421–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paro R, Hogness DS (1991) The Polycomb protein shares a homologous domain with a heterochromatin-associated protein of Drosophila. Proc Natl Acad Sci USA 88: 263–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pray-Grant MG, Daniel JA, Schieltz D, Yates JR III, Grant PA (2005) Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature 433: 434–438 [DOI] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593–599 [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032 [DOI] [PubMed] [Google Scholar]
- Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF (2001) The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J 20: 7137–7148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419: 407–411 [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bernstein BE, Karabetsou N, Morillon A, Weise C, Schreiber SL, Mellor J, Kouzarides T (2003) Methylation of histone H3 K4 mediates association of the Isw1p ATPase with chromatin. Mol Cell 12: 1325–1332 [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZW, Allis CD (2002) Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418: 104–108 [DOI] [PubMed] [Google Scholar]
- Turner BM (2002) Cellular memory and the histone code. Cell 111: 285–291 [DOI] [PubMed] [Google Scholar]
- Wood A, Krogan NJ, Dover J, Schneider J, Heidt J, Boateng MA, Dean K, Golshani A, Zhang Y, Greenblatt JF, Johnston M, Shilatifard A (2003) Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol Cell 11: 267–274 [DOI] [PubMed] [Google Scholar]
- Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, Howell S, Taylor IA, Blackburn GM, Gamblin SJ (2003) Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 421: 652–656 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1
Supplemental Figure 2 Materials and Methods
Supplemental Table