

Abstract
The ARF tumour suppressor is a central component of the cellular defence against oncogene activation. In addition to activating p53 through binding Mdm2, ARF possesses other functions, including an ability to repress the transcriptional activity of the antiapoptotic RelA(p65) NF-κB subunit. Here we demonstrate that ARF induces the ATR- and Chk1-dependent phosphorylation of the RelA transactivation domain at threonine 505, a site required for ARF-dependent repression of RelA transcriptional activity. Consistent with this effect, ATR and Chk1 are required for ARF-induced sensitivity to tumour necrosis factor α-induced cell death. Significantly, ATR activity is also required for ARF-induced p53 activity and inhibition of proliferation. ARF achieves these effects by activating ATR and Chk1. Furthermore, ATR and its scaffold protein BRCA1, but not Chk1, relocalise to specific nucleolar sites. These results reveal novel functions for ARF, ATR and Chk1 together with a new pathway regulating RelA NF-κB function. Moreover, this pathway provides a mechanism through which ARF can remodel the cellular response to an oncogenic challenge and execute its function as a tumour suppressor.
Keywords: ARF, ATR, Chk1, NF-κB, p53
Introduction
A central component of the cellular defence against oncogene activation is the ARF tumour suppressor (p14ARF in humans, p19ARF in mice) (Sherr, 2001; Lowe and Sherr, 2003). The expression of ARF, which shares a genetic locus with the p16INK4a tumour suppressor, is regulated by the action of transcription factors such as members of the E2F family (Sherr, 2001; Lowe and Sherr, 2003). Deregulation of the Rb tumour suppressor pathway, for example, will lead to aberrant activation of E2F, resulting in increased levels of ARF (Sherr and McCormick, 2002). ARF can then bind to and inhibit the Hdm2 protein (Mdm2 in mice), which functions as an inhibitor and E3 ubiquitin ligase for the p53 transcription factor (Sherr, 2001; Michael and Oren, 2002; Vousden, 2002; Lowe and Sherr, 2003). This activates p53, which then executes the final stage in this tumour suppressor pathway by inducing or repressing the expression of critical cellular proteins that regulate cell cycle progression and apoptosis. ARF is therefore a critical link in the chain from oncogene activation to p53 induction, and this is reflected by the fact that it is often inactivated by mutation or gene methylation in tumour cells (Sherr, 2001; Esteller, 2002; Lowe and Sherr, 2003). Furthermore, in model systems, tumours that retain wild-type p53 will often lose ARF expression (Eischen et al, 1999).
p53- and Hdm2-independent effects of ARF have also been reported, such as inhibition of ribosomal RNA processing and association with nucleophosmin/B23 (Sugimoto et al, 2003; Bertwistle et al, 2004), repression of E2F activity (Eymin et al, 2001; Mason et al, 2002), sequestration of hypoxia-inducible factor-1α (HIF-1α) in the nucleolus (Fatyol and Szalay, 2001) and induction of antiproliferative gene expression (Kuo et al, 2003). Furthermore, we recently discovered that ARF regulates the activity of the RelA(p65) NF-κB subunit (Rocha et al, 2003). We demonstrated that ARF regulation of RelA does not require p53 or Hdm2/Mdm2 activity and did not induce or inhibit RelA DNA binding or translocation to the nucleus (Rocha et al, 2003). Rather, it modulated the activity of pre-existing nuclear RelA. These effects occurred, at least in part, through the C-terminal RelA transactivation domain (TAD) and resulted in the increased association of RelA with histone deacetylase (HDAC) 1. This increased association resulted in the repression rather than activation of RelA-dependent gene expression. One important effect of this was to inhibit expression of the NF-κB-regulated antiapoptotic gene Bcl-xL, consistent with an observed increase in susceptibility to tumour necrosis factor α (TNF)-induced apoptosis upon ARF induction. Given the critical role aberrant activation of NF-κB is now known to play in many tumour types, the ability of ARF to keep RelA's antiapoptotic activities in check is likely to be an important component of its tumour suppressor programme (Perkins, 2004).
The exact mechanism through which ARF is able to regulate RelA was not clearly defined in our previous report. We did observe that p53 induced by ARF was phosphorylated on serine 15 (Rocha et al, 2003), a target site of the ataxia telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR) checkpoint kinases (Abraham, 2001; Vousden, 2002). Although we did not investigate the significance of this for ARF-induced p53 function, we demonstrated that ARF inhibition of RelA, in reporter gene assays, was prevented by a kinase dead dominant-negative ATR (kdATR) expression plasmid and by caffeine treatment, a relatively nonspecific inhibitor of ATM and ATR kinase activity (Rocha et al, 2003). Although regulation of p53 by ATM and ATR following DNA damage is well established, a role for any of these kinases following ARF induction has not been clearly defined. A recent report did demonstrate a requirement of ATM for ARF induction of p53 (Li et al, 2004). Although this paper is in general support of our previous observations, the effects we had previously observed were also seen in ATM null cells, ruling out this checkpoint kinase as being an effector of ARF's effects on RelA (Rocha et al, 2003). In this report, therefore, we have further investigated the ability of ARF to regulate ATR function and expanded our study to include Chk1, a kinase activated by and downstream of ATR (Bartek and Lukas, 2003). Significantly, we find that ARF activates, or engages, the ATR/Chk1 pathway and that this is required for its ability to both repress RelA and to induce p53.
Results
ATR and Chk1 are required for inhibition of NF-κB transcriptional activity by ARF
To confirm that ATR was required for ARF regulation of NF-κB, and extend our analysis to include the Chk1 kinase, two sets of two siRNA oligonucleotides targeting ATR and Chk1 were synthesised. Experiments validating these siRNAs were performed in NARF2 or NARF2-E6 cells (Supplementary Figure 1), which are derived from the U-2 OS human osteosarcoma line and contain an integrated, IPTG-inducible, p14ARF expression plasmid (the promoter of the endogenous gene is methylated). While the NARF2 cells exhibit a wild-type p53 response upon ARF induction, the NARF2-E6 cells also express the papillomavirus E6 protein, which inhibits the expression and function of the wild-type p53 protein normally found in this cell type (Rocha et al, 2003). Therefore, NARF2 cells undergo a strong G1 cell cycle arrest as a consequence of p53 activation and induction of p21, but no cell cycle effects are seen upon ARF induction in NARF2-E6 cells (Supplementary Figure 2).
We have previously demonstrated that ARF induction inhibits the expression of the NF-κB-regulated Bcl-xL gene (Rocha et al, 2003). Using RNA interference, we found that depletion of ATR abolishes the inhibition of Bcl-xL gene expression seen upon induction of ARF in NARF2-E6 cells (Figure 1A). Significantly, knockdown of Chk1 also abolished ARF-mediated repression of Bcl-xL (Figure 1A). In contrast, siRNAs directed to ATM and Chk2, although functional (Figure 4H, Supplementary Figure 1; data not shown), did not block inhibition of Bcl-xL expression (Figure 1A). As an additional specificity control, we found that the ATR and Chk1 siRNAs did not prevent ultraviolet (UV-C) light-mediated repression of Bcl-xL expression (Figure 1B). Although apparently counter-intuitive, given the role of both kinases following UV-induced DNA damage (Abraham, 2001; Bartek and Lukas, 2003), this result is consistent with our previous observations where we demonstrated RelA NF-κB-dependent repression of Bcl-xL gene expression following UV treatment, but demonstrated that this was mechanistically distinct to repression by ARF (Campbell et al, 2004) (also data not shown).
Figure 1.
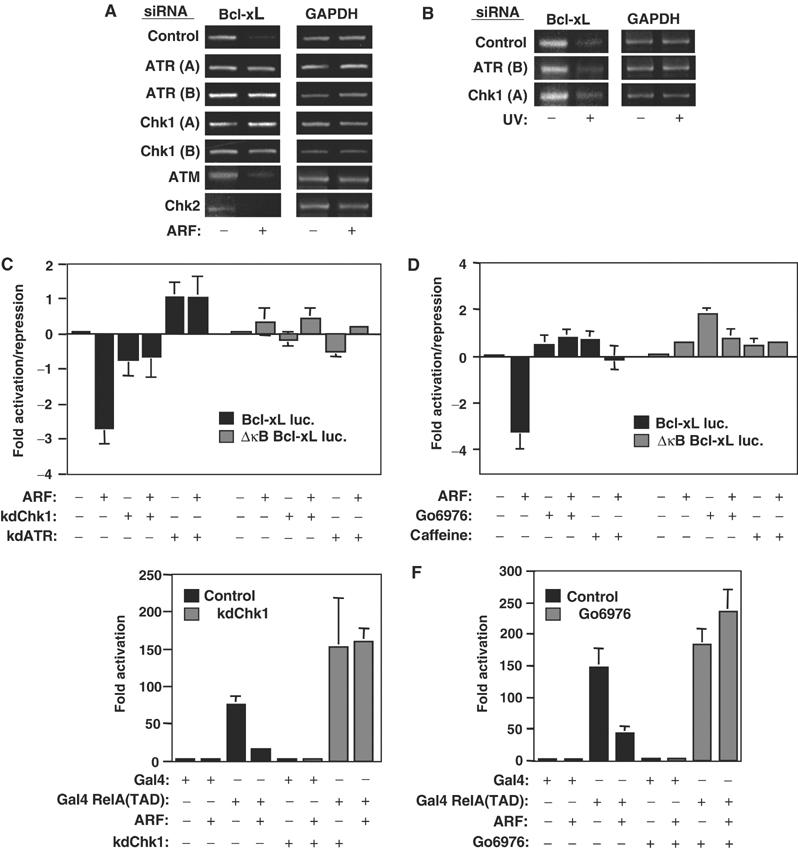
ARF repression of NF-κB activity is ATR and Chk1 dependent. (A, B) siRNAs targeting ATR and Chk1 abolish ARF-induced repression of Bcl-xL. PCR analysis of Bcl-xL expression was performed following treatment of NARF2-E6 cells with the indicated siRNAs. Cells were either treated with IPTG to induce ARF expression (A) or stimulated with UV-C (40 J/m2) (B). (C, D) Inhibition of ATR or Chk1 activity abolishes ARF-mediated repression of the Bcl-xL promoter. In all, 1.5 μg of the Bcl-xL or Bcl-xL ΔκB luciferase reporter plasmids was transfected into NARF2-E6 cells. Either (C) 1 μg of kdATR or kdChk1 expression plasmids were cotransfected as indicated or (D) cells were treated with 2 mM caffeine or 1 μM Gö6976 as indicated at the time of ARF induction. In this and subsequent luciferase assays, results are expressed as fold activation or repression, relative to levels seen in the relevant untreated cell controls. The absence of error bars for some data points, in this and subsequent figures, results from them being below the resolution of the graph. All data points in this and subsequent figures result from at least three independent experiments. (E, F) Chk1 is required for ARF-mediated repression of the RelA transcriptional activation domain. The Gal4 E1B luciferase reporter plasmid (1 μg) was transfected into NARF2-E6 cells together with expression plasmids encoding either Gal4 alone (0.75 ng) or Gal4 RelA(TAD) (0.75 ng). Where indicated, cells were also cotransfected with 1 μg of kinase dead dominant-negative Chk1 expression plasmid (E) or treated with 1 μM Gö6976 at the time of ARF induction (D).
Figure 4.

ATR is required for ARF-induced p53 activation. (A) ARF induces the association of ATR with p53. ATR was immunoprecipitated with 200 μg of whole-cell lysate prepared from NARF2 cells treated as indicated. The immunoprecipitated complex was resolved by SDS–PAGE and immunoblotted with an anti-p53 and anti ATR antibodies. (B) ARF-induced p53 transcriptional activity requires ATR. In all, 1.5 μg of the p53-dependent PG13 luciferase and p21WAF1 luciferase reporter plasmids were transfected into NARF2 cells. Where indicated, cells were cotransfected with 1 μg kdATR expression. (C, D) Caffeine treatment abolishes ARF induction of p53. Western blot analysis of whole-cell lysates showing the induction of ARF, p53 and p21 following treatment of NARF2 cells with IPTG or Hs68 E2F1–ER cells with Tamoxifen in the presence or absence of 2 mM caffeine. (E, F) ATR siRNA treatment inhibits ARF induction of p53 activity. Western blot analysis of whole-cell lysates from NARF2 (E) or Hs68 E2F1–ER (F) cells showing the induction of ARF, p53 and p21. Cells were transfected with either ATR or control siRNAs. (G) ATM siRNA treatment does not abolish ARF induction of p53 in NARF2 cells. Western blot analysis of NARF2 cell whole-cell lysates showing the induction of ARF, p53 and phospho-serine 15-modified p53 following treatment with either ATM or control siRNAs. In the top panel, PCR analysis of ATM RNA levels is shown following treatment with ATM siRNAs. Inhibition of ATM protein expression by ATM siRNA is shown in Supplementary Figure 1G. (H) ATM siRNA inhibits p53 activation following DNA damage. Western blot analysis of whole-cell lysates showing the induction of p53 and phospho-serine 15-modified p53 following treatment with 2 μM doxorubicin for 4 h in NARF2 cells treated with either ATM or control siRNAs.
While the reported off-target effects of siRNAs seemed an unlikely explanation for our data, we nonetheless wished to confirm these results using other means. Similar to our previous results (Rocha et al, 2003), we found that expression of kdATR blocked the κB site-dependent, ARF-mediated, repression of the Bcl-xL promoter (Figure 1C). In parallel and consistent with our siRNA data, we also found that a kinase dead Chk1 (kdChk1) expression plasmid similarly prevented this effect of ARF induction (Figure 1C). Furthermore, both caffeine, an inhibitor of ATR, and Gö6976, a UCN-01-related inhibitor of Chk1 kinase activity (Ishimi et al, 2003; Kohn et al, 2003), had effects very similar to overexpression of the kinase dead proteins (Figure 1D). We had previously demonstrated that ARF repressed the C-terminal RelA TAD when fused to Gal4 and that this effect was also blocked by kdATR and caffeine (Rocha et al, 2003). We also found that both kdChk1 and Gö6976 blocked RelA TAD inhibition, thus confirming that Chk1 is an integral component of the pathway through which ARF regulates RelA (Figure 1E and F). Previously we had shown that ARF repression of RelA results in sensitivity to TNF-induced apoptosis (Rocha et al, 2003). Importantly, we found that ATR and Chk1 siRNAs also significantly inhibited this ARF-dependent effect (Figure 2). Taken together, these significantly extend our original observations: both ATR and Chk1 are required for ARF-mediated repression of RelA NF-κB function.
Figure 2.
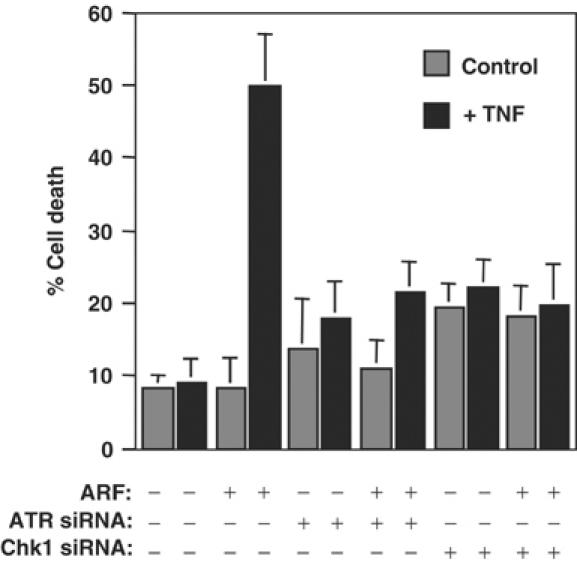
ARF-induced sensitivity to TNF-induced cell death requires ATR and Chk1. NARF2 E6 cells were transfected once with control, ATR and Chk1 siRNAs (oligos A and B combined). At 48 h after siRNA transfection, cells were treated with IPTG to induce ARF. After a further 24 h, cells were treated with TNF (30 ng/ml) and, 24 h later, percentage cell death was determined as described previously (Rocha et al, 2003).
ARF induces Chk1-dependent RelA threonine 505 (Thr505) phosphorylation
Previously, we found that a single mutation of Thr505 to alanine within the RelA TAD was sufficient to abolish ARF-mediated repression of this NF-κB subunit (Rocha et al, 2003). Furthermore, this mutation also abolished repression by cotransfected HDAC1 (Rocha et al, 2003). We proposed that this region of RelA represents a Thr505 phosphorylation-dependent RelA corepressor docking site. However, since this site had not previously been implicated in the regulation of RelA, the reagents were not available to demonstrate that this amino acid was actually phosphorylated at the time of our previous study. To address this, a phospho-specific antibody to this site was raised. Using this antibody, a strong increase in endogenous RelA Thr505 phosphorylation was observed following ARF induction which was not seen, in these cells, with TNF and ultraviolet light, both well-characterised inducers of NF-κB activity (Figure 3A and B). Furthermore, incubation with the Chk1 inhibitor Gö6976 abolished ARF induction of phosphorylation at this site (Figure 3C). This is consistent both with the requirement for Thr505 identified previously and the requirement for Chk1 activity seen in Figure 1. Moreover, analysis of the Thr505 site revealed that it conforms to the previously described Chk1 consensus phosphorylation sequence (Figure 3D) (Hutchins et al, 2000). Chk1 has not previously been identified as a RelA kinase and therefore we investigated the ability of recombinant, purified Chk1 to phosphorylate the RelA TAD in vitro. Using the RelA Thr505 antibody, specific Chk1-dependent phosphorylation was observed at this site (Figure 3E). No signal was observed with a GST-RelA TAD substrate where Thr505 had been mutated to alanine. The phospho-specificity of this antibody was further confirmed by peptide ELISA analysis (Supplementary Figure 3). Immunofluorescence analysis demonstrated that Thr505-modified RelA is predominantly nuclear in ARF-induced cells (Figure 3F).
Figure 3.
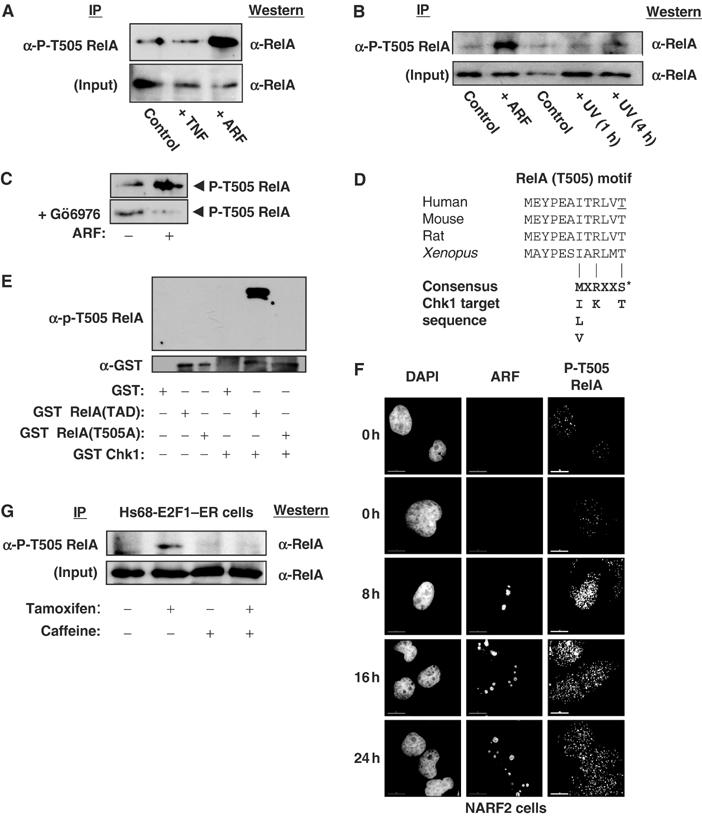
Phosphorylation of RelA at Thr505. (A) ARF induces Thr505 phosphorylation. Whole-cell lysates were prepared from NARF2 cells either treated with IPTG to induce ARF for 24 h, stimulated with TNF (10 ng/ml for 30 min) or left untreated. Phosphorylated RelA was then immunoprecipitated by incubating 150 μg of lysate with purified phospho-RelA T505A antibody. The immunoprecipitate was resolved by SDS–PAGE before Western blotting with an anti-RelA antibody (sc-372 Santa Cruz). (B) UV light does not induce Thr505 phosphorylation. The experiment was performed as in (A), except that immunoprecipitations were performed with 100 μg of nuclear protein extract. Input lanes represent 10 μg of nuclear extract. As indicated, some cells were treated with UV-C (40 J/m2) and harvested after the times shown. (C) ARF-induced Thr505 phosphorylation is prevented by the Chk1 inhibitor Gö6976. The experiment was performed as in (A), except that that indicated cells were incubated with 1 μM Gö6976 at the same time as IPTG addition. (D) Schematic diagram showing species conservation of the RelA Thr505 region and similarity to the Chk1 consensus phosphorylation sequence. The human Thr505 residue is underlined. (E) The RelA Thr505 motif is phosphorylated by Chk1 in vitro. Purified GST, GST RelA TAD (amino acids 428–551) and GST RelA TAD T505A were incubated with recombinant purified GST-Chk1 and unlabelled ATP. Proteins were then resolved by SDS–PAGE and immunoblotted with the anti-phospho RelA Thr505 antibody (upper panel). Immunoblotting with anti-GST antibody demonstrated equivalent loading of GST-RelA (TAD) and GST-RelA TAD (T505A) proteins (lower panel). (F) ARF induces nuclear phospho-Thr505-modified RelA. NARF2 cells were plated onto coverslips and fixed at the indicated times after IPTG treatment. Cells were stained with mouse anti-p14ARF and rabbit anti-P-T505 RelA antibodies. In this and subsequent experiments, cells were analysed and images were acquired using a DeltaVision microscope. (G) Induction of RelA Thr505 phosphorylation in Hs68 E2F1–ER cells. The experiment was performed as in (A), except that immunoprecipitations were performed with 300 μg of whole-cell lysates from Hs68 E2F1–ER cells. Input lanes represent 30 μg of extract. As indicated, cells were treated with Tamoxifen and 2 mM caffeine and harvested 24 h later.
Although NARF2 cells do not express ARF at the high levels seen following many transient transfection experiments, it is still higher than that seen with the endogenous protein (S Rocha, N Perkins, unpublished observation). To address this, we utilised Hs68 E2F1–ER cells, which are human neonatal foreskin fibroblasts containing an E2F1–ER fusion protein that upon activation with tamoxifen induces endogenous ARF expression (Brookes et al, 2002) (see also Figure 8D). Tamoxifen treatment induced Thr505 phosphorylation of endogenous RelA in these cells that was prevented by the ATR inhibitor caffeine (Figure 3G). This result is consistent with our previous report where we also demonstrated that endogenous ARF regulates RelA transactivation (Rocha et al, 2003). Together, these results are consistent with the RelA Thr505 motif being an ARF-dependent target of Chk1 phosphorylation.
Figure 8.
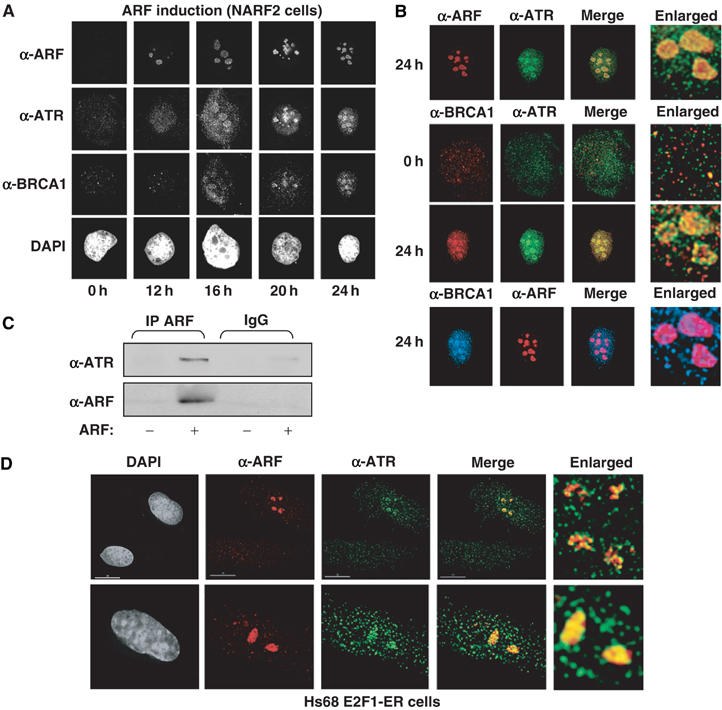
Recruitment of ATR and BRCA1 to ARF-containing nucleolar complexes. (A, B) Immunofluorescence analysis of ARF, ATR and BRCA1 following ARF induction. NARF2 cells were plated onto coverslips and fixed at the indicated times after IPTG treatment. Cells were stained with rabbit anti-p14ARF, goat anti-ATR and mouse anti-BRCA1 antibodies as indicated. In (B), selected data were further analysed and colocalisation of the indicated proteins was determined by examination of merged and enlarged images. Since these are digital images, colour designation is arbitrary in this and all subsequent images. (C) ATR coimmunoprecipitates with ARF. ARF was immunoprecipitated from 150 μg of whole-cell lysate prepared from NARF2 cells. The immunoprecipitated complex was resolved by SDS–PAGE and immunoblotted with anti-ARF or ATR antibodies. (D) E2F-induced ARF colocalises with ATR. Immunofluorescence analysis of ARF and ATR in Hs68 E2F-ER cells. Cells were plated onto coverslips and fixed 24 h after Tamoxifen treatment. Cells were stained with rabbit anti-p14ARF and goat anti-ATR antibodies. An enlarged section of the merged ATR/ARFs image is also shown.
ATR is also required for ARF induction of p53
ATR is also known to regulate the p53 tumour suppressor and transcription factor following DNA damage (Abraham, 2001; Vousden, 2002). Therefore, we became interested in whether ATR might also have a role regulating p53 following ARF induction. Consistent with this hypothesis, we found that ATR coimmunoprecipitated with p53 following ARF induction in NARF2 cells (Figure 4A). This interaction was also seen in the presence of ethidium bromide, indicating it did not occur due to DNA binding by either protein (not shown) (Lai and Herr, 1992). Furthermore, cotransfection of the kdATR expression plasmid abolished induction of the p21 promoter and PG13 p53 reporter plasmids by endogenous, ARF-induced, p53 (Figure 4B). In addition, caffeine and ATR siRNA not only inhibited ARF-induced p53 serine 15 phosphorylation, but also abolished efficient induction of p53 protein in both NARF2 and Hs68 E2F1–ER cells (Figure 4C–F). In Figure 4E, with NARF2 cells and an ATR siRNA, while complete inhibition of p53 serine 15 phosphorylation and p21 induction was observed, only a partial inhibition of induced p53 protein levels was seen. More complete inhibition can be seen in other experiments (see Figure 9), but this observation suggests that serine 15 phosphorylation might be the primary site of ATR action following ARF induction or that higher levels of ARF can induce p53 protein solely by binding Hdm2. Serine 15 phosphorylation of p53 is known to stimulate its transcriptional activity and enhance its interaction with coactivators (Dumaz and Meek, 1999; Vousden, 2002). Inhibition of this modification alone would therefore be expected to have a significant effect on p53 function.
Figure 9.

ARF-induced p53 activation requires BRCA1. Western blot analysis of NARF2 whole-cell lysates showing the induction of ARF, p53, phospho-serine 15 p53 and p21. Cells were doubly transfected with either ATR (oligos A and B combined), BRCA1 or control siRNAs as indicated. BRCA1 siRNA validation is shown in Supplementary Figure 1I.
Although we did not find that ATM was required for ARF-mediated repression of RelA (Figure 2; Rocha et al, 2003), a recent report indicated that it was required for ARF induction of p53 (Li et al, 2004). In our hands, however, downregulation of ATM had no effect on ARF induction of p53 in NARF2 cells (Figure 4G). Using the same siRNA, complete inhibition of p53 induction by doxorubicin could be observed (Figure 4H). Therefore, our data indicate that ATR and not ATM is the critical kinase regulating p53 following ARF induction. It is possible that these differences reflect cell type differences or levels of ARF expression.
The functional significance of these effects was further confirmed by analysing ARF-induced growth arrest in NARF2 cells. Inhibition of p53 by E6 protein expression (Supplementary Figure 2A) or a p53 siRNA (Figure 5) inhibits ARF-induced growth arrest. Importantly, downregulation of ATR, but not ATM, also inhibits ARF-induced growth arrest to a level similar to that seen with the p53 siRNA (Figure 5).
Figure 5.

ARF-induced inhibition of cell proliferation requires p53 and ATR. NARF2 cells were transfected with the indicated siRNA oligonucleotides and, 48 h later, ARF was induced with IPTG. Cell proliferation was measured every 24 h, as indicated, by alamarBlue assay. Results are obtained from four separate experiments performed in triplicate.
ARF activates the ATR/Chk1 pathway
All these results suggested that ARF must be activating or engaging the ATR/Chk1 pathway. To investigate this, we utilised an anti-phospho-SQ/TQ antibody that specifically binds phosphorylated ATR and ATM substrates. Western blot analysis demonstrated that ARF induction did indeed result in a strong increase in ATR/ATM substrate phosphorylation (Figure 6A). A number of proteins phosphorylated were distinct to those seen following stimulation of the same cells with UV-C, which also activates ATR. Although this suggests that ARF-induced ATR might phosphorylate different substrates, without further analysis the significance of this result is not known. A previous study had found that this phospho-SQ/TQ antibody recognised specific nuclear complexes (Stojic et al, 2004). Deltavision microscopy demonstrated that these ARF-induced SQ/TQ phosphorylations were also predominantly nuclear (Supplementary Figure 4A). Further analysis demonstrated that ARF induced SQ/TQ phosphorylation in both NARF2 and NARF2-E6 cells (Supplementary Figure 4B). Importantly, an increase in SQ/TQ phosphorylation was also seen in Hs68 E2F1–ER cells upon tamoxifen treatment (Figure 6B). This effect was abolished by treatment with either ATR or ARF siRNAs. This demonstrates that induction of ATR activity in these cells is dependent upon ARF and does not occur due to E2F1 itself, as has been reported in other circumstances (Lindstrom and Wiman, 2003).
Figure 6.
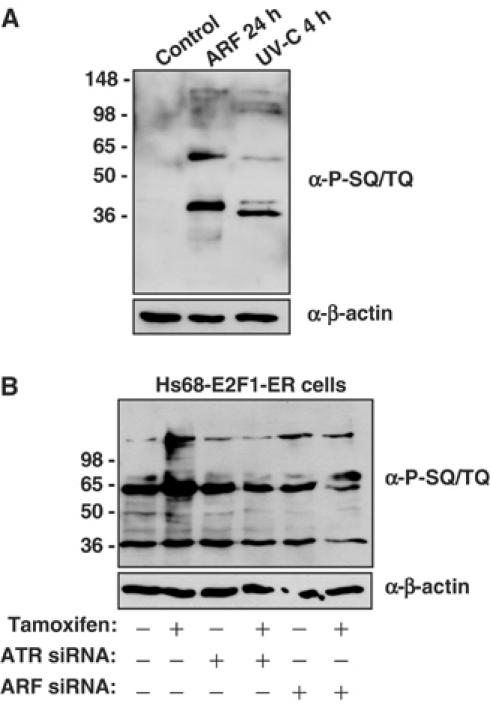
ARF induces ATR activity. (A) ARF induces the phosphorylation of ATM/ATR substrates. Whole-cell lysates were prepared from NARF2 cells either treated with IPTG to induce ARF for 24 h, stimulated with UV-C (40 J/m2) or left untreated. The lysate (20 μg) was then resolved by SDS–PAGE and Western blotted with the anti-phospho-SQ/TQ antibody. (B) Induction of ATR activity in Hs68 E2F1–ER cells by endogenous ARF. Western blot analysis of whole-cell lysates using the anti-phospho-SQ/TQ antibody following treatment of Hs68 E2F1–ER cells with Tamoxifen for 24 h. Cells were transfected twice with either ATR (A, B combined), ARF (A, B combined) or control siRNAs as indicated. ARF siRNA validation is shown in Supplementary Figure 1H.
ATR activation typically leads to Chk1 phosphorylation and activation. Since the latter kinase is required for ARF's effects on RelA, we next investigated phosphorylation of Chk1 at two sites modified by ATR and ATM, serines 317 and 345 (Bartek and Lukas, 2003). In both NARF2 and NARF2 E6 cells, both residues became rapidly phosphorylated upon ARF induction (Figure 7). These effects were not as pronounced as that seen with UV light but persisted over many hours, consistent with the phosphorylation of RelA at Thr505 we observed earlier (Figure 3F).
Figure 7.
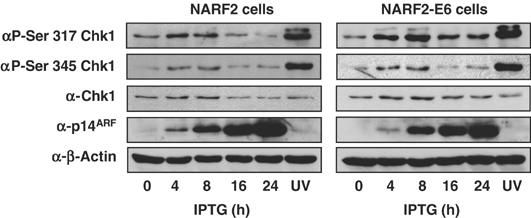
ARF induces phosphorylation of Chk1. Whole-cell lysates were prepared from NARF2 cells either treated with IPTG to induce ARF for the indicated times, stimulated with UV-C (40 J/m2) or left untreated.
It was conceivable that ARF activates ATR as a result of replication arrest or inadvertent DNA damage. The observation that the rate of proliferation of NARF2-E6 cells is unaffected by ARF induction (Supplementary Figure 2A) while still inducing ATR activity (Supplementary Figure 4B) suggests that this is not the case. Furthermore, FACS analysis of NARF2 cells following ARF induction reveals a G1/G2 arrest and an almost total loss of S-phase cells (Supplementary Figure 2B). This indicates an absence of an S-phase arrest and suggests that these effects do not result from stalled replication forks. Further supporting this hypothesis, analysis of cells with a γ-H2AX antibody revealed no significant increase in signal upon ARF induction (Supplementary Figure 5A). Phosphorylation of the histone variant H2AX, to produce γ-H2AX, is a marker for ATM and ATR activity following DNA damage and stalled replication forks (Ward and Chen, 2001; Furuta et al, 2003; Hammond et al, 2003; Ward et al, 2004).
Recruitment of ATR and BRCA1 to nucleoli following ARF induction
ARF is a nucleolar protein and can sequester proteins to nucleoli (Lowe and Sherr, 2003). Interestingly, we found that, upon ARF induction in NARF2 cells, there is a dramatic relocalisation of endogenous ATR to the nucleolus of ARF-expressing cells (Figure 8A and B). In contrast, ATM localisation is not affected by ARF (data not shown).
Consistent with these observations, ATR also coimmunoprecipitated with ARF (Figure 8C). Indeed, gel filtration analysis revealed ARF to be present in a >1 MDa complex which cofractionated with ATR (not shown). ARF has been previously described as being in a 2–5 MDa complex that also contains nucleophosmin/B23 (Bertwistle et al, 2004). Due to the presence of this high-molecular-weight complex, it is hard to determine whether ATR binds ARF directly or does so through an intermediary protein such a nucleophosmin. Furthermore, ARF is a ‘sticky' protein and for this reason we have not investigated whether ARF directly binds ATR in vitro since any results obtained would be hard to interpret.
We also verified that relocalisation of ATR to nucleolar sites did not result from ARF overexpression. Induction of ARF in Hs68 E2F ER cells demonstrated that ATR also becomes localised with endogenous ARF in the nucleolus (Figure 8D). In addition, in the top image, a cell where ARF has failed to induce is also shown. Here there is no nucleolar relocalisation of ATR. With our Hs68 E2F1–ER cells, we find that only 25% of cells induce ARF upon tamoxifen addition (not shown), possibly reflecting selection against E2F1–ER or ARF expression during prolonged cell culture.
Previously, it has been shown that ATR/ATM phosphorylation of a subset of targets (including p53 and Chk1/Chk2) is dependent on the breast cancer susceptibility gene, BRCA1 (Yarden et al, 2002; Foray et al, 2003), which colocalises with ATR following genotoxic stress (Tibbetts et al, 2000). Furthermore, BRCA1 has been previously found to stabilise and coactivate p53 in an ARF-dependent manner (Somasundaram et al, 1999). Examination of BRCA1 revealed that it is also recruited to nucleoli following ARF induction coincidentally with ATR (Figure 8A) and merged images revealed coassociation of BRCA1 with both ATR and ARF at the nucleolus (Figure 8B). Some nucleoplasmic association of BRCA1 and ATR can also be seen. Consistent with these previous studies of BRCA1 and ATR function, knockdown of BRCA1 expression also inhibited ARF induction of p53 to a level similar to that seen upon loss of ATR (Figure 9). Although, in this experiment, protein levels are lower in the ATR siRNA-treated samples, inhibition of ARF-induced p53 levels can clearly be seen.
In contrast to these results, no relocalisation of replication protein A (RPA) was seen to nucleolar sites upon ARF induction (Supplementary Figure 5B). In DNA-damaged cells, RPA binds single-stranded DNA and interacts with ATR interacting protein (ATRIP), thus recruiting ATR (Zou and Elledge, 2003).
Chk1 does not localise to nucleoli but shows increased association with ATR
Investigation of Chk1 localisation revealed that it did not form the same defined ARF-associated nucleolar structures as ATR (Figure 10A). Coassociation of ATR and Chk1 could be seen, however, throughout the nucleus. Immunoprecipitation analysis revealed that, surprisingly, a significant increase in ATR and Chk1 association could be observed upon ARF induction (Figure 10B). Since Chk1 is a substrate of ATR, it is known that these proteins interact. But such a stable association is typically not seen following DNA damage. It is possible that the interaction seen here derives from the continuous expression of ARF protein. This result indicates that ARF may function, directly or indirectly, by facilitating the interaction of ATR and Chk1, resulting in Chk1 phosphorylation (Figure 7) and activation of Chk1 kinase activity.
Figure 10.

Analysis of Chk1 localisation and ATR association after ARF induction. (A) ARF does not recruit Chk1 to nucleoli. NARF2 cells were fixed 24 h after ARF induction and stained with goat anti-ATR, rabbit anti-ARF and sheep anti-Chk1. Enlarged sections from merged Chk1/ATR images are shown. (B) ATR coimmunoprecipitates with Chk1 following ARF induction. ATR was immunoprecipitated from 150 μg of whole-cell lysate prepared from NARF2 cells with or without ARF induction. The immunoprecipitated complex was resolved by SDS–PAGE and immunoblotted with anti-Chk1 and anti-ATR antibodies.
Serine 15 phosphorylated p53 is selectively located at nucleoli following ARF induction
Since p53 is an ATR substrate, we also investigated its subnuclear localisation. No effect of ARF on total distribution of p53 was observed, apart from an increase in overall protein level, (Figure 11A). But when an anti-phospho-serine 15 p53 monoclonal antibody was used, a different result was seen. Following UV-C treatment, a general increase in serine 15-phosphorylated p53 was observed, distributed evenly throughout the nucleus (Figure 11B). However, following ARF induction, serine 15-phosphorylated p53 was not only distributed throughout the nucleoplasm, but was also specifically associated with nucleoli (Figure 11C; Supplementary Figure 5C). This result suggests that ATR/BRCA1 nucleolar complexes represent active sites of p53 serine 15 phosphorylation in cells where ARF is induced. A nucleolar function for p53 is possible since a role inhibiting pol I-dependent gene expression has been previously described (Budde and Grummt, 1999; Zhai and Comai, 2000). Inhibiting rRNA synthesis would have the effect of reducing cell growth and proliferation, consistent with the tumour suppressor properties of ARF and p53. However, further experiments will be required to confirm how widespread the observation of nucleolar serine 15-phosphorylated p53 is, and also its functional significance.
Figure 11.

Serine 15-phosphorylated p53 localises to ARF and ATR-containing nucleolar complexes. (A) Immunofluorescence analysis of p53 following ARF induction. NARF2 cells were stained for p53 with mouse anti-p53 DO1 antibody. (B) Imaging of serine 15-phosphorylated p53 following UV-C treatment. Immunofluorescence analysis of serine 15-phosphorylated p53 in U-2 OS cells 4 h after treatment with UV-C (40 J/m2). Cells were stained with mouse 16G8 monoclonal anti-phospho serine 15 p53 antibody. (C) Serine 15-phosphorylated p53 associates with ARF and ATR in the nucleolus. Immunofluorescence analysis of ARF, ATR and serine 15-phosphorylated p53 following induction of ARF in NARF2 cells. Cells were stained with rabbit anti-p14ARF, goat anti-ATR and mouse 16G8 monoclonal anti-phospho-serine 15 p53 antibodies. Additional images can be seen in Supplementary data.
Discussion
In this report, we demonstrate that ARF activates the ATR/Chk1 pathway and that this is required for ARF's ability not only to regulate RelA but also to function as an inducer of p53 (Figure 12). We propose that engagement of ATR/Chk1 represents a fundamental requirement for ARF to efficiently fulfil its function as tumour suppressor. There appear to be two distinct mechanisms through which ARF regulates ATR (Figure 12). The most visually dramatic of these occurs at the nucleoli, where we see recruitment of ATR and BRCA1 together with an increased concentration of serine 15-phosphorylated p53. But since Chk1 does not localise to the nucleoli and is required for phosphorylation and repression of RelA, we propose that there are distinct nucleoplasmic effects of ARF. In support of this, we observe colocalisation of ATR and Chk1 throughout the nucleus and ARF-induced immunoprecipitation of an ATR/Chk1 complex. Furthermore, we do not see RelA localising to nucleoli following ARF induction with either generic RelA or phospho-Thr505 RelA antibodies (Figure 3F; data not shown).
Figure 12.
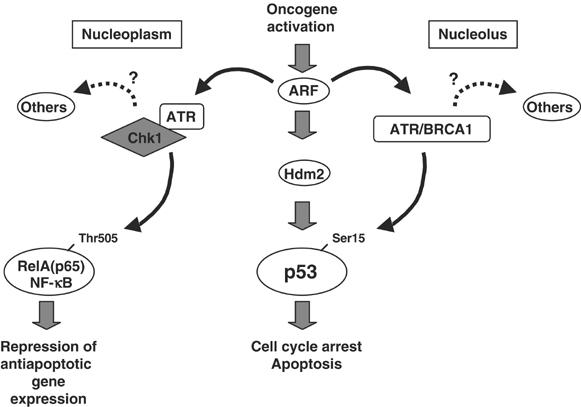
Schematic model depicting engagement of the ATR/Chk1 pathway by ARF. Data presented in this report demonstrate that ARF engages ATR through two pathways distinct to those following DNA damage. In the nucleoplasm, Chk1 is activated by ATR and is required for Thr505 phosphorylation of the RelA NF-κB subunit. This inhibits RelA transactivation resulting in decreased Bcl-xL levels, sensitising cells to apoptotic stimuli. At the nucleoli, ARF induces the assembly of an ATR/BRCA1 complex that appears to be required for activation of p53 at serine 15. It is probable that nucleoplasmic ATR/BRCA1 will also contribute to p53 activation. In addition, both pathways are likely to regulate many other cellular processes and make a wider contribution to ARF-mediated tumour suppression.
Although ARF often has the appearance of being an exclusively nucleolar protein, other studies have shown that it can execute its effects on p53 while being exclusively nucleoplasmic (Llanos et al, 2001). Furthermore, it has been proposed that a significant quantity of ARF is naturally nucleoplasmic but that it is rapidly turned over due to proteolysis (Llanos et al, 2001; Rodway et al, 2004). Therefore, the nucleolar and nucleoplasmic effects that we see are entirely consistent with previous reports of ARF function. It is possible that there is some functional redundancy between the effects of ARF on ATR in the nucleoplasm and nucleoli. This might, in part, explain why there exists some disagreement as to the primary site of ARF function within the nucleus.
Although there are some similarities, such as the induction of p53, phosphorylation of Chk1 and the requirement for BRCA1, there are also key differences between ARF activation and DNA-damage-mediated activation of the ATR/Chk1 pathway. Firstly, recruitment of ATR to the nucleolus is something not previously described for this protein. Furthermore, the pattern of ARF-induced ATR substrate phosphorylation appears distinct to that seen following UV stimulation (Figure 6A). Although this represents only a single time point and further investigation of this observation is required to confirm the significance of this effect, this result suggests that ARF-activated ATR might associate with scaffold proteins distinct to those following UV-C stimulation. These differences are also reflected by the observation that, while ATR is required for ARF-mediated repression of RelA function and Bcl-xL expression (Figure 1), it is not required for repression of these same targets following UV-C stimulation (Figure 1B; Campbell et al, 2004; data not shown). We also see phosphorylation of RelA at Thr505 following ARF induction but not after UV-C treatment, suggesting that ARF-induced or -regulated adaptor proteins are also required to target Chk1 to specific substrates under these conditions (Figure 3B). Consistent with these findings, we see no induction of γ-H2AX phosphorylation or localisation of RPA to the sites of ATR complex formation at the nucleoli (Supplementary Figure 5A and B), both of which would be expected if ARF-induced ATR activation was similar to that seen following DNA damage. Taken together, our data indicate that ARF activation of ATR and Chk1 is mechanistically and functionally distinct to that seen following DNA damage.
This pathway also provides a further mechanism to co-ordinately regulate NF-κB and p53 function. A number of pathways describing crosstalk between NF-κB and p53 have been described (discussed in Perkins, 2004). These include descriptions of RelA being required for induction of p53 (Ryan et al, 2000; Fujioka et al, 2004), as well as inhibiting its activity (Webster and Perkins, 1999; Tergaonkar et al, 2002). We have, however, previously established that the effects of ARF on RelA are p53 and Hdm2/Mdm2 independent. Whether ARF-regulated NF-κB plays a role in p53 function is not something we have pursued at this time. However, given the previous role describing the NF-κB pathway as regulating both p53 and Hdm2 expression (Tergaonkar et al, 2002; Fujioka et al, 2004), such effects cannot be ruled out.
From our results, we conclude that an ability to engage the ATR checkpoint pathway will prove to be an important component of ARF's tumour suppressor activity. We envisage that this pathway provides a mechanism through which ARF can activate and reprogramme many cellular functions in order to fully counter the effects of oncogenes. By engaging ATR, ARF ensures that p53 is appropriately activated. This pathway also ensures that the antiapoptotic effects of RelA are neutralised and even co-opted into an antitumour role (Perkins, 2004). It is probable that this pathway will target many other proteins. These could include Hdm2, a known ATR/ATM substrate (Khosravi et al, 1999; de Toledo et al, 2000; Maya et al, 2001; Shinozaki et al, 2003), and other transcription factors or proteins with proliferative or antiapoptotic functions. In addition, the activity of this pathway in tumour cells will be of great interest. Since there is great selective pressure on developing tumour cells to inactivate the link between ARF and p53, it is possible that the ability of ARF to engage ATR and Chk1 will also be targeted during tumorigenesis in many cell types. For example, mutations in BRCA1, which we have shown to play a role in this pathway, might disrupt ARF function and contribute to BRCA1-induced tumours.
Materials and methods
Cells
NARF2 and NARF2-E6 and Hs68 E2F1–ER cell lines were provided by Dr Gordon Peters (CR-UK London Research Institute) and have been described previously (Stott et al, 1998; Brookes et al, 2002; Rocha et al, 2003). Hs68 E2F1–ER human neonatal foreskin fibroblast cells contain a tamoxifen-regulated E2F1–ER fusion protein (Brookes et al, 2002). ARF expression was induced in NARF2 and NARF2-E6 cells by the inclusion of 1 mM IPTG and, except where indicated, was for 24 h. Hs68 E2F1–ER cells were induced by 0.1 μM Tamoxifen (Sigma).
Phospho-specific antibody production
The RelA Thr505 phospho-specific antibody was raised in rabbits by Moravian Biotechnology. The peptide used had the sequence EAITRLVpTGAQRPPD (phospo-Thr505 is indicated). The antibody was purified using peptide affinity chromatography with the phospho- and non-phospho-peptides. ELISA analysis confirmed that the purified antibody was specific for the phosphorylated epitope.
Chk1 in vitro kinase assay
In all, 1 μg of purified GST, GST-RelA(TAD) (amino acids 428–551) and GST-RelA(TAD T505A) protein were incubated for 30 min at 30°C, with recombinant Chk1 kinase (5 U/ml final concentration) in kinase buffer (50 mM Hepes/KOH, pH 7.4, 10 mM MgCl2, 2% glycerol, 0.1% NP40, 0.1 M NaCl final concentration) in the presence of 50 μM ATP. Reactions were stopped by addition of SDS loading buffer and proteins were then resolved by SDS–PAGE before Western blotting. Purified, recombinant GST-Chk1 protein was provided by Dr John Rouse (University of Dundee).
Oligonucleotides, siRNAs and antibodies
Oligonucleotides and siRNAs not described previously (Rocha et al, 2003; Campbell et al, 2004) as well as antibodies are listed in Supplementary Material. siRNA validation and controls are shown in Supplementary Figure 1.
Microscopy
For immunofluorescence, cells grown on coverslips were fixed after washing once in PBS by incubation in 3.7% formaldehyde/PBS (pH 6.8) for 15 min. Cells were permeabilised in PBS–0.1% Triton X-100 for 15 min and then blocked in PBS–0.05% Tween supplemented with 1% normal donkey serum for 30 min. The dilutions of the antibodies used were: anti-p53 monoclonal antibody, 1:500; mouse anti-phospho-serine 15 p53, 1:1000; mouse anti-BRCA1, 1:50; mouse anti-RPA, 1:50; rabbit and mouse anti-p14ARF, 1:100; sheep anti-Chk1, 1:100 mouse monoclonal anti-Chk1, 1:200; rabbit anti-claspin, 1:500; goat anti-ATR antibody, 1:1000; rabbit anti-γ-H2AX (phospho-ser 139), 1:1000; rabbit anti-P-T505 RelA antibody, 1:50. All secondary antibodies (labelled with either FITC, TRITC, X-Red or Cy5) were purchased from Jackson Immunoresearch and used at 1:500 dilution. For anti-P-T505 RelA experiment, the antibody was used with 1 μM microcystin phosphatase inhibitor and 10 μg/ml non-phospho-peptide. For all antibodies used in this study, appropriate controls were performed. Cells were either stained with primary antibodies without secondary antibodies to control for auto-fluorescence or stained with secondary antibodies alone to control for background staining. Cells were analysed and images were acquired using a DeltaVision microscope. Images were deconvoluted using SoftWorx (Applied Precision).
Other assays
Proliferation assays, cell death assays, transient transfections, luciferase assays, siRNA knockdown, RNA extraction, semiquantitative RT–PCR, protein extracts and Western blots were all performed as described previously (Rocha et al, 2003; Campbell et al, 2004; Roche et al, 2004). All transfections were performed a minimum of three times before calculating means and standard deviations as shown in figures and contained appropriate levels of RSV or CMV control plasmid such that each dish received the same amount of DNA.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Material
Acknowledgments
We would like to thank all the members of the NDP laboratory and the Division of Gene Regulation and Expression at the University of Dundee for their help and assistance, particularly Jason Swedlow for help with microscopy. We also thank Gordon Peters (CR-UK London Research Institute) for providing reagents and advice and Paul Harkin (Queen's University, Belfast) for providing the BRCA1 siRNA sequence. We thank Julian Blow and John Rouse for reading this manuscript and their helpful comments. NDP is funded by a Royal Society University Fellowship, SR is funded by a grant from Cancer Research UK, and KJC and KS are funded by Wellcome Trust 4 year PhD studentships. MDG is funded by the Institute of Cancer Research and Cancer Research UK (C309/A2187).
References
- Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–429 [DOI] [PubMed] [Google Scholar]
- Bertwistle D, Sugimoto M, Sherr CJ (2004) Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol 24: 985–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes S, Rowe J, Ruas M, Llanos S, Clark PA, Lomax M, James MC, Vatcheva R, Bates S, Vousden KH, Parry D, Gruis N, Smit N, Bergman W, Peters G (2002) INK4a-deficient human diploid fibroblasts are resistant to RAS-induced senescence. EMBO J 21: 2936–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde A, Grummt I (1999) p53 represses ribosomal gene transcription. Oncogene 18: 1119–1124 [DOI] [PubMed] [Google Scholar]
- Campbell KJ, Rocha S, Perkins ND (2004) Active repression of antiapoptotic gene expression by ReIA(p65) NF-κB. Mol Cell 13: 853–865 [DOI] [PubMed] [Google Scholar]
- de Toledo SM, Azzam EI, Dahlberg WK, Gooding TB, Little JB (2000) ATM complexes with HDM2 and promotes its rapid phosphorylation in a p53-independent manner in normal and tumor human cells exposed to ionizing radiation. Oncogene 19: 6185–6193 [DOI] [PubMed] [Google Scholar]
- Dumaz N, Meek DW (1999) Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J 18: 7002–7010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL (1999) Disruption of the ARF–Mdm2–p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev 13: 2658–2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M (2002) CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene 21: 5427–5440 [DOI] [PubMed] [Google Scholar]
- Eymin B, Karayan L, Seite P, Brambilla C, Brambilla E, Larsen CJ, Gazzeri S (2001) Human ARF binds E2F1 and inhibits its transcriptional activity. Oncogene 20: 1033–1041 [DOI] [PubMed] [Google Scholar]
- Fatyol K, Szalay AA (2001) The p14(ARF) tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1 α (HIF-1 α) and inhibits HIF-1-mediated transcription. J Biol Chem 276: 28421–28429 [DOI] [PubMed] [Google Scholar]
- Foray N, Marot D, Gabriel A, Randrianarison V, Carr AM, Perricaudet M, Ashworth A, Jeggo P (2003) A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein. EMBO J 22: 2860–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka S, Schmidt C, Sclabas GM, Li ZK, Pelicano H, Peng B, Yao A, Niu JG, Zhang W, Evans DB, Abbruzzese JL, Huang P, Chiao PJ (2004) Stabilization of p53 is a novel mechanism for proapoptotic function of NF-κB. J Biol Chem 279: 27549–27559 [DOI] [PubMed] [Google Scholar]
- Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT, Nussenzweig A, Aladjem MI, Bonner WM, Pommier Y (2003) Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem 278: 20303–20312 [DOI] [PubMed] [Google Scholar]
- Hammond EM, Dorie MJ, Giaccia AJ (2003) ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J Biol Chem 278: 12207–12213 [DOI] [PubMed] [Google Scholar]
- Hutchins JRA, Hughes M, Clarke PR (2000) Substrate specificity determinants of the checkpoint protein kinase Chk1. FEBS Lett 466: 91–95 [DOI] [PubMed] [Google Scholar]
- Ishimi Y, Komamura-Kohno Y, Kwon HJ, Yamada K, Nakanishi M (2003) Identification of MCM4 as a target of the DNA replication block checkpoint system. J Biol Chem 278: 24644–24650 [DOI] [PubMed] [Google Scholar]
- Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, Shkedy D (1999) Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc Natl Acad Sci USA 96: 14973–14977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn EA, Yoo CJ, Eastman A (2003) The protein kinase c inhibitor Go6976 is a potent inhibitor of DNA damage-induced S and G(2) cell cycle checkpoints. Cancer Res 63: 31–35 [PubMed] [Google Scholar]
- Kuo M, Duncavage EJ, Mathew R, den Besten W, Pei D, Naeve D, Yamamoto T, Cheng C, Sherr CJ, Roussel MF (2003) Arf induces p53-dependent and -independent antiproliferative genes. Cancer Res 63: 1046–1053 [PubMed] [Google Scholar]
- Lai JS, Herr W (1992) Ethidium-bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc Natl Acad Sci USA 89: 6958–6962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wu D, Chen B, Ingram A, He L, Liu L, Zhu D, Kapoor A, Tang D (2004) ATM activity contributes to the tumor-suppressing functions of p14(ARF). Oncogene 23: 7355–7365 [DOI] [PubMed] [Google Scholar]
- Lindstrom MS, Wiman KG (2003) Myc and E2F1 induce p53 through p14ARF-independent mechanisms in human fibroblasts. Oncogene 22: 4993–5005 [DOI] [PubMed] [Google Scholar]
- Llanos S, Clark PA, Rowe J, Peters G (2001) Stabilization of p53 by p14(ARF) without relocation of MDM2 to the nucleolus. Nat Cell Biol 3: 445–452 [DOI] [PubMed] [Google Scholar]
- Lowe SW, Sherr CJ (2003) Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev 13: 77–83 [DOI] [PubMed] [Google Scholar]
- Mason SL, Loughran O, La Thangue NB (2002) p14(ARF) regulates E2F activity. Oncogene 21: 4220–4230 [DOI] [PubMed] [Google Scholar]
- Maya R, Balass M, Kim ST, Shkedy D, Leal JFM, Shifman O, Moas M, Buschmann T, Ronai Z, Shiloh Y, Kastan MB, Katzir E, Oren M (2001) ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev 15: 1067–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael D, Oren M (2002) The p53 and Mdm2 families in cancer. Curr Opin Genet Dev 12: 53–59 [DOI] [PubMed] [Google Scholar]
- Perkins ND (2004) NF-κB: tumor promoter or suppressor? Trends Cell Biol 14: 64–69 [DOI] [PubMed] [Google Scholar]
- Rocha S, Campbell KJ, Perkins ND (2003) p53- and Mdm2-independent repression of NF-κB transactivation by the ARF tumor suppressor. Mol Cell 12: 15–25 [DOI] [PubMed] [Google Scholar]
- Roche KC, Wiechens N, Hughes TO, Perkins ND (2004) The FHA domain protein SNIP1 is a regulator of the cell cycle and cyclin D1 expression. Oncogene 23: 8185–8195 [DOI] [PubMed] [Google Scholar]
- Rodway H, Llanos S, Rowe J, Peters G (2004) Stability of nucleolar vs non-nucleolar forms of human p14(ARF). Oncogene 23: 6186–6192 [DOI] [PubMed] [Google Scholar]
- Ryan KM, Ernst MK, Rice NR, Vousden KH (2000) Role of NF-κB in p53-mediated programmed cell death. Nature 404: 892–897 [DOI] [PubMed] [Google Scholar]
- Sherr CJ (2001) The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol 2: 731–737 [DOI] [PubMed] [Google Scholar]
- Sherr CJ, McCormick F (2002) The RB and p53 pathways in cancer. Cancer Cell 2: 103–112 [DOI] [PubMed] [Google Scholar]
- Shinozaki T, Nota A, Taya Y, Okamoto K (2003) Functional role of Mdm2 phosphorylation by ATR in attenuation of p53 nuclear export. Oncogene 22: 8870–8880 [DOI] [PubMed] [Google Scholar]
- Somasundaram K, MacLachlan TK, Burns TF, Sgagias M, Cowan KH, Weber BL, El-Deiry WS (1999) BRCA1 signals ARF-dependent stabilization and coactivation of p53. Oncogene 18: 6605–6614 [DOI] [PubMed] [Google Scholar]
- Stojic L, Mojas N, Cejka P, di Pietro M, Ferrari S, Marra G, Jiricny J (2004) Mismatch repair-dependent G(2) checkpoint induced by low doses of S(N)1 type methylating agents requires the ATR kinase. Genes Dev 18: 1331–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, Palmero I, Ryan K, Hara E, Vousden KH, Peters G (1998) The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J 17: 5001–5014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M, Kou M, Roussel MF, Sherr CJ (2003) Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol Cell 11: 415–424 [DOI] [PubMed] [Google Scholar]
- Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I (2002) p53 stabilization is decreased upon NFκB activation: a role for NFκB in acquisition of resistance to chemotherapy. Cancer Cell 1: 493–503 [DOI] [PubMed] [Google Scholar]
- Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston D, Elledge SJ, Abraham RT (2000) Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev 14: 2989–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH (2002) Activation of the p53 tumor suppressor protein. BBBA Rev Cancer 1602: 47–59 [DOI] [PubMed] [Google Scholar]
- Ward IM, Chen JJ (2001) Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 276: 47759–47762 [DOI] [PubMed] [Google Scholar]
- Ward IM, Minn K, Chen JJ (2004) UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J Biol Chem 279: 9677–9680 [DOI] [PubMed] [Google Scholar]
- Webster GA, Perkins ND (1999) Transcriptional cross talk between NF-κB and p53. Mol Cell Biol 19: 3485–3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC (2002) BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet 30: 285–289 [DOI] [PubMed] [Google Scholar]
- Zhai WG, Comai L (2000) Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol Cell Biol 20: 5930–5938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Material