

Abstract
The ability to translocate virulence proteins into host cells through a type III secretion apparatus (TTSS) is a hallmark of several Gram-negative pathogens including Shigella, Salmonella, Yersinia, Pseudomonas, and enteropathogenic Escherichia coli. In common with other types of bacterial secretion apparatus, the assembly of the TTSS complex requires the preceding formation of its integral outer membrane secretin ring component. We have determined at 1.5 Å the structure of MxiM28–142, the Shigella pilot protein that is essential for the assembly and membrane association of the Shigella secretin, MxiD. This represents the first atomic structure of a secretin pilot protein from the several bacterial secretion systems containing an orthologous secretin component. A deep hydrophobic cavity is observed in the novel ‘cracked barrel' structure of MxiM, providing a specific binding domain for the acyl chains of bacterial lipids, a proposal that is supported by our various lipid/MxiM complex structures. Isothermal titration analysis shows that the C-terminal domain of the secretin, MxiD525–570, hinders lipid binding to MxiM.
Keywords: lipid binding, secretin pilot, TTSS, type III secretion, X-ray crystallography
Introduction
Shigella flexneri invades colonic epithelial cells and causes bacillary dysentery, a severe form of gastroenteritis, which leads to over 1 million deaths per year worldwide (Philpott et al, 2000). This cell invasion process requires a functional type III secretion system (TTSS), encoded by the mxi and spa genes in the Shigella virulence plasmid (Parsot et al, 1995). The secretion and subsequent translocation of proteins (known as effectors) into the cytoplasm of host cells by the TTSS is a process essential for virulence of Shigella as well as many other Gram-negative bacterial pathogens of mammals and plants. While much is known about the type and function of virulence effector proteins delivered to host cells by the TTSS, the detailed molecular mechanisms of secretion and translocation remain poorly understood (Tampakaki et al, 2004).
Protein secretion in general is required for different aspects of the bacterial life cycle including organelle biogenesis, nutrient acquisition, as well as delivery of virulence effectors into the host cell (Thanassi and Hultgren, 2000). The latter is a particularly challenging task for Gram-negative bacteria because secreted proteins must cross three barriers: the inner or cytoplasmic membrane, the periplasmic space, and the outer membrane (OM). Multiple protein secretory pathways have evolved in these microorganisms to carry out this difficult yet important process. These pathways are found to differ not only in the means by which the secreted proteins reach the cell surface, but also in the types and organization of the structural components involved in the secretion process. In spite of this diversity, the secretin superfamily of proteins appears to be the one, if not the only, structural component shared among several apparently unrelated protein secretion pathways: TTSS, the type II secretion system, and the type IV pilus biogenesis pathway (Thanassi and Hultgren, 2000).
Secretins are integral membrane proteins that are synthesized in the bacterial cytoplasm and exported to the periplasm by the sec-dependent pathway. They are found to associate into large and highly stable oligomers of 12–14 subunits in the bacterial OM (Linderoth et al, 1997; Burghout et al, 2004; Collins et al, 2004). A proposed role as an OM protein-conducting channel has been supported by electron microscopy (EM) studies of PulD (Nouwen et al, 1999) and several other representative secretins (Koster et al, 1997; Crago and Koronakis, 1998; Tamano et al, 2000; Blocker et al, 2001), which showed that they adopt a ring-like structure with a central pore ranging from 2 to 10 nm in diameter, wide enough to accommodate completely unfolded or partially folded proteins (Koster et al, 1997; Nouwen et al, 2000; Blocker et al, 2001).
Construction of the various bacterial multiprotein secretory complexes is believed to occur in a highly regulated manner, and accessory proteins are often present to ensure specific structural components are correctly localized and adopt their functional conformation. In particular, a specialized class of proteins known as ‘pilots' mediate the assembly of the homo-oligomeric secretin ring. Previous studies have demonstrated that these pilot proteins bind and directly affect the stability, localization, and/or multimerization states of their associated secretins (Drake et al, 1997; Crago and Koronakis, 1998; Daefler and Russel, 1998; Shevchik and Condemine, 1998; Schuch and Maurelli, 1999; Burghout et al, 2004). EM studies of the type II secretion apparatus, PulS-PulD, indicate that the membrane-embedded PulD secretin complex is flanked by protein spokes (Nouwen et al, 1999). These radial spokes have been attributed to the secretin pilot protein, PulS, implying a key structural role in anchoring PulD to the OM. To date, PulS of Klebsiella oxytoca, OutS of Erwinia chrysanthemi, PilP of Pseudomonas aeruginosa, InvH of Salmonella, YscW of Yersinia, and MxiM of Shigella have been identified as pilots for the secretins PulD, OutD, PilQ, InvG, YscC, and MxiD, respectively. Although these pilots are all relatively small (∼15 kDa) and are all predicted to be covalently attached to a lipid moiety at their N-termini, very little sequence identity is observed (typically in the range of 17–19%). This is perhaps not surprising given that pilots typically bind to the C-terminal tail of their associated secretin, the most sequence-divergent region among the otherwise well-conserved family (Shevchik et al, 1997; Daefler and Russel, 1998; Schuch and Maurelli, 2001).
The pilot protein MxiM from the Shigella TTSS is a 142-residue lipoprotein, which binds and affects several features of the secretin MxiD, including its stability in the periplasm, OM association, as well as assembly into multimeric structures (Schuch and Maurelli, 2001). A deficiency in MxiM results in a complete loss of TTSS function and virulence, presumably due to the lack of and/or improper assembly of the key secretin component MxiD (Schuch et al, 1999). Previous lipid labeling and fractionation studies have indicated that MxiM is anchored to the inner leaflet of the OM via its N-terminal lipid moiety (Schuch and Maurelli, 1999). Its peripheral membrane localization suggests that MxiM assists in secretin ring formation by anchoring the MxiD to the OM.
In this work, we present the crystal structure of MxiM from S. flexneri, the first high-resolution structural data for a secretin pilot protein from any system. The structure reveals a pseudo-β-barrel fold that presents a narrow and deep hydrophobic cavity for the binding of specific lipids. Our isothermal titration calorimetry (ITC) experiments show that MxiM binds lipids and its cognate secretin MxiD. Collectively, our findings provide a foundation for understanding the structural and functional roles of secretin pilots in assembly of the secretin complex, an essential component in the secretion apparatus from various protein secretion pathways.
Results and discussion
Purification, biochemical characterization, and crystallization of MxiM28–142
To provide sufficient quantities of soluble MxiM necessary for crystallization, a recombinant form excluding the N-terminal signal sequence (residues 1–23) but including a cleavable N-terminal hexahistidine tag was used. Mass spectrometry analysis of the purified protein after thrombin-mediated cleavage of the histidine tag reported a mass of 12 888.0 Da, indicating that further processing had occurred resulting in the N-terminal deletion mutant MxiM28–142 (calculated molecular mass of 12 886.9 Da).
MxiM and other secretin pilots encode at their N-termini the classic LXGC sequence motif characteristic of the lipidation site of many bacterial lipoproteins (covalently bound to the cysteine residue) (Wu and Tokunaga, 1986). Biochemical studies have shown that MxiM is both lipid modified at Cys24 of the LXGC motif and is membrane associated (Allaoui et al, 1992; Schuch and Maurelli, 1999). Importantly for this study, binding and assembly of the secretin/pilot complex occurs even in the absence of the lipid anchor (Schuch and Maurelli, 2001). While the lipidation of MxiM has been shown to be important for specifically targeting MxiM to the OM as well as stability to heat denaturation of the membrane-embedded pilot/secretin complex (Schuch and Maurelli, 1999), studies with an unlipidated G23R mutant show that this periplasmic form also associates with MxiD oligomers although the resulting complex is less heat resistant than those formed with lipidated MxiM (Schuch and Maurelli, 2001). Recently, studies of the Yersinia secretin pilot, YscW, have shown that a putatively unlipidated construct (YscWΔlip) putatively still binds to YscC (as evidenced by altered expression levels in coexpression systems with YscW) but apparently fails to promote YscC secretin oligomer formation and localization to the OM (Burghout et al, 2004). While these apparently contrasting results may suggest fundamental differences in pilot function between the two species, we note that in the Yersinia study, no evidence was provided for the translocation of the unlipidated YscW construct to the periplasm (which would obviously be a critical step for subsequent YscC oligomerization and OM localization).
Static light scattering experiments in the presence of 0.5 mM dodecylmaltoside (DDM) show that MxiM28–142 has a measured mass of 15 kDa, indicating that the protein is predominately monomeric in solution. These results are consistent with size exclusion chromatography experiments where the elution position relative to molecular weight standards on a superdex 75 column (Amersham) also suggests a monomeric state.
MxiM could only be crystallized in the presence of maltoside-based detergents or 1-monohexanoyl-2-hydroxy-sn-glycero-3-phosphate as described in Materials and methods. The crystals are monoclinic (C2) with a monomer of MxiM observed in the asymmetric unit also consistent with our static light scattering and size exclusion results.
Unique fold of Shigella MxiM
We have determined the crystal structure of the MxiM secretin pilot from S. flexneria to 1.5 Å. The structure was solved using the method of multiwavelength anomalous diffraction (MAD) with a single crystal of selenomethionine (Se-Met)-labeled protein. Data collection and refinement statistics are presented in Table I.
Table 1.
Data collection and refinement statistics
| Se MAD |
Native | +Lipida | |||
|---|---|---|---|---|---|
| Data set | Peak | Remote | Inflection | ||
| Data collection | |||||
| Wavelength (Å) | 0.9794 | 0.8981 | 0.9796 | 1.00 | 1.5418 |
| Resolution range (Å)b | 20–1.7 (1.76–1.70) | 20–1.7 (1.76–1.70) | 20–1.7 (1.76–1.70) | 20–1.50 (1.53–1.50) | 42–1.87 (1.94–1.87) |
| Total observations | 46 694 | 46 586 | 47 370 | 73 722 | 75 315 |
| Unique reflections | 23 037 | 23 083 | 23 220 | 18 209 | 9566 |
| Completeness (%)b | 90.0 (65.6) | 90.3 (65.7) | 90.3 (62.8) | 96.5 (100.0) | 97.6 (94.4) |
| Rmerge (%)b,c | 4.8 (20.9) | 5.0 (24.3) | 4.8 (22.2) | 4.4 (42.1) | 6.4 (32.7) |
| 〈I/σI〉 | 24.1 (3.5) | 20.7 (2.9) | 25.7(3.5) | 16.5 (3.0) | 21.5 (4.4) |
| Refinement | |||||
| Resolution range (Å)b | 20–1.5 (1.52–1.50) | 42–1.87 (1.89–1.87) | |||
| Rworkb,d | 22.0 (25) | 20.6 (26) | |||
| Rfreeb,e | 25.1 (32) | 24.9 (31) | |||
| Average B-factor (Å2) | |||||
| Main chain | 23.0 | 31.9 | |||
| Side chain | 25.2 | 33.7 | |||
| R.m.s. deviation | |||||
| Bond lengths (Å) | 0.013 | 0.0014 | |||
| Angles (deg) | 1.5 | 1.5 | |||
| aAnionic lipid, 1-hexanoyl-2-hydroxy-sn-glycerol-3-phosphate. | |||||
| bValues in parentheses correspond to the highest resolution shell. | |||||
| cRmerge=∑∣(Ihkl)−〈I〉∣/∑(Ihkl), where Ihkl is the integrated intensity of a given reflection. | |||||
| dRwork=(∑∣Fo−Fc∣)/(∑Fo), where Fo and Fc are observed and calculated structure factors. | |||||
| eA total of 5% of reflections were excluded from the refinement to calculate Rfree. | |||||
MxiM is a conical-shaped structure with average dimensions of 40 × 30 × 30 Å (Figure 1A). The 10 β-strands present in the structure curve to form a β-sheet (Figure 1B). This curved β-sheet resembles a β-barrel structure except that the barrel is discontinuous due to the presence of the α-helix, F1. In addition, this pseudo-barrel structure is closed at one end in part due to the presence of a disulfide bridge between loops L2 and L4 that tethers the N-terminus of helix F1 to the loop L2 (Figure 1). The fold we observe for MxiM is considered unique based on a DALI search of the existing structural database (Holm and Sander, 1995).
Figure 1.
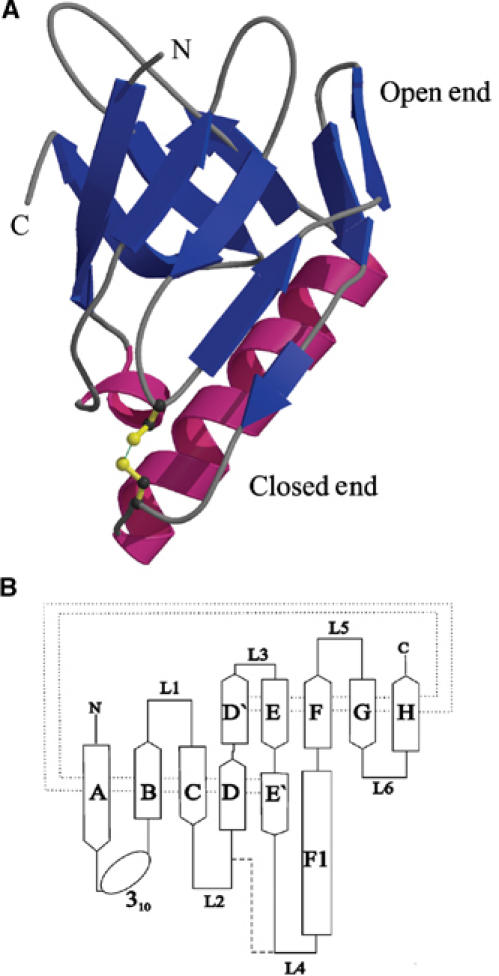
Structure of MxiM. (A) A ribbon representation of the protein with the β-structure shown in blue and the helical domains shown in magenta. (B) The fold topology of MxiM is shown where the β-strands of the antiparallel β-sheet are shown as arrows and labeled A–H. The α-helix is shown as a rectangle and labeled F1. The dotted line represents the hydrogen bonding interactions between the β-strands and the dashed line represents the single disulfide bond in the structure.
It is difficult to predict if pilots from other species or from other systems will have a similar fold as that observed for MxiM. The lack of sequence identity among secretin pilots was found to extend to a lack of similarity in predicted secondary structure. A number of algorithms were utilized, including Jpred (Cuff et al, 1998), PredictProtein (Rost and Liu, 2003), and nnPredict (Zhang and Zhang, 2000), to analyze the sequences of the known secretin pilots, resulting in a wide variety of predicted secondary structures. MxiM and the type III secretin pilot YscW from Yersinia (Burghout et al, 2004) are predicted to be composed of primarily β-structure in accordance with our structural observations, while sequences of other pilots generate a variety of secondary structure predictions. There is clearly significant precedent for proteins with similar function to adopt strikingly similar structural folds despite very low or undetectable sequence identity and despite misleading secondary structure prediction profiles (the type III secretion chaperones are a case in point; Luo et al, 2001; Birtalan et al, 2002). Structural characterization of functional orthologs of MxiM will enable us to understand fully the similarities among this sequence diverse family of proteins.
Hydrophobic ligand-binding cavity and membrane association
The ‘cracked barrel' motif of MxiM creates a dramatic cleft in the center of the protein, ∼8 Å wide and 20 Å deep (Figure 2A and B). The cavity is entirely hydrophobic in nature and is notably rich in aromatic groups nearer the surface of the protein. Aromatic groups are commonly observed at the protein/membrane interface (Saez-Cirion et al, 2003; Gambhir et al, 2004) and suggest a possible role of membrane association for these conserved amino acids at the MxiM surface. Other observations that suggest that the hydrophobic cavity of MxiM is likely in close proximity to the OM include the observed charge localization surrounding this area of the protein. Although the overall electrostatic surface of the MixM structure is for the most part evenly distributed with positive and negative charges, there is one exception in a concentration of positively charged surfaces (involving residues K34, K82, K121, K118, and R140) observed near the mouth of the hydrophobic cavity (Figure 2C). These basic amino acids could plausibly interact with negatively charged lipid phosphate groups present on the inner leaflet of the OM.
Figure 2.
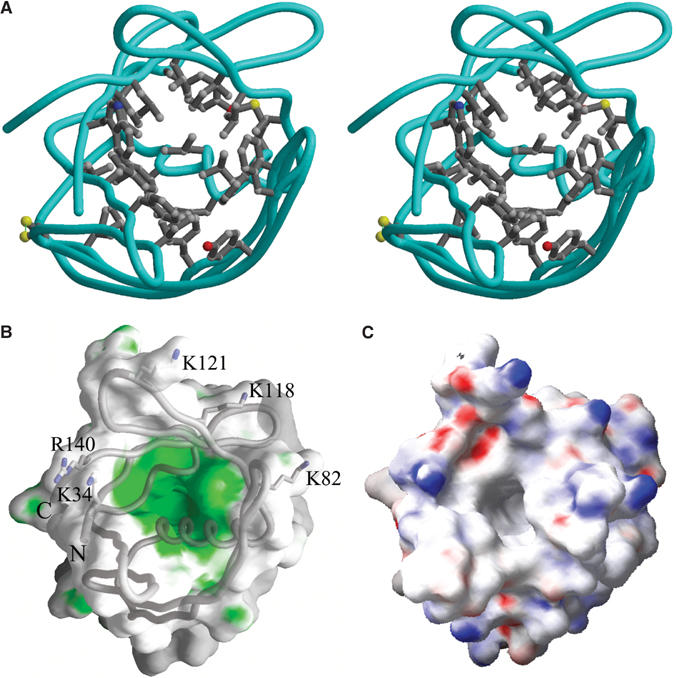
Hydrophobic cavity of MxiM. (A) Stereoscopic representation showing the side chains of the residues that line the hydrophobic cavity. Atoms are colored according to type: carbon atoms are shown in gray, nitrogen atoms are colored blue, oxygen atoms are colored red, and sulfur atoms are colored yellow. The protein backbone trace is represented by the cyan colored worm. (B) GRASP (Nicholls et al, 1991) surface representation of MxiM. Polar and hydrophobic areas are in gray and green, respectively. (C) The electrostatic surface of MxiM is mapped on to the molecular surface as calculated using the program GRASP. The basic residues at the lip of the pore are labeled. Positive and negatively charged areas are blue and red, respectively. All three panels are shown with a similar view.
Given the dimensions and nature of the hydrophobic pore, its functional role could serve to bind a single fatty acid tail of a membrane phospholipid. Within the putative lipid-binding cavity of MxiM, we observe additional Fo−Fc difference density at 3σ that is not accounted for after the amino-acid sequence had been traced in the electron density maps. Contouring the electron density map at a slightly lower cutoff (2.5σ) indicated that this density was continuous (Figure 3A) and can be accommodated by the lipid tail of the DDM detergent required for ordered crystal formation of MxiM. When the lipid tail of DDM (11 carbon acyl chain) was included in the structure, both the R and Rfree crystallographic factors were lower than the model refined without the lipid tail. The lipid tail of DDM forms several hydrophobic interactions with the side chains of residues lining the pore including W36, I38, W54, F83, L111, I128, and L138. There is no evidence for the maltoside head group of DDM at the top of the cavity, indicating that it does not form stabilizing interactions with the protein surface and thus is likely highly mobile (disordered) in our electron density maps (Figure 3A). Supporting our structural data of DDM binding to MxiM, we observed that the diffraction quality of MxiM crystals was dramatically affected by the presence of detergent. Crystals soaked in precipitant solutions lacking DDM retained their crystal habit (no observed cracking) but lost all diffraction. As there is no other evidence for the presence of ordered detergent in this high-resolution crystal structure (1.5 Å), it is plausible that the presence of the detergent tail in the cavity is important for stabilizing the unique fold of MxiM by maximizing hydrophobic packing within the core of the protein. It is tempting to speculate that in vivo, the natural N-terminal lipidation on MxiM could serve an analogous stabilizing role for the protein structure before association of the pilot with the MxiD secretin and OM. Furthermore, this intramolecular interaction could provide a regulatory mechanism to prevent the premature association of unbound pilot monomers to the membrane. Such a lipid-switch mechanism has been previously characterized for the myristoylated protein recoverin (Ames et al, 1999). Although our construct lacks the lipidation site, our structure suggests that the N-terminus is localized sufficiently near the lipid-binding cavity to potentially facilitate such an interaction.
Figure 3.
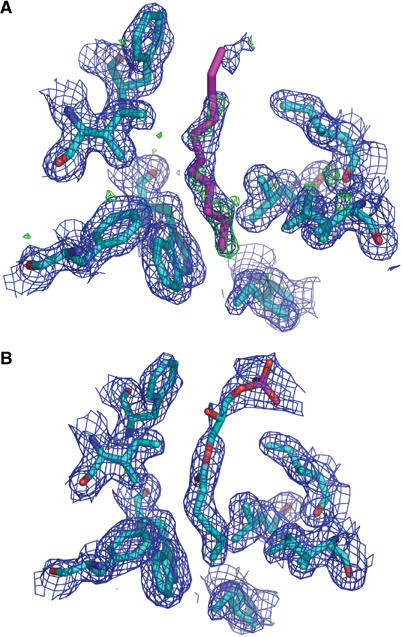
Representations of the electron density within the hydrophobic core of MxiM. (A) Electron density for the MxiM structure excluding the detergent acyl chain in the refinement. Green contours represent the Fo−Fc map contoured at 2.5σ and blue represents the weighted 2Fo−Fc contoured at 1σ. The Fo−Fc map represents electron density present in the structure that is not accounted for by the model. The refined position of the acyl chain is shown in the figure. (B) Refined MxiM–lipid (1-monohexanoyl-2-hydroxy-sn-glycerol-3-phosphate) crystal structure. Blue contours represent the weighted 2Fo−Fc contoured at 1σ. These figures are orientated such that the top is at the opening of the cavity.
The proposed role of MxiM interacting directly with phospholipids on the inner leaflet of the OM (via the observed hydrophobic cavity) is an attractive hypothesis. The proposal is consistent with the observation that both lipidated and unlipidated MxiM can stabilize MxiD oligomers (Schuch and Maurelli, 1999, 2001). Further, one can envisage that disruption of the membrane via the extraction and binding of lipid molecules by MxiM could facilitate the subsequent insertion of the associated secretin MxiD. Our observations also provide the basis for an additional anchoring role of MxiM (along with the N-terminal lipidation) to optimally stabilize the large secretin ring once inserted into the membrane.
Lipid binding to MxiM
To investigate further the role of lipid binding to MxiM, co-crystallization experiments with MxiM and a variety of lipids were carried out. The size and quality of the MxiM crystals improved with the presence of the negatively charged single-chain lipid 1-hexanoyl-2-hydroxy-sn-glycerol-3-phosphate (Avanti #857119) in the crystallization buffer. High-resolution data (1.87 Å) were obtained for these lipid–MxiM cocrystals using a rotating anode X-ray source. The crystal structure of the lipid/pilot complex unambiguously identifies the presence of the short-chain (C-5) lipid in the hydrophobic cavity (Figure 3B). The structure shows that the binding cavity selectively holds a single chain of lipid, with the close packing of hydrophobic groups around the lipid tail sterically prohibiting the binding of a second lipid chain or bound water molecules (Figure 4A and B). The lipid makes close contacts with the same residues as that observed in the complex with DDM, but the geometry of the aliphatic chain differs slightly due to the shorter length of the lipid, which results in the inclusion of its carbonyl group deep within the nonpolar cavity (Figure 4B).
Figure 4.
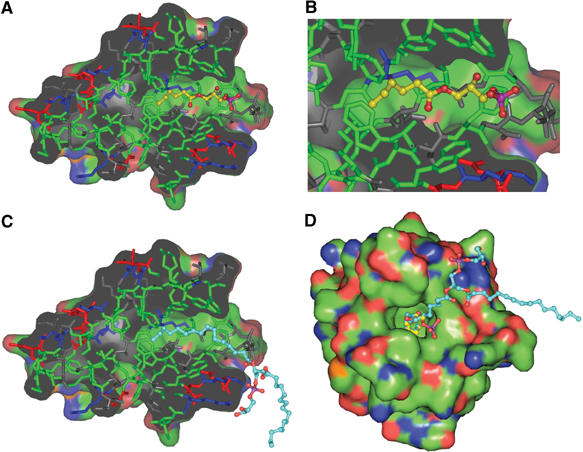
Molecular surface representations performed using PyMol (DeLano, 2004), where the surface is colored according to atom type: nitrogen atoms are in blue, oxygen atoms in red, and carbon atoms in green. Stick representation of the protein is gray with coloring to emphasize the different residue types: polar residues (gray), hydrophobic residues (green), acidic residues (red), and basic residues (blue). (A) The refined position of the lipid (1-monohexanoyl-2-hydroxy-sn-glycerol-3-phosphate) is represented in ball and stick where the carbon atoms are shown in yellow and the polar atoms are colored according to atom type. (B) Zoomed view of the hydrophobic cavity in (A). (C) Shown in cyan are the carbon atoms of 1-palmitoyl-2-palmitoleoyl-glycerol-3-phospho-rac-1-glycerol (PG) which is modeled into the cavity. (D) Top view of the cavity opening showing the differing positions of the head groups of the bound lipid (yellow) and the modeled PG (cyan).
In order to investigate the binding of MxiM28–142 with various OM lipids in solution, we performed ITC studies where MxiM28–142 (in 20 mM HEPES, pH 7.5) was titrated with four different negatively charged lipids ((1) 1,2-dioctanoyl-sn-glycero-3-phosphate (Figure 5); (2) 1,2-dioctanoyl-sn-glycero-3-phospho-rac-(1-glycerol); (3) 1,2-dihexanoyl-sn-glycero-3-phosphate; (4) 1,2-dihexanoyl-sn-glycero-3-phospho-rac-(1-glycerol)). The binding constants obtained from these studies range in value from 2 × 104 to 1 × 106 M−1 at 25°C, indicating that MxiM28–142 does indeed bind lipids with varying specificity (where lipid (1) K=7 (±3) × 105 M−1, n=0.6 (±1), ΔH=−5.0 (±0.6); (2) K=2 (±1) × 105 M−1, n=0.60 (±0.05), ΔH=−6 (±1); (3) K of <104 M−1, n and ΔH indeterminate; (4) K=2 (±1) × 104 M−1, n=0.40 (±0.05), ΔH=−4.9 (±0.9)). Due to the uncertainty regarding the presence of residual bound detergent in the lipid-binding cavity of MxiM, the reported binding constants are either Ka or K′ (apparent equilibrium constants). Nevertheless, these ITC results confirm that longer octyl chain lipids bind to MxiM with a more favorable binding constant: K between 105 and 106 M−1 at 25°C. Shorter hexyl chain lipids displayed weaker binding, with the binding affinity of 1,2-dihexanoyl-sn-glycero-3-phosphate being below the detection limit of the instrument. In our structure, the most extensive hydrophobic interactions observed between the lipid tail and MxiM originate from the five carbons at the terminal end of either one of the two acyl tails of the lipid. The second acyl tail of the lipid is therefore free to interact with a second MxiM molecule. This may explain our regressed binding stoichiometry, which was less than unity for all lipids studied. The hydrophobic interaction between the alkyl tail and MxiM would likely be quite similar among the various lipids studied, and thus the observed differences in binding are likely a result of the ability to position simultaneously the lipid head groups in an optimal manner to interact with polar and charged amino acids at the surface of the lipid-binding cavity, an ability that is obviously a direct consequence of varying lipid tail lengths. The negatively charged head group of the lipid used in our structural study (1-hexanoyl-2-hydroxy-sn-glycerol-3-phosphate) has the weakest electron density in the lipid/pilot complex, likely due to the positioning of the polar head group within the hydrophobic neck of the cavity, a restriction imposed by the shorter length of the lipid chain (Figure 4A and B). Although the extremely hydrophobic and aggregative nature of the longer chain lipids prohibited analysis in solution and in our structures, further modeling based on our current lipid complexes indicates that optimal length of acyl chain for the observed binding cavity would be ∼15 carbon atoms, in line with that of the lipids that make up the Gram-negative bacterial OM (the naturally abundant phosphatidylglycerol (PG) lipids such as 1-palmitoyl-2-palmitoleoyl-glycerol-3-phospho-rac-1-glycerol modeled in Figure 4C and D). This length of acyl chain allows for the optimal positioning of the negatively charged head group with the adjacent positively charged surfaces in the MxiM pilot.
Figure 5.
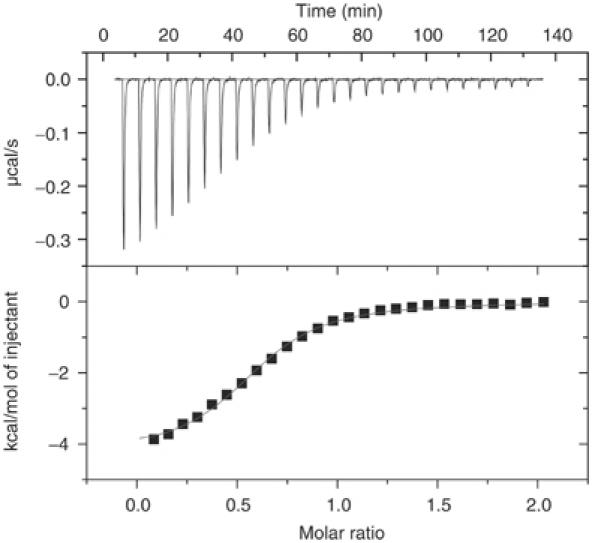
Analysis of the lipid, 1,2-dioctanoyl-sn-gylcero-3-phosphate, binding to MxiM using ITC, at 298 K. (Top) Raw titration data showing the heat response resulting from each 10 μl injection of 0.24 mM lipid into the ITC cell containing 24.6 μM MxiM in 20 mM HEPES at pH 7.5. (Bottom) Peak area normalized to the moles of lipid added and corrected for the heat of dilution (squares), and nonlinear least squares fit (line) to a single-site bimolecular interaction model.
The presence of the deep hydrophobic cavity in the secretin pilot is reminiscent of that observed in the lipocalin family. The lipocalin family is structurally conserved as an intact eight-stranded β-barrel that typically enclose around a hydrophobic ligand-binding domain (Flower, 1996). It is conceivable that the structure of MxiM was derived from a lipocalin as the insertion of helix F1 in between β-strands E′ and F could cause the distortion and splitting of the two previous β-strands into the smaller strands, D, D′, E, and E′ (Figure 1B). Although there is no detectable sequence identity between MxiM and any lipocalin family member, there are several similarities between these proteins. For example, the bacterial lipocalin, Blc, which also contains an N-terminal secretory signal peptide, is targeted to the periplasm, is membrane-anchored through a lipidated cystine residue (Bishop et al, 1995; Bishop, 2000), has a comparable pore cross-section and thus a similar pore diameter to MxiM (Figure 6). No crystallographic structures of lipocalins bound to fatty acid lipids currently exist. A detailed comparison of the binding cavity of MxiM and Blc indicates that ligand specificity is likely dependant on the diverse distribution of residues lining the interior. Blc has been suggested to play an important role in membrane biogenesis and repair, as has been observed for its eukaryotic homologs, apolipoprotein D (ApoD) and lazarillo (Bishop, 2000). These similarities between Blc and MxiM support our hypothesis that the presence of the hydrophobic cavity in MxiM plays an important role in altering the bacterial OM to facilitate the assembly and insertion of its secretin MxiD.
Figure 6.
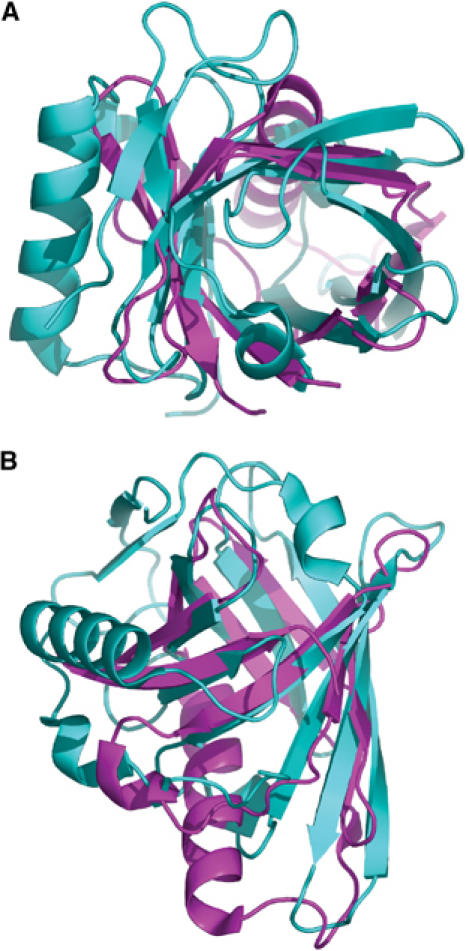
Superposition of MxiM (magenta) and bacterial lipocalin Blc (cyan; PDB code: 1QWD) carried out using PyMol (DeLano, 2004). The C-terminal helix of Blc, which is positioned at the side of the β-barrel, was excluded from the figure for clarity. (A) View looking down the internal cavities. (B) Side view rotated 90° from view (A). Blc had the highest structural similarity to MxiM based on a DALI search (Holm and Sander, 1995) where the root mean square deviation (r.m.s.d.) was calculated to be 3.1 Å (86 common Cα atoms). While there is β-strand overlap between the two structures for all eight strands of Blc, the proposed insertion of the α-helix in MxiM divides two of its β-strands resulting in two additional β-strands.
The bacterial integral OM binding protein, PagP (Ahn et al, 2004), showed a similar β-barrel topology to their soluble lipocalin counterparts and an extended hydrophobic cleft similar in dimensions to that observed in our secretin pilot MxiM. PagP is an acyltransferase enzyme that binds a palmitate chain from a phospholipid for transfer to lipid A. Interestingly, an acid–alcohol pair of residues (D76 and S77) have been identified as important for catalytic acyltransferase activity of PagP (Hwang et al, 2002). Similarly, MxiM has a structurally conserved pair of residues (D124 and T125) on a loop at the mouth of the lipid-binding cavity presenting the possibility that MxiM is a soluble acyltransferase that may act to lipidate components of the TTSS including its cognate secretin.
MxiM28–142 binding with its cognate secretin MxiD525–570
Secretin pilots are known to bind an approximately 50-residue C-terminal region of their respective secretins (Daefler et al, 1997; Shevchik and Condemine, 1998). Yeast two-hybrid experiments have previously suggested that there is a strong interaction between MxiM and a 46-residue C-terminal fragment of MxiD (Schuch and Maurelli, 2001). In order to probe directly the presence and strength of the unlipidated MxiM28–142 interaction with MxiD525–570, we performed ITC studies where MxiM28–142 was titrated with the MxiM-binding domain (MBD) of MxiD (MxiD525–570) in solution (Figure 7A). The results show that MxiM28–142 binds to MxiD525–570 in a 1:1 ratio (n=0.79 (±0.01)) with a binding constant Ka=1.1 (±0.3) × 107 M−1 at 25°C. The binding is driven by a small enthalpy change (ΔH=−2.79 (±0.006) kcal/mol). This confirms that there is a protein–protein binding interaction in the absence of the lipid anchor of MxiM. In order to study the effect of MxiD525–570 binding on lipid binding, ITC studies titrating 1,2-dioctanoyl-sn-glycero-3-phosphate into MxiM28–142 in the presence of MxiD525–570 were carried out. With the C-terminal fragment of MxiD prebound, there was no indication of lipid binding to MxiM (K<103, data not shown). However, for the reverse experiment where MxiD525–570 is titrated into MxiM28–142 in the presence of 1,2-dioctanoyl-sn-glycero-3-phosphate, a similar binding affinity of the lipid-free MxiD525–570–MxiM28–142 interaction was observed (Ka=1.2 (±0.1) × 107 M−1, n=0.90 (±0.01) at 25°C) (Figure 7B). Again the binding is driven by a small favorable enthalpy change (ΔH=−4.65 (±0.002) kcal/mol). These results indicate that the C-terminal fragment, MxiD525–570, blocks the lipid from binding to MxiM28–142, but the lipid under similar conditions does not exclude MxiD525–570 from binding. Furthermore, the size and hydrophobic nature of the cavity in MxiM exclude the possibility that an elongated C-terminus of MxiD525–570 binds directly into the cavity.
Figure 7.
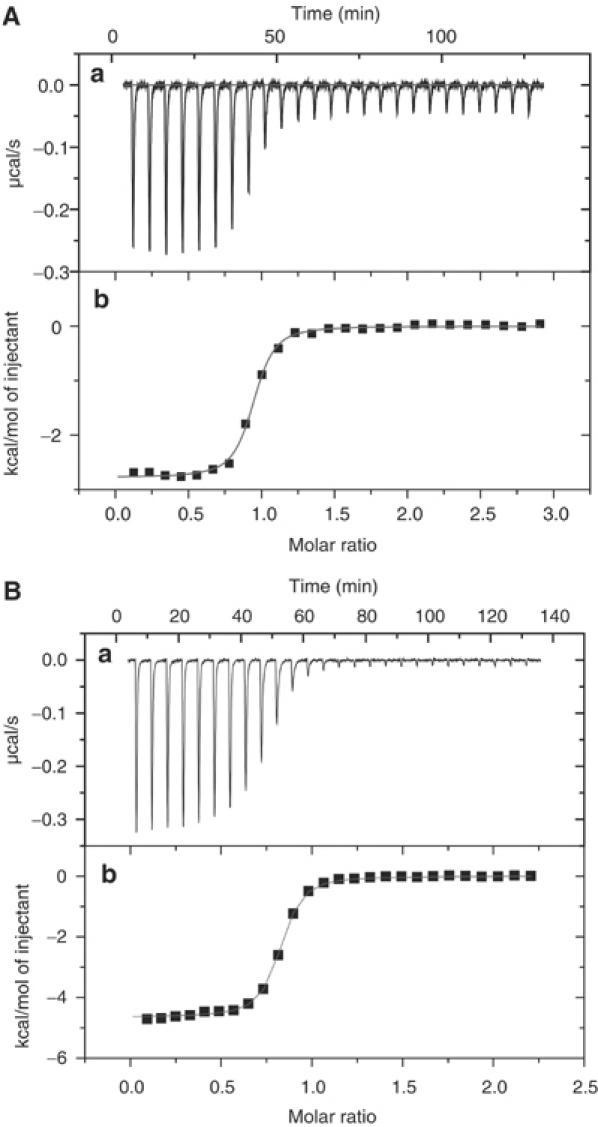
(A) Analysis of MxiD525–570 binding to MxiM using ITC, at 298 K. (Top) Raw titration data showing the heat response resulting from each 10 μl injection of 280 μM MxiD into the ITC cell containing 20 μM MxiM in 20 mM HEPES at pH 7.5 and 0.5 mM DM. (B) Analysis of MxiD binding to MxiM incubated with the lipid 2-dioctanoyl-sn-glycero-3-phosphate. (Top) Raw titration data showing the heat response resulting from each 10 μl injection of 220 μM MxiD into the ITC cell containing 20 μM MxiM in 20 mM HEPES at pH 7.5 and 43 μM of the lipid. (Bottom of A, B) Peak area normalized to the moles of MxiD added and corrected for the heat of dilution (squares), and nonlinear least squares fit (line) to a bimolecular interaction model.
A proposed model for the role of the pilot protein in secretin assembly on the bacterial OM
Based on both the binding data and the structural characteristics of MxiM, we propose that MxiD525–570 controls access to the lipid-binding cavity on the MxiM secretin pilot. Since MxiM is known to interact with MxiD in the periplasm prior to membrane insertion (Schuch and Maurelli, 2001) and we observe that the C-terminal portion of MxiD obstructs lipid binding to MxiM, a change of the MxiM–MxiD525–570 interaction would be necessary for MxiM to subsequently interact with phospholipid at the OM surface. Such an allosteric effect could result from conformational or steric differences associated with the multimerization of MxiD, which would alter specific secretin/pilot interactions such that the lipid-binding cavity would become accessible for lipid exchange with the OM. This proposal is compelling in that it would provide a regulatory component preventing preformed MxiM–MxiD heterodimers (in which the lipid-binding cavity is blocked) from competing with fully oligomerized MxiD/M rings for attachment to the membrane. Purification and concentration of our coexpressed MxiM24–142 and MxiD525–570 did not indicate the formation of larger order oligomers by static light scattering analysis and gel filtration chromatography (data not shown), indicating that oligomerization is not a consequence of interactions between MxiM and the C-terminal portion of MxiD. However, OM ring structures were prominently detected during the overexpression of intact MxiD in the presence of an unlipidated G23R mutant of MxiM (Schuch and Maurelli, 2001), yet only trace levels were detected when MxiD was overexpressed in the absence of MxiM. Together, these results indicate that both the MxiM24–142 and MxiD525–570 interaction and an MxiD oligomerization involving residues between 1 and 525 are critical to the formation of the OM ring structure. These data also support the proposal that presence of the N-terminal lipidation site on MxiM is not essential for formation of the secretin ring and likely plays a role in further stabilization of these ring structures once embedded in the OM (as seen through increased heat resistance of the lipidated complex) (Schuch and Maurelli, 2001).
Conclusions
Our structural analysis of MxiM, the secretin pilot of MxiD in S. flexneri, provides the first atomic picture of this essential family of proteins critical to the assembly of a variety of bacterial secretion apparatus. All secretin pilots have similar functional roles: creation and stabilization of the characteristic secretin oligomeric ring as well as subsequent membrane insertion. Given the customized lipid-binding domain we observe in MxiM, its roles in the formation of the TTSS could ostensibly involve membrane disruption through binding of lipid, allowing for the proper insertion of the large, multimeric MxiD secretin ring, and perhaps provide a regulatory mechanism preventing premature MxiM and MxiM–MxiD heterodimers from interacting with the OM. One might expect that pilot proteins that have conserved functional roles would either share the same fold or maintain critical structural features such as the observed lipid-binding cavity within a different fold. The crystal structure of MxiM provides the necessary foundation for further mutagenesis studies where the hydrophobic nature or the accessibility of the hydrophobic cavity can be disrupted in order to identify its role(s) in MxiD stabilization, multimerization, and membrane association. The identification of this exposed hydrophobic cavity also presents a new target for the design of specific inhibitors that could block the critical functions of the secretin pilot in the formation of the TTSS in Shigella and other pathogenic bacteria.
Materials and methods
Protein expression, purification, and mass spectrometry of MxiM28–142
Standard procedures were used for DNA manipulation and for Escherichia coli transformations. The DNA encoding MxiM24–142 was excised from a Novagen pet19b-derived vector containing the plasmid and ligated into the NdeI/BamHI sites of a pET28a (Novagen). BL21 (λDE3) cells transformed with the pet28a-derived plasmid were grown in LB media containing 25 μg/ml kanamycin at 37°C to an OD600 of 0.6–0.8 and then cooled to 20°C. Protein expression was induced by the addition of isopropyl β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM and the cells were harvested 12 h postinduction. Cells were resuspended in 15 ml of lysis buffer (20 mM HEPES pH 7.5 and 150 mM NaCl) per wet gram of cells. The cells were lysed in two passes using an Avestin™ cell homogenizer at 20 000 psi. Decylmaltoside (DM) was added to the lysate to a final concentration of 20 mM, and then the mixture was gently stirred for 1 h at 4°C followed by centrifugation at 44 000 g, 4°C for 35 min. The supernatant was applied to a 10 ml column of fast-flow chelating Sepharose (Amersham) charged with nickel and equilibrated in buffer A (20 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM DM, and 50 mM imidazole). The column was washed with 100 ml of buffer A plus 50 mM imidazole before the protein was eluted in a step from buffer A to buffer B (20 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM DM, and 300 mM imidazole). Fractions containing MxiM were pooled and thrombin (Sigma) was added at a mass ratio of 1000:1. The protein was then dialyzed overnight against buffer C (20 mM HEPES pH 7.5 and 5 mM DDM). The thrombin-cleaved material was applied to a MonoS (5/5) column (Pharmacia) equilibrated in buffer C, washed with 10 column volumes to exchange the detergent, and eluted with buffer D (20 mM HEPES, pH 7.5, 5 mM DDM and 1 M NaCl) at a flow rate of 0.1 ml/min to achieve concentration of the protein. The peak fraction containing MxiM was typically 15–25 mg/ml and subsequently dialyzed against buffer C to remove NaCl. The mass of the eluted MxiM (12 888.02 Da) was determined with an Applied Biosystems Voyager MALDI-TOF instrument using sinapinic acid as matrix in a saturated solution of 3:1:2 formic acid:water:isopropanol. The spectra were calibrated using the singly and doubly charged species of horse apomyoglobin (ABI). This mass corresponds to a fragment of the thrombin cleavage product MxiM28–142.
Crystallization and structural determination of MxiM28–142
In order to use MAD phasing techniques to solve the crystal structure, Se-Met-substituted protein was prepared using previously described methods (Doublie, 1997) and using an analogous purification protocol as the native protein. X-ray diffraction quality crystals of native (DDM bound) and Se-Met MxiM28–14 were grown in a 1:1 mixture of protein (13 mg/ml: 20 mM HEPES, pH 7.5, 5 mM DDM) and reservoir solution (100 mM NaCH3CO2, pH 4–4.5, 50–200 mM LiSO4, 2.0 M (NH4)2SO4) via hanging drop vapor diffusion method at 20°C. 1-Monohexanoyl-2-hydroxy-sn-glycero-3-phosphate (Avanti cat# 857119P) lipid cocrystals with MxiM28–14 were grown as above (excluding DDM in protein buffer) with 1:1.5 mol ratio of MxiM to lipid in the protein solution.
All forms crystallized in the C2 space group with isomorphous cell dimensions. The unit cell dimensions for the MxiM/DDM complex were a=82.84 Å, b=49.68 Å, c=29.49 Å, and β=102.27° with a single copy per asymmetric unit. A single crystal of the MxiM28–142/DDM complex with cell dimensions 500 × 500 × 300 μm was flash frozen in gaseous N2 at −100°C directly from the crystallization experiment and a highly redundant 1.5 Å resolution data set was collected using an ADSC Q-4 charge-coupled device (CCD) detector on beamline X8C at the National Synchrotron Light Source (NSLS, Upton, NY). A smaller Se-Met derivative crystal with dimensions 100 × 100 × 50 μm was used to obtain a 1.7 Å resolution MAD data set on beamline 8.2.1 of the Advanced Light Source (ALS, Berkeley, CA) using an ADSC Q-315 CCD detector. The lipid–MxiM cocrystal with the dimensions 300 × 300 × 200 μm yielded a 1.87 Å resolution data set using a Mar345 image plate detector with a Rigaku RU200 rotating anode X-ray generator. All the X-ray data sets were processed with the HKL suite of programs (Otwinowski and Minor, 1997) and the data collection statistics are provided in Table I.
The programs SOLVE and RESOLVE (Terwilliger, 2004) were used to phase the initial MxiM electron density maps from protein crystals containing incorporated Se-Met. The high quality of the electron density maps at 1.5 Å enabled the automatic building of over 70% of the structure using the program ARP/WARP (Perrakis et al, 2001; Morris et al, 2003). The refinement was started using the CNS suite of programs incorporating annealing techniques (Brunger et al, 1998) and then the final refinement stages were carried out with REFMAC5 (Winn et al, 2001). Model building was carried out using the program Xfit of the XtalView suite of programs (McRee, 1999). The final native model was refined to values R of 0.220 and Rfree of 0.251 and the lipid–MxiM structure was refined to values R of 0.206 and Rfree of 0.249 (see Table I). Five residues at the N-terminus of this cleavage product were not observed in the electron density for both structures and therefore are not included in the crystal structures. Figures were prepared with MolScript (Kraulis, 1991), Raster3D (Merritt and Murphy, 1994), PyMol (DeLano, 2004), GRASP (Nicholls et al, 1991), and XtalView (McRee, 1999).
Coordinates
The coordinates and structure factors for the crystallographic model of MxiM28–142 and lipid–MxiM28–142 have been deposited in the Protein Data Bank (PDB entry codes: 1Y9L and 1Y9T, respectively).
Protein expression and purification of MxiD525–570
The MBD of the secretin MxiD is its C-terminus. The MBD construct, MxiD525–570, was inserted into Nde/BamHI sites of Novagen pet21 (Novagen). BL21 (DE3) cells carrying the resulting expression plasmid were grown and induced as described previously for MxiM. After harvesting the cells, the pellet was resuspended in 20 ml of 20 mM HEPES pH 7.5 and 150 M NaCl (buffer A) per gram of wet cell paste. The cells were lysed via two passes through an Avestin cell homogenizer. Inclusion bodies (IB) were harvested by centrifuging at 12 000 g for 15 min and washed three times in 25 ml of buffer A plus 1% Triton X-100, 5 mM EDTA, and 2 mM DTT. The final IB pellet was solubilized in 20 ml of 6 M guanidine-HCl and then centrifuged at 12 000 g for 30 min to remove the insoluble material, The supernatant was dialyzed against 2 × 2 l of 20 mM HEPES pH 7.5. The soluble refolded protein was incubated with thrombin at a mass ratio of 1000:1 for 8 h at 4°C to remove the hexahistidine tag. The tagless MBD was applied to a MonoQ (5/5) column (Amersham) equilibrated in 20 mM Tris pH 8.0 and eluted in a linear NaCl gradient from 0 to 0.5 M. Protein-containing peaks were combined and dialyzed into 20 mM HEPES pH 7.5 and 5 mM DM buffer.
Static light scattering
The oligomerization state of MxiM28–142 was analyzed using static light scattering with a Superdex 75 (Amersham) gel exclusion column connected to a WYATT Technology MiniDawn light scattering unit with an in-line Optilab DSP interferometric refractometer. All runs were preformed in the presence of 20 mM HEPES pH 7.5, 150 mM NaCl, and 0.5 mM DDM.
Isothermal titration calorimetry
ITC was performed using a VP ITC (MicroCal Inc., Northampton, MA). All lipid samples were pH 7.5 in 20 mM HEPES. Titrations were performed by injecting consecutive 5–10 μl aliquots of lipid solution (0.20–1.39 mM) into the ITC cell (volume=1.3528 ml) containing MxiM (20.1–98 μM) at pH 7.5 in 20 mM HEPES. The ITC data were corrected for the heat of dilution of the titrant by subtracting mixing enthalpies for 5–10 μl injections of lipid solution into protein-free buffer. Two to four independent titration experiments were performed at 25°C to determine the binding constant of lipid to MxiM. Binding stoichiometry, enthalpy, and equilibrium association constants were determined by fitting the corrected data to a bimolecular (one type of binding site) interaction model (software provided with instrument). Parameters fit were n, the number of binding sites on MxiM; K, the association constant; and ΔH, the enthalpy of association.
ITC experiments with MxiD525–570 were at pH 7.5 in 20 mM HEPES and 5 mM DM. These titrations were performed by injecting 26 consecutive 10 μl aliquots of MxiD525–570 solution (280 μM) into the ITC cell (volume=1.3528 ml) containing MxiM28–142 (20 μM). Two independent titration experiments were performed at 25°C to determine the binding constant of MxiD525–570 to MxiM28–142. Binding stoichiometry, enthalpy, and equilibrium association constants were determined by fitting the corrected data to a bimolecular interaction model (software provided with instrument).
ITC experiments titrating MxiD525–570 into MxiM preincubated with lipid (1) (2-dioctanoyl-sn-glycero-3-phosphate, ∼30 min, 43 μM) were performed at pH 7.5 in 20 mM HEPES. These titrations were performed by injecting 26 consecutive 10 μl aliquots of MxiD525–570 solution (220 μM) into the ITC cell (volume=1.3528 ml) containing MxiM28–142 (20 μM). ITC experiments titrating lipid (1), 2-dioctanoyl-sn-glycero-3-phosphate, into MxiM and MxiM preincubated with MxiD525–570 (∼30 min, 50 μM) were performed at pH 7.5 in 20 mM HEPES. These titrations were performed by injecting 26 consecutive 10 μl aliquots of the lipid solution (250 μM) into the ITC cell (volume=1.3528 ml) containing MxiM28–142 (25 μM). The ITC data were corrected for the heat of dilution of the titrant by subtracting mixing enthalpies for 10 μl injections of MxiD525–570 solution into protein-free buffer. Two independent titration experiments were performed at 25°C to determine the binding constant of MxiD525–570 to MxiM28–142. Binding stoichiometry, enthalpy, and equilibrium association constants were determined by fitting the corrected data to a bimolecular interaction model (software provided with instrument).
Acknowledgments
We acknowledge Dr Andrew Lovering and Calvin Yip for helpful discussions. We thank the US Department of Energy, the HHMI, and the staffs at beamlines X8C (NSLS) and 8.2.1 (ALS) for access and technical assistance in data collection. This work was supported by research grants awarded to NS by the Canadian Institutes of Health Research, the Howard Hughes Medical Institute, and the Canadian Bacterial Diseases Network of Excellence. PL is a CIHR Fellow and NS is a CIHR Investigator and HHMI International Scholar.
References
- Ahn VE, Lo EI, Engel CK, Chen L, Hwang PM, Kay LE, Bishop RE, Prive GG (2004) A hydrocarbon ruler measures palmitate in the enzymatic acylation of endotoxin. EMBO J 23: 2931–2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaoui A, Sansonetti PJ, Parsot C (1992) MxiJ, a lipoprotein involved in secretion of Shigella Ipa invasins, is homologous to YscJ, a secretion factor of the Yersinia Yop proteins. J Bacteriol 174: 7661–7669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames JB, Dizhoor AM, Ikura M, Palczewski K, Stryer L (1999) Three-dimensional structure of guanylyl cyclase activating protein-2, a calcium-sensitive modulator of photoreceptor guanylyl cyclases. J Biol Chem 274: 19329–19337 [DOI] [PubMed] [Google Scholar]
- Birtalan SC, Phillips RM, Ghosh P (2002) Three-dimensional secretion signals in chaperone-effector complexes of bacterial pathogens. Mol Cell 9: 971–980 [DOI] [PubMed] [Google Scholar]
- Bishop RE (2000) The bacterial lipocalins. Biochim Biophys Acta 1482: 73–83 [DOI] [PubMed] [Google Scholar]
- Bishop RE, Penfold SS, Frost LS, Holtje JV, Weiner JH (1995) Stationary phase expression of a novel Escherichia coli outer membrane lipoprotein and its relationship with mammalian apolipoprotein D. Implications for the origin of lipocalins. J Biol Chem 270: 23097–23103 [DOI] [PubMed] [Google Scholar]
- Blocker A, Jouihri N, Larquet E, Gounon P, Ebel F, Parsot C, Sansonetti P, Allaoui A (2001) Structure and composition of the Shigella flexneri ‘needle complex', a part of its type III secreton. Mol Microbiol 39: 652–663 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Burghout P, Beckers F, de Wit E, van Boxtel R, Cornelis GR, Tommassen J, Koster M (2004) Role of the pilot protein YscW in the biogenesis of the YscC secretin in Yersinia enterocolitica. J Bacteriol 186: 5366–5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RF, Frye SA, Kitmitto A, Ford RC, Tonjum T, Derrick JP (2004) Structure of the Neisseria meningitidis outer membrane PilQ secretin complex at 12 Å resolution. J Biol Chem 279: 39750–39756 [DOI] [PubMed] [Google Scholar]
- Crago AM, Koronakis V (1998) Salmonella InvG forms a ring-like multimer that requires the InvH lipoprotein for outer membrane localization. Mol Microbiol 30: 47–56 [DOI] [PubMed] [Google Scholar]
- Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ (1998) JPred: a consensus secondary structure prediction server. Bioinformatics 14: 892–893 [DOI] [PubMed] [Google Scholar]
- Daefler S, Guilvout I, Hardie KR, Pugsley AP, Russel M (1997) The C-terminal domain of the secretin PulD contains the binding site for its cognate chaperone, PulS, and confers PulS dependence on pIVf1 function. Mol Microbiol 24: 465–475 [DOI] [PubMed] [Google Scholar]
- Daefler S, Russel M (1998) The Salmonella typhimurium InvH protein is an outer membrane lipoprotein required for the proper localization of InvG. Mol Microbiol 28: 1367–1380 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2004) The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific LLC [Google Scholar]
- Doublie S (1997) Preparation of selenomethionyl proteins for phase determination. Methods Enzymol 276: 523–530 [PubMed] [Google Scholar]
- Drake SL, Sandstedt SA, Koomey M (1997) PilP, a pilus biogenesis lipoprotein in Neisseria gonorrhoeae, affects expression of PilQ as a high-molecular-mass multimer. Mol Microbiol 23: 657–668 [DOI] [PubMed] [Google Scholar]
- Flower DR (1996) The lipocalin protein family: structure and function. Biochem J 318: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambhir A, Hangyas-Mihalyne G, Zaitseva I, Cafiso DS, Wang J, Murray D, Pentyala SN, Smith SO, McLaughlin S (2004) Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys J 86: 2188–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C (1995) Dali: a network tool for protein structure comparison. Trends Biochem Sci 20: 478–480 [DOI] [PubMed] [Google Scholar]
- Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CR, Prive GG, Bishop RE, Kay LE (2002) Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc Natl Acad Sci USA 99: 13560–13565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster M, Bitter W, de Cock H, Allaoui A, Cornelis GR, Tommassen J (1997) The outer membrane component, YscC, of the Yop secretion machinery of Yersinia enterocolitica forms a ring-shaped multimeric complex. Mol Microbiol 26: 789–797 [DOI] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Linderoth NA, Simon MN, Russel M (1997) The filamentous phage pIV multimer visualized by scanning transmission electron microscopy. Science 278: 1635–1638 [DOI] [PubMed] [Google Scholar]
- Luo Y, Bertero MG, Frey EA, Pfuetzner RA, Wenk MR, Creagh L, Marcus SL, Lim D, Sicheri F, Kay C, Haynes C, Finlay BB, Strynadka NC (2001) Structural and biochemical characterization of the type III secretion chaperones CesT and SigE. Nat Struct Biol 8: 1031–1036 [DOI] [PubMed] [Google Scholar]
- McRee DE (1999) XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J Struct Biol 125: 156–165 [DOI] [PubMed] [Google Scholar]
- Merritt EA, Murphy MEP (1994) Raster3D version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr D 50: 869–873 [DOI] [PubMed] [Google Scholar]
- Morris RJ, Perrakis A, Lamzin VS (2003) ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol 374: 229–244 [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296 [DOI] [PubMed] [Google Scholar]
- Nouwen N, Ranson N, Saibil H, Wolpensinger B, Engel A, Ghazi A, Pugsley AP (1999) Secretin PulD: association with pilot PulS, structure, and ion-conducting channel formation. Proc Natl Acad Sci USA 96: 8173–8177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouwen N, Stahlberg H, Pugsley AP, Engel A (2000) Domain structure of secretin PulD revealed by limited proteolysis and electron microscopy. EMBO J 19: 2229–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology, Carter J, Sweet RM (eds) Vol. 276, pp 307–326. New York: Academic Press [DOI] [PubMed] [Google Scholar]
- Parsot C, Menard R, Gounon P, Sansonetti PJ (1995) Enhanced secretion through the Shigella flexneri Mxi–Spa translocon leads to assembly of extracellular proteins into macromolecular structures. Mol Microbiol 16: 291–300 [DOI] [PubMed] [Google Scholar]
- Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS (2001) ARP/wARP and molecular replacement. Acta Crystallogr D 57: 1445–1450 [DOI] [PubMed] [Google Scholar]
- Philpott DJ, Edgeworth JD, Sansonetti PJ (2000) The pathogenesis of Shigella flexneri infection: lessons from in vitro and in vivo studies. Philos Trans R Soc London B 355: 575–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B, Liu J (2003) The PredictProtein server. Nucleic Acids Res 31: 3300–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Cirion A, Gomara MJ, Agirre A, Nieva JL (2003) Pre-transmembrane sequence of Ebola glycoprotein. Interfacial hydrophobicity distribution and interaction with membranes. FEBS Lett 533: 47–53 [DOI] [PubMed] [Google Scholar]
- Schuch R, Maurelli AT (1999) The mxi–Spa type III secretory pathway of Shigella flexneri requires an outer membrane lipoprotein, MxiM, for invasin translocation. Infect Immun 67: 1982–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuch R, Maurelli AT (2001) MxiM and MxiJ, base elements of the Mxi–Spa type III secretion system of Shigella, interact with and stabilize the MxiD secretin in the cell envelope. J Bacteriol 183: 6991–6998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuch R, Sandlin RC, Maurelli AT (1999) A system for identifying post-invasion functions of invasion genes: requirements for the Mxi–Spa type III secretion pathway of Shigella flexneri in intercellular dissemination. Mol Microbiol 34: 675–689 [DOI] [PubMed] [Google Scholar]
- Shevchik VE, Condemine G (1998) Functional characterization of the Erwinia chrysanthemi OutS protein, an element of a type II secretion system. Microbiology 144: 3219–3228 [DOI] [PubMed] [Google Scholar]
- Shevchik VE, Robert-Baudouy J, Condemine G (1997) Specific interaction between OutD, an Erwinia chrysanthemi outer membrane protein of the general secretory pathway, and secreted proteins. EMBO J 16: 3007–3016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamano K, Aizawa S, Katayama E, Nonaka T, Imajoh-Ohmi S, Kuwae A, Nagai S, Sasakawa C (2000) Supramolecular structure of the Shigella type III secretion machinery: the needle part is changeable in length and essential for delivery of effectors. EMBO J 19: 3876–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampakaki AP, Fadouloglou VE, Gazi AD, Panopoulos NJ, Kokkinidis M (2004) Conserved features of type III secretion. Cell Microbiol 6: 805–816 [DOI] [PubMed] [Google Scholar]
- Terwilliger T (2004) SOLVE and RESOLVE: automated structure solution, density modification and model building. J Synchrotron Radiat 11: 49–52 [DOI] [PubMed] [Google Scholar]
- Thanassi DG, Hultgren SJ (2000) Multiple pathways allow protein secretion across the bacterial outer membrane. Curr Opin Cell Biol 12: 420–430 [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D 57: 122–133 [DOI] [PubMed] [Google Scholar]
- Wu HC, Tokunaga M (1986) Biogenesis of lipoproteins in bacteria. Curr Top Microbiol Immunol 125: 127–157 [DOI] [PubMed] [Google Scholar]
- Zhang CT, Zhang R (2000) A graphic approach to evaluate algorithms of secondary structure prediction. J Biomol Struct Dyn 17: 829–842 [DOI] [PubMed] [Google Scholar]