

Abstract
In this study, we used deoxyribozyme (DNAzyme) functionalized gold nanoparticles (AuNPs) to catalytically silence tumor necrosis factor-α (TNF-α) in vivo as a potential therapeutic for myocardial infarction (MI). Using primary macrophages as a model, we demonstrated 50% knockdown of TNF-α, which was not attainable using Lipofectamine-based approaches. Local injection of DNAzyme conjugated to gold particles (AuNPs) in the rat myocardium yielded TNF-α knockdown efficiencies of 50%, which resulted in significant anti-inflammatory effects and improvement in acute cardiac function following MI. Our results represent the first example showing the use of DNAzyme AuNP conjugates in vivo for viable delivery and gene regulation. This is significant as TNF-α is a multibillion dollar drug target implicated in many inflammatory-mediated disorders, thus underscoring the potential impact of DNAzyme-conjugated AuNPs.
Keywords: nanoparticles, gene regulation, deoxyribozyme (DNAzyme), myocardial infarction, tumor necrosis factor-α (TNF-α)
1. Introduction
Ischemic heart disease is considered to be the leading cause of death in the world today.[1] Myocardial infarction (MI), most commonly known as a “heart attack”, is the initial occurrence, which involves a thrombotic occlusion of a coronary artery and subsequent depletion of oxygen and nutrients to a segment of the left ventricular wall. This lack of blood flow initiates an inflammatory response as a defense mechanism to prevent terminal damage to the myocardium. An early and crucial step in the process of cardiac repair following acute MI is the inflammatory reaction that begins with the influx of neutrophils and macrophages that absorb the necrotic tissue.[5-8] However, unbridled inflammation fueled by continuous up-regulation of pro-inflammatory cytokines can exacerbate tissue damage, thus contributing to adverse cardiac remodeling and non-contractile scar formation that ultimately culminates in HF.[9-12] Much interest has been devoted to studying the role of inflammation following acute MI, and interestingly, many pharmacological therapies with demonstrated efficacy for the management of heart disease exhibit anti-inflammatory properties distinct from their perceived primary mechanism of action, suggesting modulation of inflammation as a contributing factor to the clinical benefit ascribed to these medications.[4, 9]
The pro-inflammatory cytokine Tumor Necrosis Factor-α (TNF-α) is an extensively studied potential therapeutic target implicated in many of the pathological processes involved irreversible damage caused to the heart post-MI. The pathological mechanism for TNF-α mediated myocardial dysfunction is thought to occur through a combination of factors that includes induction of reactive oxygen and nitrogen species, initiation of pro-inflammatory cytokine cascades and down-regulation of contractile proteins.[13-18] However, several reports have also indicated that TNF-α involvement in the early repair process as a double-edged sword in that physiological levels of TNF-α can be cardioprotective.[19-21] Therefore, selection of an optimal therapeutic window as well as duration of action are critical in targeting TNF-α as an anti-inflammatory therapy in order to suppress the elevated inflammatory response immediately following MI.
Despite the volumes of encouraging pre-clinical data for TNF-α antagonism, human trials employing a soluble TNF-α receptor (etanercept) or a monocolonal antibody (infliximab) designed to neutralize TNF-α have failed to demonstrate long-term clinical benefit in patients with chronic HF post MI.[22] Therefore designing alternative approaches to deliver transient, but potent suppression of TNF-α during the critical acute inflammatory stage is an important step towards successfully translating TNF-α antagonism as a treatment strategy for acute MI. Oligonucleotide-based gene silencing therapeutics offer potent yet transient target gene regulation, and are thus excellent candidates for treating acute MI that may circumvent many of the limitations of protein-based strategies. However, delivery of therapeutic nucleic acids is an immense challenge due to the difficulties in transporting highly negatively charged oligomers across a cell membrane and protecting nucleic acids from degradation.[25, 26] Current delivery methods include complexing of nucleic acids to cationic lipids. However, the inherent toxicity and the positively charged surface characteristics that elicit immune responses deem such vehicles unsuitable for treating inflammatory pathologies.[27] A powerful, yet emerging approach for transfection entails spatially organizing therapeutic oligonucleotides into a compact spherical nanostructure.[28, 29] Work by Mirkin and colleagues has shown that these spherical DNA structures are internalized by virtually all nucleated cells.[31] Internalization is mediated through scavenger receptor A and proceeds rapidly and without the aid of a transfection agent.[32, 33] DNA-modified gold particles escape from endosomal compartments within a few hrs of incubation with cells. DNA-modified AuNP are then distributed within the cytoplasm but not the nucleus. In fact, transfection is so efficient that spherical nucleic acids have been shown to readily enter a mouse's epithelial layer to regulate EGFR expression and even cross the blood brain barrier to treat glioblastoma.[34, 35] In order to address the issues of achieving efficient transfection of therapeutic oligonucleotides with minimal immune response, we hypothesized that functional DNA conjugated to spherical gold nanoparticles would provide the combination of highly efficient and non-toxic delivery of oligonucleotides along with potent and transient knockdown of TNF-α. We specifically investigated functional DNA enzymes (or DNAzymes) as a novel catalytic modality of gene silencing molecules to inhibit TNF-α mRNA.
The 10-23 DNAzyme is composed of a 15 base catalytic core and flanked by two recognition arms that can selectively degrade target mRNA independent of RNAi, thus importantly avoiding the need to hijack the host cell's machinery for catalysis. Compared to siRNA mediated gene silencing, DNAzymes offer the added advantages of enhanced stability (DNA versus RNA), cost-effective synthesis and limited immune response, which is typically triggered by double stranded RNA in siRNA approaches. DNAzyme therapies have shown great potential in pre-clinical trials in treating a wide variety of ailments ranging from cancer to spinal cord injury including myocardial infarction, notably targeting transcription factors c-Jun and Egr-1 for knockdown.[36-40] DNAzymes are also currently in human trials for treating nasopharyngeal carcinoma.[41] In previous work, we have demonstrated enhanced catalytic gene silencing activity in vitro demonstrated by DNAzymes when functionalized in similar spherical nucleic acid structures[42]. In this report, we tested whether DNAzyme functionalized gold nanoparticles could be effective anti-inflammatory agents in the heart by regulating TNF-α expression in a rodent model of acute MI and confer therapeutic benefits to the damaged heart.
2. Materials and methods
2.1. Materials
All chemicals were purchased from Sigma-Aldrich unless otherwise noted. Stock solutions were made using Nanopure water (Barnstead Nanopure system, resistivity = 18.2 MΩ). The DNA fluorescence assay kit (Quant-iT™ OliGreen® ssDNA kit) was acquired from Life Technologies (formerly Invitrogen, Carlsbad, CA) and was used to quantify DNA density on the particle surface. All oligonucleotides were custom synthesized by Integrated DNA Technologies (IDT) and are summarized in SI Table 1.
2.2. Gold-DNAzyme nanoparticle synthesis
Citrate-stabilized gold nanoparticles (AuNP; 14±3 nm) were prepared using published procedures.[43] A 500 mL solution of 1 mM hydrogen tetrachloroaurate(III) trihydrate solution was brought to a vigorous boil, and once boiling, 50 mL of a 38.8 mM sodium citrate tribasic dihydrate solution was added and allowed to reflux for 15 min. The reaction mixture was filtered using a 0.45 μm acetate filter, producing monodisperse AuNPs. The surface plasmon resonance wavelength of the gold nanoparticles was determined using UV-vis absorbance spectrometry, and particle size was determined using transmission electron microscopy (TEM).
2.3. Preparation of DNAzyme-functionalized gold nanoparticles
Disulfide-modified oligonucleotides at the 3′ terminus were purchased from Integrated DNA Technologies (IDT). The disulfide was reduced to a free thiol by incubating 50 nmols of lyophilized oligonucleotide with 1.0 mL of disulfide cleavage buffer (0.1 M dithiothreitol (DTT), 170 mM phosphate buffer at pH 8.0) for 3 hours at room temperature. The reduced oligonucleotides were purified using a NAP-25 column (GE Healthcare, Piscataway, NJ) with Nanopure water as the eluent. Subsequently, 40 nmol of DNA was added to 10 mL of 14 nm gold nanoparticles (10 nM), bringing the final concentration of oligonucleotide and gold nanoparticles to ∼3.0 μM and ∼7.0 nM, respectively. The pH of the solution was adjusted to pH 7.4 by adding 1/10th the total volume (∼133 μL) of 100 mM phosphate buffer, thus bringing the phosphate buffer concentration to 9 mM. The particles were stabilized by adding sodium dodecyl sulfate (SDS) to the solution and bringing the SDS concentration to 0.1% (g/mL) by using a stock solution of 10% SDS. The particles were successively salted with eight NaCl additions that were spaced 20 min apart using a stock solution of 2.0 M NaCl and 10 mM phosphate buffer. The final NaCl concentration of the DNA AuNP solution was increased to 0.7 M. The first two NaCl additions increased the concentration by 0.05 M increments, while the remaining six NaCl additions increased the NaCl concentration by 0.1 M increments. The particles were immediately sonicated for 10 s after each salt addition to maximize DNA packing, as indicated in literature.[44] Fully salted particles were then incubated overnight, in the dark at room temperature. We found that the 10-23 active catalytic core of the DNAzyme had a tendency to drive the formation of nanoparticle aggregates at 0.7 M NaCl due to partial self-complementarity of sequences. The formation of these aggregates did not result in any observable reduction in the quality of the particles (as measured by UV-vis absorbance, TEM, DNA density, and ultimately TNF-a knockdown). The particles were stored as a stock solution (under high salt and excess DNA) at 4°C until needed, for a maximum duration of 1 month. Note that Dz-conjugated particles that were washed and stored in Nanopure water for more than one week displayed a decrease in their gene-regulation activity, Error! Reference source not found.S9 (no salt or excess DNA). Therefore, particles were only washed prior to knockdown studies, where particles were centrifuged four times at 13,500 RPM, filtered using a 0.2 μm syringe filter, and reconstituted in PBS.
2.4. Quantification of DNAzyme loading density
The commercial Quant-iTTM OliGreen® ssDNA kit was used to determine the total DNAzyme density per particle. The Quant-iTTM OliGreen® ssDNA kit required preparation of a calibration curve by diluting a DNA stock solution (4 μg/mL) composed of the same thiolated oligonucleotide used during particle functionalization to 0.1, 0.2, 0.5, 0.75, 1.0, and 2.0 μg/mL at a final volume of 100 μL in 1× TE buffer. DNAzyme particle solutions were prepared by diluting a ∼10 nM stock solution to 0.4, 0.6, and 0.8 nM with TE buffer. The oligonucleotides were then released from the particle by oxidizing/dissolving the gold with potassium cyanide (KCN). This was performed by adding 1 μL of a 5 M stock solution of KCN to each well, including the calibration wells to ensure an accurate calibration curve and blank subtraction. The samples were incubated with KCN for 30 min to ensure complete dissolution. After complete dissolution of the gold nanoparticles, 100 μL of the freshly prepared 1× Quant-iTTM OliGreen® solution was added to each well and fluorescence intensities (485/528 nm excitation/emission) of each well were then measured using a Bio-Tek Synergy HT plate reader to determine the total DNA density.
2.5. Cell Culture
RAW 264.7 macrophages obtained from American Type Culture Collection (ATCC number: TIB-71) were maintained at 37°C under a humidified atmosphere of 5% CO2 in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS), supplemented with penicillin (100 U/mL) and streptomycin (100 mg/mL) and 2 mM L-Glutamine. For primary macrophage culture, peritoneal macrophages were isolated from Sprague Dawley rats as described previously [45]. Briefly, cold PBS was infused into the peritoneal cavity of rats and cellular lavage was collected, spun down at 1000 × g at 4°C for 10 minutes, following which cell solution was resuspended in media described above. The cell solution was plated in 24-well plates (3×106 cells/well) and left to adhere overnight for treatment the following day. Primary single ventricular myocytes culture from Sprague Dawley rat hearts is described in supporting information.
2.6. Particle uptake by macrophages and myocytes
To confirm the intracellular distribution of DNA delivered by DNA modified gold nanoparticles, RAW264.7 macrophages or adult rat cardiomyocytes were treated with Cy5 labeled DNAzyme functionalized gold particles for 1 hour (5 nM particle concentration equivalent to 666.7 nM DNA concentration) and then washed in PBS three times, fixed in 4% PFA and stained with DAPI for nuclei and calcein for macrophages or FITC Maleimide for cardiomycocytes to delineate the cell surface, and imaged using laser scanning confocal microscopy (Zeiss LSM 510 META) to identify Cy5 fluorescence within the cells to indicate internalization of Cy5-DNA modified gold particles. Further, particle uptake was compared with commercially available Lipofectamine® RNAiMAX reagent (Life Technologies) mediated transfection at 1, 4, 18 and 24 hours using fluorescence microscopy in live cells for macrophages, and in fixed cells at 18 hours for cardiomyocytes.
2.7. In vitro silencing of TNF-α with Dz particles
For TNF-α knockdown studies, peritoneal macrophages were treated with three types of particles, active (Dz), inactive (i-Dz) or active but nonspecific (NS-Dz) at a particle concentration of 10 nM/well (1.3 mM DNA). After 20 hours of treatment, the media was removed and the cells were re-treated with the same concentration of particles while at the same time receiving stimulation with LPS (5 ng/ml) to induce TNF-α production. After 4 hours of stimulation, 100 uL of media was collected from each sample and analyzed for secreted TNF-α via ELISA according to the manufacture's protocol (eBioscience).
2.8. Cytotoxicity
Cytotoxicity of DNAzyme particles was compared with a commercially available, commonly used transfection reagent, using pro-inflammatory cytokine TNF-α itself as a surrogate cytotoxicity indicator. Primary peritoneal macrophages were transfected with either Lipofectamine® RNAiMAX containing scrambled DNA or NS-Dz for 20 hours. Extracellular medium was collected and TNF-α production quantified by ELISA.
2.9. Myocardial infarction and particle injection
A randomized and blinded study was conducted using adult Sprague Dawley rats. The animals were assigned to treatment groups (n=8-12) using a random number generator and the animal surgeon was only given letter codes to identify groups. While one group was subjected to a sham surgery, the other four groups received myocardial infarction via permanent ligation of the left descending artery. Briefly, the animals were anesthetized (1-3% isoflurane) and following tracheal intubation, the heart was exposed by separating the ribs. Myocardial infarction was performed by ligation of the left anterior descending coronary artery. For groups receiving particle injections, immediately after coronary artery ligation, 100 μL of one of the following DNAzyme modified particles were injected into the cyanotic ischemic zone (3 locations) through a 25-gauge needle while the heart was beating: Dz, i-Dz or NS-Dz. The dose of DNAzyme injected was 0.07 mg/kg (100 nM particle concentration). Following injection, the chests were closed and animals were allowed to recover on a heating pad until functional assessments were made 3 days following MI surgery using echocardiography (Acuson Sequoia 512 with a 14 MHz transducer) and invasive pressure-volume hemodynamics (Millar Instruments). These studies conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and all animal studies were approved by Emory University Institutional Animal Care and Use Committee.
2.10. In vivo imaging
Rats were subjected to MI as described above and Cy5 labeled NS-Dz gold particle conjugates were injected in 3 areas in the border zone surrounding the infarct. Rats were sacrificed and the hearts, liver, spleen and kidneys were harvested and imaged on days 0, 1, 2, and 3 post-injection (n=1 per time point) using Bruker In-Vivo Xtreme. The Cy5 fluorescent intensity in these organs was plotted as arbitrary fluorescence over time to demonstrate retention of the particles up to 3 days in the heart.
2.11. Echocardiography and invasive pressure-volume hemodynamics analysis
In a separate study, rats were subjected to MI surgery and injections as described above, and 3 days post-surgeries and treatment anesthetized rats were subjected to echocardiography (Acuson sequoia 512) and invasive pressure-volume hemodynamics (Millar MPVS instruments) analyses to assess the functional effects of treatment. From echocardiography, short axis values of left ventricular end systolic (ES) and end diastolic (ED) dimensions were obtained. An average of 3 consecutive cardiac cycles was used for each measurement and was performed three times in by a blinded investigator. Fractional shortening was calculated as [(end-diastolic diameter – end-systolic diameter)/(end-diastolic diameter)] and expressed as a percentage. For invasive hemodynamics, the pressure-volume probe was inserted into the left ventricle. After stabilization, baseline left ventricular pressure-volume loops were recorded for at least 10 cardiac cycles and data averaged to get mean values for each animal. Data extracted include +dP/dT, -dP/dT, end-systolic and end-diastolic pressures and volumes, and left-ventricular Ejection Fraction (%EF).
2.12. In vivo gene expression and plasma cytokine analysis
Following functional measurements at 3 days, animals were sacrificed and the left ventricle infarct tissue was harvested and homogenized, which the total RNA contents were isolated using Trizol (Invitrogen) according to the manufacturer's protocol. Complementary DNA (cDNA) was synthesized using SuperScript III kit (Invitrogen) and the quantitative real-time PCR (qRT-PCR) was performed using Power SYBR Green (Invitrogen) master mix with Applied Biosystems StepOnePlusTM real time PCR system. Absolute quantification method was employed to analyze gene expression levels of each of the target genes. Standard curves for each of the primer sets (Supplemental Table 1) were constructed with serial dilutions of input DNA templates and validated comparable amplification efficiencies (curve slopes: -3.32 to -3.64). Relative mRNA levels were obtained by extrapolation of Ct values from the slopes of the standard curve for each primer set. Gene expression levels were then normalized to the endogenous housekeeping gene GAPDH and further, expressed as fold changes compared to expression levels in sham animals in order to minimize batch-to-batch variability in tissue processing. Prior to sacrificing the animals, blood was collected via cardiac puncture, plasma separated from blood cells and stored in -70°C until cytokine analysis using the Luminex multiplex analyzer system (Luminex LX100). A custom cytokine bead panel from eBioscience was used to analyze cytokines IL-12β, IL-1 β, IL-6 and IL-10 in the plasma.
2.13. Immunohistochemistry
In a separate set of animals (n=3), hearts were immediately snap frozen following sacrifice in OCT compound and transferred to liquid nitrogen, before frozen sections (10μm) were made for staining. To stain for inflammatory cells, sections were fixed with acetone and incubated with anti-CD68 antibody (Abcam) followed by incubation with Alexafuor-647 (Invitrogen). Sections were counterstained with DAPI and imaged on a Zeiss fluorescent microscope with equal exposure times for each sample. TUNEL staining was performed according to manufacturer's protocol (Abcam) following fixation with 4% paraformaldehyde.
2.14. Ca2+ sparks imaging
Ca2+ sparks are elementary SR Ca2+-release events through a cluster of ryanodine receptors (RyR2s) that occur spontaneously during diastole and summate in time and space during systole to form the global Ca2+ transient during E-C coupling. To monitor rapidly changing phenomena such as fluorescence signals from Ca2+ sparks, laser scanning confocal microscopy can be utilized and these line scans enable visualization of rapid fluorescence intensity changes with time. Confocal microscopy (FV1000, Olympus) was used to image Ca2+ sparks, with excitation at 488nm and emission collected at >500nm. Cardiac myocytes were loaded with 20 μm fluo-4/AM for 20 min at room temperature (RT), followed by a 20 min wash in 2 mM Ca2+ Tyrode's at RT. Ca2+ spark measurements were acquired from intact myocytes perfused with 2 mM Ca2+ Tyrode's solution during rest after 1 Hz stimulation in line scan mode at 2 ms/line with a pixel size of 0.155 μm. All fluorescent signals were background subtracted. Changes in [Ca2+]i are expressed as ΔF/F0, where ΔF is the change in fluorescence [measured fluorescence (F) – F0] and F0 is resting baseline fluo-4 fluorescence. For all subsequent Ca2+ imaging experiments, cells were placed on laminin-coated coverslips. Action potentials and global Ca2+ transients were elicited by electrical field stimulation using a pair of platinum electrodes. Experiments were conducted at room temperature (22-24°C).
2.15. Statistics
All statistical analyses were performed using Graphpad Prism 5 software. Quantitative results were presented as means ± SEM. Statistical comparisons were performed by one way analysis of variance (ANOVA) followed by the appropriate post-test as described in the figure legends. P values of less than 0.05 were considered significant.
3. Results and discussions
3.1. Synthesis and characterization of gold-DNAzyme nanoparticles (DzNPs)
DNAzyme functionalized nanoparticles were synthesized via self-assembly through mixing thiolated DNA with citrate stabilized gold nanoparticles and performing a salt titration (Fig. 1A).[46] Particles were functionalized with catalytically active (Dz), inactive (i-Dz) or active but nonspecific (NS-Dz) DNAzymes with a loading of 99±3, 109±7, and 65±2 oligonucleotides per nanoparticle, respectively. This equates to a high surface density ranging from 6 – 9 nm2/DNA, sufficiently high to ensure optimal activity and equivalent particle uptake.[46, 47] It should be noted that NS-Dz has a lower DNA density on the particle due to possessing a stable secondary hairpin structure. The resulting particles were very homogeneous and monodisperse with a mean particle diameter of 14± 3 nm as determined by TEM analysis and ∼80 nm as determined by dynamic light scattering. The extinction spectra showed a peak at 525 nm corresponding to the nanoparticle's surface plasmon resonance (Fig. S1).
Fig. 1. DzNP synthesis and In vitro Cy5-NS-DzNP particle uptake by macrophages.
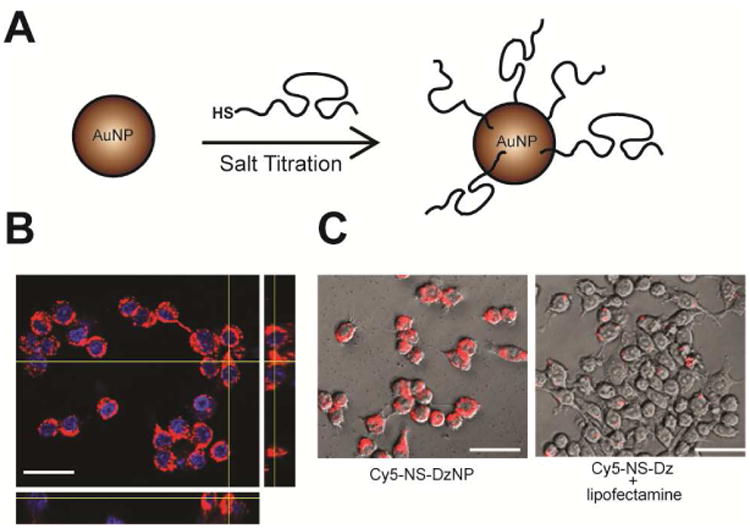
(A) Schematic showing the approach used to synthesize DNAzyme modified gold nanoparticles. Thiolated DNA was mixed with gold nanoparticles, and the salt concentration was gradully increased. (B) RAW 264.7 macrophages were treated with 5 nM Cy5-NS-DNA modified AuNPs and imaged using confocal microscopy following 1 hour of treatment to visualize cytoplasmic localization of Cy5-DNA. The representative Z-stack image confirms internalization of nanoparticles within macrophages (scale bar, 30 μm). (C, left) Uptake efficiency was compared in macrophages treated with equivlant cocnentraions of DNA containing Dz-particles at 1 hour to (C, right) Lipofectamine® transfection reagent for 24 hours. DNA (red, Cy5), Nucleus (blue, DAPI). (Scale bar, 30 μm).
3.2. Internalization of DzNPs by macrophages and myocytes in vitro
For in vitro and in vivo imaging studies, Cy5-labeled nonspecific DNAzyme modified particles (Cy5-NS-Dz) were synthesized in a similar manner. To determine cellular uptake, primary RAW 264.1 macrophages were treated with 5 nM Cy5-NS-Dz particles (300 nM DNA) and fluorescence was measured. We observed non-punctate, diffuse fluorescence intracellularly (Fig. 1B & Fig. S2) in macrophages treated with the Dz-NPs at the 1 hour time point. This indicates that the DNA is diffusely localized to the cytoplasm, in agreement with literature precedent for other cell types and what we have previously observed.[28, 48] Most likely, Dz-NPs remain intact since macrophages were only treated for 1 hour and DNA-NPs have been shown to be stable up to 16 hours.[33] We qualitatively compared the particle uptake with the most commonly used commercial transfection reagent Lipofectamine® RNAiMAX complexed with equivalent DNA concentration (300 nM) through a time course of 24 hours. While the DzNPs were taken up by macrophages as early as 1 hour (Fig. 2B, left) and are continually taken up through 24 hours, Lipofectamine®-mediated transfection was only detected inside cells at the 24 hour time point and in punctate signals indicating possible entrapment within the endosomal compartments (Fig. 1C, right). Phagocytic cells such as macrophages are a particulalry challenging target for transfection and gene silencing, as they are programmed to take up and degrade foreign particles. However, the mechanism of particle internalization is dependent on particle size. Particles below 500 nm in size are usually taken up via endocytosis in phagocytic and non-phagocytic cells,[49] which is preferable in the context of therapeutic delivery. Accordingly, we further show that these particles are taken up by non-phagocytic adult rat cardiomyocytes at 18 hours following transfection (Fig. S3). Notably, there was no observable fluorescence in cardiomyocytes transfected with Cy5-DNA utilizing Lipofectamine® RNAiMAX (Fig. S3).
Fig. 2. Biocompatibility of DNAzyme-gold particles and in vitro knockdown of TNF-α in rat peritoneal macrophages.
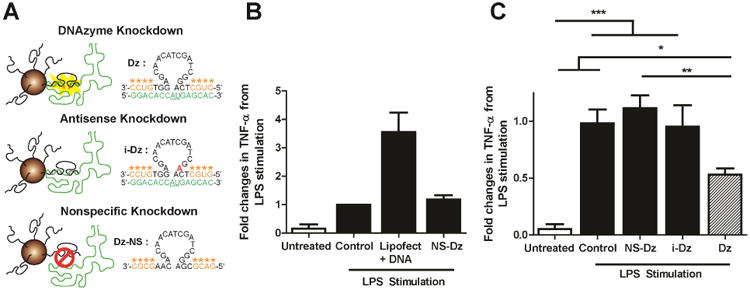
(A) Schematic showing the sequences used for Dz, iDz, and Dz-NS AuNP conjugates. Green indicates the mRNA sequence, underlined bases denotes the location of phosphodiester hydrolysis. The red base shows the location of the mutation that leads to inhibition of Dz catalytic function. (B) Plot showing the fold change in TNF-α levels in untreated and LPS-stimulated macrophages that were also treated with Lipofectamine® as well as NS-Dz AuNPs. TNF-α levels were measured using an ELISA kit at the 24 hour time point and were normalized to untreated LPS stimulated cells and reported as fold changes in TNF-α expression levels. Grouped data (mean ± SEM; n=3). (C) Plot of TNF-α expression levels when macrophages are treated with NS-Dz, i-Dz, and Dz AuNP conjugates at the 24 hour time point. Grouped data (mean ± SEM; n=6) (*p<0.05; **p<0.01; ***p<0.001; One-way ANOVA followed by Tukey' Multiple Comparison post-test).
3.3. DzNPs do not elicit cytotoxicity in vitro
We next investigated whether DNAzyme functionalized AuNPs are a biocompatible delivery vehicle and whether we could deliver catalytically active Dz into macrophages and inhibit TNF-α expression in vitro. In order to establish an in vitro model to demonstrate knockdown of TNF-α, we stimulated primary peritoneal cavity isolated macrophages with bacterial endotoxin LPS following particle treatment to upregulate the production of TNF-α. LPS stimulates TNF-α expression via activation of the Toll Like Receptor (TLR), orchestrating a potent inflammatory response, and therefore TNF-α expression additionally serves as a surrogate assay to evaluate the cytotoxicity profile of particle uptake compared to the commonly used transfection reagent Lipofectamine® RNAiMAX. Upon treatment with Lipofectamine®, macrophages exhibited ∼4 fold higher TNF-α levels compared to the basal LPS stimulation, illustrating the cytotoxic response elicited by cationic lipids, which are widely used for nucleic acid delivery. The nonspecific DNAzyme particle (NS-Dz) treatment caused no significant additional cytotoxicity over the control group (Fig. 2B), thus indicating that DNAzyme functionalized particles do not elicit cytotoxicity and are suitable for applications in an inflammatory disease context such as the post-MI myocardium.
3.4. In vitro TNF-α knockdown
To determine the intracellular catalytic activity of DNAzyme AuNP conjugates within the in vitro macrophage model, active and inactive DNAzymes modified particles were synthesized targeting the start codon (AUG) of the TNF-α mRNA displaying minimal secondary structure. The recognition arms were 7 and 10 bases in length, which was designed based on literature precedent defining DNAzyme design rules as shown in Fig. S4.[37] A single base mutation was introduced in the 15 base catalytic core to inhibit DNAzyme activity of the i-Dz sequence, thus allowing one to distinguish antisense knockdown from DNAzyme mediated knockdown (Fig. 2A). To minimize DNAzyme degradation, four 2′-methyl ether modifications were added to the termini of the recognition arms. Surprisingly, initial tests using DNAzymes with 10 mer long arms showed limited activity, and Dz and i-Dz treated macrophages showed only 14% and 2% knockdown, respectively (Fig. S4). We hypothesized that this was caused by product inhibition due to high stability of the 10 base recognition arms. To address this concern, the recognition sequence for one of the arms was shortened from 10 to 7, in addition to changing the two terminal thymidine bases in the T10 spacer region to two cytosines in order reduce the affinity to the mRNA target. In the optimized design (Fig. 2A), compared to control LPS stimulated cells, only Dz particles showed significant (∼50%) reduction in TNF-α levels while NS-Dz and i-Dz particles showed no effect (Fig. 2C). Together, these results show that spherical nucleic acids can efficiently deliver catalytically active DNAzymes to macrophages and decrease the expression of TNF-α in activated macrophages.
3.5. Ex vivo fluorescence imaging to determine biodistribution of DzNPs
We next investigated the ability of these particles to deliver DNAzymes to the myocardium in a rat model of MI. Cy5-NS-Dz modified gold particles were injected intramyocardially to 3 different regions of the infarct following MI surgery. This route of delivery is clinically relevant in the context of MI as routine interventional procedures such as primary percutaneous interventions (PCI) post-MI require catheterization and can be leveraged for site-specific therapeutic administration.[50] The time-course ex-vivo bioimaging revealed strong Cy5 fluorescence emitting from the heart for all 3 days tested indicating efficient particle uptake and retention in the myocardium following injection for at least up to 3 days (Fig. 3B-C). Due to the pulsatile nature of the heart, as anticipated, particles are introduced to the blood circulation to a certain extent, and secondary to the heart, they predominantly biodistributed to the kidney and liver, suggesting renal and/or hepatobilary clearance.
Fig. 3. Ex-vivo fluorescence imaging of organs from animals injected with Cy5-NS-DzNP.
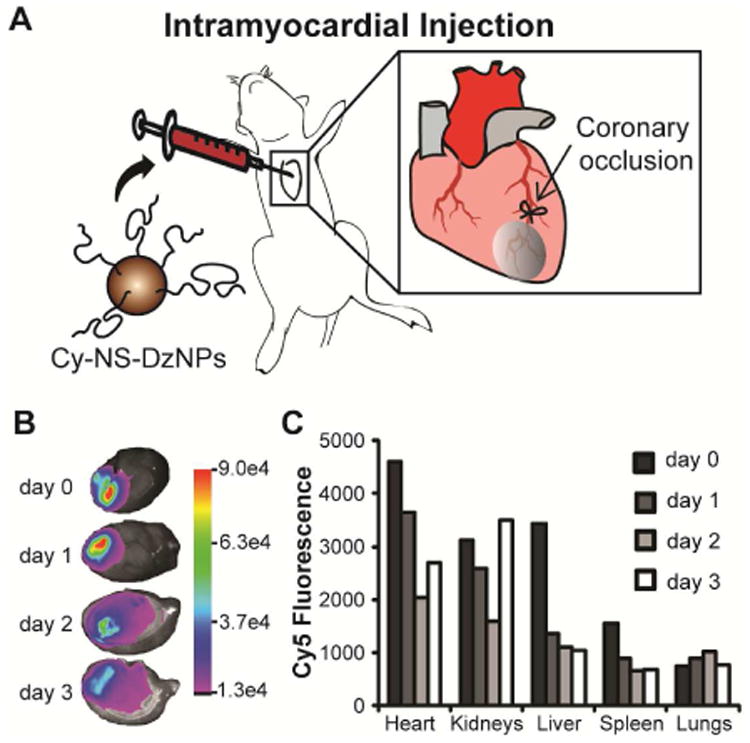
(A) Schematic of ligation of the coronary artery to cause permanent ligation MI in rats followed by particle injection. (B) Fluorescence images of hearts from each day following injection. The heat map indicates the Cy5 emission intensity in arbitrary units. (C) Fluorescence intensity of all organs imaged from days 0, 1, 2 and 3. The Cy5 fluorescence intensity is expressed in arbitrary units. A total of four rats were used where each time point consists of fluorescence from an individual rat.
3.6. Silencing TNF-α improves acute cardiac function and elicits an anti-inflammatory response
To test the potential of DNAzyme functionalized AuNPs as an anti-inflammatory agent for the treatment of acute MI, we investigated the effect of injecting these particles to the myocardium of rats immediately following experimental MI. We used cardiac echocardiography, invasive hemodynamics measurements at a 3-day acute time point, and also measured the inflammatory gene expression profile in the left ventricular infarct tissue. As shown in Fig. 4, delivery of Dz significantly reduced the TNF-α mRNA levels in LV tissue compared to all controls and further, exhibited no statistical significance from sham controls.
Fig. 4. Effect of in vivo delivery of Dz particles on TNF-α gene expression in a rat model of MI.
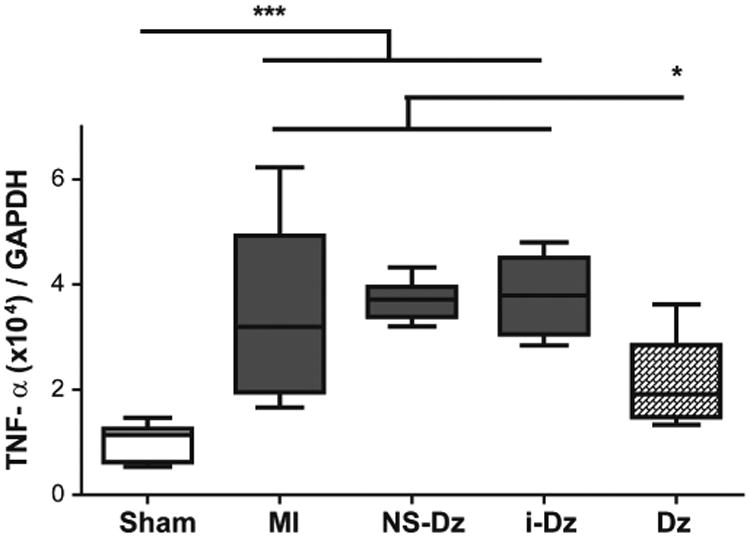
Plots represent grouped data (mean ± SEM; n=5-9) from 3 days. A significant decrease in mRNA expression in the LV infarct issue only occurs in the Dz treated rats compared with all controls. Gene expression was evaluated by qRT-PCR using the quantitative standard curve method, and the results were normalized to GAPDH levels and reported as fold changes in copy number of mRNA levels compared to sham animals (*p<0.05; ***p<0.001; one-way ANOVA followed by Tukey's Multiple Comparison post-test).
Several studies have illustrated the central role that TNF-α plays in myocardial dysfunction following MI by propagating pro-inflammatory cytokines, such as IL-6 and IL-1β, that acutely regulate myocyte survival/apoptosis and trigger further cellular inflammation, including oxidative stress and recruitment of inflammatory cells to the infarct.[12, 51-53] Therefore, to determine the downstream effect of down regulating TNF-α, we measured the gene expression profile of commonly upregulated pro-inflammatory mediators IL-12β, IL-1 β, IL-6 as well as iNOS, an inducible form of Nitric Oxide (NO) synthase stimulated by cytokines such as TNF-α and IL-1b (Fig. 5).[54] Silencing TNF-α gene expression resulted in a strong anti-inflammatory effect that disrupted the gene expression of these pro-inflammatory markers.
Fig. 5. Downstream effects of inhibiting TNF-α on pro-inflammatory cytokine and iNOS gene expression.
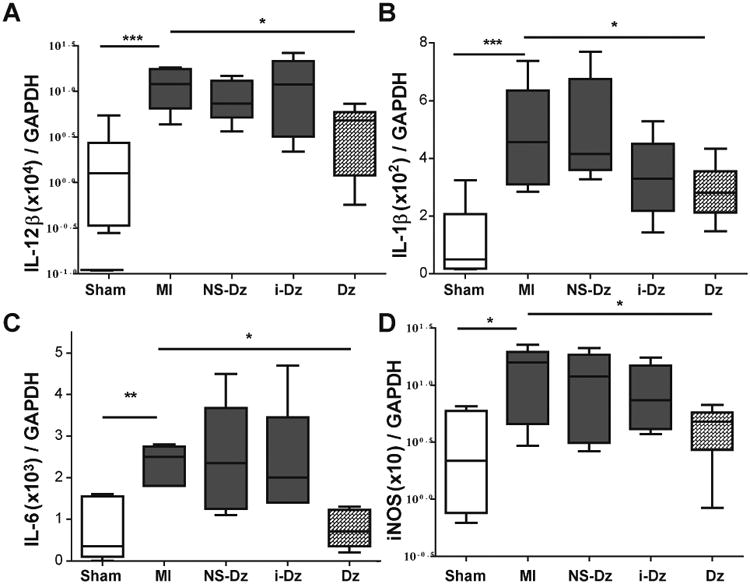
Grouped data (mean ± SEM; n=5-9) from LV infarct tissue at 3 days following MI and injection. There was a significant upregulation in gene expression of pro-inflammatory cytokines (A) IL-12β, (B) IL-1β, (C) IL-6 as well as (D) iNOS enzyme in infarct LV tissue compared to sham animals and was significantly reduced in Dz treated animals. Gene expression was evaluated by qRT-PCR using the quantitative standard curve method, and the results were normalized to GAPDH levels reported as fold changes in copy number of mRNA levels compared to sham animals (*p<0.05; **p,0.01; *** p<0.0001 vs. MI; one-way ANOVA followed by Dunnett's Multiple Comparison post-test).
Systemic levels of these cytokines measured from plasma partially reflected the local gene expression patterns and exhibited a trend of Dz treatment mediated anti-inflammatory effect (Fig. S6 A-C), though this was not statistically significant. An endogenous anti-inflammatory cytokine IL-10, although did not show any statistically significant changes in local gene expression (Fig. S7), did exhibit a trend of upregulation in plasma levels with Dz treatment (Fig. S6 D). Combined with the trend for downregulation of IL-12β only in the Dz treatment group (Fig. S6 A), these results suggest a lower ratio of IL-12/IL-10 in systemic circulation, which is a surrogate measure of anti-inflammatory response employed in several studies due to the apparent opposing relationship of these cytokines and positive implications in resolution of inflammation.[55] It is important to note that these differences were not statistically significant and could indicate the systemic changes do not necessarily reflect what may occur in the local environment, or that changes could diluted in the blood as compared to local tissue RNA.
Functional changes were determined by both echocardiography and pressure-volume catheter analysis. All groups receiving MI demonstrated significantly reduced cardiac function (p < 0.001) as measured in absolute change in fractional shortening compared to sham animals. While there was no effect for NS-Dz or i-Dz particle treated groups, Dz particle treatment significantly restored function by ∼40% compared to the untreated MI group (p < 0.01; Fig. 6A). The invasive hemodynamics measurements also demonstrated a significant improvement in several indices of cardiac function (Fig 6B-F). Ejection fraction, the parameters that measure the rate of change of pressure in the left ventricle chamber (dp/dt), end-systolic volume, and both end-systolic and end-diastolic pressures were all negatively altered in MI mice. While treatment with NS-Dz or i-Dz had no effect, treatment with Dz improved all of these measurements. There were no changes in end-diastolic volume in this study, most likely due to the acute time-point.
Fig. 6. Effect of in vivo delivery of Dz particles on cardiac function.
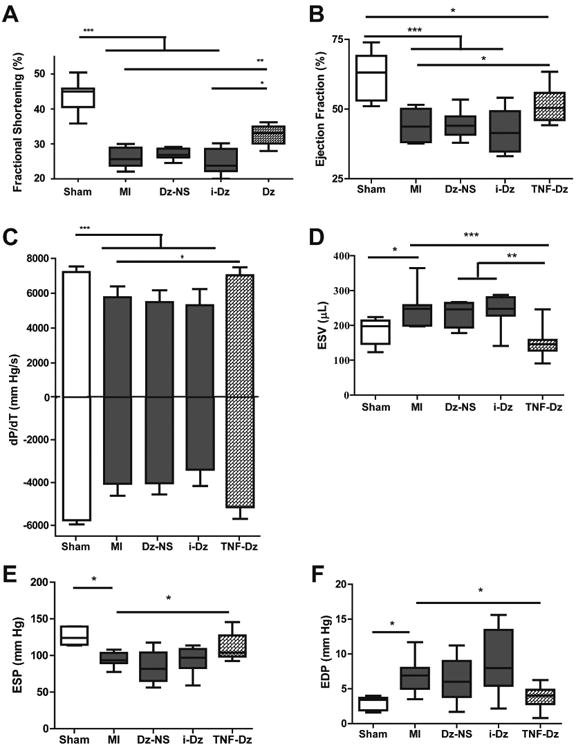
Plots represent grouped data (mean ± SEM; n=8-12). (A) There was a significant decrease in cardiac function in animals that received an MI compared to sham as demonstrated by % FS. Among treatment groups, there was a significant restoration in function only in the Dz treated rats compared with untreated MI and i-Dz groups. Similar effects were seen for (B) ejection fraction, (C) dP/dT, (D) end-systolic volume (ESV), (E) end-systolic pressure (ESP), and (F) end-diastolic pressure (EDP). (*p<0.05; ***p<0.001; one-way ANOVA followed by Tukey's Multiple Comparison post-test).
To determine whether Dz treatment improved acute inflammation and prevented cell death, immunohistochemistry was performed. Representative images are shown in Fig. 7 for both CD68 and TUNEL staining. Qualitatively, there was reduced cellular infiltration and CD68 in Dz treated mice compared with MI alone and mice treated with i-Dz. Similarly, there was extensive TUNEL staining in MI mice and mice treated with i-Dz that was largely absent in Dz treated mice. While the exact cell types were not identified in the study, these data demonstrate that treatment with TNF-α-specific DzNPs reduced local inflammatory responses and improved overall cell survival.
Fig. 7. Histological measurements of inflammation and survival.
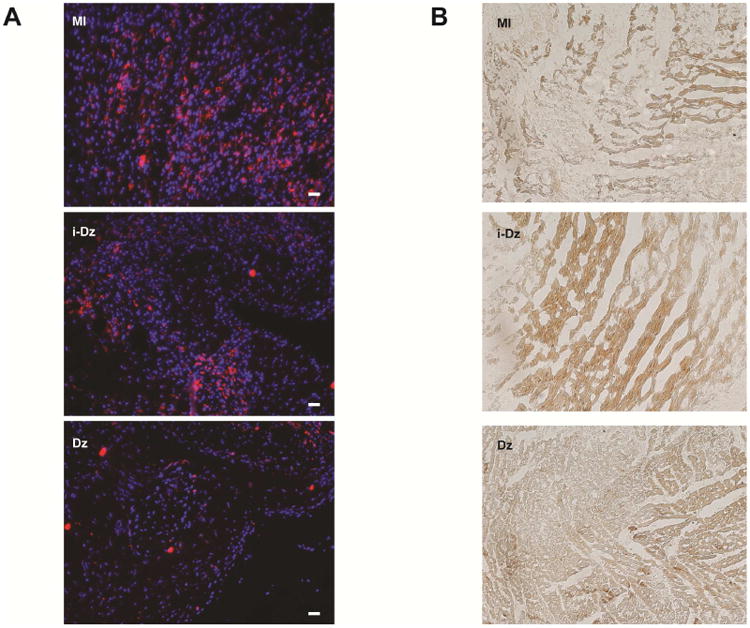
(A) Representative images from CD68 staining showing increased cellularity (DAPI=blue) and increased CD68 staining (red) in MI and i-Dz treated animals (scale bar = 50μm). (B) Representative TUNEL staining showing increased cell death in MI and i-Dz treated animals (images taken in 20× magnification).
Finally, as another potential mechanism, there is substantial evidence showing that TNF-α and subsequently induced pro-inflammatory cytokines IL-6 or IL-1β can attenuate myocyte contractility through the reduction of systolic cytosolic Ca2+ levels via alterations in sarcoplasmic reticulum function,[12, 56] down-regulation of contractile proteins or β-adrenergic receptor uncoupling.[57] TNF-α can decrease myocyte contractility indirectly via oxidative effects on the ryanodine receptor RyR2 [58, 59] or through nitric oxide-dependent attenuation of myofilament Ca2+ sensitivity.[54, 60] It is also well established that induced NO production has a negative inotropic effect on cardiac myocytes and that high levels of NO produced by iNOS are cytotoxic.[60, 61] Reduction in gene expression of pro-inflammatory markers and iNOS resulting from Dz treatment in our study suggests that a combination of direct and indirect mechanisms as a consequence of knocking down TNF-α could have partially contributed to the improvement in cardiac contractility we observe (improvements in dP/dT and ejection fraction). In order to substantiate this proposed mechanism in vitro, we determined whether TNF-α silencing had an effect on myocyte function by recording spatially resolved dyssynchronous Ca2+ sparks via line scan confocal imaging upon stimulating primary adult rat cardiomyocytes with LPS to induce TNF-α production followed by treatment with Dz particles. Our data (Fig. S8) shows reduction in inhomogeneous subcellular Ca2+ spark frequency in cardiomyocytes treated Dz particles compared to the untreated LPS stimulated myocytes, thus demonstrating that suppression of TNF-α in cardiomyocytes may improve calcium handling, and that may be a potential mechanism for future evaluation.
4. Conclusions
Taken together, we demonstrate the first example of using catalytic Dz-AuNPs as an in vivo therapeutic. This likely represents the tip of iceberg, and given the broad range of therapeutic targets that have been proposed and tested for soluble DNAzymes, we expect that DNAzyme-AuNP conjugates to present an important and viable route for re-examining their efficacy in clinical settings. Specifically, this work shows potent gene silencing in vivo via catalytic DNA particles, and identifies significant anti-inflammatory effects brought about as a direct consequence of suppressing the pro-inflammatory cytokine TNF-α in an acute MI setting. While this study shows promising acute functional improvement likely brought about by the anti-inflammatory effect of TNF-α mRNA specific Dz treatment at the initial inflammatory stage post-MI, follow up studies evaluating the chronic effects of this treatment on cardiac remodeling and chronic function are needed to determine whether repeated administrations are necessary to sustain the benefits of transient knockdown of TNF-α. High concentrations of TNF-α has been shown to contribute to structural alterations in the failing heart and are often used as an indicator of chronic HF as described earlier. Therefore, TNF-α gene regulation by DNAzyme-AuNPs merits further investigation beyond acute modulation. [12, 14, 16] Despite these limitations, this study presents a strong case for early anti-inflammatory intervention and the potential to revive TNF-α blockade as a viable therapeutic avenue for the treatment of acute cardiac dysfunction.
Supplementary Material
Acknowledgments
This work has been funded in part with federal funds from the National Heart, Lung and Blood Institute, National Institute of Health, Department of Health and Human Services, under Contract No. HHSN268201000043C to MED. K.S. would like to acknowledge the Alfred P. Sloan Research Fellowship and the Camille-Dreyfus Teacher-Scholar Award for support.
Abbreviations
- TNF-α
Tumor Necrosis Factor-α
- MI
myocardial infarction
- HF
heart failure
- DNAzyme
deoxyribozyme
- Dz
deoxyribozyme
- CHF
chronic heart failure
- TLR
Toll Like Receptor
- PCI
percutaneous interventions
- NO
Nitric Oxide
- IL-12β
Interleukin 12β
- IL-1β
Interleukin 1β
- IL-6
Interleukin 6
- iNOS
Nitric Oxide Synthase
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- LV
left ventricle
Footnotes
Supporting Information: A detailed description of materials and methods along with supplemental figures is available as a separate file. Figures S1-S8 consist of particle characterization, intracellular particle uptake by macrophages and myocytes via confocal microscopy, DNAzyme sequence and optimization data, cardiac hemodynamics measurements, Ca2+ sparks imaging via confocal line scans in adult rat cardiomyocytes and plasma cytokine levels using Luminex panel.
Author Contributions: The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang F, Stouffer GA, Waxman S, Uretsky BF. Late coronary stent thrombosis: early vs. late stent thrombosis in the stent era. Catheterization and cardiovascular interventions : official journal of the Society for Cardiac Angiography & Interventions. 2002;55:142–7. doi: 10.1002/ccd.10041. [DOI] [PubMed] [Google Scholar]
- 3.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. The New England journal of medicine. 2007;357:1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 4.Kereiakes DJ. Adjunctive pharmacotherapy before percutaneous coronary intervention in non-ST-elevation acute coronary syndromes: the role of modulating inflammation. Circulation. 2003;108:III22–7. doi: 10.1161/01.CIR.0000086951.09881.51. [DOI] [PubMed] [Google Scholar]
- 5.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–47. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–6. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 8.Troidl C, Mollmann H, Nef H, Masseli F, Voss S, Szardien S, et al. Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. Journal of cellular and molecular medicine. 2009;13:3485–96. doi: 10.1111/j.1582-4934.2009.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonvini RF. Inflammatory response post-myocardial infarction and reperfusion: a new therapeutic target? Eur Heart J Suppl. 2005;7:I27–I36. [Google Scholar]
- 10.Frangogiannis NG. Targeting the inflammatory response in healing myocardial infarcts. Current medicinal chemistry. 2006;13:1877–93. doi: 10.2174/092986706777585086. [DOI] [PubMed] [Google Scholar]
- 11.Kempf T, Zarbock A, Vestweber D, Wollert KC. Anti-inflammatory mechanisms and therapeutic opportunities in myocardial infarct healing. J Mol Med (Berl) 2012;90:361–9. doi: 10.1007/s00109-011-0847-y. [DOI] [PubMed] [Google Scholar]
- 12.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–53. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 13.Bozkurt B, Kribbs SB, Clubb FJ, Jr, Michael LH, Didenko VV, Hornsby PJ, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–91. doi: 10.1161/01.cir.97.14.1382. [DOI] [PubMed] [Google Scholar]
- 14.Kleinbongard P, Schulz R, Heusch G. TNFalpha in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail Rev. 2011;16:49–69. doi: 10.1007/s10741-010-9180-8. [DOI] [PubMed] [Google Scholar]
- 15.Pagani FD, Baker LS, Hsi C, Knox M, Fink MP, Visner MS. Left ventricular systolic and diastolic dysfunction after infusion of tumor necrosis factor-alpha in conscious dogs. The Journal of clinical investigation. 1992;90:389–98. doi: 10.1172/JCI115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sack M. Tumor necrosis factor-alpha in cardiovascular biology and the potential role for anti-tumor necrosis factor-alpha therapy in heart disease. Pharmacol Ther. 2002;94:123–35. doi: 10.1016/s0163-7258(02)00176-6. [DOI] [PubMed] [Google Scholar]
- 17.Sivasubramanian N, Coker ML, Kurrelmeyer KM, MacLellan WR, DeMayo FJ, Spinale FG, et al. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2001;104:826–31. doi: 10.1161/hc3401.093154. [DOI] [PubMed] [Google Scholar]
- 18.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. The New England journal of medicine. 1990;323:236–41. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 19.Irwin MW, Mak S, Mann DL, Qu R, Penninger JM, Yan a, et al. Tissue expression and immunolocalization of tumor necrosis factor-alpha in postinfarction dysfunctional myocardium. Circulation. 1999;99:1492–8. doi: 10.1161/01.cir.99.11.1492. [DOI] [PubMed] [Google Scholar]
- 20.Jacobs M, Staufenberger S, Gergs U, Meuter K, Brandstatter K, Hafner M, et al. Tumor necrosis factor-alpha at acute myocardial infarction in rats and effects on cardiac fibroblasts. J Mol Cell Cardiol. 1999;31:1949–59. doi: 10.1006/jmcc.1999.1007. [DOI] [PubMed] [Google Scholar]
- 21.Kurrelmeyer KM, Michael LH, Baumgarten G, Taffet GE, Peschon JJ, Sivasubramanian N, et al. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci U S A. 2000;97:5456–61. doi: 10.1073/pnas.070036297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anker SD, Coats AJ. How to RECOVER from RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. International journal of cardiology. 2002;86:123–30. doi: 10.1016/s0167-5273(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 23.Bozkurt B, Torre-Amione G, Warren MS, Whitmore J, Soran OZ, Feldman AM, et al. Results of targeted anti-tumor necrosis factor therapy with etanercept (ENBREL) in patients with advanced heart failure. Circulation. 2001;103:1044–7. doi: 10.1161/01.cir.103.8.1044. [DOI] [PubMed] [Google Scholar]
- 24.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–45. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31:638–46. doi: 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- 26.Czech B, Hannon GJ. Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet. 2011;12:19–31. doi: 10.1038/nrg2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114:100–9. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 28.Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS, Mirkin CA. Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science. 2006;312:1027–30. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- 29.Cutler JI, Auyeung E, Mirkin CA. Spherical nucleic acids. Journal of the American Chemical Society. 2012;134:1376–91. doi: 10.1021/ja209351u. [DOI] [PubMed] [Google Scholar]
- 30.Choi CH, Hao L, Narayan SP, Auyeung E, Mirkin CA. Mechanism for the endocytosis of spherical nucleic acid nanoparticle conjugates. Proc Natl Acad Sci U S A. 2013;110:7625–30. doi: 10.1073/pnas.1305804110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cutler JI, Auyeung E, Mirkin CA. Spherical Nucleic Acids. Journal of the American Chemical Society. 2012;134:1376–91. doi: 10.1021/ja209351u. [DOI] [PubMed] [Google Scholar]
- 32.Choi CHJ, Hao LL, Narayan SP, Auyeung E, Mirkin CA. Mechanism for the endocytosis of spherical nucleic acid nanoparticle conjugates. Proc Natl Acad Sci USA. 2013;110:7625–30. doi: 10.1073/pnas.1305804110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu XCA, Choi CHJ, Zhang C, Hao LL, Mirkin CA. Intracellular Fate of Spherical Nucleic Acid Nanoparticle Conjugates. Journal of the American Chemical Society. 2014;136:7726–33. doi: 10.1021/ja503010a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng D, Giljohann DA, Chen DL, Massich MD, Wang XQ, Iordanov H, et al. Topical delivery of siRNA-based spherical nucleic acid nanoparticle conjugates for gene regulation. Proc Natl Acad Sci U S A. 2012;109:11975–80. doi: 10.1073/pnas.1118425109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jensen SA, Day ES, Ko CH, Hurley LA, Luciano JP, Kouri FM, et al. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci Transl Med. 2013;5:209ra152. doi: 10.1126/scitranslmed.3006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhindi R, Khachigian LM, Lowe HC. DNAzymes targeting the transcription factor Egr-1 reduce myocardial infarct size following ischemia-reperfusion in rats. J Thromb Haemost. 2006;4:1479–83. doi: 10.1111/j.1538-7836.2006.02022.x. [DOI] [PubMed] [Google Scholar]
- 37.Iversen PO, Nicolaysen G, Sioud M. DNA enzyme targeting TNF-αlpha mRNA improves hemodynamic performance in rats with postinfarction heart failure. Am J Physiol Heart Circ Physiol. 2001;281:H2211–7. doi: 10.1152/ajpheart.2001.281.5.H2211. [DOI] [PubMed] [Google Scholar]
- 38.Luo X, Cai H, Ni J, Bhindi R, Lowe HC, Chesterman CN, et al. c-Jun DNAzymes inhibit myocardial inflammation, ROS generation, infarct size, and improve cardiac function after ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2009;29:1836–42. doi: 10.1161/ATVBAHA.109.189753. [DOI] [PubMed] [Google Scholar]
- 39.Xiang G, Schuster MD, Seki T, Witkowski P, Eshghi S, Itescu S. Downregulated expression of plasminogen activator inhibitor-1 augments myocardial neovascularization and reduces cardiomyocyte apoptosis after acute myocardial infarction. J Am Coll Cardiol. 2005;46:536–41. doi: 10.1016/j.jacc.2005.04.047. [DOI] [PubMed] [Google Scholar]
- 40.Zhang L, Gasper WJ, Stass SA, Ioffe OB, Davis MA, Mixson AJ. Angiogenic inhibition mediated by a DNAzyme that targets vascular endothelial growth factor receptor 2. Cancer Res. 2002;62:5463–9. [PubMed] [Google Scholar]
- 41.Yang L, Xu Z, Liu L, Luo X, Lu J, Sun L, et al. Targeting EBV-LMP1 DNAzyme enhances radiosensitivity of nasopharyngeal carcinoma cells by inhibiting telomerase activity. Cancer biology & therapy. 2014;15:61–8. doi: 10.4161/cbt.26606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yehl K, Joshi JP, Greene BL, Dyer RB, Nahta R, Salaita K. Catalytic deoxyribozyme-modified nanoparticles for RNAi-independent gene regulation. ACS Nano. 2012;6:9150–7. doi: 10.1021/nn3034265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill HD, Mirkin CA. The bio-barcode assay for the detection of protein and nucleic acid targets using DTT-induced ligand exchange. Nat Protoc. 2006;1:324–36. doi: 10.1038/nprot.2006.51. [DOI] [PubMed] [Google Scholar]
- 44.Hurst SJ, Lytton-Jean AK, Mirkin CA. Maximizing DNA loading on a range of gold nanoparticle sizes. Analytical chemistry. 2006;78:8313–8. doi: 10.1021/ac0613582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang GW, Guo Y, Vondriska TM, Zhang J, Zhang S, Tsai LL, et al. Acrolein consumption exacerbates myocardial ischemic injury and blocks nitric oxide-induced PKCepsilon signaling and cardioprotection. J Mol Cell Cardiol. 2008;44:1016–22. doi: 10.1016/j.yjmcc.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 46.Yehl K, Joshi JP, Greene BL, Dyer RB, Nahta R, Salaita K. Catalytic deoxyribozyme-modified nanoparticles for RNAi-independent gene regulation. ACS nano. 2012;6:9150–7. doi: 10.1021/nn3034265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giljohann DA, Seferos DS, Patel PC, Millstone JE, Rosi NL, Mirkin CA. Oligonucleotide loading determines cellular uptake of DNA-modified gold nanoparticles. Nano Lett. 2007;7:3818–21. doi: 10.1021/nl072471q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yehl K, Joshi JR, Greene BL, Dyer RB, Nahta R, Salaita K. Catalytic Deoxyribozyme-Modified Nanoparticles for RNAi-Independent Gene Regulation. ACS nano. 2012;6:9150–7. doi: 10.1021/nn3034265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Champion JA, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharmaceutical research. 2008;25:1815–21. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hastings CL, Roche ET, Ruiz-Hernandez E, Schenke-Layland K, Walsh CJ, Duffy GP. Drug and cell delivery for cardiac regeneration. Adv Drug Deliv Rev. 2014 doi: 10.1016/j.addr.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 51.Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J Exp Med. 1996;183:949–58. doi: 10.1084/jem.183.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ono K, Matsumori A, Shioi T, Furukawa Y, Sasayama S. Cytokine gene expression after myocardial infarction in rat hearts: possible implication in left ventricular remodeling. Circulation. 1998;98:149–56. doi: 10.1161/01.cir.98.2.149. [DOI] [PubMed] [Google Scholar]
- 53.Moe GW, Marin-Garcia J, Konig A, Goldenthal M, Lu X, Feng Q. In vivo TNF-αlpha inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1813–20. doi: 10.1152/ajpheart.00036.2004. [DOI] [PubMed] [Google Scholar]
- 54.Habib FM, Springall DR, Davies GJ, Oakley CM, Yacoub MH, Polak JM. Tumour necrosis factor and inducible nitric oxide synthase in dilated cardiomyopathy. Lancet. 1996;347:1151–5. doi: 10.1016/s0140-6736(96)90610-8. [DOI] [PubMed] [Google Scholar]
- 55.Watson DC, Sargianou M, Panos G. Interleukin-12 (IL-12)/IL-10 ratio as a marker of disease severity in Crimean-Congo hemorrhagic fever. Clinical and vaccine immunology : CVI. 2012;19:823–4. doi: 10.1128/CVI.00030-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL. Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest. 1993;92:2303–12. doi: 10.1172/JCI116834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gulick T, Chung MK, Pieper SJ, Lange LG, Schreiner GF. Interleukin 1 and tumor necrosis factor inhibit cardiac myocyte beta-adrenergic responsiveness. Proc Natl Acad Sci U S A. 1989;86:6753–7. doi: 10.1073/pnas.86.17.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mazurek SR, Bovo E, Zima AV. Regulation of sarcoplasmic reticulum Ca(2+) release by cytosolic glutathione in rabbit ventricular myocytes. Free radical biology & medicine. 2014;68:159–67. doi: 10.1016/j.freeradbiomed.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 59.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–21. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 60.Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–9. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- 61.Song W, Lu X, Feng Q. Tumor necrosis factor-alpha induces apoptosis via inducible nitric oxide synthase in neonatal mouse cardiomyocytes. Cardiovasc Res. 2000;45:595–602. doi: 10.1016/s0008-6363(99)00395-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.