

Abstract
Bacteria are capable of performing a number of biotransformations that may activate or deactivate xenobiotics. Recent efforts have utilized metabolomics techniques to study the fate of small molecule antibacterials within the targeted organism. Examples involving Mycobacterium tuberculosis are reviewed and analyzed with regard to the insights they provide as to both activation and deactivation of the antibacterial. The studies, in particular, shed light on biosynthetic transformations performed by M. tuberculosis while suggesting avenues for the evolution of chemical tools, highlighting potential areas for drug discovery, and mechanisms of approved drugs. A two–pronged approach investigating the metabolism of antibacterials within both the host and bacterium is outlined and will be of value to both the chemical biology and drug discovery fields.
Keywords: biotransformation, metabolism, metabolomics, antibacterial, tuberculosis, small molecule
A Window into Mycobacterium tuberculosis
Mycobacterium tuberculosis causes tuberculosis (TB), which is the leading cause of death from an infectious disease worldwide, and is emblematic of the global health pandemic due to bacterial infections. The World Health Organization reported 10.4 million cases of TB in 2015 and 1.4 million deaths (http://www.who.int/tb/publications/global_report/gtbr2016_executive_summary.pdf?ua=1). Amongst HIV–infected individuals, 400,000 deaths were attributed to TB co-infection. With the emergence of multi–drug resistant and extremely drug–resistant strains, the treatment options have become limited, stressing the need for new therapies.
Various drug discovery approaches have been adopted to identify new antituberculars, but a significant issue centers on the identification and validation of novel drug targets [1]. Pioneering work by Sassetti and Rubin sought to address this in part through the identification of a set of genes required for optimal in vitro growth of M. tuberculosis [2]. Moving this work forward, we and the Lamichhane laboratory adopted a high–throughput approach for identifying essential molecules of M. tuberculosis that were products of enzymes that are encoded by these essential genes [3]. These essential metabolites could serve as a blueprint for the development of new antitubercular agents, given knowledge of the antimetabolite strategy [3]. The computational approach led to candidate metabolite mimics of L–glutamate. Since these compounds and other antitubercular agents may in many cases mimic an essential metabolite, we pondered the potential for their susceptibility to the bacterium’s metabolizing enzymes.
The importance of biotransformations within M. tuberculosis is well known with respect to the activation of current first–line and second–line drugs, i.e., isoniazid (INH), pyrazinamide (PZA), ethionamide (ETH), and aminoglycosides such as streptomycin, amikacin, and kanamycin. INH is activated by a catalase peroxidase encoded by katG and mutations in this gene can confer INH resistance to M. tuberculosis [4]. Subsequent to this discovery, a genetic strategy was used to identify the target of the activated INH complex as InhA, an enoyl reductase [5]. In the case of PZA, although its mechanism of action is not fully understood, it is known that inside M. tuberculosis, PZA is converted to pyrazinoic acid by PncA, a nicotinamidase [6–8]. ETH activation is primarily attributed to one of the six monooxygenases encoded by M. tuberculosis that contains a Baeyer–Villiger monooxygenase (BVMO) motif [9–11]. The aminoglycoside second–line drugs are also known to undergo biotransformations within M. tuberculosis. Acetyltransferases, and phosphotransferases have been shown to modify the hydroxyl or amino groups through amidation, esterification, or phosphorylation reactions, ultimately leading to the deactivation of aminoglycosides [12–17].
While many of these early studies relied on demonstration of the metabolism, or more specifically activation, of these antitubercular drugs by a purified enzyme outside of the cell, a more recent approach to track biotransformation products formed within M. tuberculosis was pioneered by the Rhee laboratory employing metabolomics technologies [18, 19]. While primarily focused on the inventory of metabolites of the cell, metabolomics methods have recently demonstrated significant promise in elucidating the intrabacterial fate of antibacterials, shedding light on their metabolism that is either activating (i.e., indicating a prodrug mechanism) and/or deactivating (i.e., signaling a detoxification pathway) [20]. Driven by the need to identify novel antitubercular agents, research laboratories are adopting metabolomics as a platform technology to evolve antibacterial chemical tools and drug discovery hit and lead compounds [21]. In this review, we will describe such efforts, grouping metabolic transformations in an early effort to generalize the types of chemical reactions of which M. tuberculosis is capable, and propose a new workflow for antitubercular chemical tools and drug discovery that involves understanding drug metabolism and biotransformations occurring within M. tuberculosis.
Biotransformation of Xenobiotics by M. tuberculosis
M. tuberculosis has the ability to perform biotransformation reactions on xenobiotics which may be to the bacterium’s benefit or detriment. Transformation reactions, such as activation of INH and sulfur oxidation in small molecules by BVMOs, have been identified and probed via genetic and biochemical approaches [4, 9, 10, 22, 23]. Amongst the known BVMOs, EthA has been extensively studied and has been shown to oxidize thioamide, thiourea, and thiophene groups [10, 22, 23]. In particular, EthA is required for the bioactivation of ETH and the second–line antituberculars thiacetazone and isoxyl [9, 10, 22, 23]. Furthermore, Hung and co–workers have identified another BVMO, MymA [24]. Most of the approaches employed to identify the bioactivation mechanism involved whole–genome sequencing of spontaneous resistant mutants, obtained by exposure of M. tuberculosis to the antitubercular, where the activating gene suffered a mutation that effectively reduced the concentration of the cidal species. The next step was typically demonstration that overexpression of this mutation conferred increased resistance to the antitubercular or that the purified gene product was responsible for bioactivation in the absence of the bacterium. Here, we discuss the use of metabolomics methods, used alone or in combination with genetic and biochemical approaches, as a powerful platform to characterize chemical transformations M. tuberculosis can perform on small molecules.
1. Hydrolysis
Ester hydrolysis is a common chemical transformation (Figure 1). We assert that its role in intrabacterial xenobiotic metabolism has been undervalued. In M. tuberculosis, esterases and lipases are suspected to play an important role in persistence [25–27].
Figure 1.
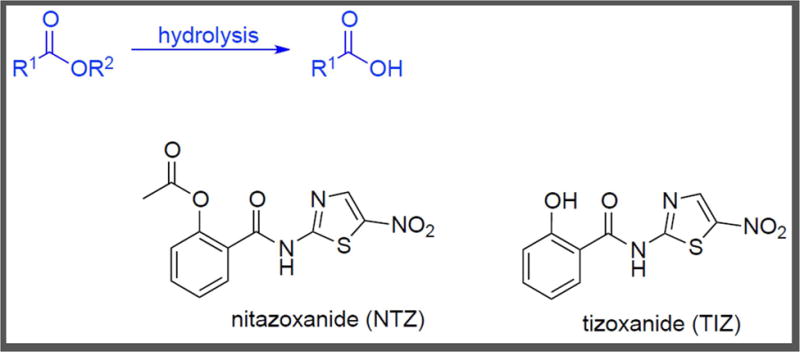
Ester Hydrolysis. Targeted metabolomic analysis of M. tuberculosis treated with NTZ showed dose–dependent accumulation of the compound and partial conversion to TIZ [38].
Tallman et al. exploited activity–based probes and fluorogenic esterase substrates to compile a comprehensive list of M. tuberculosis esterases that retained activity under replicating and non–replicating conditions [28, 29]. M. tuberculosis has the machinery to hydrolyze small molecule esters, reiterating the importance of studying this biotransformation.
Nitazoxanide (NTZ, Figure 1) is an FDA approved drug for the treatment of parasitic infections caused by Giardia and Cryptosporidium [30, 31]. It is active against anaerobic and aerobic Gram–positive bacteria in addition to anaerobic Gram–negative bacteria [32–35]. NTZ is deacetylated, or hydrolyzed, in the stomach to form tizaxonide (TIZ, Figure 1), which then inhibits pyruvate–ferredoxin oxidoreductase (PFOR) and presumably also nitroreductases and peptide disulfide isomerases [36]. de Carvalho et al. also found NTZ and TIZ to be equipotent as growth inhibitors of M. tuberculosis, and NTZ to also be active against non–replicating M. tuberculosis [37]. Leveraging metabolomics methods established in their laboratories [38, 39] to explore the mechanism of action, they studied the intrabacterial fate of NTZ and TIZ (Figure 1) [40]. M. tuberculosis was exposed to 1, 4, and 10 fold the minimum inhibitory concentration (MIC: 1 μg/mL) of NTZ and liquid chromatography/mass spectrometry (LC–MS) analysis of the cell lysate showed a dose–dependent accumulation of NTZ. Also observed was a dose–dependent conversion of NTZ to TIZ within M. tuberculosis. Moreover, an approximately 1000–fold greater accumulation of TIZ as compared to NTZ was found. Since NTZ and reactive nitrogen intermediates (RNI) have exhibited synergy in killing M. tuberculosis [37], their effect on the accumulation of the compounds was also studied. M. tuberculosis grown on plates containing NTZ and RNI did not show a significant increase in NTZ uptake nor increased conversion of NTZ to TIZ, as monitored by LC–MS analysis of the cell lysate. In summary, this work demonstrated both NTZ and TIZ (via intracellular ester hydrolysis) accumulated within NTZ–treated M. tuberculosis, and both molecules may be the subject of mechanism of action studies and molecular optimization efforts.
Nixon and co–workers [41] studied the mechanism of action of antifolates (Figure 2) such as WR99210 and the methotrexate family [42, 43]. Methotrexate is a potent dihydrofolate reductase (DHFR; encoded by dfrA) inhibitor (IC50 = 6.8 nM), but lacks significant whole–cell efficacy (MIC > 100 μM) against M. tuberculosis [43]. Two methotrexate analogs, JSF-1183, the diethyl ester, and JSF-1187, the dimethyl ester, were active against M. tuberculosis with MIC values of 0.6 μM and 0.2–0.4 μM, respectively. Also, JSF-1183 and JSF-1187 were shown to inhibit DHFR with IC50 values of 30 nM and 50 nM, respectively. Overexpression and knockdown strains of M. tuberculosis dfrA were supportive of DHFR engagement within the bacterium. To determine the fates of JSF-1183 and JSF-1187 and identify the active species formed inside M. tuberculosis, the group exposed the bacteria to the two compounds followed by LC-MS analysis of the cell lysate. Both compounds accumulated inside M. tuberculosis in approximately dose–dependent fashion. Additionally, it was noted that the parent compounds were partially converted to the corresponding mono–esters and free methotrexate within M. tuberculosis. Based on this information, the mono–esters of JSF-1183 were synthesized and tested for antitubercular whole–cell efficacy. Both mono–ethyl esters, JSF-2104 and JSF-2105, were significantly less active against M. tuberculosis with MIC values of 33 μM, while still exhibiting significant sub-micromolar IC50 values versus DHFR.
Figure 2.
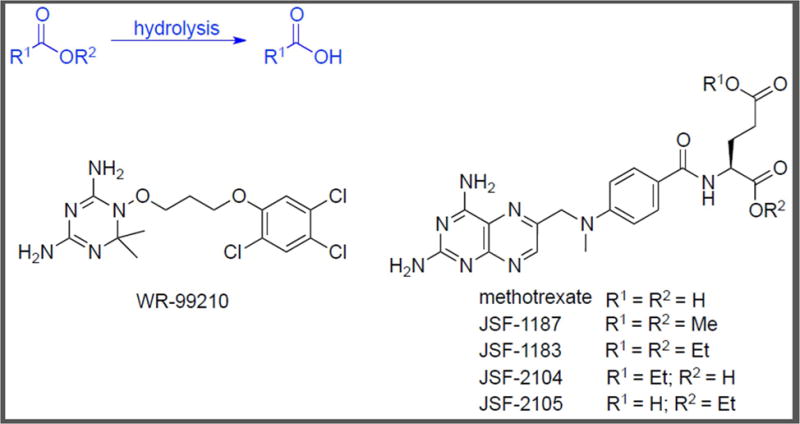
Chemical Structures of WR-99210 and Methotrexate Analogs, JSF-1187 and JSF-1183. These accumulate inside M. tuberculosis in a dose–dependent manner as confirmed by metabolomics [43].
Thus, metabolomics revealed intrabacterial metabolism of the methotrexate diester to both mono–esters as well as methotrexate. In this case, M. tuberculosis mitigated but did not abolish the whole–cell activity of JSF-1183 by hydrolysis of the ethyl ester and may also have altered the engagement of DHFR within the cell. Moreover, polypharmacology of the diester class may exist due to the generation of these multiple species within the bacterium, warranting further study of mechanism. The evolution of methotrexate analogs also has been influenced, given the option to either circumvent or embrace the intrabacterial hydrolysis of the diester parent.
An M. tuberculosis whole–cell growth inhibition screen of 300 known Plasmodium falciparum enoyl acyl carrier protein reductase inhibitors for antitubercular activity by Vilchèze and co–workers identified thienopyrimidine CD117 (Figure 3) as an active with an MIC of 0.39 μg/mL [44]. While the compound was stable in Middlebrook 7H9 medium, its poor mouse liver microsomal stability (t1/2 < 1 min) was attributed to rapid hydrolysis of the ethyl ester to afford the corresponding carboxylic acid (independently synthesized as JSF-2000; MIC > 200 μg/mL) as the major metabolite [21]. Metabolic instability of the ethyl ester was also responsible for a lack of antitubercular activity in ex vivo studies with mice. The evolved structure–activity relationship (SAR) studies demonstrated that analogs with larger ester substituents, such as α,α–dimethylphenyl and t–butyl, had significantly diminished whole–cell activity while those featuring smaller moieties (e.g., n–propyl, n–butyl) retained most of the in vitro efficacy of the parent compound. A prodrug hypothesis was consistent with these observations in that a relatively small, hydrophobic substituent could be required for permeability of M. tuberculosis (whereas the hydrophilic and charged carboxylic acid may not cross the waxy cell wall of the bacterium) and then an esterase could hydrolyze the ester to the carboxylic acid.
Figure 3.
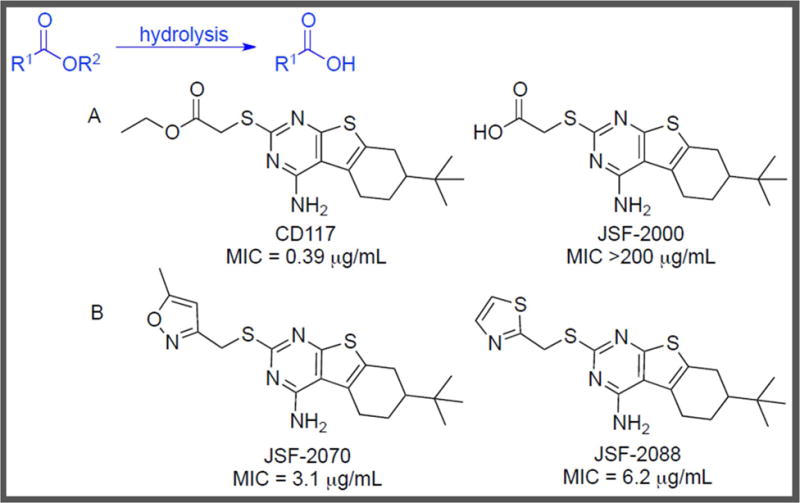
A) Ester Hydrolysis of CD117 Affords JSF-2000. While metabolomics analysis showed M. tuberculosis uptake of both CD117 and JSF-2000, CD117 is partially hydrolyzed to JSF-2000 inside the bacterium. B) Two CD117 analogs with improved metabolic stability are JSF-2070 and JSF-2088 [21].
Li et al. employed metabolomics to probe the CD117 intramycobacterial metabolism and test this hypothesis [21]. M. tuberculosis was exposed to 1x, 5x and 10x the MIC of CD117 or equimolar amounts of the carboxylic acid JSF-2000 [21, 45]. After overnight exposure, the M. tuberculosis cultures were harvested and the cell lysates were analyzed via LC–MS to monitor the uptake of CD117 or JSF-2000 as well as determine the intrabacterial fate of CD117. CD117 displayed dose–dependent accumulation inside M. tuberculosis as well as partial hydrolysis to the inactive acid. Additionally, the inactive acid JSF-2000 was shown to accumulate inside M. tuberculosis. These results were inconsistent with the prodrug hypothesis, as JSF-2000 apparently lacks whole–cell activity despite being cell permeable. Lacking knowledge of the primary biological target of CD117, the authors were unable to attribute the diminished bacterial growth inhibition of JSF-2000 to a loss of functional inhibition of a specific protein. Armed with this information, synthetic efforts were directed toward designing stable ester isosteres of CD117 to prevent formation of the inactive acid within M. tuberculosis and also to improve mouse liver microsomal stability. Two analogs, a 5–methylisoxazole (JSF-2070, MIC = 3.1 μg/mL) and a thiazole (JSF-2088, MIC = 6.2 μg/mL), showed enhanced metabolic stability compared to CD117. Thus, Li et al. leveraged knowledge of the intrabacterial and host metabolism of CD117 to direct their SAR studies and successfully identified two analogs with enhanced metabolic stability. Understanding of the metabolic transformations performed by M. tuberculosis allowed for focused medicinal chemistry efforts that led to the identification of alternate antitubercular agents.
The metabolism studies with these three different antitubercular families demonstrate the potential for M. tuberculosis to hydrolyze the ester functionality. The extent of the hydrolysis and the whole–cell efficacy of the resulting metabolite can vary widely. While not mentioned in these examples, it may be necessary to evaluate the consequence of liberating both the carboxylic acid and the alcohol.
2. N–alkylation
Amines of sufficient nucleophilicity may be alkylated in the presence of an appropriately activated alkyl group. For example, in M. tuberculosis, MamA, a DNA methyltransferase, N–methylates adenine in a six base pair recognition sequence and thereby modulates expression of certain genes [46].
Recent mechanistic studies with M. tuberculosis have uncovered N–methylation, in particular, as a means to evolve drug resistance [47]. Warrier et al. found that pyridobenzimidazole compound 14 (Figure 4) – a cidal antitubercular agent – was inactivated by Rv0560c [47]. They observed via LC–MS that co–incubation of compound 14 with purified Rv0560c and S–adenosyl–L–methionine (SAM) led to the transfer of a methyl group from SAM to the N-5 position of compound 14 to form me-14. To further understand the intrabacterial fate of the compound, M. tuberculosis overexpressing rv0560c and two resistant mutants to compound 14, namely 1A and 8A, were treated with 20x MIC of compound 14 for metabolomics analysis. As expected, the intracellular concentration of compound 14 showed a fivefold reduction in the resistant clones and a twofold reduction in rv0560c–overexpression strain when compared to wild–type M. tuberculosis. However, a corresponding increase in me-14 was not observed, suggesting that me-14 may be further modified by M. tuberculosis. In a Δrv0560c strain, levels of me-14 were twenty–fold lower and compound 14 levels were ~ten–fold higher than in wild–type M. tuberculosis. These levels reverted to wild–type metrics with constitutive expression of rv0560c in the knockout strain. The metabolomics experiments, combined with the observed N–methylation in the presence of purified Rv0560c, confirmed that Rv0560c catalyzed the N–methylation of compound 14. Furthermore, lack of inhibition of M. tuberculosis growth in the presence of increasing concentrations of N–methylated compound 14 demonstrated that this enzymatic biotransformation of compound 14 led to its deactivation inside M. tuberculosis.
Figure 4.
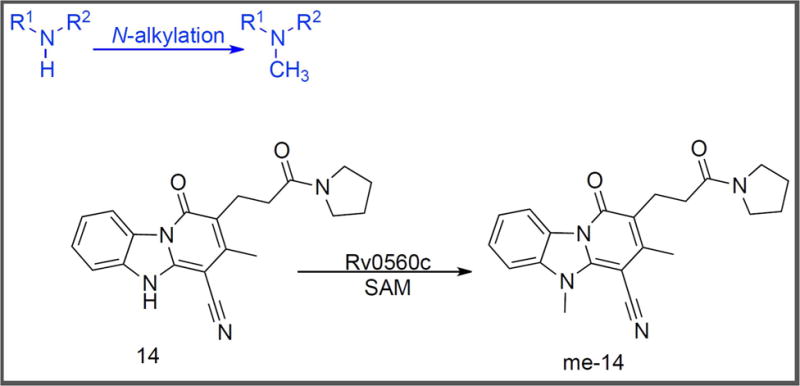
N–methylation of Compound 14 [47].
Following streptomycin, p–aminosalicylic acid was developed by Lehmann as an antitubercular [48, 49]. PAS is a close analog of p–aminobenzoate (PABA), the native substrate of dihydropteroate synthase (DHPS) and an essential metabolite in folate biosynthesis. PAS has been identified as a competitive inhibitor of DHPS with an apparent inhibition constant (Kiapp) of 1 μM [50]. In contrast to some of the more potent DHPS inhibitors like sulfamethoxazole, sulfanilamide and dapsone [51, 52], PAS possesses activity against in vitro cultured M. tuberculosis. To understand this phenomenon and further elucidate the mode of action of PAS, Chakraborty et al. performed extensive studies leveraging metabolomics methods to profile the M. tuberculosis response to PAS [53, 54]. Dose–dependent accumulation of PAS was observed within the bacteria, demonstrating cell permeability. Metabolite identification and comparison with pure synthetic standards established conversion of PAS to N–monomethylated PAS and N,N–dimethylated PAS (Figure 5A). N–monomethylated PAS was equipotent to PAS against in vitro cultured M. tuberculosis and the accumulation pattern of PABA was attributed to enzymatic transformation of PAS to N–monomethylated PAS, which also inhibited DHPS. In contrast, N,N–dimethylated PAS was not active against M. tuberculosis. Thus, in this case, amine methylation by M. tuberculosis can produce metabolites of varying whole–cell activities, including a complete loss of efficacy.
Figure 5.
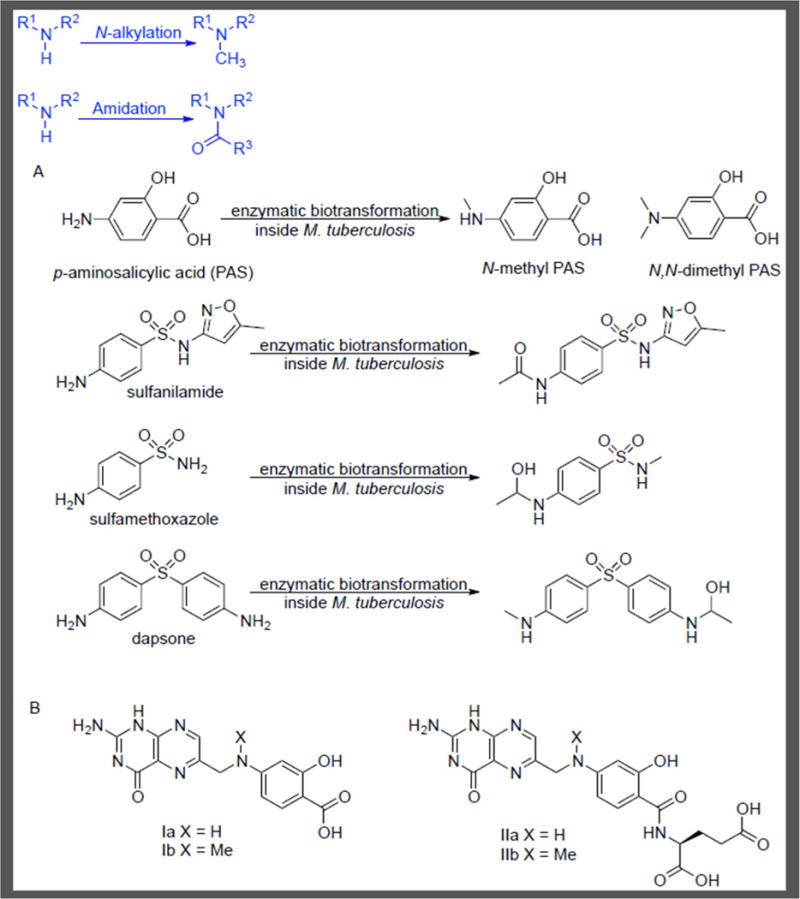
Amine Metabolism with Antifolates. A) Biotransformation of PAS, sulfanilamide, sulfamethoxazole and dapsone in M. tuberculosis. B) Metabolomics analysis showed concentration–dependent formation of PAS–derived Ia & IIa and N–methyl PAS Ib & IIb after exposure of M. tuberculosis to PAS for 18 h [53].
Further analyses showed N–monomethylated PAS and PAS–derived pteroate and folate analogs accumulated through the action of two M. tuberculosis enzymes belonging to the folate pathway, DHPS (FolP1) and dihydrofolate synthase/folyl–polyglutamyl synthase (FolC) (Figure 5B). In vitro studies with purified recombinant FolP1 and FolC corroborated these results by showing PAS to be a substrate for these two enzymes. Data also suggested that these pteroate and folate analogs can mimic the natural substrate of FolC and thereby undergo FolC catalyzed addition of L–glutamate. The subsequent L–glutamate adducts, IIa and IIb (Figure 5B), may also mimic the natural substrate of DHFR. This result is corroborated by the report from Zheng et al. wherein the antimetabolite IIa has been proposed to inhibit DHFR [55].
Metabolomics was used to monitor directly the accumulation of a drug, probe its biotransformation products, and observe their effects on folate–dependent metabolite pools. The understanding of the mode of action of a drug developed in 1944 was significantly expanded. This approach highlights how metabolomics methods can be used to elucidate how the bacterial machinery can be turned against itself to transform a drug into metabolites with inhibitory activity.
3. Amidation
Amides form the essential linkage of peptides and proteins while providing an omnipresent bond in small molecule chemical tools and drugs. Correspondingly, in bacteria numerous examples exist of amidation within biosynthetic pathways, including for example peptidoglycan biosynthesis [56, 57]. Thus, it should not be surprising that xenobiotics with the requisite chemical functionality (amine, carboxylic acid, etc.) can undergo amide bond–forming reactions within a bacterium. Chakraborty et al. also examined the metabolic fate of other DHPS inhibitors such as sulfamethoxazole (MIC = 50 μg/mL), sulfanilamide (whole–cell inactive) and dapsone (whole–cell inactive) within M. tuberculosis [53]. Metabolite identification indicated that these compounds were inactivated via biotransformations inside M. tuberculosis (Figure 5A). Sulfanilamide is amidated, or more specifically N–acetylated, inside M. tuberculosis resulting in a loss of activity. Sulfamethoxazole and dapsone in addition to N–methylation also undergo acetylation followed by apparent reduction to form a hemiaminal. Thus, their lack of whole–cell activity may be attributed to inactivation by these biotransformations and knowledge of these metabolic liabilities may form the basis of future drug discovery optimization efforts.
4. Nitro reduction
The nitro functional group, while not always the first choice for medicinal chemists due to potential toxicity issues [58, 59], may be found in numerous antibacterials of clinical utility, including nitrofurantoin and metronidazole. PA-824 (pretomanid) and delamanid are two nitroimidazoles currently in clinical trials for the treatment of M. tuberculosis infection. While evidence has existed for decades as to the importance of bioactivation of nitro–containing small molecules [59, 60], the exact chemical transformations have only recently been illuminated, particularly within M. tuberculosis. From a biological perspective, nitroreductases have been identified within M. tuberculosis although their native role is unknown [61].
The clinical stage antitubercular PA-824 (Figure 6) undergoes one or more chemical transformations within M. tuberculosis to generate reactive nitrogen species (RNS), which include nitric oxide (NO) [62, 63]. It is activated by a deazaflavin (F420)–dependent nitroreductase (Ddn) in conjunction with glucose-6-phosphate dehydrogenase Fgd1 and the F420 cofactor [62]. Dogra et al. compared the metabolites of PA-824 generated following incubation with M. tuberculosis, M. smegmatis (a non–pathogenic close relative of M. tuberculosis), human liver homogenate 9000x g supernatant (HLS9) or purified Ddn [64]. HLS9 were included to query what biotransformations may occur within the host.
Figure 6.
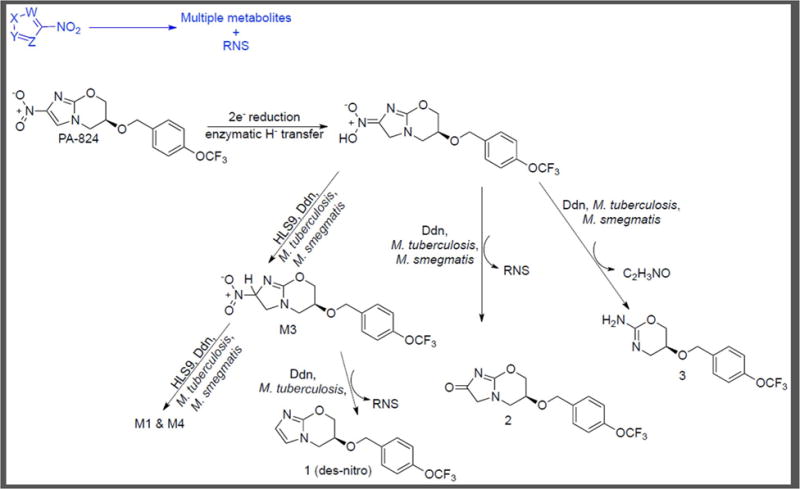
Metabolism of PA-824 [64].
Unlike the previously mentioned metabolomics procedures where filter–laden bacteria were used [45], this study utilized M. tuberculosis or M. smegmatis grown to OD650 = 0.2 in Middlebrook 7H9 broth, harvested and then resuspended in fresh media. PA-824 was added to each culture which was then incubated for 3 h [53]. Alternatively, a pre–incubated mixture of purified Ddn, Fgd1, F420 cofactor and glucose-6-phosphate in pH 7.4 phosphate buffer was treated with PA-824 for 3 h. LC–MS analysis of the extracted metabolites showed that PA-824 formed seven metabolites of which the des–nitro compound 1 was the major metabolite (Figure 6) [64]. Similarly, HLS9 were pre–incubated with NADPH and NADH cofactors in pH 7.4 phosphate buffer followed by addition of PA-824 and incubation for 3 h. HLS9 exposure resulted in formation of four previously unknown metabolites (M1, M2, M3 and M4) common with Ddn treatment but failed to show formation of the three other metabolites associated with Ddn (1, 2 and 3). M. tuberculosis exposure to PA-824 for 3 h generated the same seven metabolites formed in the presence of purified Ddn. In contrast, only six of these metabolites were observed with M. smegmatis, with no formation of the des–nitro compound 1. The structures of metabolites 1, 2, and 3 were previously reported by Singh et al. [62] and importantly shown to lack whole–cell efficacy versus M. tuberculosis, supporting the hypothesis that the formation of RNS accompanying their production is bactericidal. M3 was confirmed as the product of a two–electron reduction of PA-824 by co–elution with an authentic sample. Treatment of M3 with HLS9 resulted in the formation of M1 and M4, whereas Ddn treatment showed formation of 1 in addition to M1 and M4.
In summary, this work corroborated previous data demonstrating that the nitro reduction of metabolite M3 formed by PA-824 exposure to M. tuberculosis and Ddn results in the appearance of 1 (des–nitro PA-824) accompanied by the release of RNS. This des–nitro metabolite was not formed in the presence of HLS9 and M. smegmatis, suggesting its production may be crucial to the selective cidality of PA-824 to M. tuberculosis. The employed methods, focusing on host and bacterial systems as well as exposure to purified M. tuberculosis enzymes, have provided further insight into a clinical stage antitubercular while providing a window into the mechanism of action of other nitro–containing antituberculars [65–67].
Concluding Remarks and Future Perspectives
M. tuberculosis is known to catalyze metabolic transformations including but not limited to hydrolysis, N–alkylation, amidation, and nitro group reduction that can activate (pertinent to a prodrug) or deactivate a xenobiotic (Table 1). Given the range of chemistries operative in the biosynthetic pathways of M. tuberculosis [3], we assert that the discussed biotransformations of antituberculars are but a small subset of the reactions they may undergo. Guided by the degree of the commonality of their biosynthetic pathways with those of M. tuberculosis, other bacteria would be expected to be capable of such biotransformations on cell–permeable xenobiotics. Keeping this in mind, our strategy recommends determining the fate of a chemical tool or actual drug in the bacterium of interest through the use of metabolomics–based methods. This compliments the more typical host–focused drug metabolism studies that are a part of non–infectious disease drug discovery workflows. Drug metabolites formed in the dosed patient can have deleterious implications, influencing a drug’s efficacy, bioavailability, clearance rate and toxicity. Drugs administered to humans are subjected to phase I and phase II metabolism in the liver. To predict in vivo stability of a drug candidate, human liver microsomal (HLM) assays are widely employed. Stepping back from the administration of an antibacterial to humans, one must consider in vivo studies of efficacy in preclinical models, e.g. in the mouse, rat, guinea pig, and non–human primate. These model studies provide an opportunity to probe the metabolism of the antibacterial and may be complemented by in vitro studies with hepatocytes or microsomal fractions. Ultimately, quantification of the rate of metabolism and identification of the associated metabolite/s in these assays will highlight the structural liabilities of the antibacterial. This information can guide rational tool compound and drug optimization.
Table 1.
Known Metabolic Transformations Performed by M. tuberculosis on Small Molecule Antituberculars.
| Compound | Biotransformation reaction | Result |
|---|---|---|
| INHa | Oxidation (KatG) | Activating |
| PZAa | Hydrolysis (PncA) | Activating |
| ETHa | Oxidation (EthA, MymA) | Activating |
| Thiacetazone, isoxyla | Oxidation (EthA) | Activating |
| Aminoglycosides | Amidation, esterification, phosphorylation | Deactivating |
| NTZ | Hydrolysis | Retain potency |
| MTX esters | Hydrolysis | Deactivating |
| CD117 | Hydrolysis | Deactivating |
| Compound 14 | N–alkylation | Deactivating |
| PAS | N–alkylation |
N–methylation: equipotent N,N–dimethylation: deactivating |
| Sulfanilamide | Amidation | Deactivating |
| Sulfamethoxazole | N–alkylation, amidation (and reduction) | Deactivating |
| Dapsone | N–alkylation, amidation (and reduction) | Deactivating |
| PA-824, delamanid | Nitro reduction | Activating |
Studied via genetics/biochemical
We propose a holistic approach to the evolution of antibacterial chemical tools, to probe basic biology, as well as drug discovery entities where the workflow addresses the stability of a small molecule taking into account biotransformations occurring in the host and the target organism. Thus, novel antibacterials should be probed for metabolism within (i) the relevant bacterium via metabolomics–based methods and (ii) within pertinent hosts through studies with the actual animal or more simply with cultured hepatocytes or microsomal fractions. The simultaneous evaluation of the metabolic fate in host and bacterium will better guide the antibacterial design and optimization studies, improve the time– and cost–efficiencies of the process, and help address key questions within the antibacterials field (See Outstanding Questions).
Trends.
A shift in perspective to the antibacterial may commence with a determination of the ability of the small molecule to accumulate within the bacterium.
The antibacterial may not simply remain within the bacterium and instead suffer metabolic transformation (which may be categorized by chemical reaction type mirroring a bacterial biosynthetic reaction) that results in efficacious or inactive molecules.
The antibacterial may also be metabolized by the host, and model systems such as mouse liver microsomal preparations may be probed to discern the fate of the small molecule.
Metabolomics–based methods may inform the evolution of antibacterial chemical tools and drug discovery entities.
Outstanding Question.
What are the ultimate fates of approved antibacterial drugs within the host and bacterium that are critical to understanding their mechanism of action?
How may this knowledge be leveraged to design the next generation of antibacterials?
Acknowledgments
J.S.F. acknowledges funding from the National Institutes of Health: Centers of Excellence for Translational Research (CETR; Award Number 1U19AI109713 NIH/NIAID) for the “Center to develop therapeutic countermeasures to high–threat bacterial agents.” We thank Professor Kyu Y. Rhee (Weill Medical College of Cornell University) and Dr. Sean Ekins (Collaborations Pharmaceuticals) for critical comments during the preparation of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zumla A, et al. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov. 2013;12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 2.Sassetti CM, et al. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 3.Lamichhane G, et al. Essential metabolites of Mycobacterium tuberculosis and their mimics. MBio. 2011;2:e00301–00310. doi: 10.1128/mBio.00301-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, et al. The catalase–peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature. 1992;358:591–593. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee A, et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 6.Bonicke R, Fau - Lisboa BP, Lisboa BP. Type differentiation of tuberculosis bacteria with the aid of the nicotinamidase test. Tuberkulosearzt. 1959;13:377–384. [PubMed] [Google Scholar]
- 7.Konno K, Fau - Feldmann FM, et al. Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am Rev Respir Dis. 1967;95:461–469. doi: 10.1164/arrd.1967.95.3.461. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Mitchison D. The curious characteristics of pyrazinamide: a review. Int J Tuberc Lung Dis. 2003;7:6–21. [PubMed] [Google Scholar]
- 9.DeBarber AE, et al. Ethionamide activation and sensitivity in multidrug–resistant Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2000;97:9677–9682. doi: 10.1073/pnas.97.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fraaije MW, et al. The prodrug activator EtaA from Mycobacterium tuberculosis is a Baeyer–Villiger monooxygenase. J Biol Chem. 2004;279:3354–3360. doi: 10.1074/jbc.M307770200. [DOI] [PubMed] [Google Scholar]
- 11.Vale N, et al. Metabolism of the antituberculosis drug ethionamide. Curr Drug Metab. 2013;14:151–158. [PubMed] [Google Scholar]
- 12.Ramirez MS, Tolmasky ME. Aminoglycoside Modifying Enzymes. Drug resistance updates: reviews and commentaries in antimicrobial and anticancer chemotherapy. 2010;13:151–171. doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright GD. Aminoglycoside–modifying enzymes. Curr Opin Microbiol. 1999;2:499–503. doi: 10.1016/s1369-5274(99)00007-7. [DOI] [PubMed] [Google Scholar]
- 14.Vetting MW, et al. Aminoglycoside 2′–N–acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat Struct Biol. 2002;9:653–658. doi: 10.1038/nsb830. [DOI] [PubMed] [Google Scholar]
- 15.Magnet S, Blanchard JS. Molecular insights into aminoglycoside action and resistance. Chem Rev. 2005;105:477–498. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- 16.Ahn JW, Kim KJ. Rv3168 phosphotransferase activity mediates kanamycin resistance in Mycobacterium tuberculosis. J Microbiol Biotechnol. 2013;23:1529–1535. doi: 10.4014/jmb.1306.06048. [DOI] [PubMed] [Google Scholar]
- 17.Tsodikov OV, et al. A random sequential mechanism of aminoglycoside acetylation by Mycobacterium tuberculosis Eis protein. PLoS One. 2014;9:e92370. doi: 10.1371/journal.pone.0092370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aldridge BB, Rhee KY. Microbial metabolomics: innovation, application, insight. Curr Opin Microbiol. 2014;19:90–96. doi: 10.1016/j.mib.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Hartman TE, Rhee KY. Microbial Metabolomics: Fifty Shades of Metabolism. ACS Infect Dis. 2015;1:73–75. doi: 10.1021/id500041w. [DOI] [PubMed] [Google Scholar]
- 20.Chakraborty S, Rhee KY. Tuberculosis Drug Development: History and Evolution of the Mechanism–Based Paradigm. Cold Spring Harb Perspect Med. 2015;5:a021147. doi: 10.1101/cshperspect.a021147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li SG, et al. Evolution of a thienopyrimidine antitubercular relying on medicinal chemistry and metabolomics insights. Tetrahedron Lett. 2015;56:3246–3250. doi: 10.1016/j.tetlet.2015.02.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vannelli TA, et al. The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J Biol Chem. 2002;277:12824–12829. doi: 10.1074/jbc.M110751200. [DOI] [PubMed] [Google Scholar]
- 23.Dover LG, et al. EthA, a common activator of thiocarbamide–containing drugs acting on different mycobacterial targets. Antimicrob Agents Chemother. 2007;51:1055–1063. doi: 10.1128/AAC.01063-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grant SS, et al. Baeyer–Villiger Monooxygenases EthA and MymA Are Required for Activation of Replicating and Non–replicating Mycobacterium tuberculosis Inhibitors. Cell Chem Biol. 2016;23:666–677. doi: 10.1016/j.chembiol.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cotes K, et al. Lipolytic enzymes in Mycobacterium tuberculosis. Appl Microbiol Biotechnol. 2008;78:741–749. doi: 10.1007/s00253-008-1397-2. [DOI] [PubMed] [Google Scholar]
- 26.Saravanan P, Fau - Dubey VK, et al. Potential selective inhibitors against Rv0183 of Mycobacterium tuberculosis targeting host lipid metabolism. Chem Biol Drug Des. 2012;79:1056–1062. doi: 10.1111/j.1747-0285.2012.01373.x. [DOI] [PubMed] [Google Scholar]
- 27.Ortega C, et al. Systematic Survey of Serine Hydrolase Activity in Mycobacterium tuberculosis Defines Changes Associated with Persistence. Cell Chem Biol. 2016;23:290–298. doi: 10.1016/j.chembiol.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tallman KR, et al. Small–Molecule Probes Reveal Esterases with Persistent Activity in Dormant and Reactivating Mycobacterium tuberculosis. ACS Infect Dis. 2016;2:936–944. doi: 10.1021/acsinfecdis.6b00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tallman KR, et al. Profiling Esterases in Mycobacterium tuberculosis Using Far–Red Fluorogenic Substrates. ACS Chem Biol. 2016;11:1810–1815. doi: 10.1021/acschembio.6b00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Theodos CM, et al. Efficacy of nitazoxanide against Cryptosporidium parvum in cell culture and in animal models. Antimicrob Agents Chemother. 1998;42:1959–1965. doi: 10.1128/aac.42.8.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adagu IS, et al. In vitro activity of nitazoxanide and related compounds against isolates of Giardia intestinalis, Entamoeba histolytica and Trichomonas vaginalis. J Antimicrob Chemother. 2002;49:103–111. doi: 10.1093/jac/49.1.103. [DOI] [PubMed] [Google Scholar]
- 32.Dubreuil L, et al. In vitro evaluation of activities of nitazoxanide and tizoxanide against anaerobes and aerobic organisms. Antimicrob Agents Chemother. 1996;40:2266–2270. doi: 10.1128/aac.40.10.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Megraud F, et al. Nitazoxanide, a potential drug for eradication of Helicobacter pylori with no cross–resistance to metronidazole. Antimicrob Agents Chemother. 1998;42:2836–2840. doi: 10.1128/aac.42.11.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McVay CS, Rolfe RD. In vitro and in vivo activities of nitazoxanide against Clostridium difficile. Antimicrob Agents Chemother. 2000;44:2254–2258. doi: 10.1128/aac.44.9.2254-2258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pankuch GA, Appelbaum PC. Activities of tizoxanide and nitazoxanide compared to those of five other thiazolides and three other agents against anaerobic species. Antimicrob Agents Chemother. 2006;50:1112–1117. doi: 10.1128/AAC.50.3.1112-1117.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffman PS, et al. Antiparasitic drug nitazoxanide inhibits the pyruvate oxidoreductases of Helicobacter pylori, selected anaerobic bacteria and parasites, and Campylobacter jejuni. Antimicrob Agents Chemother. 2007;51:868–876. doi: 10.1128/AAC.01159-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Carvalho LP, et al. Nitazoxanide kills replicating and nonreplicating Mycobacterium tuberculosis and evades resistance. J Med Chem. 2009;52:5789–5792. doi: 10.1021/jm9010719. [DOI] [PubMed] [Google Scholar]
- 38.de Carvalho LP, et al. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co–catabolism of carbon substrates. Chem Biol. 2010;17:1122–1131. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 39.de Carvalho LP, et al. Activity–based metabolomic profiling of enzymatic function: identification of Rv1248c as a mycobacterial 2-hydroxy-3-oxoadipate synthase. Chem Biol. 2010;17:323–332. doi: 10.1016/j.chembiol.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Carvalho LP, et al. Nitazoxanide Disrupts Membrane Potential and Intrabacterial pH Homeostasis of Mycobacterium tuberculosis. ACS Med Chem Lett. 2011;2:849–854. doi: 10.1021/ml200157f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nixon MR, et al. Folate pathway disruption leads to critical disruption of methionine derivatives in Mycobacterium tuberculosis. Chem Biol. 2014;21:819–830. doi: 10.1016/j.chembiol.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 42.Gerum AB, et al. Novel Saccharomyces cerevisiae screen identifies WR99210 analogues that inhibit Mycobacterium tuberculosis dihydrofolate reductase. Antimicrob Agents Chemother. 2002;46:3362–3369. doi: 10.1128/AAC.46.11.3362-3369.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar A, et al. High–throughput screening and sensitized bacteria identify an M. tuberculosis dihydrofolate reductase inhibitor with whole cell activity. PLoS One. 2012;7:e39961. doi: 10.1371/journal.pone.0039961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vilcheze C, et al. Novel inhibitors of InhA efficiently kill Mycobacterium tuberculosis under aerobic and anaerobic conditions. Antimicrob Agents Chemother. 2011;55:3889–3898. doi: 10.1128/AAC.00266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nandakumar M, et al. Metabolomics of Mycobacterium tuberculosis. Methods Mol Biol. 2015;1285:105–115. doi: 10.1007/978-1-4939-2450-9_6. [DOI] [PubMed] [Google Scholar]
- 46.Shell SS, et al. DNA methylation impacts gene expression and ensures hypoxic survival of Mycobacterium tuberculosis. PLoS Pathog. 2013;9:e1003419. doi: 10.1371/journal.ppat.1003419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Warrier T, et al. N–methylation of a bactericidal compound as a resistance mechanism in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2016;113:e4523–4530. doi: 10.1073/pnas.1606590113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lehmann J. Para–aminosalicylic acid in the treatment of tuberculosis. Lancet. 1946;1:15. doi: 10.1016/s0140-6736(46)91185-3. [DOI] [PubMed] [Google Scholar]
- 49.Youmans GP, et al. The Tuberculostatic Action of para–Aminosalicylic Acid. J Bacteriol. 1947;54:409–416. doi: 10.1128/jb.54.4.409-416.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rengarajan J, et al. The folate pathway is a target for resistance to the drug para–aminosalicylic acid (PAS) in mycobacteria. Mol Microbiol. 2004;53:275–282. doi: 10.1111/j.1365-2958.2004.04120.x. [DOI] [PubMed] [Google Scholar]
- 51.Huang TS, et al. Susceptibility of Mycobacterium tuberculosis to sulfamethoxazole, trimethoprim and their combination over a 12 year period in Taiwan. J Antimicrob Chemother. 2012;67:633–637. doi: 10.1093/jac/dkr501. [DOI] [PubMed] [Google Scholar]
- 52.Nopponpunth V, et al. Cloning and expression of Mycobacterium tuberculosis and Mycobacterium leprae dihydropteroate synthase in Escherichia coli. J Bacteriol. 1999;181:6814–6821. doi: 10.1128/jb.181.21.6814-6821.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chakraborty S, et al. Para–aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science. 2013;339:88–91. doi: 10.1126/science.1228980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nandakumar M, et al. Metabolomics of Mycobacterium tuberculosis. Methods Mol Biol. 1285:105–115. doi: 10.1007/978-1-4939-2450-9_6. [DOI] [PubMed] [Google Scholar]
- 55.Zheng J, et al. para–Aminosalicylic acid is a prodrug targeting dihydrofolate reductase in Mycobacterium tuberculosis. J Biol Chem. 2013;288:23447–23456. doi: 10.1074/jbc.M113.475798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Silhavy TJ, et al. The bacterial cell envelope. Cold Spring Harb Perspect Biol. 2010;2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumar P, et al. Non–classical transpeptidases yield insight into new antibacterials. Nat Chem Biol. 2017;13:54–61. doi: 10.1038/nchembio.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boelsterli UA, et al. Bioactivation and hepatotoxicity of nitroaromatic drugs. Curr Drug Metab. 2006;7:715–727. doi: 10.2174/138920006778520606. [DOI] [PubMed] [Google Scholar]
- 59.Smith GF. Designing drugs to avoid toxicity. Prog Med Chem. 2011;50:1–47. doi: 10.1016/B978-0-12-381290-2.00001-X. [DOI] [PubMed] [Google Scholar]
- 60.Mukherjee T, Boshoff H. Nitroimidazoles for the treatment of TB: past, present and future. Future Med Chem. 2011;3:1427–1454. doi: 10.4155/fmc.11.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cellitti SE, et al. Structure of Ddn, the deazaflavin–dependent nitroreductase from Mycobacterium tuberculosis involved in bioreductive activation of PA-824. Structure. 2012;20:101–112. doi: 10.1016/j.str.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singh R, et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008;322:1392–1395. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson RF, et al. Intermediates in the reduction of the antituberculosis drug PA-824, (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1, 3]oxazine, in aqueous solution. Org Biomol Chem. 2008;6:1973–1980. doi: 10.1039/b801859f. [DOI] [PubMed] [Google Scholar]
- 64.Dogra M, et al. Comparative bioactivation of the novel anti–tuberculosis agent PA-824 in mycobacteria and a subcellular fraction of human liver. Br J Pharmacol. 2011;162(226):236. doi: 10.1111/j.1476-5381.2010.01040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ekins S, et al. Bayesian models leveraging bioactivity and cytotoxicity information for drug discovery. Chem Biol. 2013;20:370–378. doi: 10.1016/j.chembiol.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tangallapally RP, et al. Nitrofurans as novel anti–tuberculosis agents: identification, development and evaluation. Curr Top Med Chem. 2007;7:509–526. doi: 10.2174/156802607780059772. [DOI] [PubMed] [Google Scholar]
- 67.Tangallapally RP, et al. Discovery of novel isoxazolines as anti–tuberculosis agents. Bioorg Med Chem Lett. 2007;17:6638–6642. doi: 10.1016/j.bmcl.2007.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]