

SUMMARY
Postsynaptic densities (PSDs) are membrane semienclosed, submicron protein-enriched cellular compartments beneath postsynaptic membranes, which constantly exchange their components with bulk aqueous cytoplasm in synaptic spines. Formation and activity-dependent modulation of PSDs is considered as one of the most basic molecular events governing synaptic plasticity in the nervous system. In this study, we discover that SynGAP, one of the most abundant PSD proteins and a Ras/Rap GTPase activator, forms a homo-trimer and binds to multiple copies of PSD-95. Binding of SynGAP to PSD-95 induces phase separation of the complex, forming highly concentrated liquid-like droplets reminiscent of the PSD. The multivalent nature of the SynGAP/ PSD-95 complex is critical for the phase separation to occur and for proper activity-dependent SynGAP dispersions from the PSD. In addition to revealing a dynamic anchoring mechanism of SynGAP at the PSD, our results also suggest a model for phase-transition-mediated formation of PSD.
Graphical Abstract
INTRODUCTION
Eukaryotic cells need to orchestrate numerous biochemical reactions spatiotemporally. Nature solves this challenge, in part, by segregating each cell into multiple compartments using membrane bilayers so that specific chemical reactions can occur within each compartment. However, not all isolated cellular compartments are insulated by membranes. Especially for those undergoing rapid molecular exchanges with the surrounding environment, membranes can be a rate-limiting barrier. Membrane-lacking compartments, on the other hand, can be dynamic mesoscale reaction apparatuses localized at distinct regions of cells to fulfill spatiotemporal requirements of numerous cellular processes, such as cell growth, division, migration, and cell-cell communications. It is increasingly recognized that membrane-lacking compartments are ubiquitous and central for many cellular processes (Brangwynne, 2013; Hyman et al., 2014; O’Connell et al., 2012). Some of the well-recognized examples of membrane-lacking compartments include nucleoli, centrosomes, Cajal bodies, nuclear pores, mitotic spindles, and various ribonucleoprotein-enriched granules. Instead of having clearly defined molecular compositions and architectures known for cellular machineries, such as ribosomes and proteasomes, these membrane-lacking compartments usually have irregular morphologies and are enriched with specific sets of components with poorly defined stoichiometry (Brangwynne et al., 2009; Hyman et al., 2014; Jiang et al., 2015; Li et al., 2012; Molliex et al., 2015; Patel et al., 2015; Woodruff et al., 2015).
Compartmentalization is even more critical for neurons because of their large sizes and extreme polarity. In addition to membrane-enclosed organelles and membrane-lacking compartments common to other cell types, neurons contain a unique type of membrane-semi-enclosed compartments, known as synapses, which are molecular apparatuses dictating signal processing and transmissions in all nervous systems. Underneath the postsynaptic plasma membranes of each synapse resides a protein-rich subcompartment known as postsynaptic density (PSD), an assembly responsible for receiving, interpreting, and storage of signals transmitted by presynaptic axonal termini. PSDs are disc-shaped, electron-dense thickenings that contact with postsynaptic membranes on its one face and with cytoplasm on the other face, forming semi-open mesoscale cellular compartments. Biochemical and electron microscopy (EM) analyses revealed that PSDs are composed of densely packed proteins forming mega-assemblies with a few hundred nanometer in width and ~30–50 nm in thickness (Chen et al., 2008; Harris and Weinberg, 2012). Both molecular components and motions of each component within PSDs are highly dynamic (Bosch et al., 2014; Caroni et al., 2012; Choquet and Triller, 2013; Colgan and Yasuda, 2014; Nishiyama and Yasuda, 2015). The rapid and reversible structural and molecular component changes of PSD are perhaps best manifested during synaptic plasticity changes known as long-term potentiation/ depression (LTP/LTD) processes (Araki et al., 2015; Bosch et al., 2014; Huganir and Nicoll, 2013).
Since its discovery, considerable progress has been made in identifying both the compositions and copy numbers of proteins in each PSD (Cheng et al., 2006; Lowenthal et al., 2015; Sheng and Hoogenraad, 2007). Extensive studies have also illustrated numerous protein-protein interactions that organize the PSD protein network (Zhu et al., 2016). Optical and EM studies have offered rich information on the layered organizations of PSD and mobility of many PSD components (Chen et al., 2015; MacGillavry et al., 2013; Maglione and Sigrist, 2013; Nair et al., 2013; Triller and Choquet, 2008). Despite the pivotal roles of PSDs in orchestrating synaptic structure and functions, some of the most fundamental questions remain unanswered. For example, how can the submicron, semi-open PSD compartments autonomously form beneath the synaptic plasma membranes? How are the large concentration gradients of numerous proteins between PSD and spine cytoplasm maintained? How can neuronal activity alter PSD assembly?
SynGAP and PSD-95 are two very abundant proteins existing at a near stoichiometric ratio in PSD (Cheng et al., 2006). Mutations of either of SynGAP or PSD-95 are known to cause human psychiatric disorders, such as intellectual disorders (ID) and autism (Berryer et al., 2013; Hamdan et al., 2009; Parker et al., 2015). PSD-95 and its two homologous, PSD-93 and SAP102, are central scaffolding proteins in PSD, orchestrating multiple signaling cascades, as well as shaping the basic architecture of PSD (Chen et al., 2015; Levy et al., 2015; Zhu et al., 2016). SynGAP is a brain-specific GTPase-activating protein (GAP) that predominantly localizes in PSD via specifically binding to PSD- 95 (Chen et al., 1998; Kim et al., 1998). SynGAP functions as a negative regulator for excitatory synaptic strength through tuning down activities of small G proteins (Araki et al., 2015; Vazquez et al., 2004). Downregulation of SynGAP leads to premature hippocampal spine formation and overactivation of excitatory synapses in young mice (Clement et al., 2012; Vazquez et al., 2004), which may represent the underlying molecular mechanism of SynGAP mutations found in epileptic, ID, and autism patients. SynGAP has recently been shown to rapidly disperse from PSD following chemical LTP induction, and the activity-dependent dispersion of SynGAP may represent an initial step for LTP expression (Araki et al., 2015).
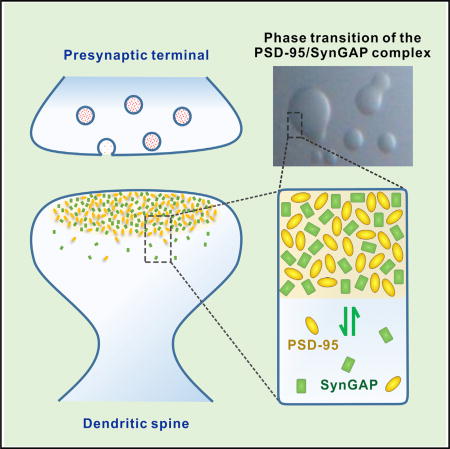
In this work, we elucidate the structural basis governing the specific interaction between SynGAP and PSD-95. We discover that SynGAP forms a parallel coiled-coil trimer capable of binding to multiple copies of PSD-95. Importantly, this multivalent SynGAP/PSD-95 interaction leads to the formation of liquidliquid phase separation, both in vitro and in living cells. The Syn-GAP/PSD-95 complex in the condensed liquid phase is densely concentrated and dynamically exchanges with the two proteins in the aqueous phase of the cytoplasm, a phenomena reminiscent of activity-dependent SynGAP dispersion from PSD in living neurons. We further show that the multivalent SynGAP/PSD-95 interaction is crucial for SynGAP stabilization in PSD and for preventing neurons from hyper-excitation. Our study indicates that the multivalent postsynaptic protein-protein interaction-induced phase transitions might be a possible mechanism for PSD formation. Our work also provides mechanistic insights into why mutations altering the SynGAP/PSD-95 interaction can contribute to various brain disorders, including autism and ID.
RESULTS
A C-Terminal α-Helix Extension of PSD-95 PDZ3 Is Required for Its Specific Binding to SynGAP
We first characterized the SynGAP/PSD-95 interaction using purified proteins. PSD-95 contains three PDZ (PSD-95, Dlg, ZO-1) domains, an SH3 (Src homology 3) domain, and a GK (guanylate kinase) domain (Figure 1A). The α1 isoform of SynGAP (referred to as “SynGAP” hereafter) (Figure 2A) contains a C-terminal PDZ binding motif (PBM), which has been reported to interact with all three PDZ domains of PSD-95 (Kim et al., 1998). Isothermal titration calorimetry (ITC)-based titration assay showed that N-terminal thioredoxin-tagged last 30 residues of SynGAP (SynGAP PBM) binds to each of the three PDZ domains with comparable and weak affinities (Kd of a few dozens of micromolars; Figure 1G, G1). Such weak bindings are unlikely to support a specific functional interaction between SynGAP and PSD-95 in vivo (Ye and Zhang, 2013; Zeng et al., 2016).
Figure 1. Structural Basis Governing the Specific Interaction between PSD-95 and SynGAP.

(A) Schematic diagram showing the domain organization of PSD-95.
(B) PSD-95 PDZ3 contains a conserved and elongated C-terminal α-helix extension (αC helix). Part of this αC helix (marked in pink) has been previously identified. In the sequence alignment, conserved residues are colored. The residues directly involved in binding to SynGAP PBM are indicated by triangles.
(C) ITC-based measurements comparing SynGAP PBM’s binding to PSD-95 PDZ3 and PDZ3-C.
(D) Ribbon diagram representation of the PSD-95 PDZ3-C/SynGAP PBM complex structure. The segment upstream of the canonical SynGAP PBM is highlighted with a dashed oval (also see Figure S1).
(E) Combined surface diagram (PSD-95 PDZ3-C) and stick-ball model (SynGAP PBM) showing the inter-molecular interaction between PSD-95 PDZ3-C and SynGAP PBM. In the surface map, hydrophobic residues are drawn in yellow; positively charged residues are in blue; and negatively charged residues are in red.
(F) Stereo view showing the detailed interactions between PSD-95 PDZ3-C and SynGAP PBM.
(G) ITC-based measurements summarizing the binding affinities between SynGAP PBM and three individual PDZ domains from PSD-95 (G1) and between various SynGAP PBM mutants and PSD- 95 PDZ3-C (G2).
Figure 2. SynGAP Forms a Parallel Coiled-Coil Trimer.

(A) Schematic diagram showing the domain organization of SynGAP.
(B) FPLC-coupled static light-scattering analysis showing that WT SynGAP CC-PBM forms a stable trimer in solution, while the “L-D&K-D” mutant is a monomer.
(C) CD spectra-based urea-induced denaturation curves comparing denaturation profiles of Syn- GAP WT CC-PBM and “L-D&K-D” mutant.
(D) Ribbon diagram representation of the SynGAP coiled-coil trimer. The SynGAP coiled-coil forms a parallel, asymmetric coiled-coil trimer (see also Figure S3).
(E) Helical net diagram of one helical strand of the SynGAP coiled-coil trimer. The two hendecad repeats are shown in the dashed boxes. The residues in the a/d positions of the heptad repeats and those in the a/d/x positions of the hendecad repeats are highlighted in yellow. The residues that contribute to charge-charge interactions are colored in blue (Lys and Arg) and red (Asp and Glu), respectively.
(F) Combined ribbon and stick-ball models showing the trimer interfaces of the SynGAP coiled-coil.
(G) Combined surface and ribbon models showing the interactions of the coiled-coil trimer. The side chains of hydrophobic and charged residues in the ribbon scheme are colored in magenta and green, respectively. The coloring code of the surface diagram is identical to that in Figure 1E.
(H) Close-up view of L1202 in the trimer interfaces. (I and J) Close-up view of the interactions between R1219 and E1224 and between K1252 and D1253, respectively.
PSD-95 PDZ3 is known to contain a short αC helix in its C terminus (amino acids [aa] E392–E398, highlighted in pink in Figure 1B), which can enhance PDZ’s target peptide binding through stabilizing the conformation of PDZ3 without directly contacting peptide ligands (Petit et al., 2009). The boundary of PDZ3 (aa R306–E398) used in our binding assay has already contained this reported αC helix, but our ITC results showed that the affinity between PDZ3 and SynGAP is still very low (Kd ~27.0 µM, Figure 1C, left). Amino acid sequence analysis reveals that a stretch of residues following αC (aa A399–N410, highlighted in yellow in Figures 1A and 1B) is highly conserved and predicted to form α-helix. We found that a longer version of PDZ3 containing the predicted long C-terminal helix extension (aa R306-S412, referred to as “PDZ3-C” hereafter) binds to SynGAP with a 15-fold higher affinity than does PDZ3 (Figure 1C), and several dozen-fold higher than does PDZ1 or PDZ2 (Figure 1G, G1). Thus, a C-terminal extension sequence beyond the canonical PDZ boundary enhances PSD-95 PDZ3’s binding specificity to SynGAP.
Structural Basis Governing the Specific PSD-95 PDZ3-C/SynGAP PBM Interaction
To elucidate the molecular mechanism underlying the specific PSD-95 PDZ3-C/SynGAP PBM interaction, we solved the complex structure by X-ray crystallography (Table S1). Crystallization of the complex was facilitated by fusing the last 15 residues of SynGAP to the C terminus of PSD-95 PDZ3-C with a flexible linker. We used NMR spectroscopy to exclude the possibility that the fusion protein used in the crystallization experiment might artificially induce the observed interaction (Figure S1C). Fully consistent with our biochemical data, PSD-95 PDZ3-C in the complex contains an extended αC helix composed of 19 residues (E392-N410), which is 12 residues longer than the extension helix observed in the previously determined PDZ3 structure (Figure S1A) (Doyle et al., 1996). Importantly, this extended αC helix, together with the PDZ3 core, forms the second binding site for SynGAP PBM (Figures 1D and 1E), explaining why the αC extension can enhance PDZ3-C’s binding to SynGAP PBM. In the complex, the last three residues of SynGAP (−2TRV°, the canonical PBM) bind to the αB/βB-groove of PSD-95 PDZ3 following the classical PDZ binding mode. The upstream residues (−9F-V−5) of SynGAP PBM interact with a hydrophobic pocket formed by the extended αC helix, βB and βC strands of PDZ3-C (Figures 1E, 1F, and S1B). The αC helix is highly amphipathic, with all of its hydrophobic residues facing βB and βC forming an elongated hydrophobic pocket accommodating the sidechains of V(−5), W(−6) and F(−9) in SynGAP PBM (Figure 1F). Substitution of V(−5) or F(−9) with a charged Glu, or replacing W(−6) with a smaller hydrophobic Ala all significantly decreased the binding of SynGAP PBM to PSD-95 PDZ3-C (Figure 1G2).
SynGAP Forms a Parallel Trimer via Its Coiled-Coil Domain
SynGAP contains a PH (Pleckstrin homology) domain, a C2 domain, and a GAP (GTPase-activating protein) domain in its N-terminal half (Figure 2A) (Chen et al., 1998; Kim et al., 1998). Detailed sequence analysis indicates presence of a possible coiled-coil domain in the C-terminal half of SynGAP. We characterized the C-terminal half of SynGAP containing the predicted coiled-coil domain and the following PBM, and identified a stable fragment (aa A1147-V1308, denoted as “CC-PBM”). Circular dichroism (CD) spectrum of SynGAP CC-PBM indicates that the protein is well-folded and enriched in α-helix (Figure S2A). Fast protein liquid chromatography (FPLC) coupled with a static light-scattering assay revealed that SynGAP CC-PBM (at the 20–150 µM range assayed) has a uniform molecular mass of ~55.5 kDa in solution, which equals three times of the theoretical monomer mass (Figures 2B and 3D), indicating that SynGAP forms a stable trimer in solution.
Figure 3. SynGAP CC-PBM Binds to PSD-95 PSG in a 3:2 Stoichiometry.

(A) ITC measurement of the interaction between SynGAP CC-PBM and PSD-95 PSG.
(B) Summary of ITC-derived binding affinities between various SynGAP CC-PBM and PSD- 95 PSG.
(C) Pull-down assay comparing PSD-95 PSG binding to various forms of the full-length SynGAP.
(D) FPLC-coupled static light-scattering analysis showing that mixing equal molar of SynGAP CC- PBM and PSD-95 PSG leads to a formation of a3:2 CC-PBM/PSG complex (black curve). The elution profiles of isolated SynGAP CC-PBM (red curve) and PSD-95 PSG (cyan curve) are also included. The calculated molecular mass and fitting error of each peak is indicated above the peak. The experiments were repeated three times using different batches of proteins.
(E) Summary of the theoretical and measured molecular weights of the SynGAP CC-PBM/PSD- 95 PSG complex using recombinant proteins without (E1) or with (E2) tags (see also Figure S5).
(F) Static light-scattering assay showing that the monomeric L-D&K-D mutant of SynGAP CC-PBM can still form complex with PSD-95 PSG, albeit that the formed complex is not homogenous.
Atomic Structure of the SynGAP Coiled-Coil Trimer
We solved the crystal structure of SynGAP coiled-coil domain (SynGAP-CC) spanning residues D1185–H1274 at 2.5 Å resolution (Table S2). The SynGAP-CC forms a parallel coiled-coil trimer (Figure 2D). Detailed structural analysis revealed that SynGAP-CC is composed of 8.5 regular heptad repeats and two hendecad repeats interspersed between heptad repeats (a hendecad repeat contains 11 residues forming three helix turns, highlighted with dashed boxes in Figure 2E). The helical diagram analysis reveals that the residues at the a and d positions of the heptad/hendecad repeats of SynGAP-CC (residues in yellow in Figure 2E) are dominated by hydrophobic residues, which are chiefly responsible for forming the folding core of the trimer (Figures 2F and 2G). A typical example is shown in Figure 2H, where L1202 at position d from three helical strands pack with each other with a 3-fold symmetry axis forming the d-layer interactions in coiled-coil structures. Chargecharge interactions between residues outside a&d positions (e.g., R1219 in the g position in one strand forming salt bridges with E1224 in the e position of the neighboring strand; Figure 2I) also stabilize the coiled-coil structure. There are also a few quite unique interactions in SynGAP-CC not common in regular coiled-coil structures. For example, the side chain of K1252 in the x position of the second hendecad repeat (equivalent to the d position in a heptad repeat) projects away from the hydrophobic core and forms a salt bridge with D1253 in the e position in the neighboring strand (Figure 2J). Although most of the sidechain interactions governing the overall folding of SynGAP-CC have a 3-fold rotational symmetry, the entire coiled-coil trimer is asymmetric (Figure S3D). This overall asymmetry is caused by several local asymmetric interactions along the coiled-coils. For example, there are two Tyr residues at the d positions along the coiled-coil (Tyr1216 and Tyr1234). The side chain of Tyr1216 from one strand (colored magenta in Figures S3A and S3B) inserts into the center core of the trimer helix, while the other two Tyr1216 sidechains point outward (Figure S3B). Similarly, Tyr1234 displays similar asymmetric packing (Figure S3C). In theory, the partially exposed Tyr is accessible for phosphorylation modifications, which may alter the stability of the SynGAP-CC trimer assembly. Additionally, SynGAP isoforms can form homo- or heterotrimers as they contain the identical coiled- coil domain (Figure 2A).
Structure-Based Design of a SynGAP Monomer
The structure of the SynGAP coiled-coil trimer allowed us to design monomeric SynGAP mutant protein containing minimal number of amino acid substitutions. Such monomeric SynGAP mutant will be valuable to evaluate functional roles of the SynGAP trimer. Single point mutation of either L1202D (Figure 2H) or K1252D (Figure 2J) weakened SynGAP trimerization (Figure S2B). When combined, the L1202D and K1252D double point mutations (referred to as “L-D&K-D” mutant hereafter) completely converted SynGAP CC-PBM into a monomer (Figure 2B), though the monomer mutant still contains significant amount of a-helical conformation (Figure S2A). Urea-induced denaturation experiment showed that the L-D&K-D mutant showed a near linear denaturation profile instead of a sigmoidal denaturation curve observed for wild-type (WT) CC-PBM (Figures 2C and S2C), indicating that the monomer mutant no longer contains a tertiary structure.
SynGAP CC-PBM Binds to PSD-95 PSG with High Affinity and Specificity
SynGAP-CC is closely adjacent to its PBM (Figure 2A). We explored whether formation of the trimer might modulate SynGAP’s binding to PSD-95. PSD-95 is a member of membrane-associated guanylate kinase (MAGUK) family, in which the PDZ-SH3-GK tandems (PSG) can function as supramodules (Li et al., 2014; McCann et al., 2012; Pan et al., 2011; Zhang et al., 2013). We thus tested whether SynGAP CC-PBM may interact with PSD-95 PSG more specifically than the interaction between SynGAP PBM and PSD-95 PDZ3-C. ITC assay showed that SynGAP CC-PBM and PSD-95 PSG (aa R306-L721) interact with each other with a Kd ~0.14 µM, which is > 10-fold stronger than PBM/PDZ3-C and ~200-fold stronger than the PBM/PDZ3 interaction (Figures 1C and 3A). The binding affinity between SynGAP CC-PBM and PSD-95 PSG is also tens- to hundreds-folds stronger than other PSD-95 PDZ and PBM-containing target interactions reported. Given the extremely high concentrations of both SynGAP and PSD-95 in PSDs, SynGAP should be the dominant binding partner of PSD-95 PDZ3.
As expected, the hydrophobic pocket of PDZ3-C accommodating the upstream residues of SynGAP PBM is important for the CC-PBM/PSG interaction. Substitution of V(−5) to E or W(−6) to A on SynGAP CC-PBM decreased their binding by several dozen folds (Figure 3B, B1 and B2), and deleting the extended αC helix (A399–N410) from PSD-95 PSG (referred to as “PSG ΔEXT”) led to > 10-fold decrease in binding to SynGAP CC-PBM (Figures 3B, B1, and S3B). Removal of the last four residues from SynGAP (denoted as “CC-PBM Δ4”) totally abolished its binding to PSD-95 PSG (Figure 3B, B2). Interestingly, formation of the SynGAP trimer does not enhance the binding affinity between SynGAP and PSD-95 (Figure 3B, B1 and B2). These data also indicate that the enhanced affinity of SynGAP binding to PSD-95 PSG, with respect to PDZ3-C, is afforded by the formation of the PSG supramodule.
We also validated the impact of the mutations on the interaction between the full-length SynGAP and PSD-95 PSG by a pulldown assay using purified GST-PSD-95 PSG to pull down Flagtagged SynGAP expressed in HEK293T cells. In agreement with our ITC data shown in Figure 3B, PSD-95 PSG interacted with the wild-type SynGAP robustly. L-D&K-D-SynGAP showed a similar binding affinity as WT-SynGAP does. Deleting the “QTRV”-motif totally eliminated SynGAP’s binding to PSD-95. The V(−5)E mutant of SynGAP showed an obviously weakened binding to PSD-95 (Figure 3C).
We further examined the SynGAP/PSD-95 interaction by overexpressing RFP-PSD-95 PSG and GFP-SynGAP CC-PBM in HeLa cells. When expressed individually, PSD-95 PSG mainly localizes in cytoplasm and SynGAP CC-PBM is exclusively enriched in nuclei (Figures S4A, A1 and A2). Mutations in SynGAP PBM (W-A, V-E, or Δ4) did not change CC-PBM nucleus localization when expressed alone (Figure S4B). Co-expression of PSD-95 and WT SynGAP can effectively re-localize SynGAP to cytoplasm (Figure S4A, A3), consistent with their robust interaction observed in biochemical assays. As to the SynGAP mutants, the amount of PSD-95-mediated cytoplasm re-localization correlates well with their binding affinities to PSD-95 PSG (Figure S4).
Trimeric SynGAP CC-PBM Recruits Two PSD-95 PSG to Form a 3:2 Complex
SynGAP CC-PBM alone exists as a trimer and PSD-95 PSG is a monomer (Figure 3D). When mixed at a 1:1 molar ratio (calculated with their respective monomer as the basic unit), a complex peak with a measured molecular mass of ~154.8 kDa was formed on a size-exclusion column, and a significant portion of PSD-95 remained in its free form (Figure 3D). The stoichiometry of the complex can be fitted as one SynGAP trimer binds to two copies of PSD-95, forming a 3:2 complex (Figure 3E1). To exclude other possibilities, we used N-terminal His-tagged PSD-95 PSG and N-terminal GB1-tagged SynGAP CC-PBM. The complex formed by mixing 1:1 molar ratio of GB1-SynGAP CC-PBM with His-PSD-95 PSG has a detected molecular mass of ~175.9 kDa, which is also fitted as a 3:2 complex composing of one SynGAP trimer and two PSD-95 monomers (Figures 3E, E2, and S5A). Consistently, mixing GB1-SynGAP CC-PBM and His-PSD-95 PSG at a 3:2 ratio produced a single peak on the size exclusion column corresponding to a 3:2 complex (Figure S5B).
We tested the impact of several SynGAP mutants on their binding to PSD-95 PSG. As expected, deletion of the “QTRV” from SynGAP (“Δ4”) completely eliminated the complex formation (Figure S5C). Curiously, we found that the coiled-coil-mediated SynGAP trimer assembly is crucial for the 3:2 SynGAP/ PSD-95 complex formation, as the complex formed between L-D&K-D-SynGAP CC-PBM and PSD-95 PSG has a lower and inhomogeneous molecular mass distribution on the FPLC column (Figure 3F). Since the binding affinities of L-D&K-D- and WT-SynGAP CC-PBM to PSD-95 PSG are similar, the failed 3:2 SynGAP/PSD-95 complex formation must be a result of the disruption of the SynGAP trimer formation.
SynGAP CC-PBM/PSD-95 PSG Complex Undergoes a Concentration-Dependent Phase Transition
During the sample preparations for light-scattering experiments, we found that mixing purified SynGAP CC-PBM and PSD-95 PSG above certain concentrations caused the sample solutions to become opalescent immediately. In contrast, solutions containing each individual component were always clear. We examined the solution of the SynGAP CC-PBM and PSD-95 PSG mixture shortly after mixing by light microscope and observed numerous small spherical droplets spanning various diameters, a phenomenon characteristic of liquid-to-liquid phase separation (Figure 4A). Within minutes, small droplets gradually coalesced into larger ones (Figure 4B). Isolated solutions of SynGAP CC-PBM and PSD-95 PSG, our controls, remained clear aqueous solutions at the same concentration (Figure 4A).
Figure 4. Phase Transition of the SynGAP CC-PBM/PSD-95 PSG Complex.

(A) Isolated SynGAP CC-PBM and PSD-95 PSG solutions are stable and homogeneous at 100 µM concentration under light microscope at room temperature (RT). Mixing the two proteins, each at a concentration of 100 µM, resulted in formation of numerous droplets. The images shown in the figure were acquired 3 min and onward after mixing. The dashed box is the region of zoomed-in analysis in (B).
(B) The small droplets underwent time-dependent coalescence into larger ones.
(C) Schematic diagram illustrating the sedimentation assay to separate the condensed liquid phase and the aqueous phase of the SynGAP CC-PBM/PSD-95 PSG mixtures in the rest of the study.
(D) Representative SDS-PAGE analysis and quantification data showing the distribution of proteins between aqueous-solution/supernatant (S) and condensed liquid phase/pellet (P) fractions for various SynGAP CC-PBM/PSD-95 PSG mixtures. The concentration of each protein in all of the experiments is at 100 µM, calculated as their monomer units.
(E) Sedimentation assay showing that the phase transition of the SynGAP/PSD-95 complex is concentration dependent. SynGAP and PSD-95 were mixed at a 1:1 ratio at various concentrations, and the proteins were quantified by their band intensities.
(F) Time-lapse DIC images of SynGAP CC-PBM/ PSD-95 PSG mixture (1:1 at 100 µM) showing numerous droplets at RT in a coverslip chamber. Droplets are rapidly dispersed after adding the 15AA peptide (see also Movie S1). The arrow refers to the time point of adding the 15AA peptide to the mixture.
(G) SDS-PAGE analysis and quantification results showing pre-formed SynGAP/PSD-95 droplets can be reversed to aqueous phase by the competing 15AA peptide. The indicated concentrations of the peptide or a peptide-free buffer was added to a 1:1 SynGAP/PSD-95 mixture at 100 µM.
(H) DSG-mediated cross-linking reveals PSD-95 PSG undergoes a SynGAP peptide binding-induced dimerization. The CRIPT peptide does not induce dimer formation of PSD-95 PSG. The peptide to PSD-95 molar ratio and reaction time are indicated in the figure.
All statistic data in this figure represent the results from three independent batches of experiments and are expressed as mean ± SD.
To analyze the molecular compositions of the condensed liquid phase, we performed a sedimentation assay to separate the condensed liquid phase from the bulk aqueous solutions by centrifugation and assessed protein components in each fraction by SDS-PAGE with Coomassie blue staining (Figures 4C and 4D). Neither SynGAP CC-PBM nor PSD-95 PSG alone could form condensed liquid phase (Figure 4D). For the 1:1 molar ratio mixture of SynGAP CC-PBM and PSD-95 PSG at 100 µM, approximately half of SynGAP and PSD-95 were recovered from the condensed liquid phase (Figure 4D), indicating that it contains both SynGAP CC-PBM and PSD-95 PSG. Considering that the condensed liquid phase droplet pellet volume is much smaller than that of the supernatant aqueous solution, protein concentrations for both SynGAP and PSD-95 in the condensed liquid phase are much higher (estimated to be at least 100-fold higher based on the volumes of the two fractions) than those in the aqueous solution.
We have also observed the liquid-to-liquid phase separation and condensed liquid phase droplets fusion of the SynGAP/ PSD-95 complex using Alexa488-labeled SynGAP CC-PBM and Cy3-labeled PSD-95 PSG by fluorescence microscopy (Figure S6A). Fluorescence recovery after photobleaching (FRAP) analysis of Cy3-labled PSD-95 droplets demonstrated that PSD-95 molecules constantly exchange between droplets and the surrounding aqueous solution (Figure S6B). To rule out the possibility that the highly concentrated proteins in the condensed liquid phase form immobile aggregates, we denatured the fluorescence labeled proteins in the flow chamber with a quick heat pulse and performed the same FRAP experiment. Heating of the flow chamber led to formation of clusters of Cy3-PSD-95 with amorphous shapes presumably due to denaturation-induced aggregation, and the fluorescence signals of these clusters do not recover at all after photobleaching (Figure S6C).
We found that the phase separation of the SynGAP CC-PBM/ PSD-95 PSG complex is sharply concentration-dependent (Figure 4E) and can occur at a wide temperature range (4~37°C assayed). Lower temperature can slightly promote phase transition (Figures S7A and S7B). As controls, 100 µM SynGAP or PSD-95 alone in all assayed temperatures remained in aqueous supernatant (Figures S7C and S7D). Since the CC-PBM/PSG complex formation is an exothermic reaction (Figure 3A), its binding is favored by decreasing the reaction temperature. This is different from intrinsically disordered protein interaction-mediated phase transitions, which are mainly govern by hydrophobic interactions and favored by higher temperatures.
Phase Transition Requires the Multivalent Interaction between SynGAP and PSD-95
Next, we dissected the role of the SynGAP CC-PBM/PSD-95 PSG interaction in the phase transition of the complex. First, we found that deleting the “QTRV”-motif of SynGAP prevented phase transition (“Δ4,” Figure 4D), indicating that the PDZ domain-mediated binding is required for the phase transition to occur. Second, the phase transition of the SynGAP CC-PBM/ PSD-95 PSG complex can be blocked or reversed by a PSD- 95 PDZ3 binding peptide. The addition of a 15-residue SynGAP peptide, which binds to PSD-95 PDZ3-C with the same affinity as the SynGAP PBM does (Figure 1C), led to immediate dispersion of the condensed liquid phase into homogenous aqueous solution (Figure 4F; Movie S1) in a peptide concentration-dependent manner (Figure 4G). The SynGAP peptide-mediated dispersion of pre-formed liquid phase of the CC-PBM/PSG complex suggests that formation of the SynGAP trimer is required for the phase transition to occur upon binding to PSD-95. Consistent with this hypothesis, the L-D&K-D-SynGAP CC-PBM mutant, which is a monomer but retains the same binding affinity to PSD-95 PSG (Figures 3B and 3C), could not induce phase transition after mixing with PSG (Figure 4D).
A prerequisite for liquid-liquid phase transition to occur is that the SynGAP CC-PBM/PSD-95 PSG complex needs to form oligomer. We demonstrated earlier that the PSG tandem of another MAGUK protein, Pals1, can form a domain-swapped dimer following ligand binding (Li et al., 2014). This triggered us to explore the possibility of ligand-induced dimerization of PSD-95 PSG. Using a chemical cross-linking approach, we could detect that binding of the 15-residue SynGAP peptide induces PSD-95 PSG to form dimer/multimer (Figure 4H). Importantly, binding of excess amount of the CRIPT peptide (the PBM motif of CRIPT that does not require the extended aC-helix of PDZ3 for binding [Petit et al., 2009]) does not induce PSD-95 PSG dimer formation (Figure 4H), indicating that PSD-95 PSG can undergo specific SynGAP binding-induced dimerization/ multimerization.
The SynGAP/PSD-95 Complex Forms Condensed Liquid Phase in Living Cells
Next, we tested whether the SynGAP CC-PBM/PSD-95 PSG complex can undergo phase transition in living cells. When GFP-SynGAP CC-PBM and RFP-PSD-95 PSG were co-expressed in HeLa cells, we observed many bright puncta and containing both GFP and RFP signals (Figures 5A and 5B). Within these puncta, both GFP and RFP signal intensities are much brighter than the surrounding cytoplasm, indicating the puncta are enriched with SynGAP and PSD-95 (Figure 5A). No puncta were observed in cells when only PSD-95 PSG or SynGAP CC-PBM was expressed. Nicely correlated with the in vitro phase transition experiment shown in Figure 4D, no or very little puncta could be detected when PSD-95 PSG was co-expressed individually with various SynGAP CC-PBM mutants (Figure 5B). Similarly, co-expression of a PSD-95 PSG mutant (“ΔEXT”) with WT SynGAP CC-PBM also significantly decreased the puncta formation (Figure 5B). The above data indicate that the SynGAP CC-PBM- and PSD-95 PSG-enriched puncta formation also depends on the specific and multivalent interaction between the two proteins.
Figure 5. The SynGAP/PSD-95 Complex Forms Condensed Liquid Phase in Living Cells.

(A) Representative images showing co-expression of GFP-SynGAP CC-PBM and RFP-PSD-95 PSG in HeLa cells produce multiple bright puncta containing both fluorophores. Dashed boxes show the zoomed-in regions.
(B) Trimer-disruption mutant (“L-D&K-D”) or PSD- 95 PDZ binding mutants (“Δ4,” “W-A” and “V-E” on SynGAP or “ΔEXT” on PSD-95) showing significantly decreased puncta formation when compared to the WT proteins (n = number of batch of cultures with >600 cells counted for each batch). Data are presented as mean ± SEM; ns, not significant, *p < 0.05, **p < 0.01, and ***p < 0.001 using one-way ANOVA with Tukey’s multiple comparison test.
(C) Representative time-lapse FRAP images showing that GFP-SynGAP signal within the puncta recovered within a few minutes (see also Movie S2).
(D) Quantitative results for FRAP analysis of GFP-SynGAP CC-PBM in puncta and cytoplasm of HeLa cells. The red curve represents the averaged FRAP data of 24 puncta from 13 cells, and the black curve is the averaged FRAP data of 12 cytoplasmic regions from six cells. Time 0 refers to thetime point of the photobleaching pulse. All data are represented as mean ± SD.
(E) Representative time-lapse images showing that the GFP-SynGAP CC-PBM and RFP-PSD-95 PSG-positive puncta undergo time-dependent fusion (see also Movie S3).
The SynGAP CC-PBM- and PSD-95 PSG-enriched puncta observed in HeLa cells could be condensed liquid phase droplets as in Figure 4B, or could be immobile aggregates of the two proteins. To address this issue, we monitored the exchange rate of GFP-SynGAP in the puncta with the proteins in the surrounding cytoplasm using FRAP. After bleaching, the GFP-Syn-GAP signals within the puncta were recovered within a few minutes and the recovery speed of the protein signals in the cytoplasm is much faster and within a few seconds (Figures 5C and 5D; Movie S2), indicating that GFP-SynGAP in the puncta exchanges rapidly with those in the surrounding cytoplasm. This result strongly indicates that the SynGAP CC-PBM- and PSD-95 PSG-enriched puncta formed in cells are also condensed liquid phase droplets. This conclusion is further supported by the observation that the SynGAP CC-PBM- and PSD-95 PSG-en-riched puncta were originally rather small and underwent time-dependent fusion into larger puncta (Figure 5E; Movie S3). Consistent with earlier reports (Brangwynne et al., 2009; Li et al., 2012), the speed of the small droplets fusion is slower than the rate of the molecular exchange between droplets and cytoplasm. Taken together, the above imaging analysis indicates that the phase transition of the SynGAP/PSD-95 complex can also occur in living cells.
SynGAP Trimer-Mediated Binding to PSD-95 Is Required for Synaptic Localization and Activity-Dependent Dispersion of SynGAP
To investigate the cellular functions of SynGAP trimer-mediated PSD-95 interaction in neurons, we used a molecular replacement approach described previously (Araki et al., 2015), where we knocked down endogenous SynGAP by using a short hairpin RNA (shRNA) and replaced it with an shRNA-resistant WT or mutant SynGAP cDNAs (Figures 6A and 6B). We found that synaptic localizations of the “L-D&K-D” mutant and the three PSD-95 PDZ binding mutants of SynGAP were all significantly reduced (Figures 6A and 6B). These results underscore the importance of both the formation of the SynGAP trimer and the SynGAP C-terminal extended PBM-mediated binding to PSD-95 for the synaptic localization of SynGAP.
Figure 6. The Multivalent SynGAP/PSD-95 Interaction Is Critical for Synaptic Localization and Proper Activity-Induced Dispersion of SynGAP from Synapses.

(A and B) Endogenous SynGAP was replaced with GFP-SynGAP WT or its mutants using an shRNA molecular replacement strategy. mCherry was cotransfected as cell morphology marker. Average synaptic enrichment levels of SynGAP were quantified (n = 31 pairs of spines and dendrites from 6 independent experiments/neurons for each group were analyzed).
(C and D) Activity-dependent synaptic dispersion of replaced SynGAP WT and its mutants during chemLTP (n = 31 pairs of spines and dendrites from six independent experiments/neurons for each group were analyzed). Spines were analyzed before and after chemLTP using live-imaging techniques. Data are presented as mean ± SEM; *p < 0.05 and ***p < 0.001 using one-way ANOVA with Tukey’s multiple comparison test.
Next, we investigated whether these mutations would affect the activity- dependent SynGAP dispersion from synapses (Figures 6C and 6D). Similar to the experiments described in Figure 6A, we replaced endogenous SynGAP either with “L-D&K-D” mutant or with PDZ binding mutants. The “L-D&K-D” mutant has less enrichment on basal state as described in Figures 6A and 6B. Furthermore, upon chemLTP induction, a larger fraction of the SynGAP mutants were dispersed from synapses compared to WT-SynGAP (Figures 6C and 6D), suggesting that the trimer formation not only facilitates synaptic localization of SynGAP at basal conditions, but also regulates proper levels of SynGAP dispersion upon synaptic activations. Similarly, the PSD-95 PDZ binding mutants also displayed larger LTP-induced dispersions when compared to WT-SynGAP (Figures 6C and 6D). The levels of LTP-induced dispersions of the three mutants are correlated with the degrees of their reduction in PSD-95 binding affinity (Figure 3B).
Next, we examined if these SynGAP mutations might affect synaptic plasticity. We replaced endogenous SynGAP with the “L-D&K-D” mutant or PSD-95 PDZ binding mutants, and also co-transfected mCherry as a cell morphology tracer and SEP-GluA1 for monitoring AMPAR trafficking (Figures 7A and 7B). As to WT-SynGAP, following chemLTP induction, some synaptic spines were structurally enlarged, with newly inserted AMPARs (marked with arrows in yellow). When compared to the WT group, neurons replaced with the “L-D&K-D” mutant have enlarged spines and more SEP-GluA1 even at the basal state (“L-D&K-D”: Basal), indicating a hyper-excitation status. Interestingly, upon chemLTP induction, these neurons have further enlarged spines and more SEP-GluA1 recruitment (“L-D&K-D”: chemLTP) compared to the WT group in the chemLTP state, an observation consistent with the results in Figures 6C and 6D that show less enrichment of the mutant, both at the basal and chemLTP states. Again, neurons replaced with the PSD-95 PDZ binding mutants of SynGAP also displayed larger spines and more GluA1 recruitment upon chemLTP inductions (Figures 7A and 7B).
Figure 7. The Role of Trimer Formation and PSD-95 Binding on SynGAP-Regulated Synaptic Plasticity.

(A and B) Endogenous SynGAP was replaced by SynGAP WT or its various mutants. SEP-GluA1 and mCherry were transfected for monitoring AMPA receptor trafficking and synaptic spine morphology during chemLTP, respectively (n = 86 spines from 16 independent experiments/ neurons for each group were analyzed). Spines were analyzed before and after chemLTP using live imaging.
(C and D) In threshold LTP experiments, weak LTP stimulus (10 µM glycine/0 Mg2+) was given to neurons with WT or L-D&K-D SynGAP replacement (n = 16 spines from three independent experiments/ neurons for each group were analyzed). Spines were analyzed before and after chemLTP using live imaging. Data are presented as mean ± SEM; ns, not significant, *p < 0.05, **p < 0.01 and ***p < 0.001 using one-way ANOVA with Tukey’s multiple comparison test.
Since the “L-D&K-D” mutant of SynGAP is easier to be dispersed from synapses than the WT counterpart (Figure 6C), we reasoned that “L-D&K-D” mutant-containing synapses might be more sensitive in responding to weak chemLTP stimuli. To test this hypothesis, we used a weaker LTP protocol (10 µM glycine stimulation instead of 200 µM Glycine for chemLTP inductions) to investigate whether neurons replaced with the “L-D&K-D” Syn- GAP mutant would respond to such weak stimuli. Neurons rescued with the WT SynGAP do not respond to the weaker stimulation. In contrast, neurons with the “L-D&K-D” SynGAP replacement were responsive to this weaker stimulus both with enlarged spine volumes and with increased GluA1 recruitment (Figures 7C and 7D). This result indicates that the “L-D&K-D” mutant of SynGAP is more sensitive and more responsive to weak stimuli. Taken together, the formation of SynGAP trimer plays a critical role in anchoring SynGAP to the synapse with proper dynamic modulation and thereby regulates synaptic strength under the normal physiological conditions. Our results also provide a molecular explanation to why SynGAP mutations leading to the disruption of its trimerization or alterations of its PSD-95 binding will lead to neuronal hyperexcitation because of mutation-induced excessive dispersions of SynGAP from synapses.
DISCUSSION
Phase Transition of the SynGAP/PSD-95 Complex and Its Implication for PSD Formation
We have demonstrated that SynGAP and PSD-95, two of the most abundant proteins in the PSD, following mixing can spontaneously undergo phase transition, forming condensed, membrane-lacking compartments both in vitro and in living cells. Analogous to PSD assemblies, the SynGAP/PSD-95 complex in the condensed liquid phase is highly concentrated compared to the aqueous phase in cytoplasm and can rapidly exchange with the corresponding proteins in the aqueous phase and thus is mobile, and can be regulated by competing binding ligands in vitro or synaptic activity changes in living neurons. We postulate that organization of PSD mega-assemblies may also be driven by phase transitions induced by the SynGAP/ PSD-95 complex and its associated proteins.
We have demonstrated that the specific and multivalent interaction between the SynGAP trimer and PSD-95 is essential for the phase transition of the SynGAP/PSD-95 complex to occur. This observation is consistent with recent examples of protein-protein or protein-nucleic acid interaction-induced phase transitions in other cellular systems showing that multivalent interactions are hallmarks for essentially all these membrane-lacking compartmental systems characterized (Banjade and Rosen, 2014; Brangwynne, 2013; Hyman et al., 2014; Li et al., 2012). In PSD, the multivalent interactions are further manifested by several PSD-95-associated proteins, including SAPAP, Shank, and Homer, all of which exist in concentrations comparable to those of PSD-95 and SynGAP (Cheng et al., 2006). For example, the C-terminal SAM domain of Shank can self-oligomerize (Baron et al., 2006). The N termini of SAPAPs contain 2–5 repeat sequences capable of binding to PSD-95 GK domain (Kim et al., 1997; Zhu et al., 2011). The coiled-coil domain of Homer can form a homotetramer positioning two EVH1 domains in each side of the tetramer for binding to Shank (Hayashi et al., 2009). One might envision that both the valency and the branching points for binding to additional proteins for the SynGAP/PSD-95/SAPAP/Shank/Homer assembly will be much higher than those for the SynGAP/PSD-95 complex alone. Accordingly, the phase transition-mediated PSD formation, when it occurs, would be more complicated than the phase separation observed for the SynGAP/PSD-95 complex alone. It is also likely that the threshold concentrations for the SynGAP/ PSD-95/SAPAP/Shank/Homer assembly to undergo phase transition would be considerably lower than that of the SynGAP/ PSD-95 complex.
Functional Implications of Phase-Transition-Mediated PSD Assembly on Synaptic Functions
One of the most obvious functions of forming a gel-like PSD is to enrich numerous synaptic proteins into a relatively small region of the dendritic spine beneath the postsynaptic membranes. Compared to most of other cells types, neurons have much larger cytoplasmic volumes and are with extremely long processes of axons and dendrites. As such, neurons face additional physical challenges in properly distributing and maintaining their cellular components in different regions in order to keep their morphological and functional polarities. If freely diffusible, each neuron will have to synthesize tens to hundreds fold more of synaptic proteins in order to allow these proteins to reach their required concentrations in synapses. Such high levels of protein synthesis will add huge metabolic burdens to neurons. Neurons solve this problem, at least in part, by employing molecular motor-mediated transports to selectively deposit different proteins to specific sub-compartments of cells (e.g., dendritic spines or axonal boutons). In each sub-compartment such as PSDs, these proteins, once surpassing certain threshold concentrations, may form even higher density assemblies via the phase separation mechanism. Additionally, because PSDs are in direct contact with the aqueous cytoplasmic phase of each synapse, selected synaptic proteins (e.g., SynGAP) can be rapidly dispersed from or incorporated into the PSD responding to neuronal activity changes without need of active transport machineries used in membrane-enclosed compartments. Such activity-dependent, rapid protein flux in and out of the PSD is critical for synaptic plasticity. As exemplified in this study, several SynGAP mutants defective in phase separations in vitro display decreased synaptic enrichments and enhanced dispersions from spines. These mutant SynGAP-harboring neurons are accordingly hyper-sensitive to stimulation (Figures 7C and 7D).
In addition to concentrations of each component, the affinities of the interactions between molecular components are also critical for phase transition to occur (Li et al., 2012; this study). This implies that phase transition can be regulated by tuning protein-protein interaction strengths via rapid post-translational modifications in addition to protein synthesis and degradations occurring at longer timescales. We demonstrated earlier that phosphorylation of SynGAP can weaken its binding to PSD-95 and thus increase its dispersion from PSD and subsequently promote LTP induction (Araki et al., 2015). Phosphorylation-mediated regulations of synaptic protein-protein interactions appear to be a common strategy in synapses (Huganir and Nicoll, 2013; Trinidad et al., 2008; Zhu et al., 2011), and many protein kinases are known to enrich in PSDs (Sheng and Hoogenraad, 2007).
Phase Transition and Human Diseases Associated with Mutations of SynGAP
Mutations of one allele of SynGAP in human can lead to severe diseases, such as seizure, autism and ID, likely because of hyper-excitation and subsequent E/I imbalance of neuronal circuits (Berryer et al., 2013; Hamdan et al., 2009; Parker et al., 2015). At a first glance, it is hard to rationalize the exquisite dosage sensitivity of mammalian central nervous systems to such an abundant enzyme, as decrease of its catalytic capacity due to loss of one SynGAP allele in theory can be compensated by a prolong of reaction time. The findings presented in this work provide a possible explanation to the dosage sensitivity of SynGAP (and possibly other synaptic proteins) to synaptic functions. The phase transition of the SynGAP/PSD-95 complex is a sharply concentration-dependent process. Only when the concentration of SynGAP reaches a certain threshold can the formation of the condensed SynGAP/PSD-95 liquid phase occur. It is possible that loss of 50% of SynGAP may lower the concentration of the protein below its threshold concentration for integrating into PSD via PSD-95 (i.e., the effective concentration decrease of SynGAP in PSDs of SynGAP+/− neurons can be much greater than 50% reduction of the protein). It is noted that a large portion of the mutations of SynGAP found in human patients are de novo nonsense mutations lacking the critical trimer formation coiled-coil domain. These SynGAP variants would not be effectively integrated into PSD because of loss of its capacity in binding to PSD-95. Therefore, formation of the SynGAP trimer and its interaction with PSD-95, via a liquid-to-liquid phase separation, can serve as a stable but dynamic anchoring mechanism for SynGAP in PSD and for maintaining proper E/I balance in excitatory synapses.
STAR*METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
- EXPERIMENTAL MODEL AND SUBJECT DETAILS
- Primary Neuronal Culture
- HeLa Cells and HEK293T Cells Culture
- METHOD DETAILS
- Constructs and Protein Expression
- Isothermal Titration Calorimetry Assay
- Crystallization, Data Collection, and Processing
- NMR Experiments
- Fast Protein Liquid Chromatography Coupled with Static Light Scattering
- Circular Dichroism Measurements
- GST Pull-Down Assay
- In Vitro Phase Transition Assay
- Chemical Cross-Linking Assay
- HeLa Cell Imaging and Data Analysis
- Fluorescence Recovery after Photo-Bleaching Assay
- Neuronal Cultures, Imaging, and LTP Induction
QUANTIFICATION AND STATISTICAL ANALYSIS
- DATA AND SOFTWARE AVAILABILITY
- Data Resources
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-FLAG (clone M2) | Sigma-Aldrich | Cat#F1804; RRID: AB_262044 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Synthesized SynGAP 15AA PBM peptide (AQRGSFPPWVQQTRV) | Synthesized by ChinaPeptides | N/A |
| Synthesized CRIPT PBM peptide (MCGKKVLDTKNYKQTSV) | Synthesized by ChinaPeptides | N/A |
| DSG | ThermoFisher | Cat#20593 |
| Alexa Fluor 488 NHS ester | ThermoFisher | Cat#A20000 |
| Cy3 NHS ester | AAT Bioquest | Cat#141 |
| Uridine | Sigma-Aldrich | Cat#U3003 |
| 5-Fluro-2′-deoxyuridine | Sigma-Aldrich | Cat#F0503 |
| Neurobasal media | GIBCO | Cat#21103-049 |
| B27 | GIBCO | Cat#17504044 |
| Horse serum | Hyclone | Cat#SH30074 |
| Recombinant protein: PSD-95 PSG WT (aa R306-L721, ref#NP_001122299) | This paper | N/A |
| Recombinant protein: SynGAP CC-PBM WT (aa A1147-V1308, ref#J3QQ18, lacking 1192V-1193K and 1293E-1295G) | This paper | N/A |
| Recombinant protein: PSD-95 PDZ3-C-GSGS-SynGAP PBM (PSD-95 R306-S419-GSGS-AQRGSFPPWVQQTRV) | This paper | N/A |
| Recombinant protein: PSD-95 PDZ3-C-thrombin-SynGAP PBM (PSD-95 R306-S419-SGLVPRGS- AQRGSFPPWVQQTRV) | This paper | N/A |
| Critical Commercial Assays | ||
| Lipofectamine2000 transfection kit | Invitrogen | Cat#11668019 |
| NanoJuice transfection kit | Novagen | 71902-4 |
| Deposited Data | ||
| PSD-95 PDZ3-C/SynGAP PBM complex structure | This paper | PDB: 5JXB |
| SynGAP coiled-coil domain structure | This paper | PDB: 5JXC |
| Experimental Models: Cell Lines | ||
| Human: HeLa cells | ATCC | CCL-2 |
| Human: HEK293T cells | ATCC | CRL-3216 |
| Rat: embryonic day 18 hippocampal primary neuron culture | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| Escherichia coli: BL21 (DE3) | Invitrogen | Cat#C600003 |
| Recombinant DNA | ||
| Plasmid: Flag-SynGAP | Araki et al., 2015 | N/A |
| Plasmid: GFP-SynGAP res#5 | Araki et al., 2015 | N/A |
| Plasmid: pSUPER shSG#5-CMV:mCherry | Araki et al., 2015 | N/A |
| Plasmid: GFP-SynGAP CC-PBM | This paper | N/A |
| Plasmid: RFP-PSD-95 PSG | This paper | N/A |
| Software and Algorithms | ||
| Origin7.0 | OriginLab | http://www.originlab.com/ |
| HKL2000 | Otwinowski and Minor, 1997 | http://www.hkl-xray.com/ |
| PHASER | McCoy et al., 2007 | https://www.phenix-online.org/ |
| phenix.refinement | Adams et al., 2010 | https://www.phenix-online.org/ |
| phenix.xtriage | Adams et al., 2010 | https://www.phenix-online.org/ |
| Coot | Emsley et al., 2010 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| phenix.model_vs_data validation tools | Adams et al., 2010 | https://www.phenix-online.org/ |
| PyMOL | PyMOL | http://pymol.sourceforge.net/ |
| ASTRA6 | Wyatt | http://www.wyatt.com/products/software/astra.html |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism | GraphPad Software Inc | http://www.graphpad.com/scientific-software/prism/ |
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Mingjie Zhang (mzhang@ust.hk).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary Neuronal Culture
Hippocampal neurons from embryonic day 18 (E18) rats were seeded on 25-mm poly-L-lysine coated coverslips. The cells were plated in Neurobasal media (GIBCO) containing 50U/ml penicillin, 50mg/ml streptomycin and 2mM GlutaMax supplemented with 2% B27 (GIBCO) and 5% horse serum (Hyclone). At DIV6 cells were thereafter maintained in glia-conditioned NM1 (Neurobasal media with 2mM GlutaMax, 1% FBS, 2% B27, 1 × FDU (5mM Uridine (SIGMA U3003), 5mM 5-Fluro-2′-deoxyuridine (SIGMA F0503)).
HeLa Cells and HEK293T Cells Culture
HeLa and HEK293T cells (both from ATCC) were cultured in DMEM media supported by fetal bovine serum.
METHOD DETAILS
Constructs and Protein Expression
The full-length PSD-95 gene was PCR amplified from a human cDNA library (reference sequence NCBI: NP_001122299, 721 aa). SynGAP CC-PBM construct was PCR amplified from a mouse cDNA library (reference number UniProt: J3QQ18, 1308 aa). Note that, the amplified sequence does not contain 1192V-1193K or 1293E-1295G. Various mutations or shorter fragments of PSD-95 and SynGAP were generated using standard PCR-based methods and confirmed by DNA sequencing. Recombinant proteins were expressed in Escherichia coli BL21 (DE3) cells in LB medium at 16°C and purified using a nickel-NTA agarose (for Trx-His6-/ GB1-His6-/His6-tagged proteins) or a glutathione Sepharose (for GST-tagged protein) affinity column followed by size-exclusion chromatography with a column buffer containing 50mM Tris pH7.8, 100mM NaCl, 1mM EDTA, 5mM DTT (for CC-PBM/PSG) or 1mM DTT (for other proteins). Uniformly 15N-labeled PDZ3-C-PBM fusion protein was prepared by growing bacteria in M9 minimal medium using 15NH4Cl as the sole nitrogen source. When needed, tags were cleaved by HRV 3C protease and separated by another step of size-exclusion chromatography. SynGAP 15AA PBM peptide (AQRGSFPPWVQQTRV) and CRIPT PBM peptide (MCGKKVLDTKNYKQTSV) were commercially synthesized. The full length Flag-tagged SynGAP construct used was described in our earlier work (Araki et al., 2015).
Isothermal Titration Calorimetry Assay
ITC measurements were carried out on a Microcal VP-ITC calorimeter at 25°C. Proteins used for ITC measurements were dissolved in an assay buffer composed of 50mM Tris pH7.8, 100mM NaCl, 1mM EDTA, and 5mM DTT. High concentration of proteins (400 µM for Trx-SynGAP PBM and 200 µM for PSD-95 PSG) was individually loaded into the syringe and titrated into the cell containing low concentration of corresponding interactors (40 µM for Trx-PSD-95 PDZ and 20 µM for SynGAP CC-PBM). For each titration point, a 10 µl aliquot of a protein sample in the syringe was injected into the interacting protein in the cell at a time interval of 2–3min. ITC titration data were analyzed using the Origin7.0 software and fitted with the one-site binding model.
Crystallization, Data Collection, and Processing
The PSD-95 PDZ3-C/SynGAP PBM Complex
Crystals of PSD-95 PDZ3-C fused with a GSGS linker and SynGAP PBM peptide were obtained by the hanging drop vapor diffusion method at 16°C. Freshly purified fusion protein was concentrated to 10mg/ml. Crystals were grown in solution containing 0.1M HEPES sodium pH7.5, 10% v/v 2-Propanol, 20% w/v Polyethylene glycol 4,000. Glycerol (25%) was added as the cryoprotectant. A 2.9 Å resolution X-ray dataset was collected at the beam-line BL17U1 of the Shanghai Synchrotron Radiation Facility. The diffraction data were processed and scaled by HKL2000 (http://www.hkl-xray.com/).
Using the structure of the PSD-95 PDZ3 domain (PDB id: 1BE9) as the search model, the initial structural model was solved using the molecular replacement method in PHASER (https://www.phenix-online.org/). The model was then refined by phenix.refinement (https://www.phenix-online.org/). The dataset was twinned with a twin fraction of 0.3 as indicated by phenix.xtriage (http://www.phenix-online.org/). Twin refinement restraints were applied during the refinement. Coot (http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/) was used for peptide modeling and model adjustments. The final structure was validated by the phenix.model_vs_data validation tools (https://www.phenix-online.org/). The final refinement statistics of the complex structure are listed in Table S1.
The SynGAP Coiled-Coil Trimer
Crystals of the SynGAP coiled-coil domain (D1185-H1274) were also obtained by the hanging drop vapor diffusion method at 16°C. Freshly purified protein was concentrated to 9mg/ml. The crystal was grown in solution containing 0.1M HEPES sodium pH7.5, 0.1M L-proline, 20% w/v Polyethylene glycol 1,500. Polyethylene glycol 400 (25%) was added as the cryoprotectant. To obtain phase information, Se-Met-labeled SynGAP coiled-coil protein was purified and concentrated to 9mg/ml. Se-Met SynGAP coiled-coil crystals were grown in solution containing 0.1M HEPES sodium pH7.5, 16% w/v Pentaerythritol propoxylate 426 (5/4 PO/OH) at 16°C. The concentration of Pentaerythritol propoxylate 426 (5/4 PO/OH) in the reservoir was further increased to 26% w/v to be used as the crystal cryoprotectant. Both the native and MAD datasets were collected at the beam-line BL17U1 of the Shanghai Synchrotron Radiation Facility. The diffraction data were processed and scaled by HKL2000 (http://www.hkl-xray.com/).
Using the SeMet derivative dataset, the MAD diffraction phase was determined and a partial structural model was traced using phenix.autosol (https://www.phenix-online.org/). Using this partial structural model, the initial structural model for the coiled-coil was solved against the native dataset using the molecular replacement method. The model was then extended and refined using phenix.refinement (https://www.phenix-online.org/). Coot was used for model building and adjustments (http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/). TLS refinement was applied at the final refinement stage. The final structure was validated by phenix.model_vs_data (https://www.phenix-online.org/). The final refinement statistics of the SynGAP coiled-coil trimer are listed in Table S2. All structure Figures were prepared using the program PyMOL (http://pymol.sourceforge.net/).
NMR Experiments
15N labeled PDZ3-C-SynGAP PBM fusion protein samples at a concentration of 200 µM in a buffer containing 50mM PBS pH6.5, 50mM NaCl, 1mM EDTA and 1mM DTT were prepared for NMR measurements. 1H,15N-HSQC spectra of the samples with and without thrombin digestion were acquired at 30°C on a Varian Inova 800 MHz spectrometer.
Fast Protein Liquid Chromatography Coupled with Static Light Scattering
The analysis was performed on an AKTA FPLC system (GE Healthcare) coupled with a static light scattering detector (miniDawn, Wyatt) and a differential refractive index detector (Optilab, Wyatt). Protein samples (concentration of 20 µM for SynGAP trimer in Figures 2B and S2B and 100 or 150 µM for 3:2 complex detection in Figures 3 and S5) were filtered (condensed liquid phase was removed) and loaded into a Superose 12 10/300 GL column pre-equilibrated by a column buffer composed of 50mM Tris pH7.8, 100mM NaCl, 1mM EDTA, and 5mM DTT. Data were analyzed with ASTRA6 (Wyatt).
Circular Dichroism Measurements
The CD spectra of the proteins were acquired on a Jasco J-815 CD spectrometer at the room temperature. SynGAP CC-PBM proteins were purified, tag-cleaved and diluted into 20 µM in a buffer containing 25mM Tris pH7.8, 50mM NaCl, 0.5mM EDTA and 0.5mM DTT. For urea-induced denaturation assay, the fraction of remain folded at each urea concentration was calculated by comparing the ellipticity value of the sample at 222nm with that in the absence of urea.
GST Pull-Down Assay
Flag-tagged full-length SynGAP proteins were expressed in HEK293T cells. An aliquot of 25 µl glutathione Sepharose beads charged with 0.2nmol purified GST-tagged PSD-95 PSG or GST were used to pull down the WT or mutant SynGAP proteins in each reaction. After extensive washing, bound SynGAP proteins were eluted and detected by Western blotting using anti-Flag antibody (Sigma F1804).
In Vitro Phase Transition Assay
Both SynGAP CC-PBM and PSD-95 PSG (WT or mutants with purification tags cleaved and removed) were prepared in buffer containing 50mM Tris pH7.8, 100mM NaCl, 1mM EDTA, 5mM DTT and pre-cleared via high-speed centrifugations. Typically, the two proteins were mixed at a 1:1 stoichiometry at final concentrations spanning 10–125 µM. Formations of phase transition were assayed either directly by imaging-based methods or by sedimentation-based separation of the condensed liquid phase from the aqueous phase.
For imaging, droplets were observed either in solution drops by light microscope or by injecting mixtures into a homemade flow chamber comprised of a glass slide sandwiched by a coverslip with one layer of double-sided tape as a spacer for DIC (Nikon eclipse 80i) or fluorescent imaging (Zeiss LSM 880). For the sedimentation assay, samples were subjected to centrifugation at 16,873 g for 10min at indicated temperatures (20°C if not specified) on a table-top temperature-controlled micro-centrifuge. Supernatant and pellet were separated into two tubes immediately after centrifugation. The pellet fraction was washed once with the assay buffer and thoroughly re-suspended with the same buffer to the equal volume as supernatant fraction. Proteins from both fractions were analyzed by 15% SDS-PAGE with Coomassie blue staining. Band intensities were quantified using the ImageJ software.
Chemical Cross-Linking Assay
PSD-95 PSG (without affinity tag), SynGAP 15AA PBM peptide and CRIPT PBM peptide were prepared in buffer with 50mM PBS pH7.4, 100mM NaCl, 5mM DTT. The DSG to PSD-95 molar ratio was set as 20:1. The cross-linking reaction was carried out at room temperature and quenched by 200mM Tris pH 8.3. Proteins were analyzed by 10% SDS-PAGE with Coomassie blue staining.
HeLa Cell Imaging and Data Analysis
For each well in a 12-well plate, 0.5 mg RFP-PSD-95 PSG plasmids with 0.25 µg GFP-SynGAP CC-PBM plasmids were co-transfected into HeLa cells using NanoJuice transfection kit (Novagen). Cells were imaged using a Leica sp8 or Zeiss LSM 880 confocal microscope by a 40X oil-immersion lens without immunostaining, and acquired images were processed with ImageJ. For nuclear localization, nuclei of cells were defined by DAPI staining and the percentage of SynGAP nuclear localization was calculated as GFPnuclear intensity/ (GFPnuclear intensity + GFPcytoplasmic intensity).For puncta counting assay, data were collected from 3–6 independent batches of cultures as indicated in the figure. In each batch, at least 600 fluorescence-positive cells were counted for each group of experiments. A cell with more than three bright fluorescent puncta was counted as a puncta-positive cell. Experiments were conducted in a blinded fashion. Live cell imaging was performed using a Nikon Ti-E-PFS microscope supported with a Chamlide TC temperature, humidity, and CO2 chamber. Images were collected by a 40X air lens for up to 24 hr at 5 min/frame and processed with ImageJ.
Fluorescence Recovery after Photo-Bleaching Assay
HeLa cells were cultured in glass-bottom dishes (MatTek) and transfected as described above. FRAP assay was performed on a Leica sp8 or Zeiss LSM 880 confocal microscope at 37°C. GFP signal was bleached using a 488-nm laser beam. Puncta with diameters around 0.6–1.1 µm were assayed. The fluorescence intensity difference between pre-bleaching and at time 0 (the time point right after photobleaching pulse) was normalized to 100%. The experimental control is to quantify fluorescence intensities of similar puncta/ cytoplasm regions without photobleaching.
Neuronal Cultures, Imaging, and LTP Induction
E18 rat hippocampal neurons were transfected at DIV17–19 with Lipofectamine2000 (Invitrogen) in accordance with manufacture’s manual. After 2 days, we placed coverslips on a custom-made perfusion chamber with basal ECS (143mM NaCl, 5mM KCl, 10mM HEPES pH 7.42, 10mM Glucose, 2mM CaCl2, 1mM MgCl2, 0.5mM TTX, 1mµM Strychnine, 20µM Bicuculline) and time-lapse images were captured with either LSM510 (Carl Zeiss) or Spinning disk confocal system controlled by axiovision software (Carl Zeiss). Following 5–10min of basal recording, cells were perfused with 10ml of Glycine/0Mg ECS (143mM NaCl, 5mM KCl, 10mM HEPES pH 7.42, 10mM Glucose, 2mM CaCl2, 0mM MgCl2, 0.5mµM TTX, 1µM Strychnine, 20µM Bicuculline, 200mM (or 10µM for threshold LTP) Glycine) for 10min followed 10ml of basal ECS. To stabilize focus during long term imaging, we used definite focus (Zeiss) system. For quantification, we selected all spines on a secondary dendrite (25–45 microns) branching immediately after a primary dendrite. We used the mCherry channel to select the spine region that is well separated from dendritic shaft. These regions of interest (ROIs) in the mCherry channel were transferred to the Green channel to quantify total SynGAP content in spines. SynGAP content in each spine was calculated as: (Average Green signal at ROI – Average Green signal at Background region) * (Area of ROI) as described in detail previously (Araki et al., 2015).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the definitions and exact values of n (e.g., number of experiments, number of cells, number of spines, etc), distributions and deviations are reported in the Figures and corresponding Figure Legends. Data of in vitro phase transition sedimentation assay and FRAP assay were expressed as mean ± SD. Data of HeLa cell and primary neuron culture were expressed as mean ± SEM; ns, not significant, *p < 0.05, **p < 0.01 and ***p < 0.001 using one-way ANOVA with Tukey’s multiple comparison test. Data are judged to be statistically significant when p < 0.05 by one-way ANOVA with Tukey’s multiple comparison test. None of the data were removed from our statistical analysis as outliers. Statistical analysis was performed in GraphPad Prism. All experiments related to cell cultures and imaging studies were performed in blinded fashion.
DATA AND SOFTWARE AVAILABILITY
Data Resources
The atomic coordinates of the PSD-95 PDZ3-C/SynGAP PBM complex and the SynGAP coiled-coil trimer are deposited to the Protein Data Bank under the accession codes PDB: 5JXB and PDB: 5JXC, respectively.
Supplementary Material
In Brief.
The interaction between two major components of postsynaptic densities induces phase separation of the newly formed complex into liquid-like droplets, suggesting a mechanism for the formation and activity-dependent modulation of synaptic complexes.
Highlights.
SynGAP forms a coiled-coil trimer, and each binds to two molecules of PSD-95
SynGAP/PSD-95 complex undergoes liquid-liquid-phase separation
SynGAP/PSD-95 phase separation suggests a possible PSD formation mechanism
Phase-separation-mediated SynGAP PSD enrichment is correlated with synaptic activity
Acknowledgments
The authors thank the Shanghai Synchrotron Radiation Facility (SSRF) BL17U and B19U1 for X-ray beam time. Dr. Fei Ye helped with NMR analysis. This work was supported by grants from RGC of Hong Kong (664113, 16103614, AoE-M09-12, and T13-607/12R) and a 973 program grant from the Minister of Science and Technology of China (2014CB910204) (to M.Z.). The work was also supported by a grant from the NIH (R01NS036715) (to R.L.H.). M.Z. is a Kerry Holdings Professor in Science and a Senior Fellow of IAS at HKUST.
Footnotes
Supplemental Information includes seven Figures, two tables, and three movies and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.07.008
AUTHOR CONTRIBUTIONS
M. Zeng, Y.S., Y.A., and T.G. performed experiments; M. Zeng, Y.S., Y.A., T.G., R.L.H., and M. Zhang analyzed the data; M. Zeng, Y.S., Y.A., R.L.H., and M. Zhang designed the research; M. Zeng and M. Zhang drafted the manucsript and all authors commented on it; Y.S. and Y.A. made equal contributions to the study; and M. Zhang coordinated the project.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Y, Zeng M, Zhang M, Huganir RL. Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron. 2015;85:173–189. doi: 10.1016/j.neuron.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banjade S, Rosen MK. Phase transitions of multivalent proteins can promote clustering of membrane receptors. eLife. 2014;3:e04123. doi: 10.7554/eLife.04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron MK, Boeckers TM, Vaida B, Faham S, Gingery M, Sawaya MR, Salyer D, Gundelfinger ED, Bowie JU. An architectural framework that may lie at the core of the postsynaptic density. Science. 2006;311:531–535. doi: 10.1126/science.1118995. [DOI] [PubMed] [Google Scholar]
- Berryer MH, Hamdan FF, Klitten LL, Møller RS, Carmant L, Schwart-zentruber J, Patry L, Dobrzeniecka S, Rochefort D, Neugnot-Cerioli M, et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum. Mutat. 2013;34:385–394. doi: 10.1002/humu.22248. [DOI] [PubMed] [Google Scholar]
- Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M, Hayashi Y. Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron. 2014;82:444–459. doi: 10.1016/j.neuron.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP. Phase transitions and size scaling of membrane-less organelles. J. Cell Biol. 2013;203:875–881. doi: 10.1083/jcb.201308087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Jülicher F, Hyman AA. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- Caroni P, Donato F, Muller D. Structural plasticity upon learning: regulation and functions. Nat. Rev. Neurosci. 2012;13:478–490. doi: 10.1038/nrn3258. [DOI] [PubMed] [Google Scholar]
- Chen H-J, Rojas-Soto M, Oguni A, Kennedy MB. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron. 1998;20:895–904. doi: 10.1016/s0896-6273(00)80471-7. [DOI] [PubMed] [Google Scholar]
- Chen X, Winters C, Azzam R, Li X, Galbraith JA, Leapman RD, Reese TS. Organization of the core structure of the postsynaptic density. Proc. Natl. Acad. Sci. USA. 2008;105:4453–4458. doi: 10.1073/pnas.0800897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Levy JM, Hou A, Winters C, Azzam R, Sousa AA, Leapman RD, Nicoll RA, Reese TS. PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc. Natl. Acad. Sci. USA. 2015;112:E6983–E6992. doi: 10.1073/pnas.1517045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D, Hoogenraad CC, Rush J, Ramm E, Schlager MA, Duong DM, Xu P, Wijayawardana SR, Hanfelt J, Nakagawa T, et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteomics. 2006;5:1158–1170. doi: 10.1074/mcp.D500009-MCP200. [DOI] [PubMed] [Google Scholar]
- Choquet D, Triller A. The dynamic synapse. Neuron. 2013;80:691–703. doi: 10.1016/j.neuron.2013.10.013. [DOI] [PubMed] [Google Scholar]
- Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, Almonte AG, Miller BH, Wiltgen BJ, Miller CA, et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell. 2012;151:709–723. doi: 10.1016/j.cell.2012.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan LA, Yasuda R. Plasticity of dendritic spines: subcom-partmentalization of signaling. Annu. Rev. Physiol. 2014;76:365–385. doi: 10.1146/annurev-physiol-021113-170400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R. Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell. 2010;85:1067–1076. doi: 10.1016/s0092-8674(00)81307-0. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, Dobrzeniecka S, Côté M, Perreau-Linck E, Carmant L, et al. Synapse to Disease Group Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N. Engl. J. Med. 2009;360:599–605. doi: 10.1056/NEJMoa0805392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Weinberg RJ. Ultrastructure of synapses in the mammalian brain. Cold Spring Harb. Perspect. Biol. 2012;4:4. doi: 10.1101/cshperspect.a005587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi MK, Tang C, Verpelli C, Narayanan R, Stearns MH, Xu R-M, Li H, Sala C, Hayashi Y. The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell. 2009;737:159–171. doi: 10.1016/j.cell.2009.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, Jülicher F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014;30:39–58. doi: 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- Jiang H, Wang S, Huang Y, He X, Cui H, Zhu X, Zheng Y. Phase transition of spindle-associated protein regulate spindle apparatus assembly. Cell. 2015;163:108–122. doi: 10.1016/j.cell.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Naisbitt S, Hsueh Y-P, Rao A, Rothschild A, Craig AM, Sheng M. GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J. Cell Biol. 1997;136:669–678. doi: 10.1083/jcb.136.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Liao D, Lau L-F, Huganir RL. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron. 1998;20:683–691. doi: 10.1016/s0896-6273(00)81008-9. [DOI] [PubMed] [Google Scholar]
- Levy JM, Chen X, Reese TS, Nicoll RA. Synaptic Consolidation Normalizes AMPAR Quantal Size following MAGUK Loss. Neuron. 2015;87:534–548. doi: 10.1016/j.neuron.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Banjade S, Cheng H-C, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–340. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wei Z, Yan Y, Wan Q, Du Q, Zhang M. Structure of Crumbs tail in complex with the PALS1 PDZ-SH3-GK tandem reveals a highly specific assembly mechanism for the apical Crumbs complex. Proc. Natl. Acad. Sci. USA. 2014;111:17444–17449. doi: 10.1073/pnas.1416515111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenthal MS, Markey SP, Dosemeci A. Quantitative mass spectrometry measurements reveal stoichiometry of principal postsynaptic density proteins. J. Proteome Res. 2015;14:2528–2538. doi: 10.1021/acs.jproteome.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGillavry HD, Song Y, Raghavachari S, Blanpied TA. Nanoscale scaffolding domains within the postsynaptic density concentrate synaptic AMPA receptors. Neuron. 2013;78:615–622. doi: 10.1016/j.neuron.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglione M, Sigrist SJ. Seeing the forest tree by tree: super-resolution light microscopy meets the neurosciences. Nat. Neurosci. 2013;76:790–797. doi: 10.1038/nn.3403. [DOI] [PubMed] [Google Scholar]
- McCann JJ, Zheng L, Rohrbeck D, Felekyan S, Kühnemuth R, Sutton RB, Seidel CAM, Bowen ME. Supertertiary structure of the synaptic MAGuK scaffold proteins is conserved. Proc. Natl. Acad. Sci. USA. 2012;109:15775–15780. doi: 10.1073/pnas.1200254109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–133. doi: 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair D, Hosy E, Petersen JD, Constals A, Giannone G, Choquet D, Sibarita JB. Super-resolution imaging reveals that AMPA receptors inside synapses are dynamically organized in nanodomains regulated by PSD95. J. Neurosci. 2013;33:13204–13224. doi: 10.1523/JNEUROSCI.2381-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama J, Yasuda R. Biochemical Computation for Spine Structural Plasticity. Neuron. 2015;87:63–75. doi: 10.1016/j.neuron.2015.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell JD, Zhao A, Ellington AD, Marcotte EM. Dynamic reorganization of metabolic enzymes into intracellular bodies. Annu. Rev. Cell Dev. Biol. 2012;28:89–111. doi: 10.1146/annurev-cellbio-101011-155841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography. 1997;276(Pt A):307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pan L, Chen J, Yu J, Yu H, Zhang M. The structure of the PDZ3-SH3-GuK tandem of ZO-1 protein suggests a supramodular organization of the membrane-associated guanylate kinase (MAGUK) family scaffold protein core. J. Biol. Chem. 2011;286:40069–40074. doi: 10.1074/jbc.C111.293084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MJ, Fryer AE, Shears DJ, Lachlan KL, McKee SA, Magee AC, Mohammed S, Vasudevan PC, Park SM, Benoit V, et al. De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am. J. Med. Genet. A. 2015;167A:2231–2237. doi: 10.1002/ajmg.a.37189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Petit CM, Zhang J, Sapienza PJ, Fuentes EJ, Lee AL. Hidden dynamic allostery in a PDZ domain. Proc. Natl. Acad. Sci. USA. 2009;106:18249–18254. doi: 10.1073/pnas.0904492106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu. Rev. Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Triller A, Choquet D. New concepts in synaptic biology derived from single-molecule imaging. Neuron. 2008;59:359–374. doi: 10.1016/j.neuron.2008.06.022. [DOI] [PubMed] [Google Scholar]
- Trinidad JC, Thalhammer A, Specht CG, Lynn AJ, Baker PR, Schoepfer R, Burlingame AL. Quantitative analysis of synaptic phosphorylation and protein expression. Mol. Cell. Proteomics. 2008;7:684–696. doi: 10.1074/mcp.M700170-MCP200. [DOI] [PubMed] [Google Scholar]
- Vazquez LE, Chen H-J, Sokolova I, Knuesel I, Kennedy MB. SynGAP regulates spine formation. J. Neurosci. 2004;24:8862–8872. doi: 10.1523/JNEUROSCI.3213-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff JB, Wueseke O, Viscardi V, Mahamid J, Ochoa SD, Bunkenborg J, Widlund PO, Pozniakovsky A, Zanin E, Bahmanyar S, et al. Centrosomes. Regulated assembly of a supramolecular centrosome scaffold in vitro. Science. 2015;348:808–812. doi: 10.1126/science.aaa3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Zhang M. Structures and target recognition modes of PDZ domains: recurring themes and emerging pictures. Biochem. J. 2013;455:1–14. doi: 10.1042/BJ20130783. [DOI] [PubMed] [Google Scholar]
- Zeng M, Shang Y, Guo T, He Q, Yung WH, Liu K, Zhang M. A binding site outside the canonical PDZ domain determines the specific interaction between Shank and SAPAP and their function. Proc. Natl. Acad. Sci. USA. 2016;113:E3081–E3090. doi: 10.1073/pnas.1523265113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lewis SM, Kuhlman B, Lee AL. Supertertiary structure of the MAGUK core from PSD-95. Structure. 2013;21:402–413. doi: 10.1016/j.str.2012.12.014. [DOI] [PubMed] [Google Scholar]
- Zhu J, Shang Y, Xia C, Wang W, Wen W, Zhang M. Guany-late kinase domains of the MAGUK family scaffold proteins as specific phospho-protein-binding modules. EMBO J. 2011;30:4986–4997. doi: 10.1038/emboj.2011.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Shang Y, Zhang M. Mechanistic basis of MAGUK-organized complexes in synaptic development and signalling. Nat. Rev. Neurosci. 2016;17:209–223. doi: 10.1038/nrn.2016.18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.