

Abstract
Deletion of Ca2+/calmodulin-dependent protein kinase II delta (CaMKIIδ) has been shown to protect against in vivo ischemia/reperfusion (I/R) injury. It remains unclear which CaMKIIδ isoforms and downstream mechanisms are responsible for the salutary effects of CaMKIIδ gene deletion. In this study we sought to compare the roles of the CaMKIIδB and CaMKIIδC subtypes and the mechanisms by which they contribute to ex vivo I/R damage. WT, CaMKIIδKO, and mice expressing only CaMKIIδB or δC were subjected to ex vivo global ischemia for 25 minutes followed by reperfusion. Infarct formation was assessed at 60 minutes reperfusion by triphenyl tetrazolium chloride (TTC) staining. Deletion of CaMKIIδ conferred significant protection from ex vivo I/R. Re-expression of CaMKIIδC in the CaMKIIδKO background reversed this effect and exacerbated myocardial damage and dysfunction following I/R, while re-expression of CaMKIIδB was protective. Selective activation of CaMKIIδC in response to I/R was evident in a subcellular fraction enriched for cytosolic/membrane proteins. Further studies demonstrated differential regulation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling and tumor necrosis factor alpha (TNF-α) expression by CaMKIIδB and CaMKIIδC. Selective activation of CaMKIIδC was also observed and associated with NF-κB activation in neonatal rat ventricular myocytes (NRVMs) subjected to oxidative stress. Pharmacological inhibition of NF-κB or TNF-α significantly ameliorated infarct formation in WT mice and those that re-express CaMKIIδC, demonstrating distinct roles for CaMKIIδ subtypes in I/R and implicating acute activation of CaMKIIδC and NF-κB in the pathogenesis of reperfusion injury.
Keywords: CaMKII, NF-κB, TNF-α, Ischemia/Reperfusion, Heart
Subject codes: Signal Transduction, Inflammation, Myocardial Infarction
1.3 INTRODUCTION
The calcium/calmodulin dependent protein kinase II (CaMKII) is a dodecameric enzyme consisting of subunits encoded by four different genes known as CaMKIIα, β, γ, and δ. The predominant cardiac isoform is CaMKIIδ and it is alternatively spliced in the heart to generate CaMKIIδB and CaMKIIδC as well as other minor subtypes [1–5]. We have generated global and cardiac specific knockouts of CaMKIIδ and demonstrated that deletion of this protein ameliorates heart failure development in response to pressure overload, Gαq expression, and isoproterenol infusion [6–8]. We further reported that deletion of CaMKIIδ diminishes infarct development in response to in vivo ischemia/reperfusion [9]. Other studies using CaMKII inhibitory peptides or knock-ins of activation-deficient CaMKII have similarly concluded that CaMKII activation by a range of cardiac insults, including myocardial infarction, is deleterious [10–13]. In all of the aforementioned studies both the CaMKIIδB and CaMKIIδC subtypes of CaMKIIδ were genetically deleted or inhibited. Accordingly it is not yet known which subtype is responsible for the protective effect of ablating CaMKIIδ activity in the heart.
The studies reported here use mice in which CaMKIIδ deletion has been restored by crossing CaMKIIδKO mice with transgenic lines expressing either the CaMKIIδB (δBTG/δKO) or CaMKIIδC (δCTG/δKO) subtype [14]. This approach allowed us to examine the unique roles of CaMKIIδB and CaMKIIδC in cardiomyocyte survival and infarct formation in response to I/R. We demonstrate that CaMKIIδC expression reverses and exacerbates the diminished I/R damage observed in CaMKIIδKO mouse hearts whereas CaMKIIδB expression further attenuates I/R damage. The difference in infarct development observed in δBTG/δKO and δCTG/δKO mice is associated with greater I/R-induced inhibitor of kappa B kinase (IKK) and NF-κB activation in δCTG/δKO mice. CaMKIIδC-mediated NF-κB activation is recapitulated in NRVMs exposed to oxidative stress, and selective activation of CaMKIIδC in a cytosol/membrane fraction is observed in NRVMs exposed to oxidative stress and in hearts exposed to I/R. TNF-α expression is also selectively increased in hearts from δCTG/δKO mice following I/R. Blocking either IKK activation or TNF-α signaling diminished infarct development in δCTG/δKO as well as in WT mice. These data suggest that selective activation of the CaMKIIδC subtype in cardiomyocytes regulates cardiac-autonomous pro-inflammatory signaling events that contribute to ischemia/reperfusion injury.
1.4 METHODS
Transgenic Animals
Transgenic Black Swiss mice in which the predominant cardiac subtypes of CaMK, CaMKIIδB and CaMKIIδC, are over expressed were generated in our laboratory and characterized as described [15, 16]. Conventional CaMKIIδKO mice were generated and characterized as previously described [6]. CaMKIIδB and CaMKIIδC transgenic mice were crossed with conventional CaMKIIδKO mice to generate mice that express only CaMKIIδB or CaMKIIδC. We refer to these animals as CaMKIIδBTG/δKO and CaMKIIδCTG/δKO. All mice used in the present study were male at 8 weeks of age, unless otherwise noted. Animal studies were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committees of University of California at San Diego.
CaMKII activity assay
CaMKII activity was measured in ventricular homogenate using Syntide-2, a synthetic CaMKII-specific substrate peptide. Hearts were isolated and ventricles homogenized in lysis buffer (50 mmol/L HEPES, 10% ethylene glycol, 2 mg/ml BSA, 5 mmol/L EDTA, pH 7.5), and assayed immediately without freezing. The assay buffer contained 50 mmol/L HEPES, 10 mmol/L magnesium acetate, 1 mg/ml BSA, 20 μmol/L Syntide-2, 1 mmol/L DTT, 400 nM [γ-32P]ATP, pH 7.5 and either 1 mmol/L EGTA (for autonomous activity) or 500 μmol/L CaCl2, plus 1 μmol/L calmodulin (for maximal activity). The reaction was carried out at 30 °C for 10 min and blotted onto Whatman P81 phosphocellulose paper.
Heart tissue fractionation procedure
Mice were killed by cervical dislocation, and hearts were quickly removed and placed in ice-cold PBS. Following cannulation, hearts were perfused with PBS to remove blood, frozen in liquid N2, and pulverized. Tissue was then homogenized in isolation buffer (70mmol/L sucrose, 190mmol/L mannitol, 20mmol/L HEPES, 0.2mmol/L EDTA) that was supplemented with various inhibitors: sodium vanadate, leupeptin, aprotinin, p-nitrophenyl phosphate, and phenylmethylsulfonyl fluoride. Homogenate was centrifuged at 600g for 10m at 4°C, and the supernatant transferred to another tube. The supernatant was centrifuged at 5,000g for 15m at 4°C to yield a cytosol/membrane fraction. The pellet from the initial 600g centrifugation was resuspended in nuclear lysis buffer (20mmol/L NaCl, 1.5mmol/L MgCl2, 20 mmol/L HEPES, 200 nmol EDTA, 25% glycerol) and centrifuged at 600g for 10m at 4°C to yield a nuclear fraction.
Neonatal Rat Ventricular Myocyte (NRVM) isolation and adenoviral infection
Neonatal rat ventricular myocytes (NRVMs) were isolated from 1–2-day-old Sprague-Dawley rat pups, digested with collagenase, plated at density of 3.5×104/cm2 and maintained overnight in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin), Prior to adenoviral Infection, isolated NRVMs were transferred to serum-free medium and infected with CaMKII δB or δC adenovirus at 10, 30, 50, 100, 300 multiplicity of infection (MOI) for 3 h. Cells were washed and maintained in serum-free medium for an additional 21 hrs prior to treatment with H2O2 or vehicle. To assess changes in nuclear and cytosolic/membrane fractions, NRVMs were fractionated according to a previously reported protocol [17]. Purity was determined using the cytosolic marker Rho GDP dissociation inhibitor (RhoGDI) and the nuclear marker Lamin A/C.
Transthoracic echocardiography
Echocardiography was performed using the VisualSonics VeVo 770 Imaging System (VisualSonics, Toronto, Canada) equipped with high-frequency 30 MHz probe, as described [18]. Body temperatures were maintained between narrow ranges (37.0 ± 1.0 °C) to avoid confounding effects of hypothermia.
Isolated perfused (ex vivo) I/R
Mice were killed by cervical dislocation, and hearts were quickly removed and placed in ice-cold Ca2+-free Krebs-Henseleit buffer. Aortas were cannulated and hearts perfused by gravity flow on a Langendorff perfusion system (Radnoti LLC) at 37 °C and aconstant pressure of 80 mmHg with a modified Krebs-Henseleit buffer solution containing (in mmol/L); 2.0 CaCl2, 130 NaCl, 5.4 KCl, 11 dextrose, 2 pyruvate, 0.5 MgCl2, 0.5 NaH2PO4, and 25 NaHCO3 and aerated with 95% oxygen and 5% carbon dioxide, pH 7.4. To measure infarct size, hearts were subjected to 25-minute global ischemia and 1-hour reperfusion; the ventricles were then frozen and cut transversely into 5 slices of equal thickness. The slices were then incubated in 1% TTC/PBS and fixed in 10% formalin-PBS for 24 hours. Fixed slices were then scanned, and ImageJ was used to measure and calculate the size of the infarct area and the total area. For experiments utilizing BMS-345541 (Sigma-Aldrich) the drug was dissolved in Krebs-Henseleit buffer solution at a concentration of 5μmol/L and was present throughout the I/R protocol. For those using etanercept, the drug was present for the entire I/R procedure at a concentration of 5μg/ml. To assess cardiac function, a water-filled balloon connected to a pressure transducer (Gould Stathem P23 ID) was inserted into the left ventricle through the left atrium to monitor left ventricular developed pressure (LVDP); data collected using Powerlab, were processed with AD Instruments Chart 4 software (v4.12). Hearts were submersed in warm KHB (37°C) throughout the perfusion. Functional recovery was expressed as a percentage of pre-ischemic LVDP.
Immunoblotting
Ventricular tissue was homogenized in RIPA buffer (10 mmol/L Tris-Cl (pH 8.0), 1mmol/L EDTA, 0.5 EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140mmol/L NaCl) that was supplemented with various inhibitors: sodium vanadate, leupeptin, aprotinin, p-nitrophenyl phosphate, and phenylmethylsulfonyl fluoride. Western blot analysis was performed according to protocols described previously [9]. The antibodies for immunoblotting were as follows: CaMKIIδ (D.M. Bers, UC Davis); GAPDH (CST); phospho-IKKα/β at its autophosphorylation site (Ser176/180) (CST); phospho-CaMKII at its autophosphorylation site (Thr286) (Thermo); NF-κB p65 (CST); α-actinin (CST); RhoGDI (CST); Lamin A/C (CST); IκBα (CST)
RT-PCR
RNA extraction for real time analysis was performed using the solid-phase RNeasy purification kit from Qiagen (Venlo, Netherlands). First strand cDNA synthesis for Real time PCR was performed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Grand Island, NY). Gene expression was determined using Taqman® Universal PCR master mix, Cy5-labeled Taqman® probe for TNF-α and IL-6 and FAM-labeled Taqman® probe for GAPDH (Applied Biosystems).
1.5 STATISTICAL ANALYSIS
Data are presented as mean ± SEM as indicated and were analyzed by 2-tailed Student t test between 2 groups or by ANOVA when 3 or more groups were compared. P values <0.05 were considered statistically significant.
1.6 RESULTS
To examine the independent roles of the CaMKIIδB and δC subtypes in the heart we restored either δB or δC expression in a CaMKIIδ-null (KO) background. The resulting animals express only δB (δBTG/δKO) or only δC (δCTG/δKO). Survival of WT, KO, δBTG/δKO, and δCTG/δKO mice was assessed. Deletion of CaMKIIδ produced no overt phenotypic changes and did not affect survival relative to WT mice as shown previously [6]. δBTG/δKO animals also survived normally for at least 6 months. In contrast the δCTG/δKO animals, like the previously studied δCTG [16], exhibited premature death with less than 20% survival by 21 weeks (Fig. 1A). Expression of the CaMKII δB and δC subtypes also had markedly different effects on in vivo cardiac function. Echocardiography on 6–8 week old mice revealed that fractional shortening (FS) was decreased by 63% in δCTG/δKO mice compared to WT mice (Fig. 1B) while δBTG/δKO animals did not display cardiac dysfunction.
Figure 1.

CaMKIIδcTG/δKO but not CaMKIIδBTG/δKO mice display diminished survival and cardiac function. A, Kaplan-Meier analysis of survival of WT, CaMKIIδKO, δBTG/δKO, and δcTG/δKO mice. B, Representative echocardiography recordings from WT and δcTG/δKO mice and quantification of fractional shortening (FS) measured in 6–8-week-old WT, KO, δBTG/δKO, and δcTG/δKO mice. Data are mean ±SEM values from 4–6 mice. *P<0.05 vs WT.
We previously demonstrated that CaMKIIδ deletion attenuates I/R injury in response to in vivo left anterior descending coronary artery occlusion and subsequent reperfusion [9]. To examine the cardiac-intrinsic role of CaMKIIδ in I/R we performed ex vivo I/R experiments on isolated perfused hearts from 8-week-old mice. Infarct formation following 25 minutes ischemia and 1 hour reperfusion was determined by TTC staining of heart sections. In WT animals, ex vivo I/R induced infarcts comprising 36.2±2.5% of the cross-sectional area. Infarcts were significantly smaller, only 24.1±1.4%, in CaMKIIδKO mouse hearts (Fig. 2A, B). Thus regulation of infarct development by CaMKII is evident not only in vivo but also in an ex vivo I/R model.
Figure 2.

Differential effects of CaMKII subtypes on I/R injury and oxidative stress. A, Representative images hearts from WT, CaMKIIδKO, CaMKIIδBTG/δKO, and CaMKIIδcTG/δKO mice subjected to 25 minutes ischemia and 1 hour of reperfusion in the Langendroff mode. Hearts were sectioned and stained with TTC to reveal infarcted tissue. B. Infarct size was quantified from TTC stained heart sections. Data are mean±SEM values from 14–16 mice. *P<0.05 vs WT. #P<0.05 vs KO. †P<0.05 vs δcTG/δKO
Hearts from δBTG/δKO mice were then examined and found to be protected against ex vivo I/R damage, with infarcts measuring 12.2±1.9% of cross-sectional area. Conversely, in δCTG/δKO mouse hearts, the protective effect of CaMKIIδ gene deletion was lost with infarcts measuring 45.2±1.8% of the cross-sectional area, significantly larger than those of WT, KO, and δBTG/δKO (Fig. 2A, B). Assessment of left ventricular developed pressure (LVDP) recovery during reperfusion confirmed that expression of CaMKIIδC in CaMKIIδKO mice exacerbates I/R damage while expression of CaMKIIδB does not (Supplemental Fig. 1).
CaMKII is known to be activated during ex vivo I/R [19, 20], but previous experiments have not assessed subtype-specific CaMKII activation. Although we previously demonstrated that δB and δC can be equivalently activated by several pharmacological interventions [14], we wondered if I/R might lead to differential activation of the two subtypes. To assess I/R-induced activation of CaMKII we analyzed phosphorylation of CaMKII at its autophosphorylation site threonine-286. The increases in CaMKII autophosphorylation observed in whole cell lysates from hearts of δBTG/δKO and δCTG/δKO mice subjected to I/R were equivalent (Fig. 3A).
Figure 3.

CaMKIIδc is selectively activated in a subcellular fraction containing cellular membranes and cytosol. A, Lysates from the hearts of δBTG/δKO and δcTG/δKO mice were subjected to western blot analysis using an antibody specific for auto-phosphorylated CaMKII. Data are mean±SEM values from 4–6 mice. B, Subcellular fractionation of hearts from δBTG/δKO and δcTG/δKO mice was carried out as described in methods yielding a fraction that includes cytosol and cellular membranes. Data are mean±SEM values from 4–6 mice. *P<0.05 vs sham.
Many substrates of CaMKIIδC (e.g phospholamban, RyR2, and histone deacetylase 4) localize to the cytosol or the cytosolic face of the cardiac sarcoplasmic reticular (SR) membrane. Thus we carried out further studies using a subcellular fraction enriched for cytosolic and membrane proteins. Strikingly in this fraction we observed differential activation of the δB and δC subtypes in response to I/R. Indeed whereas no increases in CaMKII autophosphorylation were observed in cytosolic/membrane fractions from δBTG/δKO mouse hearts, there was a more than 3-fold increase in autophosphorylated CaMKII in cytosolic/membrane-enriched fractions from δCTG/δKO hearts (Fig. 3B).
Our earlier studies examining in vivo I/R damage linked the deleterious effects of CaMKII to activation of IKK and subsequent NF-κB nuclear accumulation [9]. To determine which CaMKIIδ subtype was responsible, and also to determine if CaMKIIδ-mediated NF-κB activation can occur in the absence of systemic factors (e.g. leukocyte infiltration), we examined regulation of IKK and NF-κB by I/R in the ex vivo heart. The phosphorylation of IKK was found to be elevated during ex vivo reperfusion in δCTG/δKO mice but not in δBTG/δKO mice (Fig. 4A). Furthermore I/R-mediated activation of IKK in δCTG/δKO mice was associated with an increased nuclear localization of the p65 subunit of NF-κB, which was not observed in δBTG/δKO animals (Fig. 4B).
Figure 4.

The NF-κB pathway is activated in CaMKIIδ cTG/δKO mice following reperfusion. A, Activation of IKKα/β was assessed in δBTG/δKO and δcTG/δKO mice using an antibody specific for auto-phosphorylated IKKα/β. B, NF-κB p65 accumulation was assessed in nuclear extracts made from hearts of δBTG/δKO and δCTG/δKO mice subjected to ex vivo I/R. Data are meant±SEM values from 6 mice. *P<0.05 vs sham.
Nuclear p65 translocation would be expected to result in transcriptional activation of NF-κB target genes. Thus as further evidence that differences in NF-κB activation in δCTG/δKO and δBTG/δKO are functionally significant, we measured mRNA levels of genes regulated by NF-κB. Ischemia/reperfusion increased interleukin 6 (IL-6) mRNA in hearts from δCTG/δKO mice to a greater extent than in those from δBTG/δKO mice (Fig. 5A). Even more striking was the robust increase in TNF-α expression in δCTG/δKO mice and the absence of upregulation of this gene in δBTG/δKO animals (Fig. 5B).
Figure 5.

Genes downstream of NF-κB are upregulated in CaMKIIδcTG/δKO mice after ex vivo I/R. NF-κB activation was assessed by measuring mRNA transcripts of genes regulated by NF-κB. A, IL-6 B, TNF-α. Data are meant±SEM values from 4 mice. *P<0.05 vs sham #P<0.05 vs δcTG/δKO
To confirm that oxidative stress can lead to selective activation of CaMKIIδC and of NF-κB in a cardiomyocyte-autonomous fashion, we infected NRVMs with adenovirus expressing CaMKIIδB or CaMKIIδC. Cells were infected with a range of MOIs of CaMKIIδB or δC virus and subsequently treated with 50μM H2O2 for 30 minutes to elicit CaMKII and NF-κB activation. In NRVM whole cell lysates we observed equivalent activation of CaMKIIδB and CaMKIIδC in response to H2O2 at all MOIs (Fig. 6A, top panel), as quantitated using an MOI of 50 (Fig. 6A lower panel). In NRVM lysates that were fractionated to enrich for cytosolic and membrane proteins, however, there was significantly greater activation of CaMKIIδC following H2O2 treatment (Fig. 6B). We further determined that expression of CaMKIIδC, but not CaMKIIδB, enhanced H2O2-mediated I kappa B alpha (IκBα) degradation (Fig. 6C) and nuclear p65 accumulation (Fig. 6D). These data, like those obtained in the isolated perfused heart (Figs. 3 and 4) suggest that oxidative stress-induced CaMKIIδC activation in the cytosolic/membrane compartment is associated with activation of NF-κB.
Figure 6.

Selective CaMKIIδc and NF-κB activation in NRVM treated with 50μM H2O2 for 30m A) CaMKII activation measured in NRVM infected with adenovirus. MOI of 50 was quantified. B) CaMKII activation measured in a cytosolic/membrane fraction isolated from NRVM expressing δB or δC (MOI 50). C) IκBα degradation assessed in NRVM infected with adenovirus. MOI of 50 was quantified. D) NF-κB p65 measured in nuclear extracts from NRVM expressing δBor δC (MOI 50). All data are mean±SEM n=3 *P<0.05 vs Veh #P<0.05 vs δC
To demonstrate that the deleterious effects of CaMKIIδC activation seen in the isolated perfused heart were mediated by IKK/NF-κB signaling we blocked IKK activation with the pharmacological inhibitor BMS-345541 (BMS) [21]. Since this drug had not, to our knowledge, been used in the ex vivo perfused heart we evaluated its efficacy and determined a dose (5μmol/L) that sequestered p65 in the cytosol/membrane fraction (Supplemental Fig. 2). Hearts from δBTG/δKO, δCTG/δKO, WT, and KO mice were then exposed to 5μmol/L BMS or vehicle prior to and throughout the I/R protocol. Infarct size was significantly reduced by BMS administration in WT and δCTG/δKO mice, while the already diminished infarct formation that was observed in δBTG/δKO animals was not affected (Fig. 7). CaMKIIδKO animals showed a modest but significant further reduction in infarct size.
Figure 7.
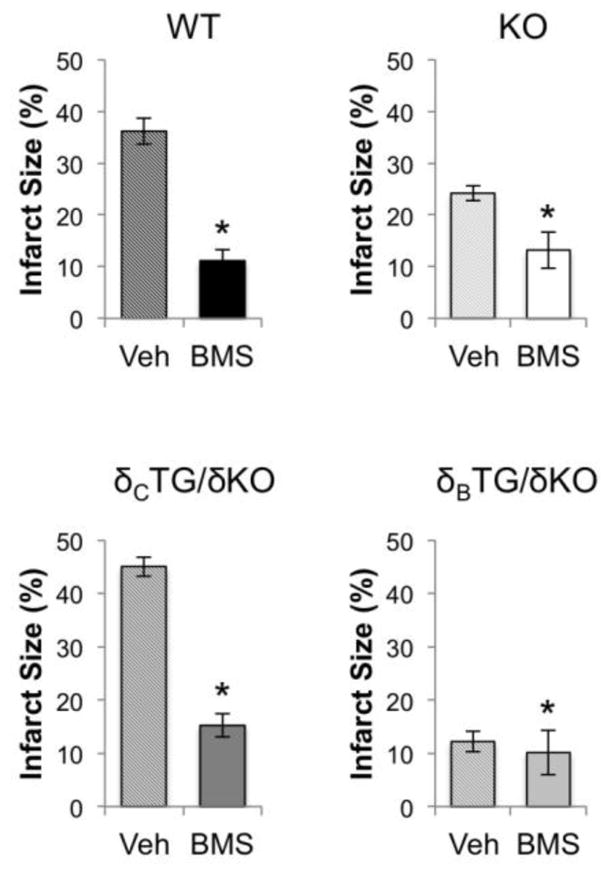
Inhibition of IKK ameliorates I/R damage. Vehicle and BMS-345541 (5μmol/L) were perfused into hearts from WT, KO, δBTG/δKO, and δcTG/δKO animals. Infarct size was measured via TTC staining following 1hr of reperfusion. Data are mean±SEM values from 4–8 mice. *P<0.05 vs veh.
In light of the selective increase in TNF-α mRNA in δCTG/δKO mice, and evidence that active TNF-α can be produced in and secreted from the isolated perfused heart [22], we asked whether TNF-α mediated the deleterious effects of CaMKIIδC. For these studies we used etanercept, which blocks the effects of TNF-α by preventing its interaction with TNF-α receptors [23, 24]. Etanercept was perfused at 5μg/ml prior to and throughout the I/R protocol. The results were similar to those obtained with IKK/NF-κB inhibition i.e. infarct size was significantly reduced in WT and δCTG/δKO mice and was not reduced further in δBTG/δKO animals. CaMKIIδKO animals showed modest but significant further reductions in infarct size in the presence of etanercept (Fig. 8). These findings reveal a surprisingly important role for autocrine and/or paracrine TNF-α signaling in the ex vivo isolated perfused heart during I/R, demonstrate that this process is regulated by CaMKIIδC, and establish an essential cardiac-intrinsic role for NF-κB in I/R injury.
Figure 8.
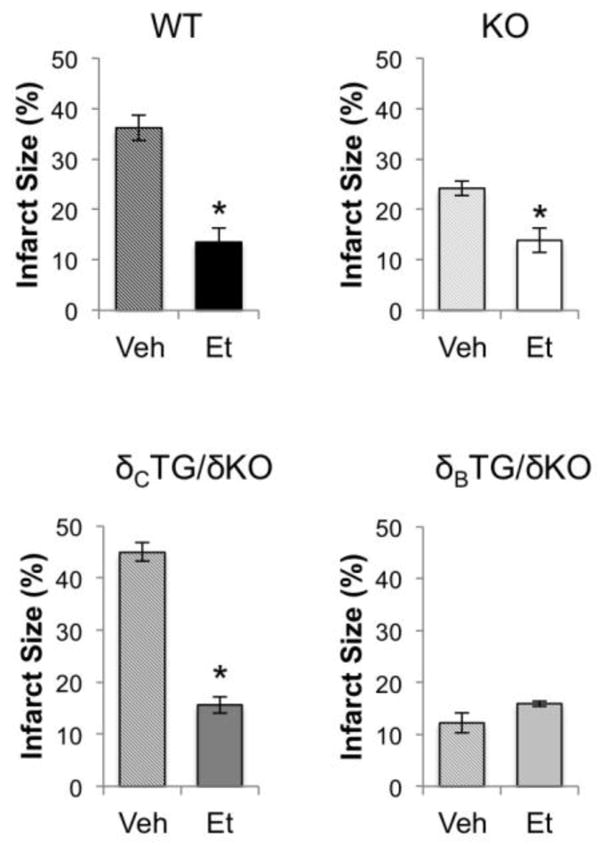
Inhibition of TNF-α ameliorates I/R damage. Vehicle or etanercept (5μg/ml) were perfused into hearts from WT, KO, δBTG/δKO, and δcTG/δKO animals. Infarct size was measured via TTC staining following 1hr of reperfusion. Data are mean±SEM values from 4–8 mice. *P<0.05 vs veh.
1.7 DISCUSSION
Our laboratory previously demonstrated that mice in which CaMKIIδ was selectively deleted from cardiomyocytes exhibited diminished infarct formation in response to in vivo I/R [9]. These experiments provided evidence that CaMKIIδ activation in cardiomyocytes mediates the deleterious effects of I/R injury but did not address the question of which subtype(s) of CaMKIIδ were responsible for cardiomyocyte death in response to I/R. Here we characterize and utilize mice in which the CaMKIIδB or CaMKIIδC isoforms are expressed in the CaMKIIδ KO animals to independently determine the role of each of these isoforms in cardiomyocyte CaMKII signaling. In addition we analyzed I/R damage in the ex vivo isolated perfused heart where the role of systemic inflammatory factors is eliminated. Our studies demonstrate that signals that are rapidly and locally generated regulate infarct development in the ex vivo heart and are significantly attenuated in the absence of CaMKIIδ. We further show that CaMKIIδC re-expression in cardiomyocytes reverses the attenuation of infarct formation seen in CaMKIIδKO mice. In contrast CaMKIIδB re-expression further limits infarct development compared to that observed in KO mice.
While total CaMKIIδ expression and maximal activity are somewhat higher in δCTG/δKO mice (Supplemental Fig. 3B) the amount of activated (Ca2+-autonomous) CaMKII is similar in δCTG/δKO and δBTG/δKO animals (Supplemental 3A). Thus it is unlikely that differential baseline levels of active CaMKIIδ explain the response of δCTG/δKO versus δBTG/δKO animals to I/R. There may, however, be a greater propensity for activation of inflammatory responses when CaMKIIδC is activated thus we determined whether I/R could lead to selective increases in NF-κB activation in δCTG/δKO mice. NF-κB has been implicated in in vivo I/R damage and myocardial infarction and our earlier studies demonstrated that CaMKIIδ activates NF-κB during in vivo I/R [9, 25, 26]. We previously assumed that in vivo I/R triggered systemic responses such as infiltration of non-cardiac inflammatory cells into the heart during the 24–48h of reperfusion, and that this was a significant contributor to infarct development. Here, however, we demonstrate that CaMKIIδC and subsequent NF-κB-mediated responses occur in the isolated perfused heart implicating cardiac-intrinsic signaling in the effects of CaMKIIδC on infarct formation.
It is possible that the deleterious effect of CaMKIIδC is initiated by induction of cardiomyocyte cell death and release of factors from necrotic cells that induce inflammatory signaling [27, 28]. Our previous work showed, however, that cyclosporine-A (CsA) treatment did not block NF-κB activation in response to in vivo I/R, indicating that this response was independent of necrosis [9] and we confirmed this in the current ex vivo study (Supplemental Fig. 4). Accordingly we suggest that there is a direct effect of CaMKII on NF-κB activation. This conclusion is further supported by our current data showing that acute activation of CaMKIIδC with H2O2 elicits NF-κB activation in NRVMs and by our previous demonstration that adenoviral expression of activated CaMKIIδC in NRVMs induces IKK activation [9].
Strikingly, we show here that NF-κB activation in response to I/R is enhanced by cardiac expression of CaMKIIδC but not CaMKIIδB. This is consistent with the finding that phosphorylation of the upstream kinase IKK is increased by I/R in δCTG/δKO but not in δBTG/δKO animals. Furthermore, we demonstrate that I/R increases activation of CaMKIIδC but not of CaMKIIδB in the cytosol/membrane, where IKK is localized. We previously reported that despite its nuclear localization sequence [29] CaMKIIδB is not strictly confined to the nucleus [14] but is in fact present in the cytosol/membrane compartment (Fig. 3B). It remains unclear why there is selective activation of CaMKIIδC and not CaMKIIδB in this location. Nevertheless our observations in NRVMs confirm that there is differential activation of equivalently expressed levels of CaMKIIδB and δC by oxidative stress and further indicate an association between cytosolic CaMKIIδC autophosphorylation and subsequent IκBα degradation and nuclear accumulation of p65. Notably another recent report confirmed the ability of CaMKIIδ subtypes to undergo differential posttranslational modification during reperfusion [30].
Previous studies using pharmacological inhibitors to demonstrate involvement of CaMKIIδ in ex vivo I/R damage linked the effects of CaMKII to phosphorylation of PLN and RyR2 and subsequent Ca2+ dysregulation [31], supporting the hypothesis that rapid phosphorylation of CaMKII targets was a driver of myocardial injury. Indeed, we originally considered it unlikely that transcriptional regulation by CaMKIIδC-mediated activation of NF-κB could play a significant role in determining cell viability over the course of only one hour of reperfusion. Surprisingly, however, we found that inhibition of either IKK or TNF-α had a profound effect on infarct formation not only in δCTG/δKO but also in WT mouse hearts subjected to 25 min ex vivo ischemia and 60 min reperfusion.
Recent studies have shown that receptor-interacting protein kinase 3 (RIP3) plays a role in TNF-α-mediated cell death through formation of the necrosome [32]. RIP3 phosphorylates and activates CaMKII during I/R [33] accordingly TNF-α could participate in a deleterious positive feedback loop leading to sustained CaMKIIδC and NF-κB activation. The experiments presented in this study clearly indicate that TNF-α signaling and myocardial NF-κB activation are mechanisms by which CaMKIIδC elicits infarct formation in the isolated perfused heart. This signaling would likely be further enhanced and sustained by Ca2+ dysregulation resulting from CaMKII-mediated phosphorylation of SR substrates and increases in reactive oxygen species (ROS) during I/R [19, 34, 35].
Paradoxically, some reports indicate that NF-κB may be protective in I/R injury [36, 37] while others demonstrate a deleterious role for NF-κB activation [25, 38]. Importantly, these studies differ in how I/R is elicited and how NF-κB is inhibited, thus resolution of these conflicting conclusions remains elusive. In the current study we have demonstrated that CaMKIIδC expressed in cardiomyocytes mediates NF-κB activation, TNF-α induction and infarct development during I/R and that these events occur rapidly and in the absence of systemic inflammatory factors. Our data contribute to the understanding of the dichotomous effects of NF-κB by clearly demonstrating an adverse cardiomyocyte-autonomous role of NF-κB activation in I/R injury.
While we demonstrate that cardiac TNF-α expression during I/R is mediated by CaMKIIδC activation in cardiomyocytes, we do not know the extent to which TNF-α is formed in and secreted from these cells. Cardiac-resident macrophages and other non-myocytes are potential sources of TNF-α and could thus act in an autocrine or paracrine fashion to affect cardiomyocyte survival. The extent to which CaMKIIδC activation in myocytes sends signals to other cells is not clear, but our studies demonstrate that the cardiomyocyte initiates signals though CaMKIIδ that lead to upregulation of TNF-α. TNF-α inhibitors such as etanercept have been used to inhibit TNF-α signaling in patients with various autoimmune disorders [39]. Effects of TNF-α inhibitors on cardiovascular disease outcomes have also been evaluated with uncertain results that could in part be due to systemic responses to sustained antibody administration [28]. Such detrimental effects would not be expected to occur during short-term treatment thus use of TNF-α inhibitors could be of value if employed at the onset of reperfusion following primary percutaneous intervention for myocardial infarction.
In summary, we demonstrate that the δC isoform of CaMKII contributes significantly to myocardial damage following ex vivo I/R. Signaling occurs through NF-κB and TNF-α and acute inhibition of the generation or function of these molecules has a very robust protective effect in WT animals and in those expressing CaMKIIδC. Importantly, we show that these events occur during a much shorter timeframe than would have been predicted by previous studies of CaMKIIδ and NF-κB signaling in in vivo I/R, and that these events occur in the absence of systemic factors such as infiltration of cells originating outside of the heart. CaMKII inhibition is predicted to be of therapeutic benefit in a number of contexts. Our results suggest that selective CaMKIIδC inhibition would confer the most benefit over blockade of all cardiac CaMKII isoforms although specific means of locally inhibiting the δC isoform do not yet exist. An alternative approach would be to acutely block events that occur downstream of CaMKIIδC activation during I/R such as IKK/NF-κB activation or TNF-α signaling.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9.

Summary of findings. NF-κB nuclear translocation and subsequent TNF-α upregulation during I/R is mediated by cytosolic CaMKIIδc activation.
HIGHLIGHTS.
CaMKIIδB and CaMKIIδC differentially regulate ischemia/reperfusion injury
CaMKIIδC is selectively activated during ischemia/reperfusion
NF-κB activation during I/R is exacerbated by CaMKIIδC expression
Inhibition of NF-κB or TNF-α blocks CaMKIIδC-mediated ischemia/reperfusion injury
Acknowledgments
1.8 SOURCES OF FUNDING
This work was supported by the National Institute of Health P01-HL080101 and HL-028143 to J.H. Brown. S. Miyamoto is supported by American heart Association 15GRNT 2297009 and NIH R56 HL097037. B.D. Westenbrink is supported by the Netherlands Heart Institute, the Dutch Heart Foundation (grant 2012T066) and the Netherlands Organisation for Scientific Research (grant 016.176.147). C.B.B. Gray and S. Mishra were supported by the NIGMS Graduate Training Program in Cellular and Molecular Pharmacology (T32 GM007752). S. Xiang was supported by the Cardiovascular Physiology and Pharmacology Training grant 5T32HL007444. C.C. Glembotski was supported by National Instituted of Health (NIH) grants R01 HL75573, R01 HL104535, and P01 HL085577. T. Suetomi is supported by the Uehara Memorial Foundation (Japan).
We thank Sarah Shires and Asa Gustaffson for their assistance with echocardiography, Melissa Barlow for animal husbandry and echocardiography, and Jeff Smith for his assistance in adenoviral synthesis and amplification.
1.2 ABBREVIATIONS
- δBTG/δKO
Animals expressing only CaMKIIδB
- δCTG/δKO
Animals expressing only CaMKIIδC
- BMS
BMS-345541
- CaMKIIδ
Ca2+/calmodulin-dependent protein kinase II delta
- CsA
Cyclosporine-A
- FS
Fractional shortening
- IKK
Inhibitor of kappa B kinase
- IL-6
Interleukin 6
- I/R
Ischemia/reperfusion
- LVDP
Left ventricular developed pressure
- NRVMs
Neonatal rat ventricular myocytes
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- ROS
Reactive oxygen species
- RIP3
Receptor-interacting protein kinase 3
- SR
Sarcoplasmic reticulum
- TTC
Triphenyl tetrazolium chloride
- TNF-α
Tumor necrosis factor alpha
Footnotes
1.9 DISCLOSURES
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
1.11 REFERENCES
- 1.Hoch B, Haase H, Schulze W, Hagemann D, Morano I, Krause EG, et al. Differentiation-dependent expression of cardiac delta-CaMKII isoforms. J Cell Biochem. 1998;68(2):259–68. [PubMed] [Google Scholar]
- 2.Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999;84(6):713–21. doi: 10.1161/01.res.84.6.713. [DOI] [PubMed] [Google Scholar]
- 3.Mayer P, Mohlig M, Idlibe D, Pfeiffer A. Novel and uncommon isoforms of the calcium sensing enzyme calcium/calmodulin dependent protein kinase II in heart tissue. Basic Res Cardiol. 1995;90(5):372–9. doi: 10.1007/BF00788498. [DOI] [PubMed] [Google Scholar]
- 4.Mayer P, Mohlig M, Schatz H, Pfeiffer A. Additional isoforms of multifunctional calcium/calmodulin-dependent protein kinase II in rat heart tissue. Biochem J. 1994;298(Pt 3):757–8. doi: 10.1042/bj2980757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schworer CM, Rothblum LI, Thekkumkara TJ, Singer HA. Identification of novel isoforms of the delta subunit of Ca2+/calmodulin-dependent protein kinase II. Differential expression in rat brain and aorta. J Biol Chem. 1993;268(19):14443–9. [PubMed] [Google Scholar]
- 6.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119(5):1230–40. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Westenbrink BD, Ling H, Divakaruni AS, Gray CB, Zambon AC, Dalton ND, et al. Mitochondrial reprogramming induced by CaMKIIdelta mediates hypertrophy decompensation. Circulation research. 2015;116(5):e28–39. doi: 10.1161/CIRCRESAHA.116.304682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grimm M, Ling H, Willeford A, Pereira L, Gray CB, Erickson JR, et al. CaMKIIdelta mediates beta-adrenergic effects on RyR2 phosphorylation and SR Ca(2+) leak and the pathophysiological response to chronic beta-adrenergic stimulation. J Mol Cell Cardiol. 2015;85:282–91. doi: 10.1016/j.yjmcc.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ling H, Gray CB, Zambon AC, Grimm M, Gu Y, Dalton N, et al. Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ Res. 2013;112(6):935–44. doi: 10.1161/CIRCRESAHA.112.276915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, et al. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491(7423):269–73. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh MV, Kapoun A, Higgins L, Kutschke W, Thurman JM, Zhang R, et al. Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. The Journal of clinical investigation. 2009;119(4):986–96. doi: 10.1172/JCI35814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, et al. Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol. 2006;291(6):H3065–75. doi: 10.1152/ajpheart.00353.2006. [DOI] [PubMed] [Google Scholar]
- 13.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. The Journal of clinical investigation. 2013;123(3):1262–74. doi: 10.1172/JCI65268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mishra S, Gray CB, Miyamoto S, Bers DM, Brown JH. Location matters: clarifying the concept of nuclear and cytosolic CaMKII subtypes. Circ Res. 2011;109(12):1354–62. doi: 10.1161/CIRCRESAHA.111.248401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang T, Kohlhaas M, Backs J, Mishra S, Phillips W, Dybkova N, et al. CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J Biol Chem. 2007;282(48):35078–87. doi: 10.1074/jbc.M707083200. [DOI] [PubMed] [Google Scholar]
- 16.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, et al. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92(8):912–9. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 17.Miyamoto S, Purcell NH, Smith JM, Gao T, Whittaker R, Huang K, et al. PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ Res. 2010;107(4):476–84. doi: 10.1161/CIRCRESAHA.109.215020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Respress JL, Wehrens XH. Transthoracic echocardiography in mice. J Vis Exp. 2010;(39) doi: 10.3791/1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Carlo MN, Said M, Ling H, Valverde CA, De Giusti VC, Sommese L, et al. CaMKII-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J Mol Cell Cardiol. 2014;74:274–83. doi: 10.1016/j.yjmcc.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, et al. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc Res. 2007;73(4):689–98. doi: 10.1016/j.cardiores.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. The Journal of biological chemistry. 2003;278(3):1450–6. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- 22.Meldrum DR. Tumor necrosis factor in the heart. Am J Physiol. 1998;274(3 Pt 2):R577–95. doi: 10.1152/ajpregu.1998.274.3.R577. [DOI] [PubMed] [Google Scholar]
- 23.Moreland LW, Schiff MH, Baumgartner SW, Tindall EA, Fleischmann RM, Bulpitt KJ, et al. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann Intern Med. 1999;130(6):478–86. doi: 10.7326/0003-4819-130-6-199903160-00004. [DOI] [PubMed] [Google Scholar]
- 24.Weinblatt ME, Kremer JM, Bankhurst AD, Bulpitt KJ, Fleischmann RM, Fox RI, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340(4):253–9. doi: 10.1056/NEJM199901283400401. [DOI] [PubMed] [Google Scholar]
- 25.Sawa Y, Morishita R, Suzuki K, Kagisaki K, Kaneda Y, Maeda K, et al. A novel strategy for myocardial protection using in vivo transfection of cis element ‘decoy’ against NFkappaB binding site: evidence for a role of NFkappaB in ischemia-reperfusion injury. Circulation. 1997;96(9 Suppl):II-280–4. discussion II-285. [PubMed] [Google Scholar]
- 26.Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME. MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol. 2012;52(5):1135–44. doi: 10.1016/j.yjmcc.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. 2008;3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circulation research. 2015;116(7):1254–68. doi: 10.1161/CIRCRESAHA.116.302317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srinivasan M, Edman CF, Schulman H. Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J Cell Biol. 1994;126(4):839–52. doi: 10.1083/jcb.126.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bell JR, Raaijmakers AJ, Curl CL, Reichelt ME, Harding TW, Bei A, et al. Cardiac CaMKIIdelta splice variants exhibit target signaling specificity and confer sex-selective arrhythmogenic actions in the ischemic-reperfused heart. Int J Cardiol. 2015;181:288–96. doi: 10.1016/j.ijcard.2014.11.159. [DOI] [PubMed] [Google Scholar]
- 31.Salas MA, Valverde CA, Sanchez G, Said M, Rodriguez JS, Portiansky EL, et al. The signalling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J Mol Cell Cardiol. 2010;48(6):1298–306. doi: 10.1016/j.yjmcc.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27(15):1640–9. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22(2):175–82. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- 34.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357(11):1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 35.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133(3):462–74. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo J, Jie W, Kuang D, Ni J, Chen D, Ao Q, et al. Ischaemia/reperfusion induced cardiac stem cell homing to the injured myocardium by stimulating stem cell factor expression via NF-kappaB pathway. Int J Exp Pathol. 2009;90(3):355–64. doi: 10.1111/j.1365-2613.2009.00659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Misra A, Haudek SB, Knuefermann P, Vallejo JG, Chen ZJ, Michael LH, et al. Nuclear factor-kappaB protects the adult cardiac myocyte against ischemia-induced apoptosis in a murine model of acute myocardial infarction. Circulation. 2003;108(25):3075–8. doi: 10.1161/01.CIR.0000108929.93074.0B. [DOI] [PubMed] [Google Scholar]
- 38.Zingarelli B, Hake PW, Denenberg A, Wong HR. Sesquiterpene lactone parthenolide, an inhibitor of IkappaB kinase complex and nuclear factor-kappaB, exerts beneficial effects in myocardial reperfusion injury. Shock. 2002;17(2):127–34. doi: 10.1097/00024382-200202000-00008. [DOI] [PubMed] [Google Scholar]
- 39.Cessak G, Kuzawinska O, Burda A, Lis K, Wojnar M, Mirowska-Guzel D, et al. TNF inhibitors - Mechanisms of action, approved and off-label indications. Pharmacol Rep. 2014;66(5):836–44. doi: 10.1016/j.pharep.2014.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.