

Abstract
MCL-1 (BCL-2 family anti-apoptotic protein) is responsible for melanoma's resistance to therapy. Cancer initiating cells also contribute to resistance and relapse from treatments. Here we examined the effects of the MCL-1 inhibitor SC-2001 in killing non melanoma-initiating-cells (bulk of melanoma), and melanoma-initiating-cells (MICs). By itself, SC-2001 significantly kills melanoma cells under monolayer conditions in vitro and in a conventional mouse xenograft model. However, even at high doses (10μM), SC-2001 does not effectively eliminate MICs. In contrast, the combination of SC-2001 with ABT-737 (a BCL-2/BCL-XL/BCL-W inhibitor) significantly decreases ALDH+ cells, disrupts primary spheres, and inhibits the self-renewability of MICs. These results were observed in multiple melanomas, including short term cultures of relapsed tumors from current treatments, independent of the mutation status of BRAF or NRAS. Using a low-cell-number mouse xenograft model, we examined the effects of these treatments on the tumor initiating ability of MIC-enriched cultures. The combination therapy reduces tumor formation significantly compared to either drug alone. Mechanistic studies using shRNA and the CRISPR-Cas9 technology demonstrated that the upregulation of pro-apoptotic proteins NOXA and BIM contribute to the combination-induced cell death. These results indicate that the MCL-1 inhibitor SC-2001 combined with ABT-737 is a promising treatment strategy for targeting melanoma.
Keywords: melanoma stem cells, cancer stem cells, drug-induced cell death, melanoma, SC-2001
INTRODUCTION
For the first time several molecular-targeted and immunotherapy drugs are significantly improving the overall survival of patients with metastatic melanoma. However, despite these new therapeutics, many patients still do not improve or eventually relapse after the initial positive response [1, 2]. Thus, there is still a pressing need for continued research especially for the patients without the mutations targeted by these new drugs or those who relapse from these treatments.
MCL-1 (Myeloid cell leukemia sequence 1) is an anti-apoptotic protein of the BCL-2 family [3, 4]. The MCL-1 locus is one of the “top ten” most amplified genomic regions in a variety of human cancers [5], correlating with an upregulation of MCL-1 activity [6–10]. This increase in MCL-1 expression is often associated with chemotherapeutic resistance and relapse from therapeutics [3]. Pharmacological inhibition or the molecular down regulation of MCL-1 via RNA-interference has shown to promote apoptosis and/or overcome drug resistance in multiple cancers [11–16]. Therefore, MCL-1 is a high-priority therapeutic target for cancer treatment [4, 6, 17]. Oncogenic activation of BRAF signaling in melanoma upregulates MCL-1 expression and contributes to increased resistance to BRAF/MEK inhibitors and overall tumor progression and survival [18]. We and others have shown that the knockdown of MCL-1 sensitizes melanoma cells to various treatments, including BRAF or MEK inhibitors [19–23] and thus, targeting MCL-1 can be a new alternative for melanoma treatment.
Cancer Initiating Cells (CICs) are a subpopulation of cancer cells which have enhanced tumor initiation, progression, and chemo-resistance [24–26]. Recently, multiple groups provided evidence of Melanoma Initiating Cells (MICs), and suggest that MICs can contribute to resistance to treatment [27–30]. Ideal cancer treatment strategies stress on eliminating the resistant subpopulations, such as CICs as well as the bulk of the tumors (non-CICs) cells to prevent relapse [29, 31].
In this study we tested the efficacy of an MCL-1 inhibitor, SC-2001, either alone or in combination with ABT-737 in killing melanoma cells and MICs. SC-2001 is a novel MCL-1 inhibitor, and it is structurally related to obatoclax, a small molecule inhibitor for multiple anti-apoptotic BCL-2 family members, including MCL-1. SC-2001 has anti-tumor activity for liver cancers and breast cancers [32–34]. ABT-737 is a small molecule BCL-2/BCL-XL/BCL-W inhibitor and has shown a promising result in cancer therapy either by itself or in combination with other chemotherapeutics in pre-clinical stage [35, 36]. However, many labs including ours have found that ABT-737 by itself is not very effective for treating melanoma as a single agent [23, 37, 38]. The results here suggest that the use of a combination of MCL-1 and BCL-2 inhibitors to induce NOXA and BIM is a promising treatment strategy for melanoma regardless of the mutation status of BRAF or NRAS, and it may overcome melanoma's resistance to current treatments.
RESULTS
MCL-1 protein expression is higher in melanomas compared with Melanocytes
MCL-1 expression is associated with chemotherapeutic resistance and relapse in various cancers [3] and its increased expression is correlated with melanoma progression [39]. However, it has not been examined whether this correlation remains consistent for the common mutations associated with melanoma. Here, the melanomas we tested include commonly used melanoma cell lines and tumor samples from melanoma patients. All tumor samples were maintained in either short term cultures in vitro or exclusively in a patient-derived xenograft model (PDX) (Figure 1). The melanomas used here include BRAF-mutant cells (HT144, 451Lu and MB2309), NRAS-mutated cells (WM852c, SKMEL-30, Hs852T), NF-null cells (Hs852T), or wild-type cells for the common mutations in BRAF, NRAS, or NF1 (MB2141). The last category has been referred to as triple-WT [40]. Most of the patient tumor samples used here have relapsed from the molecular-targeted treatment (MB2309) or treatments of multiple chemotherapies and radiation (MB2141). Figure 1 shows higher MCL-1 protein expression in multiple melanoma sphere cultures compared to normal human primary melanocytes (PIG1, HEMNMP), regardless of mutation status of these melanoma cultures. The increase in MCL-1 expression was between 4 to 21 fold. This finding provides the rationale to treat a broader range of melanomas with MCL-1 inhibitors to try to overcome resistance to current treatments. Therefore, we tested the efficacy of the MCL-1 inhibitor, SC-2001.
Figure 1. Higher MCL-1 protein expression in melanomas compared with Melanocytes.
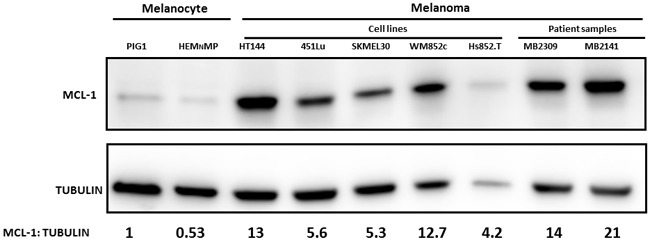
Immunoblot of MCL-1 expression in lysates from melanoma cells and melanocytes cultured in sphere condition. Melanomas include common cell lines and short term cultures of melanoma samples relapsed from current treatments, with BRAF-mutated (HT144, 451Lu, and MB2309), NRAS-mutated (SKMEL-30, WM852c and Hs852T), NF-null (Hs852T), or wild-type for common mutations in BRAF, NRAS, or NF1 (MB2141).
SC-2001 is capable of eliminating the bulk of melanoma cells in vitro and in vivo
To investigate whether SC-2001 affects cell viability, we used an ATP assay. SC-2001 significantly reduced cell viability in a dose-dependent manner in multiple melanoma cultures and only a minimal reduction for melanocytes (HEMNLP). At doses of 2.5uM or higher, SC-2001 reduced cell viability for all melanoma cell lines significantly compared to DMSO (P<0.01 or less). The melanoma cells included BRAF or NRAS mutated cell lines and tumor samples from melanoma patients that have relapsed from current treatments (Figure 2A). The IC50s for SC-2001 are 2.14μM to 4.62μM for the melanoma cell lines (Supplementary Table S1). The drug also showed increased cytotoxicity (at dose of 5 or 10 μM) in a dose-dependent manner for all four melanoma cell lines tested (P<0.001 or lesser) compared to DMSO, regardless of their mutation status (Figure 2B). Higher doses of SC-2001 (5 or 10 μM) resulted in a more rounded morphology or complete detachment from the plates (Figure 2C and Supplementary Figure S1) that is consistent with the ATP and cytotoxicity data suggesting that SC-2001 induced killing in these melanoma populations. Additionally, cleavage of PARP (Poly ADP-ribose polymerase 1) is a well-known marker of cells undergoing apoptosis [41], and we performed immunoblot assays of PARP to further assess the effects. Figure 2D shows that the higher doses of SC-2001 (5 or 10 μM) induced an increased level of cleaved PARP in all the melanoma cell lines tested. Additionally, an Annexin V assay demonstrated that SC-2001 (2.5 or 5 μM) caused dramatic apoptosis, ranging from ~35-80%, in all eight melanoma cell lines/patient samples tested relative to the DMSO control (P < 0.01 or less, Supplementary Figure S2). However, the SC-2001 treatment had only a modest effect on melanocytes (HEMNMP) (Supplementary Figure S2). Furthermore, in a conventional mouse xenograft model, SC-2001 significantly decreased the rate of tumor growth compared to the control (P <0.001) (Figure 2E).
Figure 2. SC-2001 is capable of targeting the bulk of melanoma cells in vitro and in vivo, however it did not target the MIC population.
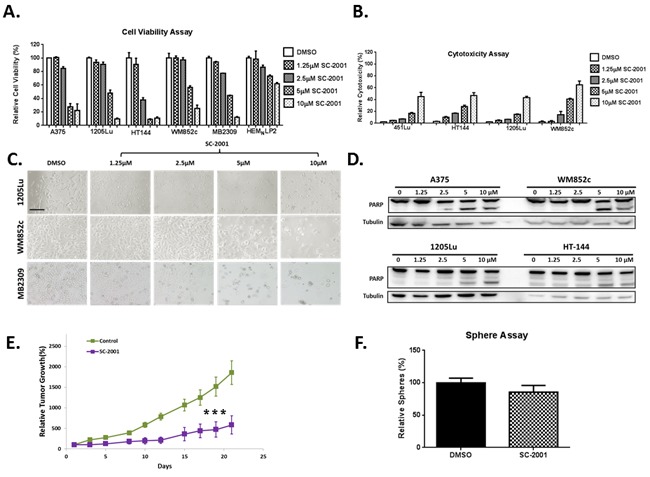
A. ATP assays with BRAF-mutated (A375, 1205-Lu, HT144 and MB2309), NRAS-mutated (WM852c) melanomas and melanocyte. At doses of 2.5uM or higher, SC-2001 reduced cell viability for all melanoma cell lines significantly compared to control (P<0.01 or less). B. Cyto-tox Glo assays with indicated melanomas. The drug, at doses of 5uM or higher, also showed increased cytotoxicity in a dose-dependent manner for all four melanoma cell lines tested compared to control (P<0.001 or lesser), regardless of their mutation status. C. Bright-field images of melanoma cells treated with increasing concentrations of SC-2001. Scale bar = 100μm. A bigger version of Figure 2C is provided in the Supplementary Figure S1. D. Immunoblot of full length and cleaved PARP for cell lysates treated with indicated doses of SC-2001. For (A) to (D), the cells were treated with the indicated concentration of SC-2001 for 48 hrs before being subjected to respective assays. E. Relative tumor volumes in a mouse xenograft model. The tumor volume at day 0 was set as 100%. SC-2001 significantly decreased the rate of tumor growth compared to the control (P <0.001). F. Sphere assays with the tumor cells collected at the end of the xenograft experiment from panel E. The tumor cells were not treated with any drugs during the sphere assay. No significant difference in number of spheres formed between the SC-2001 and DMSO treated samples. ***indicates P<0.001 or less; **indicates P<0.01.
SC-2001 does not target the MIC population
Multiple studies have identified subpopulations of melanoma cells that possess characteristics of CICs including increased resistance to treatments [28, 30, 37, 42]]. One of the best in vitro methods used to study CICs is the sphere formation assay [6] and it has been successfully used to study MICs in many studies [37, 43–46]. Melanoma-spheres display stem cell like functions including self-renewability and tumorigenicity [46]; thus they can be used as a tool to enrich the cancer cell population that exhibits stem-like features for testing the potency of cancer drugs [25, 26, 47]. The primary sphere assay helps enrich the MIC population while a secondary sphere formation assay is an assay for measuring the self-renewal capacity of MICs in vitro [37]. To determine whether SC-2001 treatment eliminated both the bulk tumor cells and MIC population in the experiment described in Figure 2E, we performed sphere-forming assays starting from single cell suspensions isolated from the aforementioned experiment. We found that there was no significant difference in number of spheres formed between the SC-2001 and DMSO treated samples indicating that although SC-2001 could shrink the tumor in a conventional xenograft model, it was unable to effectively eliminate all the MICs (Figure 2F).
Low dose SC-2001 plus ABT-737 targeted the MIC population regardless of their mutation status
Therapeutics with single molecular targets often fail in cancer therapy, and the CIC populations are thought to be reason for this. Thus, utilizing combination therapies that eliminate this resistant cell population is an emerging strategy to treat cancer [24, 48]. Many studies, including ours, have shown that targeting single anti-apoptotic BCL-2 family members is not sufficient to treat melanoma [23, 37, 38], and that targeting both MCL-1 and BCL-2 is needed to eliminate the MIC population [37]. We assessed if decreasing MCL-1 expression by shRNA can synergize with a BCL-2 inhibitor to abolish the MIC population in a sphere formation assay (Figure 3A and 3B). Knockdown of MCL-1 (shMCL-1) by itself, did not cause a significant decrease in the number of spheres compared to the shControl. However, when shMCL-1 cells were treated with the BCL-2 inhibitor ABT-737, there was a significant decrease in the number of spheres (P<0.01) (Figure 3B). This suggested that SC-2001 when combined with ABT-737 can be an effective strategy to target the MICs.
Figure 3. SC-2001 did not target the MIC population even at a high concentration (10 μM), while a lower concentration of SC-2001 (2.5 μM) combined with ABT-737 targeted the MIC population.
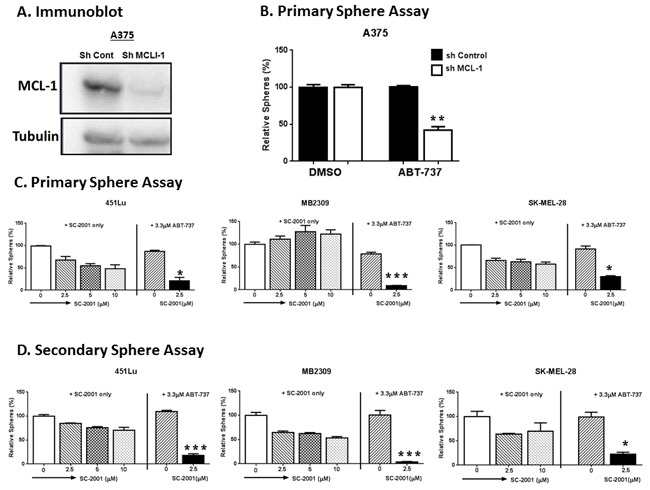
A. and B. A375 cells, stably carrying either control shRNA (sh-Control) or shRNA against MCL-1 (sh-MCL-1), were subjected to: (A) Immunoblot for MCL-1 to confirm the knockdown; or (B) Primary sphere assays. There was significant decrease in the number of spheres in sh-MCL-1 group, when treated with ABT-737 (P<0.01). C. Primary sphere assays. For primary sphere assay in (B) and (C), the spheres were treated with the indicated concentration of drug for 48 hrs before counting. D. Secondary sphere assays to test the efficacy of SC-2001 in targeting MIC by itself or in combination with ABT-737 with indicated melanoma cells. The combination inhibited the formation of both the primary (C) and secondary spheres (D) compared to DMSO, ABT-737 or SC-2001 only (2.5, 5 or 10 μM) (P < 0.05 or less). ***indicates P<0.001 or less; **indicates P<0.01; *indicates P<0.05.
We therefore examined the efficacy of SC-2001 by itself and in combination with ABT-737 against MICs using sphere forming assays on multiple melanoma cell lines (Figure 3C and 3D). SC-2001 by itself did not significantly inhibit either primary or secondary sphere formation even at high concentrations (5 or 10 μM) compared to DMSO. On the other hand, when combined with ABT-737 (3.3 μM), it was very effective even at a lower concentration (2.5 μM). The combination inhibited the formation of both the primary and secondary spheres compared to DMSO or SC-2001 only (2.5, 5 or 10 μM) (P < 0.05 or less) (Figure 3C and 3D). This decrease is comparable to the effect observed for ABT-737 treatment on sh-MCL-1 cell lines.
To further examine the effects of this combination, we extended these assays to more melanoma samples, particularly the short term cultures of tumor samples from melanoma patients (Figure 4). Again, the samples tested here include BRAF or NRAS mutated, as well as wild type for BRAF, NRAS and NF1 (triple-WT) lines. The patient samples include the ones relapsed from current treatments. The spheres were allowed to grow up to reasonable size until Day 5 after seeding before being treated with single or combination drugs for 48 hrs. In thirteen out of fourteen samples, the combination severely disrupted the primary spheres compared with the control, ABT-737 or SC-2001 treatment alone (P< 0.05 or less, Figure 4A and 4B) regardless their mutation status. The p-values for all comparisons are separately listed in Supplementary Table S2. Additionally, we performed immunoblot of PARP cleavage, and further confirmed that the combination treatment induced apoptosis in all the melanoma samples tested (Supplementary Figure S3).
Figure 4. SC-2001 combined with ABT-737 targeted the MIC population of melanoma cells regardless of the mutation status.
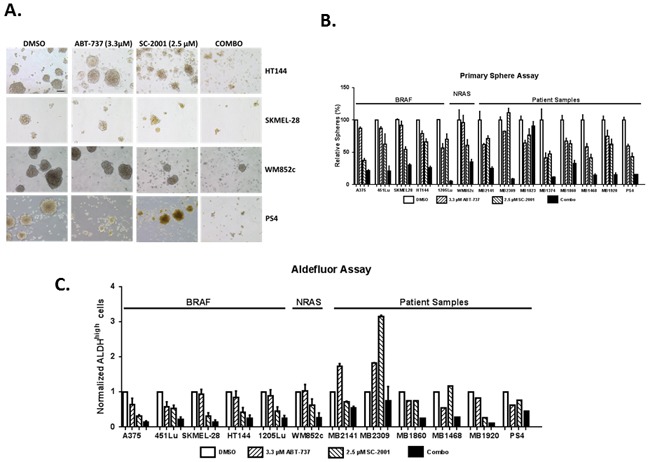
Melanoma spheres were treated with indicated compounds, and then subjected to A. Bright field analysis, Scale bar = 100μm; B. Quantification of the number of primary spheres; For primary sphere assay, the spheres were treated with the indicated concentration of drug for 48 hrs before counting. The combination severely disrupted the primary spheres compared with the control, ABT-737 or SC-2001 treatment alone (P< 0.05 or less) regardless their mutation status in thirteen out of fourteen lines. The p-values for all comparisons are separately listed in Supplementary Table S2. C. Quantification of ALDH assay. For ALDH assay, the cells were treated with the indicated concentration of drug for 48 hrs before conducting the assay. The combination of SC-2001 and ABT-737 significantly decreased the percentage of ALDHhigh cells compared with the DMSO control and ABT-737 alone regardless their mutation status (P < 0.05 or less) in seven lines. Unfortunately, we did not have enough material to do additional replicates for last four samples, so we could not statistically analyze the data. The p-values for all comparisons are separately listed in Supplementary Table S3.
Multiple groups have found a positive correlation between cancer cells expressing higher aldehyde dehydrogenase (ALDH) and tumor formation efficiency including melanoma [43, 45]. We and others have previously established that cells with higher ALDH activity are enriched with MICs [37, 43, 45]. Therefore, we used an Aldefluor assay as an additional tool to examine the effects of the combination treatment on the MIC population (Figure 4C). In seven out of eight melanoma cell lines with enough cells for statistical analyses, the combination of SC-2001 and ABT-737 significantly decreased the percentage of ALDHhigh cells compared with both the DMSO control and ABT-737 alone regardless their mutation status. (P < 0.05 or less) (Figure 4C). The p-values for all comparisons are separately listed in Supplementary Table S3. Interestingly, some of the patient samples showed an increased percentage of ALDHhigh cells compared with the DMSO control in response to the single drug treatment. Unfortunately, we did not have enough material for the last four patient samples to perform enough assay replicates for statistical analyses. However, the overall trend was similar relative to the other cell lines.
Taken together, the sphere assay and the ALDH assay demonstrate that the combination of MCL-1 with BCL-2 inhibitors is better than either single drug to kill the MICs.
SC-2001 combined with ABT-737 inhibited the self-renewability of MICs
While the primary sphere-forming assay enriches the population for stem-like cells, the secondary sphere assay measures the cell population's ability to regenerate after drug treatment [46]. This was done by conducting the primary sphere assay as described earlier, dissociating the primary spheres into single cells, and then replating the cells at the same viable cell density. However, we did not add additional drugs during the secondary sphere portion of the assay. This assay specifically assesses if any of the remaining viable cells—those that escaped chemotherapeutics—are capable of self-renewing and regenerating into a mass of tumor cells.
The combination treatment almost eliminated all secondary sphere formation in eight out of nine melanoma cell lines tested (Figure 5). Statistical analyses indicated that the combination treatment significantly decreased the number of secondary spheres formed compared with DMSO, ABT-737 or SC-2001 treatment alone (P < 0.05 or less) in eight out of nine lines tested (Figure 5A and 5B). The p-values for all comparisons are separately listed in Supplementary Table S4.
Figure 5. A low concentration of SC-2001 (2.5 μM) combined with ABT-737 inhibited the self-renewability of MICs.
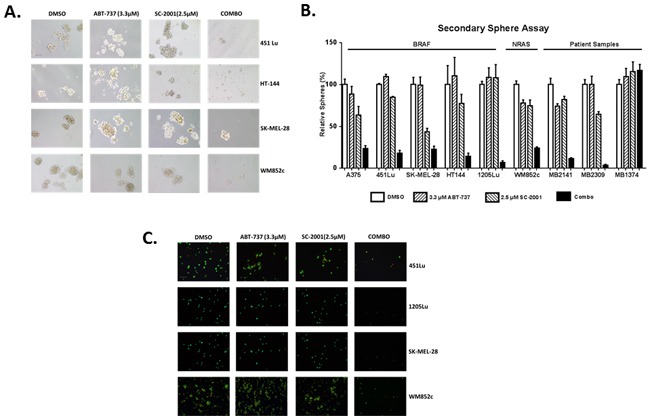
Secondary sphere assays with the melanoma cells A. Bright-field images of secondary spheres. Scale bar = 100μm B. Quantification of the number of secondary spheres. The combination treatment significantly decreased the number of secondary spheres formed compared with DMSO, ABT-737 or SC-2001 treatment alone (P < 0.05 or less) in eight out of nine lines tested. The p-values for all comparisons are separately listed in Supplementary Table S4. C. Visualization of the secondary sphere cells with the Ethidium Bromide/Acridine Orange (EtBr/AO). Scale bar = 100μm. The cells were not treated with any drugs during the secondary sphere assay.
Visualization of these cells with EtBr/AO staining indicated that the majority of the cells in the control or single drug treatments were alive, but the majority of cells in the combination treatments were dead (Figure 5C). Thus, these results showed that the combination prevented the formation of secondary spheres and therefore decreased MIC's self-renewal capability in multiple cell lines.
The combination of SC-2001 and ABT-737 inhibits the MIC-mediated tumor formation in vivo
Conventional mouse xenograft studies implant tumor cells at a very high cell number (> 1 million tumor cells) and cannot reasonably assess the efficacy of a drug in preventing the tumor initiating ability of CICs. Recently, Hirata and colleagues examined the effects of the transcription factor SphK1 on tumor initiating ability of breast CICs by injecting a small amount (100,000 viable cells) of a CIC-enriched cell population into a mouse xenograft model [49]. Similar approaches have been used previously in assessing tumor-initiating ability in other cancer stem cell studies [49–52]. Here, we employed a similar xenograft method with a low cell number implantation to test the differences in tumor-initiating-ability between the control and treatment groups. This xenograft method makes testing cancer initiation more feasible than the standard series dilution method. We also used melanoma cells derived from patients whose tumors eventually relapsed from both BRAF/MEK inhibitors and immunotherapy treatment (MB1860). These tumors were only maintained and passaged in the PDX model prior to this experiment.
We first treated sphere culture samples from a relapsed patient with DMSO, single, or combined drugs for 48 hrs in vitro. After the treatment, we implanted 50,000 surviving viable cells from each group into mice and then monitored the tumor growth in vivo as a readout for the impact of each treatment on tumor-initiation ability. Figure 6A shows that the combination group mice had the longest tumor-free survival time compared to vehicle as well as single drug group (P<0.001). Tumor incidence rate was calculated as the number of tumors generated /number of implantations expressed as percentage. Tumor incidence rate was significantly lower in the combination group compared with the vehicle or single treatment group (P < 0.05) (Figure 6B). These results suggest that the combination-treated populations contained fewer MIC-like cells. To further confirm this, we performed sphere-forming assays with the single cell suspensions isolated from the surviving tumors, and found that the combination significantly reduced the number of spheres compared with vehicle or individual treatments (P < 0.001) (Figure 6C). There was no significant difference between the control and single-drug treated groups in all the above three analyses. Taken together, results here suggest that the combination reduced the MIC population.
Figure 6. The combination of SC-2001 and ABT-737 inhibited the MIC-mediated tumor formation in vivo.
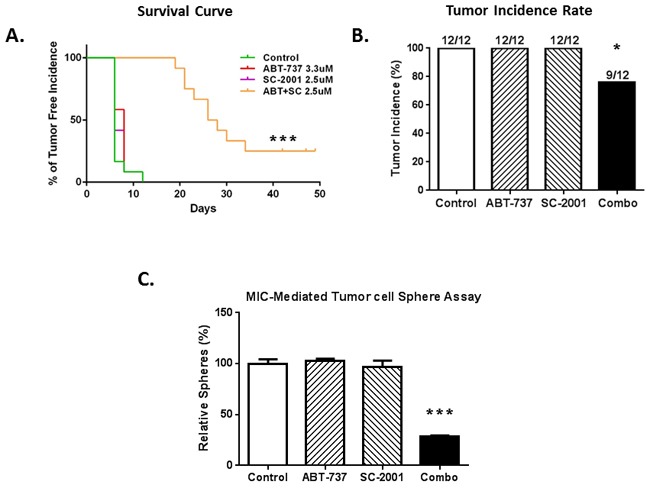
The treatment effects were tested in a xenograft model initiated with a low-number of MIC-enriched cells. For tumor injection, we used the surviving cells from the sphere cultures, upon treatments of SC-2001 and ABT-737, either alone or in combination, at the density of 50,000 viable cells per injection. A. Tumor-free survival curve shows a significantly longer tumor–free time in the combination group, compared to the vehicle or single drug group (P<0. 001). B. The percentage of tumor incidence was significantly lower in the combination group compared to control or single drug groups (P<0.05). C. Sphere assays with the tumor cells collected at the end of the animal experiment, and the number of spheres was significantly lower in the combination group compared to control or single drug groups (P< 0.001). N=3. ***indicates P<0.001 or less; *indicates P<0.05.
The SC-2001 and ABT-737 combination induces NOXA- and BIM-mediated killing of MICs
We have found previously that BH3 only pro-apoptotic proteins, NOXA and BIM, played a crucial role in inducing cell death when ABT-737 was used in combination with other agents [23, 37, 38]. These two proteins are also known to play important roles in regulating MCL-1 and BCL-2 in melanoma [37]. Thus, to evaluate the mechanisms behind the combination-induced cell death, we performed immunoblot studies to examine the expression of these two proteins. We found that the combination treatment notably increased the expression of both NOXA and BIM (Figure 7). Results were similar in BRAF mutated (A375, HT144 and 451Lu), NRAS mutated (WM852c), and patient melanoma cells (MB2309 and MB2141) (Figure 7).
Figure 7. Combination induced NOXA- and BIM-mediated killing of MICs.
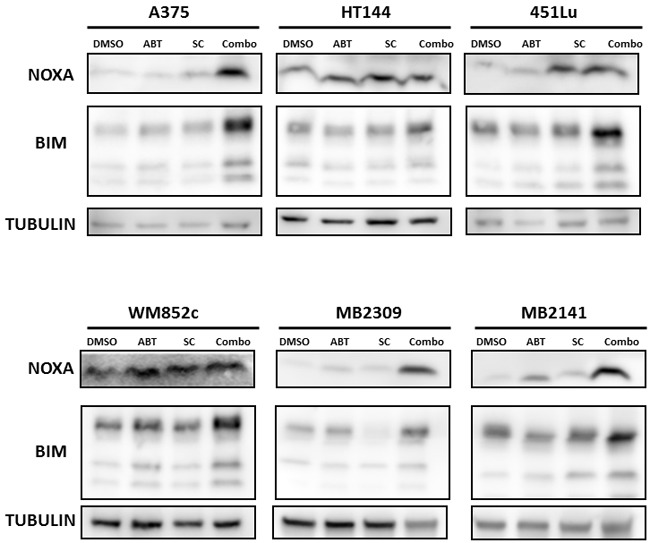
Immunoblot of the lysates harvested from cells treated with indicated drugs for 48 hrs in sphere conditions.
To further determine whether this induction of NOXA or BIM contributes to killing effects of the combination, we examined the effects of knocking-down NOXA or knocking-out BIM on the killing potency of the combination of SC-2001 and ABT-737 on cells. We have established stable cell lines with NOXA knockdown using an shRNA-mediated approach [23], and found that knockdown of NOXA significantly protected against the combination-induced disruption of spheres (P<0.05) (Figure 8A to 8D) in both BRAF and NRAS mutated melanoma cell lines.
Figure 8. Knock-Down of NOXA protected against combination-induced cell death.
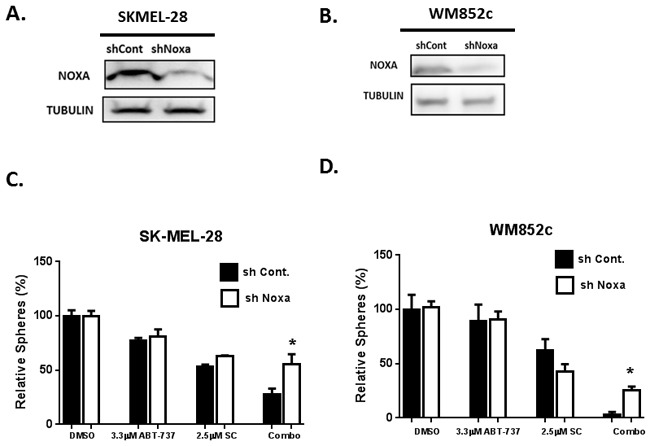
Immunoblot to confirm the NOXA knockdown for SKMEL-28 A. and WM852c B. Sphere assays were performed with melanoma cells stably carrying either control shRNA (sh control) or shRNA against NOXA (shNOXA): C. SK-MEL-28 and D. WM852c. For primary sphere assay, the spheres were treated with the indicated concentration of drug for 48 hrs before counting. Knockdown of NOXA significantly protected the cells against the combination-induced disruption of spheres (P<0.05). *indicates P<0.05.
In previous studies, we found the effects of knocking-down BIM varied depending on the cell lines used, probably due to incomplete knock-down of BIM [37]. This is one of the limitations for shRNA technology since the levels of knock down are achieved with variable efficiency in most cases. Recently, the CRISPR/Cas9 system has been successfully developed for genome engineering/editing and is one of the most innovative and revolutionary methods currently available for modifying human genome [53, 54]. This enables making modifications at the DNA level with permanent and heritable changes in the genome much more feasible [55]. Thus we decided to knockout BIM expression using the CRISPR-Cas9 technology (Figure 9). We used the same system/vectors from Dr. Zhang's group [56], which have been used in a human melanoma cell line to screen for genes that are involved in resistance to BRAF inhibitor treatment [53, 54].
Figure 9. Knock-out of BIM protected melanoma cells against combination-induced cell death.
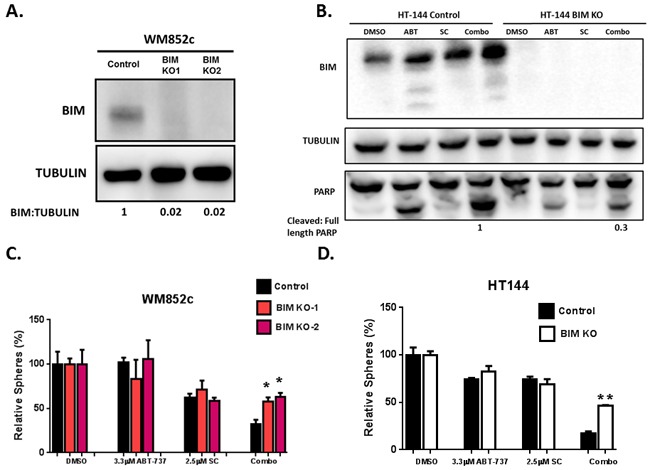
WM852c and HT144 melanoma cell lines with BIM knockout were generated with the CRISPR/Cas9 system. Knockout of BIM were determined by Immunoblots A. and B. and these cells were used for sphere assays C. and D. (B) also shows the full length and cleaved PARP from the HT144 cells, with either control or CRISPR-mediated knockout of BIM, upon indicated treatments in sphere conditions. The ratio of Cleaved/full length PARP of the cells treated with combination was quantified, and the one in the control cell was set as 1. (C) and (D) show the sphere assays with the knockout clones (KO1 and KO2) of WM852c cells with indicated drug treatments for 48 hrs. KO of BIM significantly protected the cells against the combination-induced disruption of spheres (P<0.05 or less) in both WM852c and HT144. **indicates P<0.01; *indicates P<0.05.
We successfully established multiple clones with complete reduction of BIM protein expression in melanoma cell lines with the CRISPR system (BIM KO) (Figure 9A and 9B). Sphere-forming assays demonstrated that KO of BIM also significantly protected the cells against the combination-induced disruption of spheres (P<0.05 or less) in both BRAF and NRAS mutated melanoma cell lines (Figure 9C and 9D). In addition, Immunoblot assays also indicated that the knockout (KO) of BIM reduced the drug combination-induced PARP cleavage and dramatically decreased the ratio of cleaved/full-length PARP (Figure 9B). These data suggest the combination-induced killing is both NOXA- and BIM- dependent.
DISCUSSION
This study is the first to examine the efficacy of MCL-1 and BCL-2 inhibitors in combination to induce killing of both bulk melanoma cells and MICs. Consistent with other reports we found higher endogenous MCL-1 expression in most melanomas compared to melanocytes in both cell lines and tumor samples from patients (Figure 1) [21, 39]. This data also suggests that the higher expression of MCL-1 in melanomas is independent of the mutation status for BRAF or NRAS. Therefore, the anti-apoptotic protein MCL-1 is a potential treatment target for a broad range of melanomas.
We first tested the effects of the novel MCL-1 inhibitor SC-2001 in multiple assays in vitro and in a conventional xenograft mouse model in vivo. SC-2001 by itself eradicated the bulk of melanoma cells in vitro regardless of the mutation status and inhibited tumor growth in vivo (Figure 2). In this study, we use the term “de-bulk” to describe the targeting of non-cancer-initiating cells, as in many other manuscripts in the research field of cancer stem/initiating cells [57–60]. Taken together, these results indicate that the SC-2001 could be a promising treatment option to de-bulk melanomas in vitro and in vivo.
Despite the efficacy of SC-2001, it was unable to target the MIC population (Figure 2F). This suggests that combinatorial treatments may be required to eliminate the resistant subpopulations of melanoma such as MICs. In support of this idea, we have demonstrated here that knocking down MCL-1 sensitizes the MIC population to ABT-737 (Figure 3A and 3B). This supports the hypothesis that combining MCL-1 and BCL-2 inhibitors will effectively target the MIC sub-population.
In vitro, we tested this hypothesis utilizing primary/secondary sphere formation assays and an ALDH activity assay to assess the effects of this combination on MICs from multiple melanoma cultures (Figure 4 and 5). It is well established in the cancer-stem-cell field that the primary sphere assay and ALDH assay measure the relative amount of MIC cells while the secondary sphere assay measures the capacity of self-renewal ability [43, 45, 46, 61]. The combination of SC-2001 and ABT-737 effectively eliminated the MIC population regardless of the mutation status in the primary sphere assay (Figure 4). Additionally, only the combination treatment significantly decreased self-renewal capacity by inhibiting the re-growth of tumor cells in the secondary sphere assay (Figure 5). Excitingly, these results were observed even in the melanomas that have relapsed from molecular-targeted or immune-therapies and also those with varying genetic backgrounds (wild type or mutated for BRAF, NRAS, or NF1). This data collectively suggests that this combination effectively targets the MIC population and may overcome melanoma's resistance to current therapies.
We further explored the effects of the SC-2001 and ABT-737 combination on the tumor initiating ability of the MIC population in vivo using a modified xenograft model. This was initiated with a small amount of remaining viable cells from MIC-enriched populations after being treated to test for differences in tumor initiating ability. We found that only the combination treatment significantly increased the duration of tumor-free time and decreased tumor incidence (Figure 6A and 6B). This result was further confirmed using secondary sphere-forming assays with single cell suspensions isolated from the surviving tumors (Figure 6C). These results suggest that the combination significantly decreased the MIC-mediated tumor initiating ability in vivo. This further supports that an MCL-1/BCL-2 co-targeting strategy could be beneficial for melanoma patients, especially those relapsed from other treatments.
Despite the high efficacy of this combination in the experiments reported here, not all patient samples that were tested responded to the combination treatment. Sample MB1823 was resistant to the combination in the primary sphere assay, and MB1374 was resistant in the secondary sphere assay. Further studies are needed to determine why those two tumor samples did not respond and to identify any potential biomarkers that could be predictive of the success of this combination treatment. Moreover, CICs are not the only factors contributing to tumor heterogeneity or resistance to treatment, and others may include cellular plasticity and phenotypic switching [62–65]. Future studies are needed to examine how this combination treatment may affect these aspects of melanoma.
In addition to inhibiting MCL-1, SC-2001 has been reported to induce cell death through SHP-1 dependent STAT3 inactivation in liver and breast cancer cells [33, 34]. However, we did not detect any consistent changes in SHP-1 or STAT3 upon treatment in our experiments (data not shown). Instead, one of the main mechanisms responsible for the combination induced MIC death observed here is the up-regulation of the pro-apoptotic proteins NOXA and BIM (Figure 7). In support of this idea, an increase in the expression of NOXA and BIM occurred upon treatment in both mutated BRAF and NRAS cell lines along with melanoma patient samples (Figure 7). In addition, the knock-down of NOXA (shRNA) or the knock-out of BIM (CRISPR-Cas 9 technology) significantly protected against combination-induced cell death (Figure 8 and 9). The protection we have seen for loss of either NOXA or BIM alone was 23-28%, which was commonly seen in other studies with BCL-2 family members [37, 66–68]. Multiple members in the BCL-2 family may coordinate and control the balance of life and death in the cells, therefore each protein likely only contribute partial effects.
Both NOXA and BIM are pro-apoptotic members of the BCL-2 protein family, and both can interact with MCL-1 and abolish the functions of MCL-1 [69]. Upregulation of NOXA can sensitize various cancer cell types to ABT-737 including melanoma [70–72]. TRAIL-induced MCL-1 inhibition leads to BIM-mediated apoptosis [73]. NOXA-MCL-1-BIM interplay is also important for apoptosis induced by drugs such as microtubule-targeting agents [74]. Our results also confirm the important roles of both proteins in antagonizing MCL-1 in melanoma, and that indeed NOXA-MCL-1-BIM axis can play a crucial role in the combination-induced killing of melanoma cells including the MICs.
The exciting CRISPR/Cas9 technology has advanced the gene-editing methodology tremendously, because it makes it possible to easily and inexpensively edit genetic information in virtually any organism, including human cells. This method also is more superior than shRNAs since you can knockout the gene of interest [55, 75]. We successfully used the CRISPR/Cas 9 technique to knock out BIM in our mechanistic study. Many recent studies suggest that off-target effects are rare for the CRISPR/Cas9 gene editing system [75–78]. In addition, we used two different gRNAs to delete BIM, and analyzed multiple BIM-null clones. Thus, it is very unlikely that the results obtained using these BIM-null lines are due to off-target effects.
A potentially significant anecdotal observation is that we have been unable to establish stable knockdown lines with a significant reduction in MCL-1 expression levels. Increasing the titer of the viral particles used significantly increases the level of killing during infection compared to shRNA vectors targeting other genes. We have also attempted to make MCL-1-null lines using CRISPR/Cas9 in three different melanoma cell lines using three different gRNAs against MCL-1. However, we could not find any clones lacking MCL-1 expression. Although indirect, this data further supports the idea that MCL-1 is essential for the survival of melanoma cells and is therefore an attractive therapeutic target for melanoma.
Designing a potent MCL-1 inhibitor is a huge challenge as directly inhibiting this target requires the disruption of high-affinity protein–protein interactions [79, 80]. SC-2001 is a derivative of Obatoclax, but both are small molecule BCL-2-, BCL-XL-, BCL-W-, and MCL-1 inhibitors which act on the intrinsic pathway [81]. Both compounds have been shown to inhibit MCL-1 by interrupting its interactions with other BCL-2 pro-apoptotic members [33, 82, 83]. Based on previous work and the work shown here, both compounds also act by increasing NOXA expression to antagonize MCL-1 [81, 82]. Obatoclax has been in clinical trials, but there are reports of some off-target effects [81]. Even though SC-2001 is reported to have better growth inhibition and death-inducing potential than Obatoclax in hepatocellular carcinoma cell lines [33], the study here can not completely rule out the possible contribution of off-target effects in our experiments. Recently, A-1210477 has been described as the most potent MCL-1 inhibitor available to date inducing clear on-target cellular activity. A-1210477 selectively disrupts MCL-1–NOXA and MCL-1–BIM complexes in living cells [80]. It was also demonstrated that A-121047 can synergize with the BCL-2 inhibitor ABT-263 in killing lung cancer, esophageal cancer, and multiple myeloma cell lines [80, 84]. It will be very interesting to study the clinical relevance and efficacy of this compound on killing melanoma cells and MICs.
Taken together, this work is of interest to both clinicians and researchers due to its following novelties: 1) Targeting MCL-1 and other BCL-2 family members is a promising approach to kill the bulk of melanoma cells, and most importantly, also the MICs. 2) This study also tested the effectiveness in melanoma cells from patients relapsed from current treatments, and in melanoma with wild type BRAF status which have fewer treatment options currently. 3) This study utilized the cutting-edge genomic-editing tool of CRISPR/Cas9 system to address the role of BIM in these treatments.
In summary, the data presented here suggests that MCL-1 is a promising target for treating melanomas and that SC-2001 is effective at killing non-melanoma-initiating cells. However, the combination treatment with both MCL-1 and BCL-2 inhibitors is capable of eliminating MICs which is needed to hinder relapse potential and block tumor regeneration. These treatments may be effective for melanoma regardless of the mutation status of BRAF or NRAS and may help overcome melanoma's resistance to current treatments. These results underline the need for developing potent and selective MCL-1 inhibitors that can be used in clinical trials.
MATERIALS AND METHODS
Reagents
SC2001 was synthesized as described previously in Chen et al.,[33]. ABT-737 was purchased from Selleck Chemicals.
Cell lines, patient samples and culture conditions in monolayer and sphere cultures
Human melanoma metastatic cell lines A375, 1205Lu, SK-MEL-28, 451Lu, and HT-144 were obtained from ATCC (Manassas, VA) and WM852c was kindly provided by Dr. Meenhard Herlyn. NRAS mutated melanoma lines (SK-MEL-2, Hs852T) and patient-derived melanoma cell lines were provided by the Melanoma bank at University of Colorado Cancer center. Primary melanocytes were obtained from Life Technologies (Carlsbad, CA), and immortalized melanocytes PIG1 were kindly provided by Dr. Le Poole [85].
Cells were maintained in RPMI 1640 media (Invitrogen, Grand Island, NY) with 10% fetal bovine serum (Gemini Bio-Products, Inc., West Sacramento, CA). Melanocytes were maintained in Medium 254 with Human Melanocyte Growth Supplement-2 (Life Technologies, Carlsbad, CA). To mimic melanoma culture conditions, 10% FBS was added for drug assays.
All sphere assays were performed with poly-hema (Sigma, St. Louis, MO) coated plates or dishes [86], in stem cell media as described previously [27, 87, 88]. Specifically, the media contained DME/F12 (Hyclone) supplemented with B27 (Invitrogen), 20 ng/mL EGF and 20 ng/mL bFGF (BD Biosciences), and 4 μg/mL heparin (Sigma). Cells were seeded at the density of 1-5 viable cell/μl for melanoma cell lines and 10-20 viable cell/μl for the patient derived samples for all sphere assays.
Patient samples were only cultured in vitro for a short term (within 6 month of receipt) and some of the them were only maintained in a PDX model as described in [43, 89], so that they retain original tumor characteristics such as expression and mutation profiles. Xenografted tumors of F2-F4 generations were harvested and frozen as single-cell suspensions before being used in this study, and were prepared according to the method described in [89]. These patient samples were derived from melanoma biopsy samples of patients relapsed from various treatments. These melanoma cultures were validated by the bank with Melanoma Triple Cocktail staining. Melanoma Triple Cocktail (Ventana Medical Systems, Inc, Tucson, AZ) is an antibody cocktail of anti-Melanosome (HMB45), anti-MART-1/melan A (A103), and anti-Tyrosinase (T311) mouse monoclonal antibodies. The patient samples either harbored BRAF mutation (MB2309 and MB1823), NRAS mutation (MB1920 and PS4) or were triple-WT (wild type for BRAF, NRAS and NF1) (MB2141, and MB1374). The samples were collected after patients relapsed from the following treatments: Immunotherapy (MB1823) or BRAF-MEK inhibitors (MB2309) or multiple drugs/radiation/surgery (MB2141, MB1374).
Measurement of cell proliferation, cytotoxicity, apoptosis and ALDH activity
The Cell Titer 96™ Aqueous One solution cell proliferation assay (ATP assay; Promega Corp., Madison, WI) was used to quantify cell viability, as instructed by the manufacturer. The cytotoxicity assay was conducted using the luminescent cyto-tox glo kit (Promega Corp., Madison, WI) in a 96 well format, as instructed by the manufacturer. Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, San Jose, CA) was used to quantify apoptosis according to the manufacturer's protocol. Cells were analyzed by flow cytometry using a Beckman Coulter FC500 with CXP software (Hialeah, FL) in the University of Colorado Cancer Center Flow Cytometry Core. The Aldefluor kit (Stem Cell Technologies, Vancouver, Canada) was used to detect the ALDH activity according to the manufacturer's instructions. The Aldefluor staining was detected using the FITC channel and analyzed at the University of Colorado Cancer Center Flow Cytometry Core. At least three repeats were done for each cell line. The data was normalized as the relative fold in order to visualize the change of ALDH positive cells compared to the DMSO control, with the percentage of ALDHhigh cells in DMSO condition set as “1”.
Creation of short hairpin RNA transduced cell lines
Short hairpin RNA (shRNA) expressing cell lines against various BCL2 family members were created as described previously [23]. Knockdown of genes of interest was measured by immunoblotting of cell lysates.
Creation of CRISPR mediated cell lines
BCL-2 family member BIM was knocked out by CRISPR /Cas9 technology. The protocol was followed from [54]. Briefly, the cells were first subjected to Cas-9 lentiviral transduction and then selected for Blasticydin resistance for 5 days. The Blasticydin-resistance Cas-9 transduced cell lines were then subjected to BIM gRNA lentiviral transduction. Functional Genomics Core at UC Boulder provided CRISPR/Cas9 related vectors, which were provided by Dr. Feng Zhang lab (The Broad Institute and the McGovern Institute of Brain Research at the Massachusetts Institute of Technology) [56]. Two different gRNA sequences of the lenti-guide puro-vectors are GCCCAAGAGTTGCGGCGTAT and CAACCACTATCTCAGTGCAA. After transduction, cells were selected with puromycin so that only cells transduced with a stable construct are preserved. The cells were then seeded in 96-well plate at the density of 1 cell/well using MoFlo XDP100 Cell sorter by the University of Colorado Cancer Center Flow Cytometry Core. The single cells were maintained for clonal expansion and each of the clones were expanded and tested to select for the complete knock-out, and screened and verified by immunoblotting of cell lysates.
Immunoblot
Cells, both floating and adherent, were harvested with 1x Laemmli Sample Buffer (Bio-Rad, Hercules, CA). Samples were used in the standard western blot analysis protocol as described previously [90]. The following antibodies were used at suggested dilutions from the manufacturers: PARP1 (PARP), and TUBB2A (α/β Tubulin) (Cell Signaling Technology, Danvers, MA); PMAIP1 (NOXA, EMD Biosciences, Inc. San Diego, CA); MCL1 (BD Biosciences, San Jose, CA); BIM (Millipore, Billerica, MA), β-actin (Sigma Aldrich, St. Louis, MO) and HRP-conjugated goat anti-mouse and anti-rabbit antibodies (Jackson Immuno-Research, West Grove, PA). Immunoblots were typically performed 2-3 times for each cell line, and representative examples are shown. Immunoblot data was quantified using Image Studio Ver. 2.0 (LI-COR, Lincoln, NE).
Primary and secondary sphere forming assays
Primary sphere assay
Cells were plated at a density of 1-20 viable cell/μl. Fresh media was added every 2-3 days. The spheres were allowed to grow up to reasonable size until Day 5 after seeding and then treated with indicated drugs on day 5. After 48 hrs, the numbers of spheres were counted and images captured using Nikon Eclipse TS100 scope fitted with Nikon DS-Fi2 camera.
Secondary sphere assay
Primary spheres, formed as mentioned above for indicated drug treatments, were dissociated into single cells and replated as described in [39, 41]. The procedures were the same as for the primary sphere assay, except that no drugs were added during the secondary sphere assays.
At least three repeats of both the primary and secondary sphere assays were done for each cell line. The data were normalized as the relative spheres in percentage compared to the vehicle (DMSO) control, and the number of spheres in the DMSO was set at “100”. The ethidium bromide/acridine orange stainingassay, as described previously [91, 92], was used to estimate live, dead, or apoptotic cells of the secondary spheres dissociated with PBS-EDTA [37].
In vivo mouse xenograft studies
Conventional mouse xenograft study
Female NCRNU nude mice, aged 5 weeks, were used for the study. All animal experiments are approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Colorado Denver (protocol number 88512(11)1E). Each mouse was subcutaneously injected on each flank with 1 million 1205Lu cells in a 100 μl volume consisting of 50% BD Matrigel Matrix (BD Biosciences) prepared according to the manufacturer's protocol. Drug treatments began after tumors reached approximately 100 mm3. Mice were randomly divided into two treatment groups consisting of at least 10 tumors each group: 1) vehicle only, 2) SC-2001 only. SC-2001 was administered (PO) at 10 mg/kg every alternate day, for 21 days. Mice were weighed daily and tumor volume was measured every alternate day with digital calipers.
MIC-mediated tumor xenograft
In this experiment, we employed a modified xenograft method with a low cell number implantation to test the differences in tumor initiating ability between the control and treatment groups. Melanoma sample MB1860 was used in this experiment. These cells were only maintained in the PDX model prior to this. The cells in the single-cell suspension were cultured in sphere condition, and treated with the indicated concentration of DMSO, single, or combined drug for 48hrs. Spheres were then dissociated into the single cell suspension and 50,000 viable cells were injected in each flank of nude mice and tumors were allowed to grow. Mice were weighed and tumor growth was measured as a readout for the impact of the single/combination treatment on tumor-initiation ability. Tumors were examined every alternate day. Tumor incidence was determined when the tumor was palpable and about 75 mm3 in size. Mice were sacrificed at the end of the experiment and tumors were collected and dissociated into single cell suspension to perform sphere assays. This modified xenograft method makes testing cancer initiation more feasible than the standard series dilution method. Similar approaches have been used previously in assessing tumor initiating ability in other cancer stem cell studies [49–52].
Statistical analysis
All the graphs and statistical analyses for the ATP assay, IC50, sphere-forming assays and ALDH assay were created and conducted with GraphPad Prism 6 software. Specifically, One-Way Analysis of Variance (ANOVA) was used to evaluate if there were any statistically significant differences among all the conditions within each experiment. Tukey post-hoc test were then performed to determine which comparisons among the conditions was statistic significantly different. The analyses with P-value of 0.05 and below were considered significant. Survival curve is plotted as the percentage of tumor free incidence on indicated days and we used Log rank (Mantel-Cox) test for tumor incidence with Graphpad Prism 6 software. Statistical Analysis for tumor growth data was conducted using a mixed model followed by simple effect test for pairwise comparisons of mean fold change in tumor volume between treatment groups using SPSS software (IBM, SPSS Statistics). A P-value of 0.05 and less was considered significant. Data for tumor incidence was analyzed using ELDA software (WEHI bioinformatic resources) and significance was determined by chi-square analysis as described in [93, 94]. P-values of 0.05 and less was considered significant.
SUPPLEMENTARY MATERIALS FIGURES AND TABLES
ACKNOWLEDGMENTS AND FUNDING
This work was supported in part by a Veterans Administration merit grant from the Department of Veterans Affairs (Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development) to DAN; and by a Southwestern Skin Cancer SPORE Pilot project to YGS. We would like to thank the Functional Genomics Facility at UC Boulder and Dr. Feng Zhang lab from the Broad Institute and the McGovern Institute of Brain Research at the Massachusetts Institute of Technology for providing CRISPR reagents [56]. We thank the University of Colorado Skin Cancer Biorepository for providing human melanoma samples and melanoma cell lines. We are grateful to Karen Helm, Christine Childs, and Erin Erdahl of the CU Cancer Center Flow Cytometry Core facility (supported by Cancer Center Support Grant P30CA046934 and NIAMS Skin Disease Research Core Center grant P30 AR 057212) for their expert technical assistance.
Abbreviations
- MIC
Melanoma Initiating Cells
- CSC
Cancer Stem Cells
- MCL-1
Myeloid cell leukemia sequence 1
- BCL-2
B-cell CLL/lymphoma 2
- BIM
BCL2-like 11 (apoptosis facilitator)
- PARP
Poly ADP-ribose polymerase 1
- KD
Knock Down
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Schadendorf D, Amonkar MM, Stroyakovskiy D, Levchenko E, Gogas H, de Braud F, Grob JJ, Bondarenko I, Garbe C, Lebbe C, Larkin J, Chiarion-Sileni V, Millward M, et al. Health-related quality of life impact in a randomised phase III study of the combination of dabrafenib and trametinib versus dabrafenib monotherapy in patients with BRAF V600 metastatic melanoma. Eur J Cancer. 2015;51:833–840. doi: 10.1016/j.ejca.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol. 2015;33:1889–1894. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perciavalle RM, Opferman JT. Delving deeper: MCL-1's contributions to normal and cancer biology. Trends Cell Biol. 2013;23:22–29. doi: 10.1016/j.tcb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010;584:2981–2989. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 5.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2009;66:1326–1336. doi: 10.1007/s00018-008-8637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, Bataille R, Amiot M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002;100:194–199. doi: 10.1182/blood.v100.1.194. [DOI] [PubMed] [Google Scholar]
- 8.Ding Q, He X, Xia W, Hsu JM, Chen CT, Li LY, Lee DF, Yang JY, Xie X, Liu JC, Hung MC. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007;67:4564–4571. doi: 10.1158/0008-5472.CAN-06-1788. [DOI] [PubMed] [Google Scholar]
- 9.Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, Alexander WS, Lowe SW, Robb L, Strasser A. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012;26:120–125. doi: 10.1101/gad.182980.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002;99:1885–1893. doi: 10.1182/blood.v99.6.1885. [DOI] [PubMed] [Google Scholar]
- 11.Andersson Y, Juell S, Fodstad O. Downregulation of the antiapoptotic MCL-1 protein and apoptosis in MA-11 breast cancer cells induced by an anti-epidermal growth factor receptor-Pseudomonas exotoxin a immunotoxin. Int J Cancer. 2004;112:475–483. doi: 10.1002/ijc.20371. [DOI] [PubMed] [Google Scholar]
- 12.Bolesta E, Pfannenstiel LW, Demelash A, Lesniewski ML, Tobin M, Schlanger SE, Nallar SC, Papadimitriou JC, Kalvakolanu DV, Gastman BR. Inhibition of Mcl-1 promotes senescence in cancer cells: implications for preventing tumor growth and chemotherapy resistance. Mol Cell Biol. 2012;32:1879–1892. doi: 10.1128/MCB.06214-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huelsemann MF, Patz M, Beckmann L, Brinkmann K, Otto T, Fandrey J, Becker HJ, Theurich S, von Bergwelt-Baildon M, Pallasch CP, Zahedi RP, Kashkar H, Reinhardt HC, et al. Hypoxia-induced p38 MAPK activation reduces Mcl-1 expression and facilitates sensitivity towards BH3 mimetics in chronic lymphocytic leukemia. Leukemia. 2015;29:981–984. doi: 10.1038/leu.2014.320. [DOI] [PubMed] [Google Scholar]
- 14.Murphy AC, Weyhenmeyer B, Noonan J, Kilbride SM, Schimansky S, Loh KP, Kogel D, Letai AG, Prehn JH, Murphy BM. Modulation of Mcl-1 sensitizes glioblastoma to TRAIL-induced apoptosis. Apoptosis. 2014;19:629–642. doi: 10.1007/s10495-013-0935-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pan R, Ruvolo VR, Wei J, Konopleva M, Reed JC, Pellecchia M, Andreeff M, Ruvolo PP. Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor (-)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood. 2015;126:363–372. doi: 10.1182/blood-2014-10-604975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei SH, Dong K, Lin F, Wang X, Li B, Shen JJ, Zhang Q, Wang R, Zhang HZ. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother Pharmacol. 2008;62:1055–1064. doi: 10.1007/s00280-008-0697-7. [DOI] [PubMed] [Google Scholar]
- 17.Williams MM, Cook RS. Bcl-2 family proteins in breast development and cancer: could Mcl-1 targeting overcome therapeutic resistance? Oncotarget. 2015;6:3519–3530. doi: 10.18632/oncotarget.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKee CS, Hill DS, Redfern CP, Armstrong JL, Lovat PE. Oncogenic BRAF signalling increases Mcl-1 expression in cutaneous metastatic melanoma. Exp Dermatol. 2013;22:767–769. doi: 10.1111/exd.12254. [DOI] [PubMed] [Google Scholar]
- 19.Boisvert-Adamo K, Longmate W, Abel EV, Aplin AE. Mcl-1 is required for melanoma cell resistance to anoikis. Mol Cancer Res. 2009;7:549–556. doi: 10.1158/1541-7786.MCR-08-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chetoui N, Sylla K, Gagnon-Houde JV, Alcaide-Loridan C, Charron D, Al-Daccak R, Aoudjit F. Down-regulation of mcl-1 by small interfering RNA sensitizes resistant melanoma cells to fas-mediated apoptosis. Mol Cancer Res. 2008;6:42–52. doi: 10.1158/1541-7786.MCR-07-0080. [DOI] [PubMed] [Google Scholar]
- 21.Fofaria NM, Frederick DT, Sullivan RJ, Flaherty KT, Srivastava SK. Overexpression of Mcl-1 confers resistance to BRAFV600E inhibitors alone and in combination with MEK1/2 inhibitors in melanoma. Oncotarget. 2015;6:40535–40556. doi: 10.18632/oncotarget.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller LA, Goldstein NB, Johannes WU, Walton CH, Fujita M, Norris DA, Shellman YG. BH3 mimetic ABT-737 and a proteasome inhibitor synergistically kill melanomas through Noxa-dependent apoptosis. The Journal of investigative dermatology. 2009;129:964–971. doi: 10.1038/jid.2008.327. [DOI] [PubMed] [Google Scholar]
- 23.Reuland SN, Goldstein NB, Partyka KA, Cooper DA, Fujita M, Norris DA, Shellman YG. The Combination of BH3-Mimetic ABT-737 with the Alkylating Agent Temozolomide Induces Strong Synergistic Killing of Melanoma Cells Independent of p53. PloS one. 2011;6:e24294. doi: 10.1371/journal.pone.0024294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727–738. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 25.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 26.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 27.Mukherjee N, Schwan JV, Fujita M, Norris DA, Shellman YG. Alternative Treatments For Melanoma: Targeting BCL-2 Family Members to De-Bulk and Kill Cancer Stem Cells. J Invest Dermatol. 2015;135:2155–2161. doi: 10.1038/jid.2015.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen N, Couts KL, Luo Y, Fujita M. Understanding melanoma stem cells. Melanoma Manag. 2015;2:179–188. doi: 10.2217/mmt.15.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. The Journal of clinical investigation. 2010;120:41–50. doi: 10.1172/JCI41004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shakhova O, Sommer L. Testing the cancer stem cell hypothesis in melanoma: the clinics will tell. Cancer Lett. 2013;338:74–81. doi: 10.1016/j.canlet.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Rameshwar P. Future Challenges to Target Cancer Stem Cells. Enliven: Challenges Cancer Detect Ther. 2014;1:001. [Google Scholar]
- 32.Chen KF, Lin JP, Shiau CW, Tai WT, Liu CY, Yu HC, Chen PJ, Cheng AL. Inhibition of Bcl-2 improves effect of LCL161, a SMAC mimetic, in hepatocellular carcinoma cells. Biochem Pharmacol. 2012;84:268–277. doi: 10.1016/j.bcp.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 33.Chen KF, Su JC, Liu CY, Huang JW, Chen KC, Chen WL, Tai WT, Shiau CW. A novel obatoclax derivative, SC-2001, induces apoptosis in hepatocellular carcinoma cells through SHP-1-dependent STAT3 inactivation. Cancer Lett. 2012;321:27–35. doi: 10.1016/j.canlet.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 34.Su JC, Tseng PH, Wu SH, Hsu CY, Tai WT, Li YS, Chen IT, Liu CY, Chen KF, Shiau CW. SC-2001 overcomes STAT3-mediated sorafenib resistance through RFX-1/SHP-1 activation in hepatocellular carcinoma. Neoplasia. 2014;16:595–605. doi: 10.1016/j.neo.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 36.Thomas S, Quinn BA, Das SK, Dash R, Emdad L, Dasgupta S, Wang XY, Dent P, Reed JC, Pellecchia M, Sarkar D, Fisher PB. Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther Targets. 2013;17:61–75. doi: 10.1517/14728222.2013.733001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mukherjee N, Reuland SN, Lu Y, Luo Y, Lambert K, Fujita M, Robinson WA, Robinson SE, Norris DA, Shellman YG. Combining a BCL2 inhibitor with the retinoid derivative fenretinide targets melanoma cells including melanoma initiating cells. J Invest Dermatol. 2015;135:842–850. doi: 10.1038/jid.2014.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reuland SN, Goldstein NB, Partyka KA, Smith S, Luo Y, Fujita M, Gonzalez R, Lewis K, Norris DA, Shellman YG. ABT-737 synergizes with Bortezomib to kill melanoma cells. Biol Open. 2012;1:92–100. doi: 10.1242/bio.2011035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elgendy M, Ciro M, Abdel-Aziz AK, Belmonte G, Dal Zuffo R, Mercurio C, Miracco C, Lanfrancone L, Foiani M, Minucci S. Beclin 1 restrains tumorigenesis through Mcl-1 destabilization in an autophagy-independent reciprocal manner. Nat Commun. 2014;5:5637. doi: 10.1038/ncomms6637. [DOI] [PubMed] [Google Scholar]
- 40.Cancer Genome Atlas N Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 42.Lee N, Barthel SR, Schatton T. Melanoma stem cells and metastasis: mimicking hematopoietic cell trafficking? Laboratory investigation; a journal of technical methods and pathology. 2014;94:13–30. doi: 10.1038/labinvest.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo Y, Dallaglio K, Chen Y, Robinson WA, Robinson SE, McCarter MD, Wang J, Gonzalez R, Thompson DC, Norris DA, Roop DR, Vasiliou V, Fujita M. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells. 2012;30:2100–2113. doi: 10.1002/stem.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santini R, Pietrobono S, Pandolfi S, Montagnani V, D’Amico M, Penachioni JY, Vinci MC, Borgognoni L, Stecca B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene. 2014;33:4697–4708. doi: 10.1038/onc.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santini R, Vinci MC, Pandolfi S, Penachioni JY, Montagnani V, Olivito B, Gattai R, Pimpinelli N, Gerlini G, Borgognoni L, Stecca B. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells. 2012;30:1808–1818. doi: 10.1002/stem.1160. [DOI] [PubMed] [Google Scholar]
- 46.Stecca B, Santini R, Pandolfi S, Penachioni JY. Culture and isolation of melanoma-initiating cells. Curr Protoc Stem Cell Biol. 2013:6. doi: 10.1002/9780470151808.sc0306s24. Chapter 3: Unit 3. [DOI] [PubMed] [Google Scholar]
- 47.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 48.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 49.Hirata N, Yamada S, Shoda T, Kurihara M, Sekino Y, Kanda Y. Sphingosine-1-phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand-independent Notch activation. Nat Commun. 2014;5:4806. doi: 10.1038/ncomms5806. [DOI] [PubMed] [Google Scholar]
- 50.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jokinen E, Laurila N, Koivunen P, Koivunen JP. Combining targeted drugs to overcome and prevent resistance of solid cancers with some stem-like cell features. Oncotarget. 2014;5:9295–9307. doi: 10.18632/oncotarget.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu P, Davis M, Blackwelder AJ, Bachman N, Liu B, Edgerton S, Williams LL, Thor AD, Yang X. Metformin Selectively Targets Tumor-Initiating Cells in ErbB2-Overexpressing Breast Cancer Models. Cancer Prev Res (Phila) 2014;7:199–210. doi: 10.1158/1940-6207.CAPR-13-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barrangou R. Diversity of CRISPR-Cas immune systems and molecular machines. Genome Biol. 2015;16:247. doi: 10.1186/s13059-015-0816-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6:637–645. doi: 10.1038/nrc1912. [DOI] [PubMed] [Google Scholar]
- 58.Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, Clarke MF, Hoey T, Lewicki J, Gurney AL. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One. 2008;3:e2428. doi: 10.1371/journal.pone.0002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Radvanyi L. Immunotherapy exposes cancer stem cell resistance and a new synthetic lethality. Mol Ther. 2013;21:1472–1474. doi: 10.1038/mt.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 61.Pastrana E, Silva-Vargas V, Doetsch F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell. 2011;8:486–498. doi: 10.1016/j.stem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheli Y, Giuliano S, Botton T, Rocchi S, Hofman V, Hofman P, Bahadoran P, Bertolotto C, Ballotti R. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene. 2011;30:2307–2318. doi: 10.1038/onc.2010.598. [DOI] [PubMed] [Google Scholar]
- 63.Cheli Y, Giuliano S, Fenouille N, Allegra M, Hofman V, Hofman P, Bahadoran P, Lacour JP, Tartare-Deckert S, Bertolotto C, Ballotti R. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene. 2012;31:2461–2470. doi: 10.1038/onc.2011.425. [DOI] [PubMed] [Google Scholar]
- 64.Hoek KS, Eichhoff OM, Schlegel NC, Dobbeling U, Kobert N, Schaerer L, Hemmi S, Dummer R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68:650–656. doi: 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- 65.Hoek KS, Goding CR. Cancer stem cells versus phenotype-switching in melanoma. Pigment cell & melanoma research. 2010;23:746–759. doi: 10.1111/j.1755-148X.2010.00757.x. [DOI] [PubMed] [Google Scholar]
- 66.Sheridan C, Brumatti G, Elgendy M, Brunet M, Martin SJ. An ERK-dependent pathway to Noxa expression regulates apoptosis by platinum-based chemotherapeutic drugs. Oncogene. 2010;29:6428–6441. doi: 10.1038/onc.2010.380. [DOI] [PubMed] [Google Scholar]
- 67.Weber A, Kirejczyk Z, Potthoff S, Ploner C, Hacker G. Endogenous Noxa Determines the Strong Proapoptotic Synergism of the BH3-Mimetic ABT-737 with Chemotherapeutic Agents in Human Melanoma Cells. Transl Oncol. 2009;2:73–83. doi: 10.1593/tlo.08223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou Y, Sun K, Ma Y, Yang H, Zhang Y, Kong X, Wei L. Autophagy inhibits chemotherapy-induced apoptosis through downregulating Bad and Bim in hepatocellular carcinoma cells. Sci Rep. 2014;4:5382. doi: 10.1038/srep05382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mohana-Kumaran N, Hill DS, Allen JD, Haass NK. Targeting the intrinsic apoptosis pathway as a strategy for melanoma therapy. Pigment cell & melanoma research. 2014;27:525–539. doi: 10.1111/pcmr.12242. [DOI] [PubMed] [Google Scholar]
- 70.Hauck P, Chao BH, Litz J, Krystal GW. Alterations in the Noxa/Mcl-1 axis determine sensitivity of small cell lung cancer to the BH3 mimetic ABT-737. Mol Cancer Ther. 2009;8:883–892. doi: 10.1158/1535-7163.MCT-08-1118. [DOI] [PubMed] [Google Scholar]
- 71.Lucas KM, Mohana-Kumaran N, Lau D, Zhang XD, Hersey P, Huang DC, Weninger W, Haass NK, Allen JD. Modulation of NOXA and MCL-1 as a strategy for sensitizing melanoma cells to the BH3-mimetic ABT-737. Clin Cancer Res. 2012;18:783–795. doi: 10.1158/1078-0432.CCR-11-1166. [DOI] [PubMed] [Google Scholar]
- 72.Nakajima W, Hicks MA, Tanaka N, Krystal GW, Harada H. Noxa determines localization and stability of MCL-1 and consequently ABT-737 sensitivity in small cell lung cancer. Cell Death Dis. 2014;5:e1052. doi: 10.1038/cddis.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Han J, Goldstein LA, Gastman BR, Rabinowich H. Interrelated roles for Mcl-1 and BIM in regulation of TRAIL-mediated mitochondrial apoptosis. J Biol Chem. 2006;281:10153–10163. doi: 10.1074/jbc.M510349200. [DOI] [PubMed] [Google Scholar]
- 74.Haschka MD, Soratroi C, Kirschnek S, Hacker G, Hilbe R, Geley S, Villunger A, Fava LL. The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nat Commun. 2015;6:6891. doi: 10.1038/ncomms7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harrison MM, Jenkins BV, O’Connor-Giles KM, Wildonger J. A CRISPR view of development. Genes Dev. 2014;28:1859–1872. doi: 10.1101/gad.248252.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.White MK, Khalili K. CRISPR/Cas9 and cancer targets: future possibilities and present challenges. Oncotarget. 2016;7:12305–17. doi: 10.18632/oncotarget.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beekman AM, Howell LA. Small-Molecule and Peptide Inhibitors of the Pro-Survival Protein Mcl-1. ChemMedChem. 2015 doi: 10.1002/cmdc.201500497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, Lowes KN, Kovar P, Chen J, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015;7:279ra240. doi: 10.1126/scitranslmed.aaa4642. [DOI] [PubMed] [Google Scholar]
- 81.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 82.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, Goulet D, Viallet J, Belec L, Billot X, Acoca S, Purisima E, Wiegmans A, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Perez-Galan P, Roue G, Villamor N, Campo E, Colomer D. The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood. 2007;109:4441–4449. doi: 10.1182/blood-2006-07-034173. [DOI] [PubMed] [Google Scholar]
- 84.Besbes S, Billard C. First MCL-1-selective BH3 mimetics as potential therapeutics for targeted treatment of cancer. Cell Death Dis. 2015;6:e1810. doi: 10.1038/cddis.2015.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Le Poole IC, van den Berg FM, van den Wijngaard RM, Galloway DA, van Amstel PJ, Buffing AA, Smits HL, Westerhof W, Das PK. Generation of a human melanocyte cell line by introduction of HPV16 E6 and E7 genes. In Vitro Cell Dev Biol Anim. 1997;33:42–49. doi: 10.1007/s11626-997-0021-6. [DOI] [PubMed] [Google Scholar]
- 86.Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK, Brugge JS. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nature cell biology. 2003;5:733–740. doi: 10.1038/ncb1026. [DOI] [PubMed] [Google Scholar]
- 87.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes & development. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Iwanaga R, Wang CA, Micalizzi DS, Harrell JC, Jedlicka P, Sartorius CA, Kabos P, Farabaugh SM, Bradford AP, Ford HL. Expression of Six1 in luminal breast cancers predicts poor prognosis and promotes increases in tumor initiating cells by activation of extracellular signal-regulated kinase and transforming growth factor-beta signaling pathways. Breast Cancer Res. 2012;14:R100. doi: 10.1186/bcr3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luo Y, Nguyen N, Fujita M. Isolation of human melanoma stem cells using ALDH as a marker. Curr Protoc Stem Cell Biol. 2013;26(Unit 3 8) doi: 10.1002/9780470151808.sc0308s26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ruth MC, Xu Y, Maxwell IH, Ahn NG, Norris DA, Shellman YG. RhoC promotes human melanoma invasion in a PI3K/Akt-dependent pathway. The Journal of investigative dermatology. 2006;126:862–868. doi: 10.1038/sj.jid.5700211. [DOI] [PubMed] [Google Scholar]
- 91.Ribble D, Goldstein NB, Norris DA, Shellman YG. A simple technique for quantifying apoptosis in 96-well plates. BMC biotechnology. 2005;5:12. doi: 10.1186/1472-6750-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Smith SM, Ribble D, Goldstein NB, Norris DA, Shellman YG, editors. Laboratory Methods in Cell Biology. Elsevier Inc; 2012. A Simple Technique for Quantifying Apoptosis in 96-Well Plates; pp. 361–368. PMC, ed. [Google Scholar]
- 93.Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, Yasuda H, Smyth GK, Martin TJ, Lindeman GJ, Visvader JE. Control of mammary stem cell function by steroid hormone signalling. Nature. 2010;465:798–802. doi: 10.1038/nature09027. [DOI] [PubMed] [Google Scholar]
- 94.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.