

Abstract
Pyroptosis is a form of lytic programmed cell death initiated by inflammasomes, which detect cytosolic contamination or perturbation. This drives activation of caspase-1 or caspase-11/4/5, which cleave gasdermin D, separating its N-terminal pore-forming domain (PFD) from the C-terminal repressor domain (RD). The PFD oligomerizes to form large pores in the membrane that drive swelling and membrane rupture. Gasdermin D is one of six (in humans) gasdermin family members; several other gasdermins have also been shown to form pores that cause pyroptosis after cleavage to activate their PFDs. One of these, gasdermin E, is activated by caspase-3 cleavage. We review our current understanding of pyroptosis as well as current knowledge of the gasdermin family.
Keywords: gasdermin, pyroptosis, inflammasome, programmed cell death, apoptosis
Pyroptosis defends against intracellular infection
Immune cells such as phagocytes that include macrophages and neutrophils actively survey the extracellular space and target bacteria, fungi, and parasites. Many pathogens invade host cells and, consequently, avoid detection by phagocytes and downstream cell intrinsic defenses. The continued existence of the infected cell threatens the host, and the solution is to kill the infected cell.
Killing infected cells can be accomplished through either cell-intrinsic or cell-extrinsic mechanisms. If the cell itself can identify its compromised state, it will activate cell-intrinsic death mechanisms that include cell-intrinsic apoptosis, necroptosis, and pyroptosis (Glossary). Unfortunately, many intracellular pathogens evade or inhibit cell-intrinsic programmed cell death (Glossary). Our last lines of defense are cytotoxic cells in the innate and adaptive immune systems, including natural killer cells and cytotoxic T lymphocytes that can induce cell-extrinsic apoptosis of infected cells. A comparison of intrinsic and extrinsic forms of cell death have been reviewed elsewhere [1].
Among intrinsic forms of cell death, pyroptosis has received increased attention recently. Pyroptosis can defend against intracellular infection by eliminating the compromised cell, thereby removing the pathogen’s protective niche, and simultaneously eliciting an inflammatory response. The utility of pyroptosis against intracellular pathogens can be studied by engineering normally evasive bacteria to be detected by inflammasomes (Text Box 1). For example, Salmonella Typhimurium and Listeria monocytogenes, which evade inflammasomes during systemic infection by repressing flagellin, among other strategies[1], have been engineered to force flagellin expression during the intracellular phase of infection. These modified bacteria reveal that pyroptosis kills the compromised host cell but does not kill the intracellular bacteria[1]. These bacteria remain trapped within the corpse of the pyroptotic cell in a structure we term the pore-induced intracellular trap (PIT)[2] (Glossary; Figure 1). The PIT holds bacteria in place and simultaneously increases the number of neutrophil chemoattractants[2,3] including DAMPs and eicosanoids[4]. Neutrophils, or perhaps ROS-producing macrophages[5], engulf and kill the trapped bacteria when they efferocytose (Glossary) the PIT[2,3].
Box 1. Inflammasomes survey the cytosol for contamination or perturbation.
Inflammasomes (Glossary) are innate immune sensors that survey the cytosol for contamination or cellular perturbation, and in response they trigger pyroptosis. Inflammasomes defend against numerous pathogens, although many evade detection. However, when aberrantly activated, inflammasomes drive immunopathology and sepsis. For example, the NAIP/NLRC4 inflammasome detects bacterial type III and type IV secretion systems when they aberrantly inject rod, needle, or flagellin protein into the cytosol[74]. Once activated, the NAIP/NLRC4 inflammasome activates the protease caspase-1, which in turn cleaves the inflammatory cytokines pro-IL-1β and pro-IL-18 to their mature forms and also triggers pyroptotic membrane rupture. Other caspase-1-activating inflammasomes respond to cytosolic contamination by DNA (AIM2) or proteases (NLRP1), to perturbations within the Rho GTPases (Pyrin), or to catastrophic disruption of cellular physiology (NLRP3)[74]. In parallel, murine caspase-11, and it humans orthologs caspase-4 and -5, independently activate pyroptosis but cannot directly cleave pro-IL-1β and pro-IL-18. Unlike other caspases, caspase-11/4/5 are activated by direct interaction with cytosolic LPS[74].
Figure 1. Pyroptosis converts cells into pore-induced intracellular traps (PITs).

Whereas apoptosis converts cells into apoptotic bodies, pyroptosis converts cells into PITs. The membrane rupture event that defines pyroptosis immediately disperses soluble cytosolic contents. However, although torn, the plasma membrane remains largely intact such that organelles and live intracellular bacteria remain trapped within. This is different from the image of cellular debris, where all cellular contents are dispersed. Efferocytosis is the process whereby one cell phagocytoses another. Apoptotic bodies are typically efferocytosed by macrophages. In contrast, PITs are efferocytosed by neutrophils. These neutrophils then kill the previously intracellular, now PIT-trapped, bacteria. Pictures of neutrophils (marked by Ly6G, white) that have efferocytosed macrophage PITs (marked by CD68, red) that entrap intracellular bacteria (GFP, green) after the engineered bacteria are prompted to express flagellin and trigger pyroptosis of macrophages in vivo [2].
Pyroptosis is perhaps most effective in defense against bacteria that evolved in hosts without inflammasomes. Chromobacterium violaceum and Burkholderia thailandensis are two environmental bacteria with the strongest known phenotypes of infection in inflammasome-deficient mice[6,7] (reviewed further in [8]). Pyroptosis is implicated as a major defense mechanism against these ubiquitous environmental pathogens.
Recently, gasdermin D (Glossary) was discovered to form a pore and act as the effector for pyroptosis. Gasdermin D is a member of a family of conserved proteins that includes gasdermin A, B, C, D, E, and DFNB59[9,10] (Table 1), most of which have now been shown to have pore-forming activity. Previously, gasdermin E and DFNB59 were proposed to be excluded from the gasdermin family, and instead called gasdermin-related proteins[11], however we prefer to simplify the nomenclature by including all six genes in a single gasdermin family, given recent advances[12,13]. Most family members maintain the same domain structure with significant similarity between the pore forming domain and repressor domain, but divergent linkers[10]. DFNB59 is an exception with a divergent, shorter C-terminal domain. The pore forming domain of most members can induce cell death[10,12,14,15], although this has not yet been examined for DFNB59. Whereas humans and most other mammals have one copy of each gasdermin, mice have triplicated gasdermin A (gasdermin A1-3), deleted gasdermin B, and quadruplicated gasdermin C (gasdermin C1-4). Each gasdermin member has a distinct and restricted pattern of expression.
Table 1.
Gasdermin family nomenclature and expression pattern.
| Human gene | Alternate names | Mouse gene | Expressiona | Activated by | Major disease associations |
|---|---|---|---|---|---|
| GSDMA | gasdermin (GSDM) gasdermin1 (GSDM1) |
Gsdma1 Gsdma2 Gsdma3 |
Skin, tongue, esophagus, stomach, mammary glands, and umbilical cord | Unknown | Alopecia (GOF) Asthma |
| GSDMB | PRO2521, gasdermin-like (GSDML) | Absent | Lymphocytes, esophagus, stomach, liver, and colon | Unknown | Asthma and other immune diseases |
| GSDMC | MLZE |
Gsdmc1 Gsdmc2 Gsdmc3 Gsdmc4 |
Esophagus, stomach, trachea, spleen, intestines, bladder, and skin | Unknown | Unknown |
| GSDMD | GSDMDC1, DFNA5L | Gsdmd | Immune cells, stomach, esophagus, intestines | Caspase-1/11/4/5 | Sepsis |
| DFNA5 | gasdermin E (GSDME) | Dfna5 | Placenta, brain, heart, kidney, cochlea, intestines, and IgE-primed mast cells | Caspase-3 | AD congenital deafness (GOF) |
| DFNB59 | Pejvakin, PJVK | Dfnb59 | Inner ear hair cells, auditory system, broadly expressed in other tissues | Unknown | AR congenital deafness (LOF) |
Here, we review insights into pyroptosis in light of the discovery of gasdermin D, and our current knowledge of members of the gasdermin family.
Gasdermin D pores are the effectors of pyroptosis
Cleaved gasdermin D oligomerizes to form the pyroptotic pore
Cleavage of gasdermin D was recently discovered to be the mechanism by which Caspase-1 and -11 triggers pyroptosis[10,14,16]. Gasdermin D is expressed in immune cells and in intestinal epithelial cells[11,17]. In humans, gasdermin D is composed of a 242 amino acid (aa) amino-terminal domain (also called N-domain, NT, or gasdermin domain) connected by a 43 aa linker to a 199 aa carboxy-terminal domain (also called C-domain or CT) (Figure 2). The amino-terminal domain forms a gasdermin pore (Glossary)[15,18], and thus we propose to term it the “pore-forming domain” (PFD). This pyroptotic pore forming activity has been demonstrated for most gasdermins[10,15]. However, this activity is held in check by the carboxy-terminal domain[10], which we propose to term as the “repressor domain” (RD). After the linker is cleaved, the RD separates from the PFD. The liberated PFD then integrates into the cell membrane, where an estimated 16 PFD monomers oligomerize to form a large 10–15 nm diameter pore[15,18,19]; another group suggests 21 nm[20].
Key Figure: Figure 2. Caspase-1/11 activate the gasdermin D pore to cause pyroptosis.

Caspase-1 is activated by various inflammasomes in response to contamination of the cytosol or perturbation of basic cellular homeostasis. Caspase-11 (mice) and caspase-4 and -5 (humans) activate by direct sensing of cytosolic LPS. Upon activation, these caspases cleave pro-IL-1β, pro-IL-18, and gasdermin D. Gasdermin D is composed of an amino-terminal pore-forming domain (PFD, green), a linker (red), and a carboxy-terminal repression domain (RD). The amino-terminal pore-forming domain (PFD) of gasdermin D then interacts with the plasma membrane and approximately 16 monomers oligomerize to form a gasdermin pore. The diameter of this pore is estimated in the range of 10–15 nm, which is large enough to release small proteins, including mature IL-1β (4.5 nm diameter), probably at a relatively slow rate. Simultaneously, sodium enters the cell, bringing with it water that causes the cell volume to increase. This can rapidly exceed the volume capacity of the membrane, resulting in membrane rupture that is larger in size than the gasdermin pore, but smaller or on par with most organelles and intracellular bacteria. Upon membrane rupture, all remaining soluble cytosolic contents are released so rapidly as to be essentially instantaneous.
The predicted structure of gasdermin D, based on the crystal structure of gasdermin A3, reveals an interesting mechanism of autoinhibition. The PFD and RD interact by a large surface area between these domains. The linker domain ties the two domains together at one end of the structure, while the α4 helix of the PFD extends far across the C-domain to the opposite end, where it binds to a pocket in the RD[15]. Upon activation, either caspase-1 or -11 cleaves the linker after amino acid 275 at a conserved (F/L)LTD motif[10,14]. Presumably, when the linker is cleaved, the interface dissociates, the α4 helix is released from its pocket, and the PFD is released from the RD (Figure 2). Interestingly, gasdermin D can also be cleaved by caspase-3 within the PFD[12], which should inactivate the PFD and thus prevent pyroptosis once caspase-3 initiates apoptosis.
The gasdermin D pore has affinity for liposomes containing lipids with double phosphates on the glycerol scaffold (cardiolipin) or phosphorylated head groups (phosphatidylinositol phosphate PIP1 and PIP2 species), and it may have a lower affinity for triple-phosphorylated PIP3 and the zwitterion head group phosphatidylserine (PS)[15,18]. Gasdermin D does not bind the non-charged head group lipid phosphatidylinositol (PI) or the positively charged head group lipids phosphatidylcholine (PC) and phosphatidylethanolamine (PE)[15,18]. Thus, gasdermin D has greatest affinity for lipid species whose head groups have negative charge. Since PIP species and PS are restricted to the cytosolic leaflet of the plasma membrane, gasdermin D can only form pores from the cytosolic face. Neighboring cells are thus protected from gasdermin D arising from adjacent cells[18]. Cardiolipin is a component of the inner mitochondrial membrane and bacterial membranes. It is unclear whether gasdermin D will have access to the inner leaflet of the mitochondria.
Gasdermin D pore formation mediates pyroptosis
The open gasdermin D pore breaks the normal permeability barrier of the plasma membrane (Box 2). The most catastrophic effect is disruption of the normal separation of sodium and potassium across the plasma membrane. Normally, the cytosol has a low concentration of sodium and a high concentration of potassium, which is reversed in extracellular fluid. This concentration gradient exists in the context of the electrical gradient across the plasma membrane that draws positive ions into the relatively negative cytosol. Consequently, when a pore opens in the plasma membrane, the forces created by the concentration gradient driving potassium out of the cell are roughly counterbalanced by the electrical forces that pull potassium into the cytosol, resulting in a minimal net potassium flux initially. In contrast, sodium is drawn into the cell by both its concentration gradient and the electrical gradient, resulting in a large inward sodium flux. As sodium flows into the cytosol, it brings with it hydrating water, causing the cell volume to increase.
Box 2. Considering the gasdermin pore and methods to detect pyroptosis.
With the knowledge of gasdermin D as a 10–15nm pore that drives a membrane rupture event, it is worth reevaluating the interpretation of experimental markers of caspase-1 activation and pyroptotic membrane rupture. These include propidium, IL-1β, GFP, tdRFP, and lactate dehydrogenase (LDH).
Propidium staining is often used as a proxy for membrane rupture, and indeed the two are often simultaneous[2]. Based on the concentration of propidium and sodium we expect that for every 1 propidium molecule that enters the cell, roughly 80,000 Na+ molecules enter. This means that swelling may occur faster than propidium staining by simple stoichiometry. Thus, in a cell undergoing pyroptosis without sufficient compensatory mechanisms, the membrane rupture likely occurs before significant amounts of propidium can be detected inside the cell. However, caution should be used since propidium is very small and, thus, passes freely through the gasdermin pore[28]. It is therefore formally possible that a cell can repair its membrane to remove the gasdermin pore, as was recently shown during necroptosis[24]. That propidium is not simply a marker for membrane rupture and in fact enters through gasdermin pores is illustrated by the fact that propidium can enter cells after inflammasome activation even when swelling and membrane rupture are inhibited with glycine[28] (which acts through mysterious mechanisms[75,76]).
With a predicted maximum diameter of 4.5nm, IL-1β should pass through the gasdermin pore[15,18], explaining how IL-1β can be released in the absence of pyroptosis[26–28]. With a maximum diameter of 5.4nm, GFP may also pass through the gasdermin pore prior to membrane rupture. Thus, studies using both IL-1β and GFP to evaluate pyroptosis must take into account the caveats discussed for propidium above. In contrast, the larger tandem dimer of RFP (tdRFP, also called tdimer2, whose modern variant is tdTomato [77]) and lactate dehydrogenase (LDH) seem to not pass through the gasdermin pore, and appear to only be released by the membrane rupture event. Indeed, unlike propidium entry, LDH release is impeded by glycine[28].
In summary, molecules and proteins may pass through the gasdermin pore, or through the membrane rupture, or both. Measuring proteins and small molecules of different sizes may allow experimental separation of the gasdermin pore formation from membrane rupture. Similarly, the use of glycine to delay membrane rupture facilitates evaluation of the consequences of pore formation compared to membrane rupture.
Figure I.
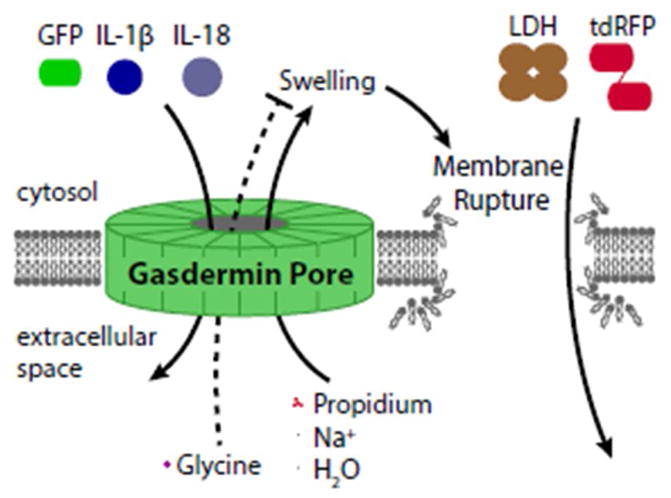
Considering the gasdermin pore and methods to detect pyroptosis
If few gasdermin pores are present, the cell should react by initiating compensatory mechanisms to decrease volume, called regulatory volume decrease. Among these are swelling-activated K+, Cl−, and organic osmolyte (e.g. taurine) channels that export these solutes and their accompanying water[21]. If the number of pores in the plasma membrane remains small, normal emergency exocytic membrane fusion events should patch the pore-containing membrane similar to how ESCRT machinery removes MLKL pores from the membrane during necroptosis[22–24]. One can also imagine a hypothetical cell where the quantity of gasdermin D is pre-set to such a low concentration that it never exceeds the regulatory volume decrease capacity and therefore never undergoes pyroptosis.
Alternatively, if gasdermin pores are present in numbers exceeding the cell’s compensatory capabilities, cell volume inevitably increases. Once the volume exceeds membrane capacity, the plasma membrane separates from the cortical cytoskeleton in large fluid-filled “balloons”. These “balloons” are distinct from apoptotic “blebs” by morphology and composition[10]. Shortly thereafter, a membrane rupture event occurs that releases soluble cytosolic contents. This tear is large enough to immediately dissipate soluble proteins like LDH (Box 2), but organelles are retained[2]. After the rupture event, we presume that osmotic pressure equalizes, stopping further volume increases.
In summary, two membrane-breaching events occur after caspase-1/11 activation: i) the gasdermin D pore opens; and soon thereafter ii) the membrane ruptures (Figure 2). We use the term “pyroptosis” as synonymous with the membrane rupture event resulting from gasdermin pores[10]. Inflammasome activation has also been implicated in expulsion of intestinal epithelial cells (IECs) from the intestinal epithelium prior to pyroptosis in response to microbial infection. While gasdermin D is required for pyroptosis but dispensable for expulsion of IECs[25], the possibility remains that other gasdermins may play a role in IEC expulsion, either alone or with redundancy.
Gasdermin D pore releases IL-1β and IL-18
During pyroptosis, caspase-1 cleaves IL-1β and IL-18 to generate mature cytokines prior to membrane rupture. As IL-1β and IL-18 lack a normal secretion signal, they are cytosol-restricted proteins. The possibility that pyroptotic membrane rupture was the release mechanism was at odds with observations of IL-1β release in the absence of pyroptosis[26–30]. Thus, whether these cytokines were actively secreted from cells or whether they were instead released by pyroptotic membrane rupture had remained unclear.
Remarkably, the 10–15 nm diameter of the gasdermin pore is large enough to allow passage of IL-1β (4.5 nm)[15,18] and IL-18 (5.0 nm), which explains the mechanism by which these proteins are released from cells prior to lysis. Importantly, the gasdermin pore is small enough to restrict larger molecules; 25–30 nm diameter ribosomes and larger organelles will not pass through the pore[2]. How can a cell release IL-1β/IL-18 through a gasdermin pore without lysing soon after? Theoretically, a cell with few gasdermin pores could shut off the inflammasome[31], remove the pores through exocytic patching mechanisms[22–24], and survive. This would release IL-1β and IL-18 through a transient gasdermin pore without killing the cell[26–30]. An additional layer of complexity arises from the membrane patching event itself, which could release a membrane-bound exosome that contains not only the gasdermin pore, but also any processed IL-1β that is nearby, perhaps explaining IL-1β observed in microparticles[32,33].
Gasdermin D pores may target other membranes
The gasdermin D pore can insert into bacterial membranes, which are rich in cardiolipin, and kill the bacteria, at least in vitro[18]. However, since bacteria survive pyroptosis[2,34], the physiologic relevance of bacterial membrane-targeting remains unclear. For vacuolar bacteria, the PFD would require passing through an existing gasdermin pore in the vacuole to oligomerize on the bacterial membrane; thus, vacuolar bacteria may have minimal exposure to the gasdermin pore. Indeed, S. Typhimurium that has been engineered to trigger pyroptosis survives in vivo. While these bacteria survive, they are damaged during the process of pyroptosis[2]. This damage might be caused by the gasdermin D pore[18], but also possibly damage from rapid volume (and presumably pressure) shifts during pyroptosis or from ROS generated from mitochondria during pyroptosis[35]. For cytosolic bacteria, the gasdermin D pore should have direct access to bacterial membranes, which has been suggested to be toxic to L. monocytogenes in vitro[18], although L. monocytogenes and another cytosol-invasive bacterium have been shown to survive pyroptosis in vitro [2]. Thus, bacteria remain alive in vivo after pyroptosis, but it remains unclear if pyroptosis damages bacteria to accelerate killing by secondary phagocytes that efferocytose these PIT-trapped bacteria.
Many organelles have cytosolic leaflets that are composed of lipids similar to the inner leaflet of the plasma membrane. If the gasdermin pore inserts into the endoplasmic reticulum, it would release calcium stores into the cytosol. This likely contributes to the calcium flux observed after caspase-1 activation but prior to pyroptosis[4,36]. The cytoplasmic leaflet of vacuoles also resembles the cytoplasmic leaflet of the plasma membrane. However, vacuolar fluid contents likely reflect the extracellular fluid. Therefore, insertion of the gasdermin pore into the vacuolar membrane would allow ionic flux that in theory should cause sodium to flow from the vacuole into the cytosol, thereby shrinking the vacuole. Thus, vacuolar bacteria may remain doubly trapped: first within the membrane-bound and compressed vacuole, and second within the PIT.
Gasdermin A
Gasdermin A is expressed in epithelial cells of the skin, tongue, esophagus, stomach, mammary glands, and umbilical cord[9,37,38]. In mice, gasdermin A1 is expressed primarily in suprabasal epidermis, hair follicles, and forestomach[11,38–40]; gasdermin A2 is expressed primarily in glandular stomach[9]; and gasdermin A3 is expressed primarily in suprabasal epidermis[11,38,40–42]. Multiple spontaneous mutations in gasdermin A3 have been associated with spontaneous alopecia and hyperkeratosis[40–45]. Histologically, inflammation and a resulting depletion of skin bulge stem cells have been observed in mice with Gsdma3 mutations[44,46]. Additionally, one Gsdma3 mutation causes defective of mammary gland development[47]. In contrast, gasdermin A3 knockout mice are phenotypically normal[38]. The alopecia phenotypes therefore likely result from gain-of-function mutations. Indeed, in vitro, these disease-causing mutations in gasdermin A3 result in an inability of the RD to effectively inhibit the PFD, causing increased spontaneous pore formation in the absence of linker cleavage[10]. These mutations therefore suggest that gasdermin A may play a role in causing inflammation within the skin. In humans, single nucleotide polymorphisms (SNPs) in GSDMA have been associated with asthma[48]. Only one study has suggests gasdermin A expression in the lung[49], consequently, the mechanism by which gasdermin A contributes to asthma remains unclear.
Gasdermin B
Like GSDMA, GSDMB SNPs are also associated with asthma in multiple populations[48,50–52]. Gasdermin B expression has been detected in lymphocytes, esophagus, stomach, liver, and colon[9,37]. While one group did not observe significant expression of gasdermin B in the lung[49], another group did observe gasdermin B in the lung and implicated it in influencing expression of genes related to airway remodeling and hyper-responsiveness[53]. Furthermore, one disease-associated SNP causes an alteration in the conformational flexibility and surface charge of a region in the RD[54]. The GSDMB SNPs and the GSDMB region of chromosome 17q21 have been associated with several other immune diseases as well[55–62]. Since GSDMA and GSDMB are genetically linked to each other as well as ORMDL3 (a gene encoding a sphingolipid biosynthesis regulator with SNPs also associated with asthma) within chromosome 17q12-21, it may be that the linkage with asthma is either due to one or some combination of these genes. The study of gasdermin B in vivo will be more difficult because mice naturally lack the gene.
A recent report shows that gasdermin B can be cleaved by caspase-3, -6, and -7 within the PFD[54], similar to PFD cleavage of gasdermin D[12]. Thus, both gasdermin B and gasdermin D may be inactivated during apoptosis to prevent pyroptosis after caspase-3 activates (although this idea is must be reconciled with caspase-3 activating gasdermin E, see below).
Gasdermin C
Little is known about the function of gasdermin C. Gasdermin C is expressed in esophagus, stomach, trachea, spleen, intestines, bladder, and skin[9,37]. Gasdermin C expression is increased in metastatic melanoma[63] but suppressed in esophageal and gastric cancer[17]; however, to date, it has not otherwise been implicated in disease of humans or mice. In mice, gasdermin C1, C2, and C4 are expressed in stomach, large and small intestines, bladder, and prostate while C3 is restricted to bladder, prostate, and large intestines[37].
Gasdermin E (DFNA5)
Mutations in GSDME are associated with development of heritable, nonsyndromal deafness (thus the original name: deafness autosomal dominant 5, DFNA5)[64–66]. Because this protein can form a pore and induce pyroptosis, Wang et al. proposed to rename it gasdermin E[13], and we adopt this nomenclature here. GSDME is expressed in placenta, brain, heart, kidney, cochlea, intestines, and IgE-primed mast cells[9,37]. Deafness associated with GSDME is characterized by an autosomal dominant inheritance and gradual loss of hearing, typically starting at high frequency by age fifteen[67]. All known mutations in GSDME associated with deafness are characterized by skipping of exon 8, resulting in frameshifts and the formation of truncated gasdermin E protein[64–66]. The truncated RD fails to repress the PFD, resulting in a gain-of-function[13]. Knocking out GSDME in mice does not result in deafness[68].
Caspase-3 was recently shown to specifically cleave gasdermin E, which then forms pores in the plasma membrane and triggers lytic cell death in response to caspase-3 activators such as chemotherapeutic drugs, TNF, and viral infection[12,13]. It was proposed that gasdermin E is the effector of secondary necrosis[12]; that is lysis of apoptotic cells after prolonged time if apoptotic cells are not efferocytosed. However, a recent study demonstrated that only specific cells express gasdermin E, and proposed that in such cells caspase-3 activation is switched from driving an apoptotic program to causing pyroptosis[13]. Thus, certain cells are programmed to undergo pyroptosis instead of apoptosis. Gsdme-knockout mice were shown to be resistant to the toxicity of chemotherapy drugs such as cisplatin[13].
It is reasonable to expect that gain-of-function mutations in gasdermin E could lead to progressive deafness by increasing susceptibility for cells within the auditory system to undergo programmed cell death. Why hair cells in the ear are uniquely susceptible to this is unclear. Transcription of GSDME increases in response to p53 activation, and its expression is silenced in colorectal, gastric, and breast cancer, suggesting a possible role of gasdermin E in suppressing cancer[67].
DFNB59
DFNB59 (also called PJVK) encodes the protein pejvakin, which, like gasdermin E, is associated with deafness[69,70]. If DFNB59 is later shown to form a pore, it may be appropriate to re-name it as “gasdermin F”. DFNB59 has been detected in hair cells of the inner ear and other cells of the auditory system, but it is also broadly expressed[69]. In humans, mutations in DFNB59 are associated with autosomal recessive nonsyndromic sensorineural hearing loss with or without cochlear dysfunction[69,70]. Unlike GSDME mutations, DFNB59 mutations are heterogeneous, including both truncations and missense mutations (p.T54I or p.R183W). In mice, functional pejvakin was necessary to allow existing peroxisomes to proliferate and protect cochlear sensory hair cells and auditory neurons from noise-induced generation of reactive oxidative species[71]. The ability of pejvakin to form pores and the possible role of pejvakin pore formation in mediating proliferation of peroxisomes remains unknown. One possibility is that pejvakin forms a pore to transport proteins into the peroxisomes necessary for peroxisome proliferation in response to increased ROS. Indeed, several PEX proteins are already known to mediate import of large proteins into peroxisomes through the formation of transient pores[72]. Another group has suggested that pejvakin instead functions through interactions with cytoskeletal proteins or through a structural role in hair cells[73].
Concluding Remarks
The involvement of gasdermins with pyroptosis is a remarkable recent discovery. That the human genome encodes six proteins with the potential to form pores in the membrane and cause lysis opens up a large landscape of study (see Outstanding Questions). At least five inflammasomes can activate caspase-1[1], driving pyroptosis via gasdermin D. Caspase-11/4/5 also cleave gasdermin D to cause pyroptosis. Are there other sensor systems with similar complexity upstream of the other gasdermins? Are these other gasdermins similarly used to defend against microbial infection, and can they contribute to immunopathology? These pore-forming cytosolic proteins are extremely hazardous to the cell, and inappropriate activation is already associated with deafness and alopecia as well as exacerbation of sepsis in animal models[14]. Do other immunopathologic diseases arise from incorrect activation of gasdermins and pyroptotic cell death? The gasdermins open up these and many other questions regarding how these six gasdermins are triggered, and of what benefit the resulting pyroptosis is to the organism.
Outstanding questions.
Does gasdermin D damage intracellular microbes in vivo?
What sensors are upstream of gasdermins?
What is the role of remaining gasdermin family members in vivo? In particular, do they play a role in defense against microbial infection and immunopathology?
How do effects of the various gasdermins differ from each other?
How do the roles and effects of gasdermins differ in non-immune cells from known effects in immune cells?
What is the benefit of DFNA5 driving apoptotic signals into membrane rupture?
Do any pathogens directly inhibit gasdermin function?
Table I.
Size and estimated diameter of molecules of relevance to pyroptosis
| Exists as | Molecular weight (Da) | Max Diameter | |
|---|---|---|---|
| H20 | Small molecule | 18 | 0.275 nm |
| Na+ | Ion | 23 | 0.277 nm |
| Glycine | Small molecule | 75 | 0.97 nm |
| Propidium | Small molecule | 562 | 1.38 nm |
| IL-1β | Mature monomer | 17,376 | 4.5 nm |
| IL-18 | Mature monomer | 18,239 | 5.0 nm |
| GFP | Monomer | 26,900 | 5.4 nm |
| tdRFP | Tandem dimer* | 56,600 | Dimer of 5.6 nm |
| LDH | Tetramer* | 146,800 | 9.6 nm |
Molecular weight and diameter of the tandem dimer and tetramer are shown. Maximum diameters were measured using MacPyMOL.
Trends Box.
Caspase-1-programmed cell death observed in 1992 was later termed pyroptosis to differentiate it from the morphologically-distinct apoptosis.
The pyroptosis effector remained unknown until 2015 when gasdermin D was discovered as a cleavage target for caspase-1 and -11.
The N-terminus of gasdermin D is a pore-forming domain that permeabilizes the plasma membrane, simultaneously releasing mature IL-1β and driving cell swelling until membrane rupture.
Membrane rupture disperses soluble cytosolic contents. Organelles and intracellular bacteria remain trapped within the torn but largely intact plasma membrane. This pore-induced intracellular trap (PIT) promotes transfer of trapped bacteria to neutrophils.
Other gasdermin superfamily proteins have homologous, pore-forming domains. DFNA5, one member of this family, is activated by caspase-3, converting apoptosis into secondary necrosis.
Glossary
- Efferocytosis
phagocytosis or endocytosis of dead cells by another cell. Apoptotic bodies are typically efferocytosed by macrophages, but can also be efferocytosed by stromal cells. We recently proposed to extend the term efferocytosis to include phagocytosis of PITs by neutrophils
- Gasdermin
a family of protein (6 in humans, 10 in mice) that have a homologous N-terminal gasdermin domain. Upon activation, this domain forms a pore in the plasma membrane (this activity has not been studied for DFNB59 at this time). Gasdermins are activated by proteolytic cleavage between their N-terminal and C-terminal domains
- Gasdermin pore
the pore formed by an activated gasdermin PFD, sometimes previously called the “pyroptotic pore”
- Inflammasome
a platform that activates caspase-1. The inflammasome includes the sensor protein (NLR, AIM2, Pyrin) which oligomerize in response to a cytosolic stimulus. In many cases the adaptor protein ASC is also included in the term “inflammasome”; inflammasomes trigger the polymerization of the entire cellular content of ASC into an ASC speck. Caspase-11 is a noncanonical inflammasome that directly detects cytosolic LPS, functioning in parallel to caspase-1
- Pore-induced intracellular trap (PIT)
The remnants of a cell after its membrane ruptures as a consequence of swelling due to a membrane pore, including the gasdermin pore that causes pyroptotic membrane rupture. PITs are composed of the ruptured, but otherwise mostly intact plasma membrane that retains (traps) organelles and intracellular bacteria
- Programmed cell death
cell death that occurs because of specific signaling events within a cell. In contrast, non-programmed cell death (necrosis) results from thermal, chemical, or other physical damage
- Pyroptosis
programmed lytic cell death by membrane rupture that is a consequence of a gasdermin pore. Cells with open gasdermin pores theoretically could avert pyroptosis by initiating regulatory volume decrease and repairing their membranes. Typically the gasdermin pore causes swelling, and in short order this results in physical rupture in the plasma membrane which marks the irreversible death of the cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jorgensen I, et al. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17:151–164. doi: 10.1038/nri.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jorgensen I, et al. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. 2016;213:2113–2128. doi: 10.1084/jem.20151613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jorgensen I, et al. IL-1β, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur J Immunol. 2016;46:2761–2766. doi: 10.1002/eji.201646647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moltke Von J, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sauer JD, et al. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc Natl Acad Sci USA. 2011;108:12419–12424. doi: 10.1073/pnas.1019041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maltez VI, et al. Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity. 2015;43:987–997. doi: 10.1016/j.immuni.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aachoui Y, et al. Canonical Inflammasomes Drive IFN-γ to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell Host Microbe. 2015;18:320–332. doi: 10.1016/j.chom.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maltez VI, Miao EA. Reassessing the Evolutionary Importance of Inflammasomes. J Immunol. 2016;196:956–962. doi: 10.4049/jimmunol.1502060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saeki N, Sasaki H. Gasdermin Superfamily: A Novel Gene Family Functioning in Epithelial Cells. In: Carrasco J, Mota M, editors. Endothelium and Epithelium. 2012. pp. 193–211. [Google Scholar]
- 10.Shi J, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 11.Tamura M, et al. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics. 2007;89:618–629. doi: 10.1016/j.ygeno.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Rogers C, et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128. doi: 10.1038/ncomms14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a Gasdermin. Nature. 2017 doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 14.Kayagaki N, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 15.Ding J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 16.He WT, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saeki N, et al. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosom Cancer. 2009;48:261–271. doi: 10.1002/gcc.20636. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aglietti RA, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sborgi L, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–1778. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffmann EK, et al. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89:193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- 22.McNeil PL, Kirchhausen T. An emergency response team for membrane repair. Nat Rev Mol Cell Biol. 2005;6:499–505. doi: 10.1038/nrm1665. [DOI] [PubMed] [Google Scholar]
- 23.Jimenez AJ, et al. ESCRT machinery is required for plasma membrane repair. Science. 2014;343:1247136–1247136. doi: 10.1126/science.1247136. [DOI] [PubMed] [Google Scholar]
- 24.Gong YN, et al. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell. 2017;169:286–300. e16. doi: 10.1016/j.cell.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rauch I, et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity. 2017;46:649–659. doi: 10.1016/j.immuni.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martín-Sánchez F, et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016;23:1219–1231. doi: 10.1038/cdd.2015.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brough D, Rothwell NJ. Caspase-1-dependent processing of pro-interleukin-1beta is cytosolic and precedes cell death. J Cell Sci. 2007;120:772–781. doi: 10.1242/jcs.03377. [DOI] [PubMed] [Google Scholar]
- 28.Russo HM, et al. Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. J Immunol. 2016;197:1353–1367. doi: 10.4049/jimmunol.1600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen KW, et al. The Neutrophil NLRC4 Inflammasome Selectively Promotes IL-1β Maturation without Pyroptosis during Acute Salmonella Challenge. Cell Rep. 2014;8:570–582. doi: 10.1016/j.celrep.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 30.Gaidt MM, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity. 2016 doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 31.Shi CS, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang JG, et al. Monocytic microparticles activate endothelial cells in an IL-1β-dependent manner. Blood. 2011;118:2366–2374. doi: 10.1182/blood-2011-01-330878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacKenzie A, et al. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity. 2001;15:825–835. doi: 10.1016/s1074-7613(01)00229-1. [DOI] [PubMed] [Google Scholar]
- 34.Miao EA, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu J, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA. 2014;111:15514–15519. doi: 10.1073/pnas.1414859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergsbaken T, et al. Coordinated host responses during pyroptosis: caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. J Immunol. 2011;187:2748–2754. doi: 10.4049/jimmunol.1100477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu C, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanaka S, et al. Functional conservation of Gsdma cluster genes specifically duplicated in the mouse genome. G3 (Bethesda) 2013;3:1843–1850. doi: 10.1534/g3.113.007393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saeki N, et al. Gasdermin (Gsdm) localizing to mouse Chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamm Genome. 2000;11:718–724. doi: 10.1007/s003350010138. [DOI] [PubMed] [Google Scholar]
- 40.Runkel F, et al. The dominant alopecia phenotypes Bareskin, Rex-denuded, and Reduced Coat 2 are caused by mutations in gasdermin 3. Genomics. 2004;84:824–835. doi: 10.1016/j.ygeno.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 41.Lunny DP, et al. Mutations in gasdermin 3 cause aberrant differentiation of the hair follicle and sebaceous gland. J Invest Dermatol. 2005;124:615–621. doi: 10.1111/j.0022-202X.2005.23623.x. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka S, et al. A new Gsdma3 mutation affecting anagen phase of first hair cycle. Biochem Biophys Res Commun. 2007;359:902–907. doi: 10.1016/j.bbrc.2007.05.209. [DOI] [PubMed] [Google Scholar]
- 43.Li J, et al. Gsdma3 is required for hair follicle differentiation in mice. Biochem Biophys Res Commun. 2010;403:18–23. doi: 10.1016/j.bbrc.2010.10.094. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Y, et al. Gsdma3 mutation causes bulge stem cell depletion and alopecia mediated by skin inflammation. Am J Pathol. 2012;180:763–774. doi: 10.1016/j.ajpath.2011.10.034. [DOI] [PubMed] [Google Scholar]
- 45.Kumar S, et al. Gsdma3(I359N) is a novel ENU-induced mutant mouse line for studying the function of Gasdermin A3 in the hair follicle and epidermis. J Dermatol Sci. 2012;67:190–192. doi: 10.1016/j.jdermsci.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 46.Shi P, et al. Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. Biochem J. 2015;468:325–336. doi: 10.1042/BJ20150204. [DOI] [PubMed] [Google Scholar]
- 47.Guo H, et al. Gsdma3 is required for mammary gland development in mice. Histochem Cell Biol. 2017;135:2162–9. doi: 10.1007/s00418-017-1542-z. [DOI] [PubMed] [Google Scholar]
- 48.Yu J, et al. Polymorphisms in GSDMA and GSDMB are associated with asthma susceptibility, atopy and BHR. Pediatr Pulmonol. 2011;46:701–708. doi: 10.1002/ppul.21424. [DOI] [PubMed] [Google Scholar]
- 49.Hao K, et al. Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genet. 2012;8:e1003029. doi: 10.1371/journal.pgen.1003029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu H, et al. Genetic variation in ORM1-like 3 (ORMDL3) and gasdermin-like (GSDML) and childhood asthma. Allergy. 2009;64:629–635. doi: 10.1111/j.1398-9995.2008.01912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moffatt MF, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 52.Moffatt MF, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Das S, et al. GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. Proc Natl Acad Sci USA. 2016;113:13132–13137. doi: 10.1073/pnas.1610433113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chao KL, et al. Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. Proc Natl Acad Sci USA. 2017;114:E1128–E1137. doi: 10.1073/pnas.1616783114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qiu R, et al. Genetic variants on 17q21 are associated with ankylosing spondylitis susceptibility and severity in a Chinese Han population. Scand J Rheumatol. 2013;42:469–472. doi: 10.3109/03009742.2013.786755. [DOI] [PubMed] [Google Scholar]
- 56.Chu AY, et al. Multiethnic genome-wide meta-analysis of ectopic fat depots identifies loci associated with adipocyte development and differentiation. Nat Genet. 2017;49:125–130. doi: 10.1038/ng.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kiani AK, et al. Genetic link of type 1 diabetes susceptibility loci with rheumatoid arthritis in Pakistani patients. Immunogenetics. 2015;67:277–282. doi: 10.1007/s00251-015-0839-0. [DOI] [PubMed] [Google Scholar]
- 58.Li X, et al. Genome-wide association studies of asthma indicate opposite immunopathogenesis direction from autoimmune diseases. J Allergy Clin Immunol. 2012;130:861–8.e7. doi: 10.1016/j.jaci.2012.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hirschfield GM, et al. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cirrhosis. Nat Genet. 2010;42:655–657. doi: 10.1038/ng.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kurreeman FAS, et al. Use of a multiethnic approach to identify rheumatoid- arthritis-susceptibility loci, 1p36 and 17q12. Am J Hum Genet. 2012;90:524–532. doi: 10.1016/j.ajhg.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Witsø E, et al. Genetic Determinants of Enterovirus Infections: Polymorphisms in Type 1 Diabetes and Innate Immune Genes in the MIDIA Study. Viral Immunol. 2015;28:556–563. doi: 10.1089/vim.2015.0067. [DOI] [PubMed] [Google Scholar]
- 62.Crosslin DR, et al. Genetic variants associated with the white blood cell count in 13,923 subjects in the eMERGE Network. Hum Genet. 2012;131:639–652. doi: 10.1007/s00439-011-1103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watabe K, et al. Structure, expression and chromosome mapping of MLZE, a novel gene which is preferentially expressed in metastatic melanoma cells. Jpn J Cancer Res. 2001;92:140–151. doi: 10.1111/j.1349-7006.2001.tb01076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bischoff AMLC, et al. A novel mutation identified in the DFNA5 gene in a Dutch family: a clinical and genetic evaluation. Audiol Neurootol. 2004;9:34–46. doi: 10.1159/000074185. [DOI] [PubMed] [Google Scholar]
- 65.Yu C, et al. A 3-nucleotide deletion in the polypyrimidine tract of intron 7 of the DFNA5 gene causes nonsyndromic hearing impairment in a Chinese family. Genomics. 2003;82:575–579. doi: 10.1016/s0888-7543(03)00175-7. [DOI] [PubMed] [Google Scholar]
- 66.Van Laer L, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet. 1998;20:194–197. doi: 10.1038/2503. [DOI] [PubMed] [Google Scholar]
- 67.de Beeck KO, et al. DFNA5, a gene involved in hearing loss and cancer: a review. Ann Otol Rhinol Laryngol. 2012;121:197–207. doi: 10.1177/000348941212100310. [DOI] [PubMed] [Google Scholar]
- 68.Van Laer L, et al. Mice lacking Dfna5 show a diverging number of cochlear fourth row outer hair cells. Neurobiol Dis. 2005;19:386–399. doi: 10.1016/j.nbd.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 69.Delmaghani S, et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat Genet. 2006;38:770–778. doi: 10.1038/ng1829. [DOI] [PubMed] [Google Scholar]
- 70.Schwander M, et al. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J Neurosci. 2007;27:2163–2175. doi: 10.1523/JNEUROSCI.4975-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Delmaghani S, et al. Hypervulnerability to Sound Exposure through Impaired Adaptive Proliferation of Peroxisomes. Cell. 2015;163:894–906. doi: 10.1016/j.cell.2015.10.023. [DOI] [PubMed] [Google Scholar]
- 72.Hettema EH, et al. Evolving models for peroxisome biogenesis. Curr Opin Cell Biol. 2014;29:25–30. doi: 10.1016/j.ceb.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harris SL, et al. Conditional deletion of pejvakin in adult outer hair cells causes progressive hearing loss in mice. Neuroscience. 2017;344:380–393. doi: 10.1016/j.neuroscience.2016.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265:130–142. doi: 10.1111/imr.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weinberg JM, et al. The role of glycine in regulated cell death. CMLS, Cell Mol Life Sci. 2016;73:2285–2308. doi: 10.1007/s00018-016-2201-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Petrat F, et al. Glycine, a simple physiological compound protecting by yet puzzling mechanism(s) against ischaemia-reperfusion injury: current knowledge. British Journal of Pharmacology. 2012;165:2059–2072. doi: 10.1111/j.1476-5381.2011.01711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shaner NC, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotech. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]