

A portable system for rapid bacterial profiling aims to rapidly diagnose health care–associated infections.
Keywords: Biosensors, fluorescence anisotropy, healthcare–associated infections, antimicrobials, antibiotics, drug resistance, superbugs, nanotechnology, nucleic acid testing
Abstract
Health care–associated infections (HAIs) and drug-resistant pathogens have become a major health care issue with millions of reported cases every year. Advanced diagnostics would allow clinicians to more quickly determine the most effective treatment, reduce the nonspecific use of broad-spectrum antimicrobials, and facilitate enrollment in new antibiotic treatments. We present a new integrated system, polarization anisotropy diagnostics (PAD), for rapid detection of HAI pathogens. The PAD uses changes of fluorescence anisotropy when detection probes recognize target bacterial nucleic acids. The technology is inherently robust against environmental noise and economically scalable for parallel measurements. The assay is fast (2 hours) and performed on-site in a single-tube format. When applied to clinical samples obtained from interventional procedures, the PAD determined the overall bacterial burden, differentiated HAI bacterial species, and identified drug resistance and virulence status. The PAD system holds promise as a powerful tool for near-patient, rapid HAI testing.
INTRODUCTION
Health care–associated infections (HAIs) and the emergence of drug-resistant pathogens are major health care issues. On any given day, 1 of 25 hospitalized patients becomes infected and as many as 1 of 9 succumb to death (1). HAIs incur a significant socioeconomic burden arising from prolonged hospital stays, lasting disability, and demand for new antimicrobials. In the United States, it is estimated that more than 600,000 patients develop HAIs every year (1), and HAI-related costs amount to $100 billion to $150 billion per year (2). Rapid, sensitive detection of pathogenic bacteria is a key to initiating timely treatment with proper antibiotics, preventing disease spread, and identifying infection sources in hospitals, homes, and other field settings (3–5).
Although bacterial culture is the clinical gold standard, it has drawbacks including long process times (up to several days), personnel cost, and the need for specialized equipment and species-specific protocols. As an alternative, nucleic acid (NA) testing has been increasingly adopted in clinical laboratories. On the basis of polymerase chain reaction (PCR) amplification of bacterial nucleotides, NA tests allow for comprehensive pathogen identification. The target sequence library is also rapidly expanding to empower these tests, aided by advances in bacterial whole-genome sequencing (6). Technical challenges, however, still remain in translating NA tests into routine clinical workflows. First, system operation can be complex, requiring trained operators. Although fully automated systems are available, they tend to be bulky and expensive. Second, assay costs are often higher than those for conventional screening. For example, in sequence-specific detection (for example, TaqMan, Molecular Beacon, and LightCycler), specialized probes for each NA target should be designed. Finally, NA tests are susceptible to false positives due to accidental contamination by amplified products. To minimize the potential for cross-contamination, it is necessary to have the pre- and post-PCR areas in different spaces or PCR workstations. These constraints limit the penetration of NA tests to centralized hospital laboratories. For prompt, effective HAI control, assay platforms that can bring NA testing to the patient level (for example, community clinics and doctor’s offices) are needed.
Here, we report a new detection system designed for rapid, cost-effective HAI diagnostics. Termed polarization anisotropy diagnostics (PAD), the system measures changes in fluorescence anisotropy when detection probes recognize target bacterial NAs (7). The detection is ratiometric, independent of fluorescence intensity, which makes the assay robust against environmental factors. We advanced the PAD to enable multilevel, on-site HAI diagnostics. Specifically, we have (i) developed a compact device with a disposable cartridge for sample preparation and multiwell detection, (ii) optimized the assay to perform NA amplification and detection without washing steps, (iii) embedded contamination control in the assay protocol, and (iv) created a library of sequence-specific probes to assess bacterial burden, pathogen types, antibiotic resistance, and virulence. As proof of concept, we applied PAD to detect clinically relevant HAI pathogens (8, 9): Gram-negative Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa and Gram-positive Staphylococcus aureus. The PAD demonstrated detection sensitivity down to the single bacterium level and determined drug resistance and virulence status. With clinical samples, the PAD achieved an accuracy comparable to that of bacterial culture; however, the PAD had a much shorter turnaround time (~2 hours) and allowed for on-site operation.
RESULTS
Polarization anisotropy diagnostics
Figure 1A shows the assay scheme. Following bacterial lysis, target NAs [for example, regions in 16S ribosomal RNA (rRNA) or mRNA] are amplified via asymmetric reverse transcription PCR (RT-PCR). Next, an all-in-one PAD mix is added. The mix consists of two reagents: (i) a detection key that is derived from an aptamer (specific to DNA polymerase) adjoined with a complementary sequence to target NAs (7) and (ii) a reporter DNA with a fluorophore. The detection key, stabilized through hybridization with the target NA, locks into DNA polymerase and deactivates its enzymatic activity. The reporter DNA then retains its structure and assumes high fluorescence anisotropy (r) due to slow diffusional motion. Conversely, the anisotropy is low in the absence of the NA targets, because unlocked DNA polymerase cleaves the reporter’s fluorophore during the extension reaction. The assay is fast (2 hours for completion) and is performed without any washing steps.
Fig. 1. PAD system.
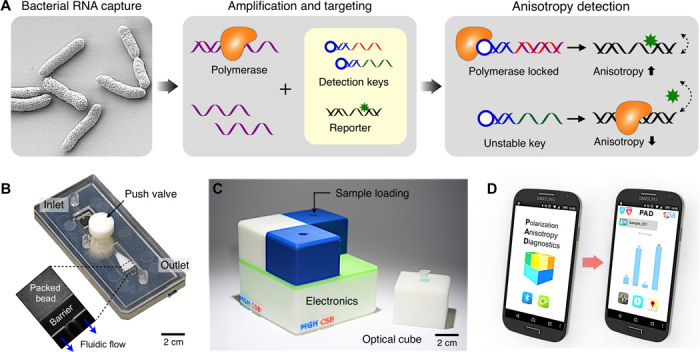
(A) Assay procedure. Bacteria are lysed, and total RNA is extracted. Following the RT-PCR amplification, samples containing amplicons and DNA polymerase are incubated with an all-in-one master mix that has the detection key and the reporter. The resulting fluorescence anisotropy of the sample is then measured. (B) Photograph of a disposable RNA extraction cartridge made in plastic. The device has an RNA extraction chamber packed with glass beads (inset). (C) Photograph of a portable system for fluorescence anisotropy detection. Four separate optical cubes can be plugged into an electronic base station. (D) PAD measurement is controlled through a custom-designed application in a smartphone. The PAD device and the smartphone communicate via Bluetooth.
PAD system
To enable on-site HAI detection, we implemented a compact PAD system (Fig. 1, B and C). The system had two major parts: a disposable sample-processing cartridge and a compact reader for fluorescence anisotropy detection. The cartridge was used to extract bacterial NAs (see fig. S1 for the device structure). It had a fluidic chamber packed with glass beads (diameter, 30 μm) to create an on-chip filter (fig. S1A) (10): negatively charged NAs could be adsorbed to the positively charged bead surface under high-salt condition and then be eluted by changing the salt concentration. We implemented the device in poly(methylmethacrylate) via injection molding (see Materials and Methods for details). When compared to a commercial column filter, the fluidic cartridge showed comparable performance, extracting RNA with good quality (fig. S2). The RNA integrity numbers (RINs), ranging from 1 (poor) to 10 (best), were 9.6 ± 0.3 (fluidic cartridge) and 9.5 ± 0.2 (column filter). By using the on-chip cartridge, however, we could remove centrifugal washing steps.
The detection system was designed for portable, parallel PAD assays. It had a modular structure consisting of a base unit for signal processing and four plug-in optical cubes (Fig. 1C and fig. S3). Each optical cube could be customized to accommodate differently sized sample containers and fluorescence optics. The packaged PAD system had a small form factor (8 × 8 × 8 cm3) and weighed ~400 g. The system wirelessly communicated with a computer or a smartphone through Bluetooth connection, which further improved system portability and simplified the assay setup. We designed a smartphone application for system operation as well as for data logging with geographic information (Fig. 1D and fig. S4).
PAD optics
Figure 2A shows the schematic of an optical cube in the PAD system. We used a light-emitting diode (LED) as an affordable light source. The illumination light passed through a linear polarizer and focused onto a sample to excite fluorophore (Fig. 2A, inset). The intensity of emission light was then measured by a pair of photodiodes. We measured both the parallel (Ix) and perpendicular (Iy) components relative to the polarized excitation light and calculated the fluorescence anisotropy (r) as r = (Ix – Iy)·(Ix + 2Iy)−1. The optics was encased into an opaque plastic body to minimize the interference from adjacent modules (fig. S3B).
Fig. 2. Optical detection design.
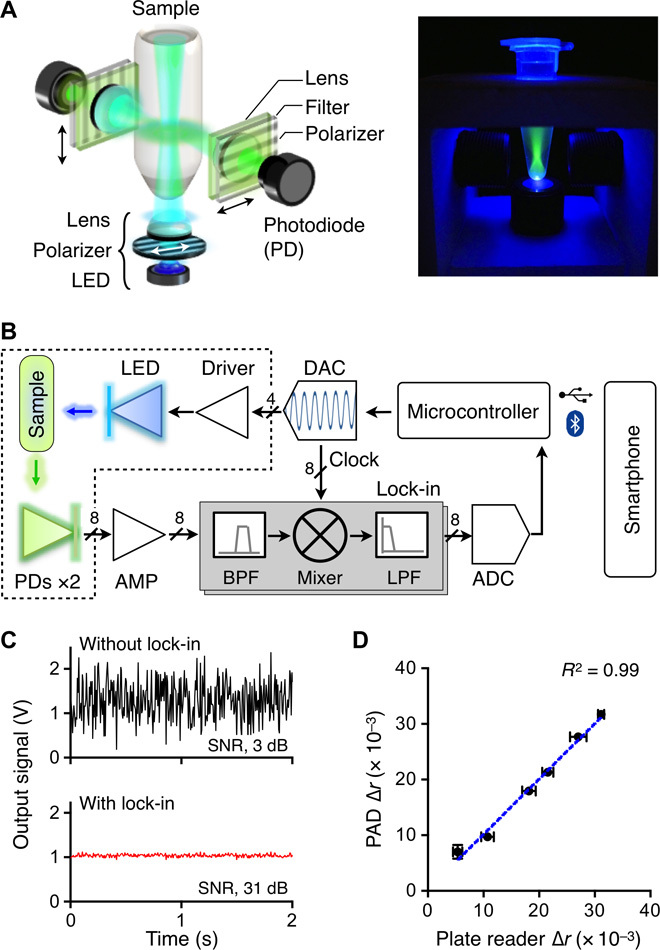
(A) Schematic of an optical cube. The optical excitation module has an LED, a linear polarizer, and a focusing lens. The emission light is measured by a pair of detector sets, each consisting of a lens, a polarization filter, and a photodiode (PD). (B) Circuit diagram. The on-board computer controls the entire system and communicates with a smartphone. To enhance the signal-to-noise ratio (SNR), the system uses the optical lock-in detection scheme. The intensity of the excitation light is amplitude-modulated, and the resulting emission intensities are mixed with the carrier frequency. The dotted box indicates an optical cube. BPF, band-pass filter; LPF, low-pass filter; AMP, amplifier. (C) The lock-in method significantly improved the signal-to-noise ratio (630 times, 28 dB). (D) The accuracy of the PAD was benchmarked against a commercial plate reader. The measured values show excellent agreements (R2 = 99%). Experiments were performed in triplicate, and the data were displayed as means ± SD. Horizontal and vertical error bars were from the plate reader and the PAD measurements, respectively.
For robust signal detection, we adopted the lock-in measurement technique. Exploiting the fact that r is intensity-independent, we modulated the intensity of the excitation light at the carrier frequency of 1 kHz. The emission signals were then frequency-locked through the homodyne signal path (Fig. 2B). The scheme significantly improved the signal-to-noise ratio (>28 dB; Fig. 2C) and allowed for reliable system operation under ambient light. Data acquisition was directed by a microcontroller unit (MCU; Arduino). A multichannel digital-to-analog converter (DAC) was used to deliver a tunable, modulating signal to the driver in each optical cube. The signal from a photodetector was amplified by a current amplifier, passed through an analog lock-in circuit, and digitized by a multichannel analog-to-digital converter (ADC). The MCU was programmed to serially poll the optical cubes, control all peripheral components, and communicate with external devices via Bluetooth (fig. S5).
We benchmarked the PAD system against a commercial plate reader (Sapphire 2, Tecan). To prepare samples of different fluorescence anisotropy, we varied the ratio between free fluorescein-labeled DNA (FAM-DNA; high fluorescence anisotropy) and template-bound FAM-DNA (low anisotropy; see Materials and Methods). The observed r values showed agreement (R2 > 99%) between the plate reader and the PAD, which served to confirm the accuracy of the PAD system (Fig. 2D).
Universal key to measure bacterial load
We first used the PAD to detect the overall bacterial burden. We designed a single, universal key (UNI key) that targets a conserved region of 16S rRNA in different bacterial species (Fig. 3A and Table 1). We also prepared a common reporter probe that was composed of FAM-DNA, its primer, and its template (Fig. 3A). The PAD output was defined as Δr = r – r0, where r0 is the fluorescence anisotropy of control samples containing DNA polymerase and the reporter only.
Fig. 3. Universal bacteria detection.
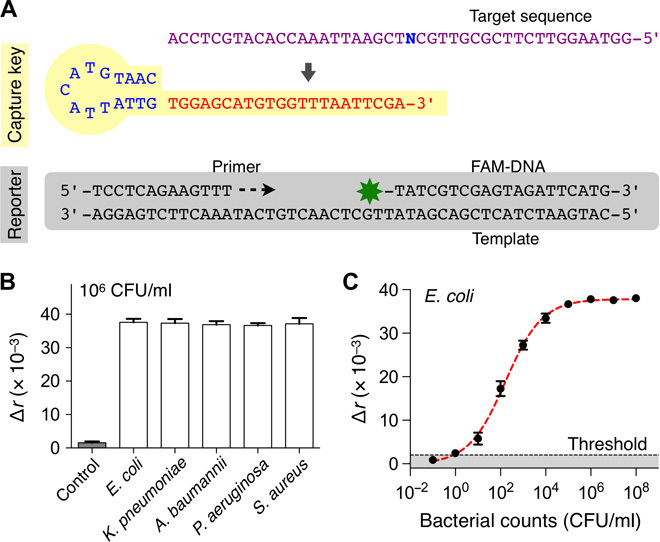
(A) The universal capture key (UNI key) detects a conserved bacterial sequence (N = A for Escherichia, Klebsiella, and Acinetobacter; N = T for Pseudomonas and Staphylococcus). The reporter is composed of a primer, a template, and FAM-DNA. (B) Five different HAI pathogens (106 CFU/ml) were detected with the PAD. The signal levels were statistically identical among concentration-matched bacterial samples. (C) Detection sensitivity. Samples with different bacterial concentrations (E. coli, 10–1 to 108 CFU/ml) were prepared through serial dilution. The limit of detection (LOD) was at the level of a single bacterium. The threshold was set at 3× SD above background signal of the sample without bacteria. All experiments were performed in triplicate and the graphs are displayed as means ± SD.
Table 1. Sequence of detection keys designed for the PAD (see table S1 for other targets).
PVL, Panton-Valentine leukocidin.
| Category | Target | DNA sequence (5′ to 3′) |
| UNI | Universal | CAATGTACAGTATTGTGGAGCATGTGGTTTAATTCGA |
| HAI | Escherichia | CAATGTACAGTATTGTGGAGGAAGGGAGTAAAGTTAAT |
| Klebsiella | CAATGTACAGTATTGTGGAGGCAAGGCGATAAGGT | |
| Acinetobacter | CAATGTACAGTATTGTTCTAGTTAATACCTAGGGATAGTG | |
| Pseudomonas | CAATGTACAGTATTGTGGAGGAAGGGCAGTAAGTTAA | |
| Staphylococcus | CAATGTACAGTATTGTGGGAAGAACATATGTGTAAGTAAC | |
| ARV | nuc | CAATGTACAGTATTGTTGCTTCAGGACCATATTTCTCTAC |
| femB | CAATGTACAGTATTGTCCAGAAGCAAGGTTTAGAATTG | |
| mecA | CAATGTACAGTATTGTCTGCTATCCACCCTCAAACAG | |
| PVL | CAATGTACAGTATTGTCGGTAGGTTATTCTTATGGTGGAG |
We applied the designed assay (UNI-PAD) to detect representative HAI pathogens (E. coli, K. pneumoniae, A. baumannii, P. aeruginosa, and S. aureus). Across different bacterial species, we observed consistent Δr values in concentration-matched samples [Fig. 3B; P = 0.8857, one-way analysis of variance (ANOVA)]; this result supported the use of the UNI-PAD in estimating total bacterial load. We next performed titration experiments with serially diluted bacterial samples (Fig. 3C; see Materials and Methods for details). The PAD assay achieved the dynamic range spanning >104 colony-forming units (CFU); the limit of detection was down to single-digit CFU.
Assay optimization for point-of-care operation
We optimized the PAD system for its point-of-care (POC) applications. One major issue in POC NA testing is to control false positives caused by sample contamination with PCR products (that is, carryover contamination) (11, 12). To minimize such effects, we adopted the uracil-DNA glycosylase (UDG) method (13, 14). By substituting deoxythymidine triphosphate (dTTP) with deoxyuridine triphosphate (dUTP) during PCR, we rendered all amplicons to have a uracil-containing DNA backbone. Applying UDG cleaved these amplicons, which selectively destroyed carryover contaminants from PCRs while keeping bona fide DNA templates (Fig. 4A). We confirmed that the method was compatible with the PAD assay. The signal level remained the same when dUTP replaced dTTP in DNA targets (fig. S6). We next tested the efficacy of this contamination control. As a carryover contaminant, dUTP-containing PCR products (107 amplicons) were spiked into samples. Without UDG treatment, the negative control with no bacterial targets showed false positives (Fig. 4B). This signal was eliminated with the addition of UDG, and only samples containing true bacterial targets yielded high Δr (Fig. 4B).
Fig. 4. Assay optimization for POC operation.
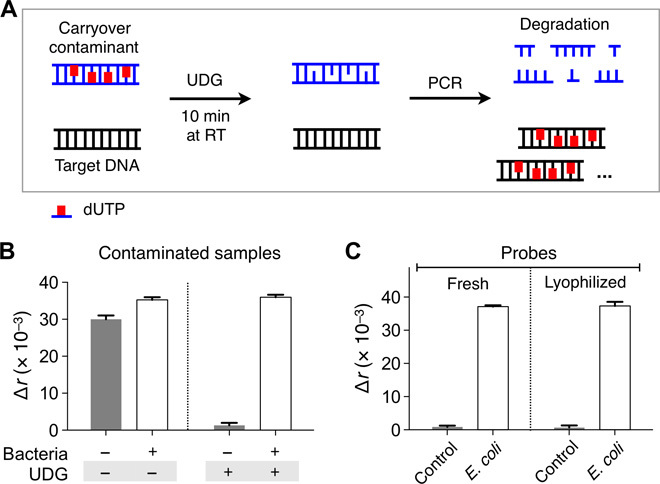
(A) Schematic illustration of UDG-mediated control on carryover contamination. Uracil-containing carryover contaminant was specifically broken down by UDG, which allows for the amplification of the true target DNA only. (B) Uracil-containing contaminants (107 copies) were added to all samples. Contaminated samples produced high signal even in the absence of target bacteria. When samples were treated with UDG, the false-positive signal was eliminated. (C) The PAD reagents were lyophilized to facilitate their transport and extend their shelf life. After 4 weeks of storage in ambient condition, the reagents were used for bacterial detection. No difference was observed between fresh and lyophilized reagents. Bacterial samples in (B) and (C) contained E. coli (106 CFU/ml). All experiments were performed in triplicate, and the data are displayed as means ± SD. RT, room temperature.
To maximize system portability, we further combined the PAD with a miniaturized thermocycler (miniPCR, Amplyus) (15). The performance of the POC system matched that of conventional benchtop equipment (fig. S7). We also lyophilized all chemical reagents (for example, detection keys and reporters) to facilitate their transport and storage (see Materials and Methods for details). The reagents retained their activity after >2 weeks of storage in ambient conditions; we observed statistically identical Δr values (P > 0.64, two-tailed t test) with fresh and stored agents (Fig. 4C and fig. S8).
Differential keys for pathogen classification
To differentiate HAI-causing pathogens, we designed a set of detection keys (HAI keys) in which each key targets the hypervariable region of 16S rRNA in different bacterial species (Table 1; see table S1 for extended targets). The sequence homology among genus types was kept <50% to minimize nonspecific binding. When tested, the HAI keys assumed high specificity. For example, the Escherichia key (Fig. 5A) showed high signal (Δr) only with its intended target, whereas off-target signals were negligible even in high biological background (106 CFU of other bacterial species; Fig. 5B). Similarly, other HAI keys displayed excellent specificity with minimal crosstalk (Fig. 5C). Electrophoretic band-shift analysis confirmed that the detection keys, in the presence of complementary target amplicon, bound to DNA polymerase and inhibited its catalytic activity (fig. S9).
Fig. 5. Bacteria typing with PAD.
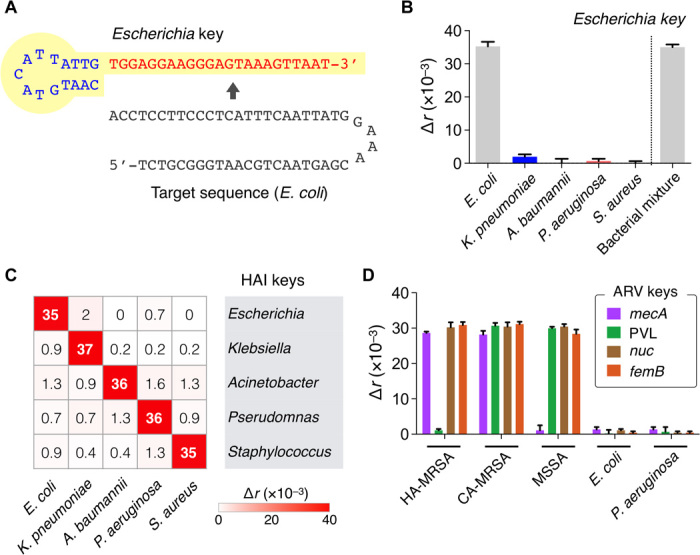
(A) A set of detection keys specific for HAI pathogens was designed (HAI keys). An Escherichia key is shown as an example. (B) The specificity of HAI keys was tested. The signal was high only in the presence of the target species even in the mixture of other bacterial species. An example of E. coli (106 CFU/ml) detection is shown. (C) Heat map of Δr values obtained for HAI detection. Bacterial concentration was 106 CFU/ml. (D) Detection keys for antibiotic resistance and virulence (ARV keys) were designed for further typing. Two types of methicillin-resistant S. aureus (MRSA; 106 CFU/ml), health care–associated MRSA (HA-MRSA) and community-acquired MRSA (CA-MRSA), were identified by targeting the specific regions in mecA and PVL genes. Three pathogens [methicillin-sensitive S. aureus (MSSA), E. coli, and P. aeruginosa; 106 CFU/ml] were included as controls. All experiments were performed in triplicate. The heat map displays mean values and the bar graphs display means ± SD.
We next prepared probes for antibiotic resistance and virulence (ARV keys) for further bacterial phenotyping. These keys targeted bacterial genes that make pathogens antibiotic-resistant or highly virulent. As a model system, we profiled samples for mecA, PVL, nuc, and femB genes. mecA is the determining factor conferring MRSA, a common multidrug-resistant HAI pathogen (16). PVL, nuc, and femB are virulence factors that contribute to the pathogenicity of S. aureus. We used two representative MRSA strains that have the following known genotypes: health care–associated MRSA (mecA+, PVL–) and community-acquired MRSA (mecA+, PVL+). Control samples were MSSA (mecA–), E. coli (mecA–, PVL–, nuc–, femB–), and P. aeruginosa (mecA–, PVL–, nuc–, femB–) (table S2) (17–20). The ARV-PAD correctly genotyped bacteria, agreeing with a quantitative real-time PCR (qPCR) (Fig. 5D and fig. S10).
Clinical application
Finally, we applied the PAD for clinical HAI diagnostics. The assay started with bacterial lysis, followed by the NA collection using the plastic cartridge. Following the NA amplification, the samples were analyzed in parallel for total bacterial burden (UNI key), HAI pathogens (HAI keys), and antibiotic resistance and virulence status (ARV keys) (fig. S11). The total assay time was ~2 hours, and the required sample volume was ~40 μl.
We acquired patient samples and aliquoted them for the PAD test (2 hours) and conventional culture (3 to 5 days) in a clinical microbiology laboratory. Test results for all detection keys (UNI, HAI, and ARV) are shown in Fig. 6A and fig. S12. Samples negative with UNI-PAD were also negative with HAI-PAD, suggesting the potential use of universal detection for sample triaging. Among six UNI-PAD–positive samples, the HAI-PAD detected HAI pathogens in five samples, and the differentiation results matched the bacterial culture readouts (Fig. 6B). One patient (no. 6) was positive with bacterial load but was negative with HAI keys; the patient was later found to be infected with Providencia rettgeri (nontargeted in the current HAI-PAD). For the sample positive for S. aureus (patient no. 2), the ARV-PAD showed mecA– status (that is, MSSA); this result matched the MRSA-negative pathology report and qPCR (Fig. 6 and fig. S13).
Fig. 6. Clinical application of PAD for HAI detection.
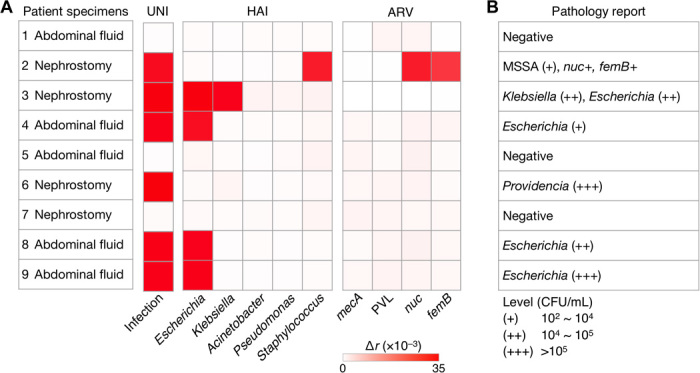
(A) Nine samples from different patients were processed by the PAD for bacterial load (UNI), presence of the HAI species (HAI), and resistance/virulence status (ARV). (B) The clinical samples were also tested by a clinical pathology laboratory (culture and qPCR). The PAD and pathology reports agreed with each other.
DISCUSSION
HAIs have become a ubiquitous, recalcitrant, and costly problem in modern health care. They increase the emergence of antibiotic resistance, cause significant morbidity and mortality, and prolong hospital stays. One of the key mandates to control this endemic is to equip local hospitals and community centers with more effective surveillance systems. The PAD platform presented here could enable rapid HAI detection in those settings. The detection system is compact and user-friendly with minimal operation complexity. The assay is comprehensive, assessing for overall bacterial burden, pathogen types, antibiotic resistance, and virulence factors.
Several features make the PAD ideal for field operation. First, the sensing scheme (fluorescence anisotropy) is inherently robust against environmental noise. We further incorporated the optical lock-in technique to significantly enhance the sensitivity. Second, the assay flow involves minimal complexity and hands-on time. A disposable plastic chip is used to collect NAs, and the remaining processes are performed in a single tube without washing steps. The assay protocol is also refined to automatically dissolve carryover contaminants, thereby minimizing false positives. Third, the platform is highly affordable. The PAD device has simple electronics and can readily be expanded for parallel detection. Making an injection-molded cartridge brings advantages including high performance reproducibility, less cross-contamination between samples, and lower cost. Finally, the PAD is scalable for comprehensive screening. Decoupling detection and signaling probes enables such assays to be cost-effective, because a common fluorescence reporter can be used for all detection targets. We have already designed detection probes for >35 targets (table S1); the incremental assay cost for additional targets is ~$0.01.
In a pilot clinical test, PAD accuracy was comparable to that of bacterial culture. In contrast to the culture, the PAD assay was fast (~2 hours), multiplexed, and cost-effective (<$2 per assay). However, we note the following limitations in the current study. First, no clinical samples were found to contain drug-resistant strains. Further studies with larger cohorts are needed to verify PAD’s capacity for drug resistance screening. Second, the PAD may present ambiguous results when target NAs overlap. For example, the current ARV keys would fail to discern community-acquired MRSA (mecA+, PVL+) from the mixture of health care–associated MRSA (mecA+, PVL–) and MSSA (mecA–, PVL+). Such incomplete classification is an inherent issue with NA-based tests. We expect that this issue would be overcome as more bacteria genomic data are accrued from whole-genome sequencing efforts.
The current prototype system could be further improved. First, we envision a self-contained, closed system in which sample preparation, thermocycling, and detection functions are all housed in a single device. Such a system would effectively eliminate erroneous results from sample contamination, user interference, or both and proffer “sample-in and answer-out” tests. Second, the current assay time could be shortened, particularly for DNA amplification. One promising direction is to adopt a photonic thermocycling system, which can complete the entire PCR in <5 min (21). Isothermal amplification is also an alternative; some isothermal reactions can be completed in 20 min, which could bring down the total assay time to <1 hour (22). Using isothermal amplification would simplify the hardware requirement and make a battery-powered device feasible. In the future, we plan to expand the test library for broader pathogen and antibiotic resistance screening. The modular nature of PAD probes should facilitate incorporating new targets such as host-response factors (for example, interleukin-4, platelet-derived growth factor B chain, monocyte chemoattractant protein-1, and C-X-C motif chemokine 10) (23), as well as other viral, fungal, and parasitic markers.
MATERIALS AND METHODS
Fabrication of the plastic cartridge
The device was made in plastic via injection molding. A metal block of the mold was first machined to have surface structures such as channels, chambers, and ports negatively shaped to those on the top and bottom parts of the plastic cartridge. To confine microbeads in the RNA capture chamber, a weir-shaped physical barrier was designed at the outlet side of the chamber. The top and bottom parts of the device were injection-molded in a foundry (Korea Institute of Machinery and Materials). More than two devices were produced per minute. The top and bottom parts were glued together and the three-way valve was inserted. Fluidic connection was made by inserting polyethylene tubes in the fluidic ports.
Device preparation
The fluidic cartridge was filled with glass beads (diameter, ~30 μm; Polysciences). Beads were suspended in 75% ethanol (Sigma) and introduced through the inlet. The beads were retained in the RNA capture chamber due to the weir-style physical barrier in the outlet side of the chamber (fig. S1A). Following bead capture, excess ethanol was collected and removed. The entire device was then flushed with cycles of RNaseZap (Life Technologies), ribonuclease (RNase)–free water (Life Technologies), and ethanol, and dried. All fluidic flow was generated by manually operating syringes.
Detection system
The illumination source in the optical cube consisted of an LED (λ = 470 nm; Thorlabs), a dichroic film polarizer (polarization efficiency, >99%; Thorlabs), and a convex lens [focal length (ƒ) = 8 mm]. The detection part had a convex lens (ƒ = 8 mm), a long-pass filter (EL0500, Thorlabs), a dichroic film polarizer, and a photodiode (S1223, Hamamatsu). A 16-bit DAC (LTC1597, Linear Technology) was used to deliver the modulated control signal to a custom-designed LED driver (LF356, Texas Instrument). The signal from the photodiode was amplified by a custom-designed current amplifier (AD549, Analog Devices). For the lock-in detection, the amplified signal was passed through a band-pass filter (center frequency, 1 kHz; bandwidth, 100 Hz), mixed with a carrier signal, and passed through a low-pass filter (time constant, 1 ms). The conditioned signal was digitized by a 16-bit ADC (LTC1867, Linear Technology). A microcontroller (Arduino MEGA 2560) was programmed to control the light sources for multiplexing, to perform real-time data recording, and to communicate with an external device (for example, computers and smartphones) via a USB 2.0 or a Bluetooth interface. The entire system was powered by a 9-V battery housed inside the base station. A typical power consumption during a single optical measurement (one cube) was ~400 mW, and each test took ~30 s.
Smartphone application
We created a custom-designed Android application to facilitate system operation and data recording. Control software was designed using MIT App Inventor 2. The application connected the smartphone to the PAD system and sent the triggering signal for the fluorescence anisotropy detection. The measured data were sent back to the phone and combined with a time stamp and Global Positioning System coordinates.
Signal detection
Fluorescence anisotropy values were measured with the excitation and emission wavelengths of 470 and 525 nm, respectively. Fluorescence anisotropy (r) was calculated using the following equation: r = (Ix – Iy)·(Ix + 2Iy)–1, where Ix and Iy are emission intensities when the emission polarizers are in parallel with and perpendicular to the excitation polarizer, respectively. The LOD was estimated by setting the threshold at 3× SD above the background signal of samples without bacteria. For comparison with the benchtop equipment (Sapphire 2, Tecan), we measured Δr = r − rFAM, where rFAM is the fluorescence anisotropy only in the presence of FAM-DNA (62.5 nM) and r is the fluorescence anisotropy in the presence of FAM-DNA (62.5 nM) along with its template. We varied the template concentrations (10, 20, 30, 40, 50, and 60 nM) to produce different amounts of hybridized FAM-DNA.
Probe design
DNA oligonucleotides were synthesized by Integrated DNA Technologies. The list of DNA sequences is summarized in Table 1 and tables S1 and S3. For the universal and species-specific detection of pathogenic bacteria, individual 16S rRNA sequences of different bacterial genera [from the National Center for Biotechnology Information (NCBI) nucleotide database] were aligned using MegAlign software (DNASTAR), and both conserved and variable regions were selected as target sequences (24). To detect ARV factors, the specific regions of nuc, femB, mecA, and PVL (from the NCBI nucleotide database) were selected as target sequences (17–20). The detection keys were designed to have a hairpin structure joined by a single-stranded capture sequence (~20 to 25 nucleotides in length) (7, 25).
Lyophilization of master mix
An all-in-one PAD mix (20 μl) was concocted by mixing a detection key (400 nM), a reporter (150 nM), a primer (150 nM), and FAM-labeled DNA (125 nM) in 20 mM tris-HCl (pH 8.3), 20 mM KCl, 5 mM (NH4)2SO4, and 6 mM MgCl2. The mixture was first frozen in liquid nitrogen and then dried in VirTis Freezemobile 25EL Freeze Dryer (SP Scientific). The lyophilized reagents were stored at room temperature and reconstituted before use by adding 20 μl of UltraPure DNase/RNase-free distilled water (Life Technologies).
Experiment with cultured bacteria
All bacteria were purchased from the American Type Culture Collection (ATCC). Bacterial cultures were grown to mid-log phase in vendor-recommended medium: E. coli (#25922) in LB medium (BD Biosciences); P. aeruginosa (#142), K. pneumoniae (#43816), and MRSA (#BAA-1720 and #BAA-1707) in tryptic soy broth (BD Biosciences); A. baumannii (#15149) in nutrient broth (BD Biosciences); and S. aureus (#25923) in Staphylococcus broth (BD Biosciences). Bacteria were collected via centrifugation (6000g, 10 min), and pellets were resuspended with the preheated TRIzol (Life Technologies). The resuspended cells were transferred to 2-ml Safe-Lock tubes (Eppendorf) containing sterilized disruptor beads (0.1 mm; Scientific Industries) and lysed using a vortex mixer. After centrifugation, the supernatant was transferred to a new tube.
RNA extraction
Bacterial lysate mixed with an equal volume of ethanol was flown through the RNA extraction chamber, in which RNA was captured by packed glass beads. Subsequent flushing with Direct-zol RNA PreWash (Zymo Research) and RNA Wash Buffer (Zymo Research) was done to remove traces of impurities and chaotropic salts. Finally, RNA was eluted in RNase-free water. For comparison of the fluidic cartridge with a commercial column (Zymo-Spin column, Zymo Research), bacterial lysate was divided into two aliquots. One sample was processed by the commercial column and the other by the fluidic cartridge. We checked the quality of the extracted RNA through an electrophoretic assay (2100 Bioanalyzer, Agilent). RNA molecular weight ladder provided in the kit (RNA 6000 Nano Chip, Agilent) was used as reference, and electrophoretic runs were performed per the manufacturer’s protocol. The analysis assigned RINs to samples, ranging from 1 to 10, where 1 indicates highly degraded RNA and 10 completely intact RNA.
PAD assay
The single-stranded complementary DNA (cDNA) was synthesized using random priming with Promega’s Reverse Transcription System as per the manufacturer’s protocol. The asymmetric PCR amplification was then carried out in a total reaction volume of 25 μl containing 2.5 μl of cDNA, 0.8 μM excess primer, 0.08 μM limiting primer (table S3), 1× PCR buffer [20 mM tris-HCl, 20 mM KCl, 5 mM (NH4)2SO4, and 2 or 3 mM MgCl2], 0.2 mM deoxyadenosine triphosphate (dATP), deoxyguanosine triphosphate (dGTP), and deoxycytidine triphosphate (dCTP), 0.4 mM dUTP (Thermo Scientific), 2 U of Antarctic Thermolabile UDG (New England Biolabs), and 2.5 U of Maxima Hot Start Taq DNA polymerase (Thermo Scientific). For the asymmetric PCR on a miniaturized thermocycler (miniPCR, Amplyus), we used the following cycling conditions: 25°C for 10 min and 94°C for 4 min; 35 cycles of 30 s at 94°C, 30 s at 56°C, and 30 s at 72°C; and an extension step of 10 min at 72°C. With a benchtop thermocycler (MasterCycler, Eppendorf), the reaction conditions were 25°C for 10 min and 94°C for 4 min; 35 cycles of 5 s at 94°C, 15 s at 56°C, and 15 s at 72°C; and a final 10 min at 72°C. The PCR solution (20 μl) was mixed with an all-in-one PAD mix composed of a detection key (200 nM) and a reporter that was preformed with a template (75 nM), a primer (75 nM), and FAM-labeled DNA (62.5 nM) at room temperature for 15 min, making a total volume of 40 μl in 20 mM tris-HCl (pH 8.3), 20 mM KCl, 5 mM (NH4)2SO4, and 4 mM MgCl2.
UDG-mediated control of carryover contamination
To mimic the carryover contamination, we spiked dUTP-containing amplification products (carryover contaminants) into new reaction samples. The copy number of carryover contaminants was ~107; a single aerosol after PCR typically contains as many as 106 amplification products (26). The dUTP-containing amplicons were prepared following the same procedures outlined above except that equal amounts of limiting and excess primers were used. The obtained amplicons were purified using the Zymoclean Gel DNA Recovery Kit (Zymo Research) and quantified by measuring the absorbance at 260 nm with NanoDrop 1000 (Thermo Scientific). The copy number was estimated on the basis of the conversion factor (26 kD per amplicon).
Clinical samples
This study was approved by the Partners Institutional Review Board. Excess and discarded samples were collected from nine subjects with clinical suspicion for infected bodily fluid or abscess and referred for drainage. Specimens were collected using routine image-guided approaches by Massachusetts General Hospital Interventional Radiology physicians and analyzed blindly using the PAD assay. Specimens (500 μl) were mixed with 1.5 ml of TRIzol LS (Life Technologies) which is more concentrated than TRIzol, and the same RNA extraction procedure was applied as in pure bacterial cultures.
Electrophoretic band-shift experiment
Solution containing 100 or 200 nM detection key, PCR products, and 12.5 U of Taq DNA polymerase (New England Biolabs) in 20 mM tris-HCl (pH 8.3), 20 mM KCl, 5 mM (NH4)2SO4, and 4 mM MgCl2 was incubated at room temperature for 20 min. The solution was mixed with 6× loading buffer and subjected to electrophoresis on a 20% polyacrylamide gel (Life Technologies). The analysis was carried out in 1× TBE (100 mM tris, 90 mM borate, 1 mM EDTA) at 150 V for 160 min at 4°C. After GelRed (Biotium) staining, gels were scanned using an ultraviolet transilluminator. DNA polymerase was neither fluorescent nor stained by the dye (27).
Quantitative real-time PCR
The cDNA derived from in vitro cultured bacteria or clinical samples was mixed with 1× PowerUp SYBR Green Master Mix (Life Technologies) and 0.4 μM specific primers used in the PAD assay. Thermal cycling was then carried out on the 7500 Fast Real-Time PCR system (Life Technologies) with the following conditions: UDG activation (50°C, 2 min), initiation (95°C, 2 min); 40 cycles of denaturation (95°C, 5 s); annealing (56°C, 15 s); extension (72°C, 30 s). The 7500 Fast software automatically calculates the Ct value, which represents the first PCR cycle at which the fluorescence signal exceeds the signal of a given uniform threshold. No-template control (NTC) remained undetected, not crossing the established threshold for 40 cycles, and was arbitrarily given a Ct value of 41. The ΔCt was generated by subtracting the Ct value of the specimen from the Ct value of NTC (28).
Supplementary Material
Acknowledgments
We thank H. Chung (Korea Advanced Institute of Science and Technology) for helpful advice on antibiotic-resistant targets and Amplyus for providing the miniPCR. Funding: This work was supported in part by NIH grants R01HL113156 (H.L.), R01EB004626 (R.W.), R01EB010011 (R.W.), and T32CA79443 (R.W.); Department of Defense Ovarian Cancer Research Program Award W81XWH-14-1-0279 (H.L.); Basic Science Research Program 2014R1A6A3A03059728 (K.S.P) through the National Research Foundation of Korea; Converging Research Center Program 2014M3C1A8048791 (Y.-E.Y) funded by the Ministry of Science, ICT and Future Planning, Korea; and Postdoctoral Research Abroad Program NSC103-2917-I-564-067 (C.-H.H.) through the Ministry of Science and Technology, Taiwan. Author contributions: K.S.P. and C.-H.H. designed the study, performed the research, analyzed data, prepared the figures, and wrote the manuscript. R.W. and H.L. designed the research, analyzed data, prepared the figures, and wrote the manuscript. C.M.C. and R.W. designed the clinical study. K.L. and Y.-E.Y. developed the plastic cartridge for RNA extraction. R.W. and H.L. provided overall guidance. All authors contributed to the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper are available from the authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/5/e1600300/DC1
table S1. Target sequences recognized by established detection keys.
table S2. Summary of a set of ARV factors targeted in this study.
table S3. DNA sequences used in this study.
fig. S1. Schematic of the plastic cartridge for RNA extraction.
fig. S2. Comparison between the fluidic cartridge and a commercial column.
fig. S3. The optical detection system in the PAD.
fig. S4. Snapshots of a PAD application.
fig. S5. The circuit diagram of the detection system.
fig. S6. The effect of dUTP on the PAD assay.
fig. S7. Portable PAD system.
fig. S8. Lyophilized probes.
fig. S9. Electrophoretic band-shift assay.
fig. S10. Detection of ARV factors.
fig. S11. Overall assay procedure for clinical samples.
fig. S12. Universal and species-specific detection of HAI pathogens in clinical samples by the PAD system.
fig. S13. Detection of ARV factors in clinical samples with PAD (top) and qPCR (bottom).
REFERENCES AND NOTES
- 1.Magill S. S., Edwards J. R., Bamberg W., Beldavs Z. G., Dumyati G., Kainer M. A., Lynfield R., Maloney M., McAllister-Hollod L., Nadle J., Ray S. M., Thompson D. L., Wilson L. E., Fridkin S. K.; Emerging Infections Program Healthcare-Associated Infections and Antimicrobial Use Prevalence Survey Team , Multistate point-prevalence survey of health care–associated infections. N. Engl. J. Med. 370, 1198–1208 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marchetti A., Rossiter R., Economic burden of healthcare-associated infection in US acute care hospitals: Societal perspective. J. Med. Econ. 16, 1399–1404 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Allegranzi B., Bagheri Nejad S., Combescure C., Graafmans W., Attar H., Donaldson L., Pittet D., Burden of endemic health-care-associated infection in developing countries: Systematic review and meta-analysis. Lancet 377, 228–241 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Polin R. A., Denson S., Brady M. T.; Committee on Fetus and Newborn; Committee on Infectious Diseases , Epidemiology and diagnosis of health care–associated infections in the NICU. Pediatrics 129, e1104–e1109 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Klompas M., Yokoe D. S., Weinstein R. A., Automated surveillance of health care–associated infections. Clin. Infect. Dis. 48, 1268–1275 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Mothershed E. A., Whitney A. M., Nucleic acid-based methods for the detection of bacterial pathogens: Present and future considerations for the clinical laboratory. Clin. Chim. Acta 363, 206–220 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Park K. S., Lee C. Y., Park H. G., Target DNA induced switches of DNA polymerase activity. Chem. Commun. 51, 9942–9945 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Mauldin P. D., Salgado C. D., Hansen I. S., Durup D. T., Bosso J. A., Attributable hospital cost and length of stay associated with health care-associated infections caused by antibiotic-resistant gram-negative bacteria. Antimicrob. Agents Chemother. 54, 109–115 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sievert D. M., Ricks P., Edwards J. R., Schneider A., Patel J., Srinivasan A., Kallen A., Limbago B., Fridkin S.; National Healthcare Safety Network (NHSN) Team and Participating NHSN Facilities , Antimicrobial-resistant pathogens associated with healthcare-associated infections: Summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009-2010. Infect. Control Hosp. Epidemiol. 34, 1–14 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Shao H., Chung J., Lee K., Balaj L., Min C., Carter B. S., Hochberg F. H., Breakefield X. O., Lee H., Weissleder R., Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat. Commun. 6, 6999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Longo M. C., Berninger M. S., Hartley J. L., Use of uracil DNA glycosylase to control carry-over contamination in polymerase chain reactions. Gene 93, 125–128 (1990). [DOI] [PubMed] [Google Scholar]
- 12.Borst A., Box A. T. A., Fluit A. C., False-positive results and contamination in nucleic acid amplification assays: Suggestions for a prevent and destroy strategy. Eur. J. Clin. Microbiol. Infect. Dis. 23, 289–299 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Taggart E. W., Carroll K. C., Byington C. L., Crist G. A., Hillyard D. R., Use of heat labile UNG in an RT-PCR assay for enterovirus detection. J. Virol. Methods 105, 57–65 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Hsieh K., Mage P. L., Csordas A. T., Eisenstein M., Soh H. T., Simultaneous elimination of carryover contamination and detection of DNA with uracil-DNA-glycosylase-supplemented loop-mediated isothermal amplification (UDG-LAMP). Chem. Commun. 50, 3747–3749 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Marx V., PCR heads into the field. Nat. Methods 12, 393–397 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Cornejo-Juárez P., Vilar-Compte D., Pérez-Jiménez C., Ñamendys-Silva S. A., Sandoval-Hernández S., Volkow-Fernández P., The impact of hospital-acquired infections with multidrug-resistant bacteria in an oncology intensive care unit. Int. J. Infect. Dis. 31, 31–34 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Pichon B., Hill R., Laurent F., Larsen A. R., Skov R. L., Holmes M., Edwards G. F., Teale C., Kearns A. M., Development of a real-time quadruplex PCR assay for simultaneous detection of nuc, Panton–Valentine leucocidin (PVL), mecA and homologue mecALGA251. J. Antimicrob. Chemother. 67, 2338–2341 (2012). [DOI] [PubMed] [Google Scholar]
- 18.McClure J.-A., Conly J. M., Lau V., Elsayed S., Louie T., Hutchins W., Zhang K., Novel multiplex PCR assay for detection of the staphylococcal virulence marker Panton-Valentine leukocidin genes and simultaneous discrimination of methicillin-susceptible from -resistant Staphylococci. J. Clin. Microbiol. 44, 1141–1144 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang K., McClure J.-A., Elsayed S., Louie T., Conly J. M., Novel multiplex PCR assay for simultaneous identification of community-associated methicillin-resistant Staphylococcus aureus strains USA300 and USA400 and detection of mecA and Panton-Valentine leukocidin genes, with discrimination of Staphylococcus aureus from coagulase-negative Staphylococci. J. Clin. Microbiol. 46, 1118–1122 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thong K. L., Lai M. Y., Teh C. S. J., Chua K. H., Simultaneous detection of methicillin-resistant Staphylococcus aureus, Acinetobacter baumannii, Escherichia coli, Klebsiella pneumoniae and Pseudomonas aeruginosa by multiplex PCR. Trop. Biomed. 28, 21–31 (2011). [PubMed] [Google Scholar]
- 21.Son J. H., Hong S., Haack A. J., Gustafson L., Song M., Hoxha O., Lee L. P., Rapid optical cavity PCR. Adv. Healthcare Mater. 5, 167–174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piepenburg O., Williams C. H., Stemple D. L., Armes N. A., DNA detection using recombination proteins. PLOS Biol. 4, e204 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buch S., Sui Y., Dhillon N., Potula R., Zien C., Pinson D., Li S., Dhillon S., Nicolay B., Sidelnik A., Li C., Villinger T., Bisarriya K., Narayan O., Investigations on four host response factors whose expression is enhanced in X4 SHIV encephalitis. J. Neuroimmunol. 157, 71–80 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Chung H. J., Castro C. M., Im H., Lee H., Weissleder R., A magneto-DNA nanoparticle system for rapid detection and phenotyping of bacteria. Nat. Nanotechnol. 8, 369–375 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park K. S., Charles R. C., Ryan E. T., Weissleder R., Lee H., Fluorescence polarization based nucleic acid testing for rapid and cost-effective diagnosis of infectious disease. Chemistry 21, 16359–16363 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aslanzadeh J., Preventing PCR amplification carryover contamination in a clinical laboratory. Ann. Clin. Lab. Sci. 34, 389–396 (2004). [PubMed] [Google Scholar]
- 27.Dittmer W. U., Reuter A., Simmel F. C., A DNA-based machine that can cyclically bind and release thrombin. Angew. Chem. Int. Ed. 43, 3550–3553 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Jordan J. A., Durso M. B., Real-time polymerase chain reaction for detecting bacterial DNA directly from blood of neonates being evaluated for sepsis. J. Mol. Diagn. 7, 575–581 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olson M. E., Nygaard T. K., Ackermann L., Watkins R. L., Zurek O. W., Pallister K. B., Griffith S., Kiedrowski M. R., Flack C. E., Kavanaugh J. S., Kreiswirth B. N., Horswill A. R., Voyich J. M., Staphylococcus aureus nuclease is an SaeRS-dependent virulence factor. Infect. Immun. 81, 1316–1324 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lina G., Piémont Y., Godail-Gamot F., Bes M., Peter M.-O., Gauduchon V., Vandenesch F., Etienne J., Involvement of Panton-Valentine leukocidin–producing Staphylococcus aureus in primary skin infections and pneumonia. Clin. Infect. Dis. 29, 1128–1132 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/5/e1600300/DC1
table S1. Target sequences recognized by established detection keys.
table S2. Summary of a set of ARV factors targeted in this study.
table S3. DNA sequences used in this study.
fig. S1. Schematic of the plastic cartridge for RNA extraction.
fig. S2. Comparison between the fluidic cartridge and a commercial column.
fig. S3. The optical detection system in the PAD.
fig. S4. Snapshots of a PAD application.
fig. S5. The circuit diagram of the detection system.
fig. S6. The effect of dUTP on the PAD assay.
fig. S7. Portable PAD system.
fig. S8. Lyophilized probes.
fig. S9. Electrophoretic band-shift assay.
fig. S10. Detection of ARV factors.
fig. S11. Overall assay procedure for clinical samples.
fig. S12. Universal and species-specific detection of HAI pathogens in clinical samples by the PAD system.
fig. S13. Detection of ARV factors in clinical samples with PAD (top) and qPCR (bottom).