

Abstract
Tamoxifen-resistant (TAMR) estrogen receptor-positive (ER+) breast cancer is characterized by elevated Erb-B2 receptor tyrosine kinase 2 (ERBB2) expression. However, the underlying mechanisms responsible for the increased ERBB2 expression in the TAMR cells remain poorly understood. Herein, we reported that the ERBB2 expression is regulated at the post-transcriptional level by miR26a/b and the RNA-binding protein human antigen R (HuR), both of which associate with the 3′-UTR of the ERBB2 transcripts. We demonstrated that miR26a/b inhibits the translation of ERBB2 mRNA, whereas HuR enhances the stability of the ERBB2 mRNA. In TAMR ER+ breast cancer cells with elevated ERBB2 expression, we observed a decrease in the level of miR26a/b and an increase in the level of HuR. The forced expression of miR26a/b or the depletion of HuR decreased ERBB2 expression in the TAMR cells, resulting in the reversal of tamoxifen resistance. In contrast, the inactivation of miR26a/b or forced expression of HuR decreased tamoxifen responsiveness of the parental ER+ breast cancer cells. We further showed that the increase in HuR expression in the TAMR ER+ breast cancer cells is attributable to an increase in the HuR mRNA isoform with shortened 3′-UTR, which exhibits increased translational activity. This shortening of the HuR mRNA 3′-UTR via alternative polyadenylation (APA) was observed to be dependent on cleavage stimulation factor subunit 2 (CSTF2/CstF-64), which is up-regulated in the TAMR breast cancer cells. Taken together, we have characterized a model in which the interplay between miR26a/b and HuR post-transcriptionally up-regulates ERBB2 expression in TAMR ER+ breast cancer cells.
Keywords: breast cancer, cancer therapy, ELAV-like protein 1 (HuR (human antigen R)), microRNA (miRNA), tumor cell biology, ERBB2, tamoxifen
Introduction
Approximately 70% of human breast cancer is estrogen receptor (ER)5-α-positive (1, 2). The use of tamoxifen (a selective ER modulator) has been shown to improve overall survival in ER+ early breast cancer and achieve a substantial objective response rate in ER+ metastatic breast cancer (3). However, many patients whose tumors are tamoxifen-responsive eventually develop resistance accompanied by tumor recurrence (4). Therefore, an improved understanding of the molecular basis of the acquisition of tamoxifen resistance and development of new strategies to improve the long-term efficacy of tamoxifen are required (4). The Erb-B2 receptor tyrosine kinase 2 (ERBB2) has been suggested as a critical mediator of resistance to hormonal therapies, including tamoxifen (3–6). ER+ breast cancers that overexpress ERBB2 showed reduced tamoxifen sensitivity (7–9). Furthermore, the acquisition of tamoxifen resistance in ER+ breast cancers is characterized by elevated ERBB2 levels (10, 11). Previous studies have shown that ERBB2 expression is regulated at the transcriptional (12, 13) and post-transcriptional levels (14–17). However, the underlying mechanisms leading to increased expression of ERBB2 in TAMR breast cancer remain poorly understood.
The post-transcriptional regulation of gene expression involves the regulation of mRNA stability and translation by non-coding RNAs, including microRNAs (miRNAs), and RNA-binding proteins (18). Increasing evidence has established the importance of miRNAs in the initiation, promotion, and progression of various human cancers (19, 20). The dysregulation of miRNAs has been demonstrated to contribute to breast cancer development and progression through promoting cell proliferation, survival, and metastasis (5, 6). In particular, several studies have reported the down-regulation of miR-26a and/or miR-26b in breast cancer, thus implicating them as tumor suppressor miRNAs (21–25). Interestingly, an increasing number of studies have also demonstrated that miRNAs are regulated by tamoxifen and are involved in tamoxifen resistance (26, 28, 45). Human antigen R (HuR) is an RNA-binding protein that belongs to the Hu/ELAV family (29). It selectively binds to AU-rich regions in the 3′-UTR of mRNAs and serves to increase the stability of the transcripts (30). Increasing evidence has supported the critical roles of HuR in breast cancer initiation, progression, metastasis, and drug resistance through its modulation of the stability of relevant transcripts (29). Importantly, the elevated expression of HuR has been reported to contribute to tamoxifen resistance (31, 32). In addition, alternative polyadenylation (APA) of the mRNA 3′-UTR also plays an important role in post-transcriptional regulation (33). Approximately 70% of human genes are characterized by multiple poly(A) sites that produce distinct transcript isoforms with variable 3′-UTR (34–37). More recent studies have shed light on the importance of APA in human cancers that favor expression of transcripts with shortened 3′-UTRs, resulting in the activation of several proto-oncogenes (33, 38–40).
Herein, we report that ERBB2 is post-transcriptionally up-regulated in TAMR breast cancer cells, which is attributable to the decreased expression of miR-26a/b and increased expression of HuR. Forced expression of miR-26a/b and/or depletion of HuR, which decreased ERBB2 expression, enhanced the response of the TAMR breast cancer cells to tamoxifen. We further elucidated a novel mechanism leading to up-regulated HuR expression in TAMR breast cancer cells, in which higher levels of CSTF2 mediate increased production of an HuR mRNA isoform with a shorter 3′-UTR through APA, thus resulting in higher HuR protein expression.
Results
ERBB2 is post-transcriptionally up-regulated in tamoxifen-resistant (TAMR) ER+ breast cancer cells
ER+ breast cancer cells (MCF7 and T47D) with acquired tamoxifen resistance were generated by chronic treatment with tamoxifen (Fig. 1A) (41, 42). These TAMR cells exhibited elevated ERBB2 protein (Fig. 1B) and mRNA levels (Fig. 1C), which is consistent with existing literature implicating increased ERBB2 expression and the ERBB2–ERα cross-talk as a key mechanism in driving tamoxifen resistance in ER+ breast cancer (3–5, 12, 41, 43, 44). It has previously been reported that ERBB2 is transcriptionally up-regulated in TAMR breast cancer (41). To determine whether the increased expression of ERBB2 in TAMR breast cancer also occurs in a post-transcriptional manner, we analyzed the ERBB2 3′-UTR–driven luciferase reporter gene activities in the control and TAMR MCF7 and T47D cells. The full-length 3′-UTR of ERBB2 was cloned downstream of the Renilla luciferase gene in the psiCHECK2 vector, and the resulting luciferase reporter constructs were transfected into the cells. The TAMR MCF7 and T47D cells exhibited increased ERBB2 3′-UTR–driven luciferase reporter gene activities as compared with the respective control cells (Fig. 1D). Hence, post-transcriptional regulation at the ERBB2 3′-UTR results in increased ERBB2 expression in TAMR ER+ breast cancer cells.
Figure 1.

The increased ERBB2 expression in TAMR ER+ breast cancer cells is ERBB2 3′-UTR–dependent. A, MCF7/TAMR and T47D/TAMR cells and their respective parental cells were treated with the indicated concentrations of tamoxifen. Cell viability was determined by MTT assay. B, ERBB2 protein levels in the MCF7 and T47D control and TAMR cells were determined by Western blotting, with β-actin as input control. C, ERBB2 mRNA levels in the MCF7 and T47D control and TAMR cells were determined by RT-qPCR, with GAPDH as input control. D, MCF7 and T47D control and TAMR cells were transfected with psi-CHECK2-Vec or psi-CHECK2-ERBB2-3′-UTR plasmids, and the resulting luciferase reporter activities were determined. *, p < 0.05; **, p < 0.01. Error bars, S.E.
ERBB2 expression is decreased post-transcriptionally by miR-26a/b binding to its 3′-UTR
We went on to determine whether ERBB2 is regulated post-transcriptionally by miRNAs targeting its 3′-UTR. We have previously demonstrated that miR-26a and miR-26b are repressed by estrogen, leading to increased estrogen-stimulated cell proliferation in ERα+ breast cancer cells (25). Moreover, low levels of miR-26 have previously been reported to be associated with tamoxifen resistance (45). In our MCF7/TAMR and T47D/TAMR cells, the levels of miR-26a/b were significantly decreased as compared with their respective control cells (Fig. 2A). The forced expression of miR-26a/b decreased ERBB2 protein levels in MCF7/TAMR and T47D/TAMR cells (Fig. 2B). Similarly, the forced expression of miR-26a/b in the TAMR cells decreased the protein levels of enhancer of Zeste homolog 2 (EZH2), which is a validated target of miR-26 (46), and was thus chosen as a positive control (Fig. 2B). Despite the observed miR-26a/b-mediated decrease in ERBB2 protein levels, the forced expression of miR-26a/b did not significantly decrease ERBB2 mRNA levels in MCF7/TAMR and T47D/TAMR cells (supplemental Fig. S1A). This suggests that ERBB2 expression is down-regulated post-transcriptionally by miR-26a/b.
Figure 2.

miR-26a/b targets ERBB2 3′-UTR and represses ERBB2 translation. A, miR-26a and miR-26b levels in MCF7 and T47D control and TAMR cells were measured by TaqMan stem-loop RT-qPCR, with U6 as input control. B, ERBB2 and EZH2 protein levels in MCF7 and T47D TAMR cells transfected with either the scrambled miRNA (NC) or miR26a/b were analyzed by Western blotting, with β-actin as input control. C, putative miR-26-binding sequence in the 3′-UTR of ERBB2 mRNA. Mutations were generated in the region of the ERBB2-3′-UTR complementary to the seed region of miR-26 (underlined). D, luciferase reporter activities were measured in T47D cells co-transfected with either wild type or mutant psi-CHECK2-ERBB2-3′-UTR, and either scrambled miRNA (NC) or miR26a/b, as indicated. E, schematic representation of biotinylated miR-26a, miR-26b, and scrambled oligomer. F, ERBB2, EZH2, ARTN, and TFF1 mRNA levels in the pulldown fractions of biotin–miR-26a, biotin–miR-26b, and biotin-scrambled oligomer. G, levels of total input mRNAs. *, p < 0.05. Error bars, S.E.
To determine whether ERBB2 mRNA is a direct target of miR-26a/b, we first performed bioinformatics analysis. A putative miR-26 binding site in the 3′-UTR of the ERBB2 transcript was predicted by RNAhybrid (47) (Fig. 2C). To further study whether miR-26a/b regulates ERBB2 expression through this predicted miR-26a/b binding site in the ERBB2 3′-UTR, we introduced mutations into this site (Fig. 2C) and compared the luciferase reporter activities controlled by either the wild-type or mutant ERBB2 3′-UTR in the presence of miR-26a/b. The forced expression of miR-26a/b markedly reduced the activity of the luciferase reporter gene fused to wild-type ERBB2 3′-UTR by >50% but did not affect the luciferase reporter activity when the predicted miR-26a/b target site in the ERBB2 3′-UTR was mutated (Fig. 2D). In addition, to verify the direct interaction of miR-26 and ERBB2 mRNA, we performed an RNA pulldown assay using biotin-labeled miR-26a or miR-26b (Fig. 2E). ERBB2 and EZH2 (positive control) mRNAs were both markedly enriched in the pulldown fractions of biotin–miR-26a or biotin–miR-26b, but not in the pulldown fraction of control scrambled miRNA (Fig. 2F). The association of biotinylated miR-26a/b with the ERBB2 and EZH2 mRNAs was specific, because biotin–miR-26 did not result in the pulldown of nonspecific mRNAs encoding ARTEMIN and TFF1 (trefoil factor 1), which are not miR-26a/b targets (Fig. 2F). In addition, the levels of total ERBB2, EZH2, ARTEMIN, and TFF1 mRNAs were not altered with the transfection of biotinylated miR-26a/b or scrambled miRNA (Fig. 2G), suggesting that the enrichment of ERBB2 and EZH2 mRNAs in the biotinylated miR-26 pulldown assay was due to interaction of these mRNAs with miR-26 rather than an increase in the total levels of these mRNAs in the cells.
These data collectively suggest that miR-26a/b acting at the 3′-UTR of ERBB2, reduces ERBB2 expression principally by inhibiting its translation. Interestingly, upon transfection of anti-miR-26a/b (antisense oligonucleotides) into MCF7 cells, the ERBB2 protein level was only slightly increased as compared with a dramatically increased EZH2 protein level (supplemental Fig. S1B). Hence, the reduction in miR-26a/b levels is probably not the only mechanism responsible for the elevated ERBB2 expression in TAMR breast cancer cells.
ERBB2 expression is increased post-transcriptionally by HuR binding to its 3′-UTR
HuR was previously shown to associate with ERBB2 3′-UTR and increase ERBB2 transcript stability (14–16). Furthermore, HuR was reported to be aberrantly expressed in breast cancer (48–50) and involved in tamoxifen resistance (31). In this study, we showed that the levels of HuR protein are increased in MCF7 and T47D TAMR cells as compared with the respective control cells (Fig. 3, A and B). To determine the potential role of HuR in the up-regulation of ERBB2 expression in the TAMR ER+ breast cancer cells, we specifically silenced HuR using specific HuR-targeted siRNAs in these cells. The siRNA-mediated depletion of HuR decreased ERBB2 mRNA levels in MCF7/TAMR cells through an increase in the rate of ERBB2 mRNA decay (i.e. destabilization of ERBB2 mRNA) (Fig. 3, C and D). Furthermore, the depletion of HuR significantly decreased the levels of ERBB2 3′-UTR luciferase reporter activity in MCF7/TAMR cells (Fig. 3E). Consistently, the depletion of HuR decreased ERBB2 protein levels in MCF7/TAMR and T47D/TAMR cells (Fig. 3F). These results suggest that the higher levels of HuR in the TAMR breast cancer cells promote the increased expression of ERBB2 in a post-transcriptional manner through increasing ERBB2 transcript stability.
Figure 3.

HuR depletion decreases ERBB2 mRNA stability and translation. A and B, the levels of HuR protein in MCF7 and T47D control and TAMR cells were determined by Western blotting, with β-actin as input control. C, MCF7/TAMR cells transfected with either HuR siRNA or control siRNA were treated with actinomycin D (10 μg/ml) 48 h after transfection and harvested at 0, 2, 4, 6, and 8 h for RNA extraction and reverse transcription. ERBB2 mRNA levels at the different time points were measured by qPCR, using GAPDH as input control. D, MCF7/TAMR cells transfected with either HuR siRNA or control siRNA were harvested after 48 h, and ERBB2 mRNA levels were measured with RT-qPCR, using GAPDH as input control. E, luciferase reporter activities were determined in MCF7/TAMR cells co-transfected with either control siRNA or HuR siRNA and either psi-CHECK2-Vec or psi-CHECK2-ERBB2-3′-UTR, as indicated. F, ERBB2 and HuR protein levels in MCF7/TAMR and T47D/TAMR cells transfected with either the scrambled siRNA (siNC) or HuR siRNA were analyzed by Western blotting, with β-actin as input control. *, p < 0.05. Error bars, S.E.
To further verify the direct association between HuR and ERBB2 mRNA in the MCF7/TAMR cells, we performed ribonucleoprotein (RNP) immunoprecipitation (IP) assays using anti-HuR antibody (16, 51, 52). The amount of ERBB2 mRNA was markedly enriched by >10-fold in the HuR antibody-precipitated samples as compared with the control IgG-precipitated samples (Fig. 4A). In contrast, HuR was shown not to interact with ARTN and TFF1 mRNAs, which are negative controls (Fig. 4A). The increase in ERBB2 mRNA levels in the HuR immunoprecipitate was solely due to the association of HuR with ERBB2 mRNA because the total levels of ERBB2 mRNA in both the anti-HuR– and the control IgG–treated samples were not significantly different (Fig. 4B). Finally, to determine the region of ERBB2 mRNA that HuR specifically binds, we employed biotinylated ERBB2 mRNAs spanning different regions, namely 5′-UTR, coding region (CR), 3′-UTR-a, and 3′-UTR-b, in the biotin pulldown assay (Fig. 4C). HuR was observed to specifically bind to the 3′-UTR-b of the ERBB2 transcript. In contrast, we did not detect any association of HuR with biotinylated RNAs containing the ERBB2 5′-UTR, CR, 3′-UTR-a, or 3′-UTR-mutant (Fig. 4C).
Figure 4.
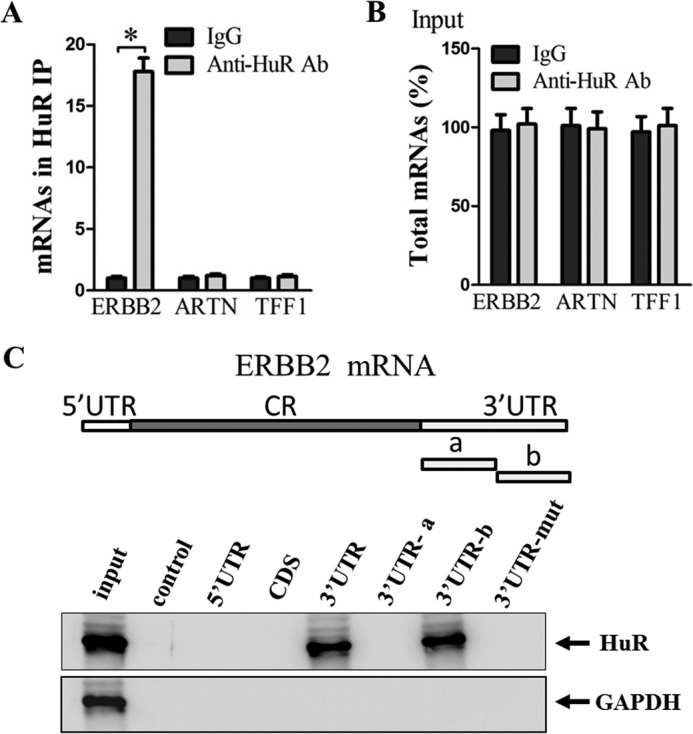
HuR binds ERBB2 3′-UTR. A, MCF7/TAMR cell lysates were subjected to RNP IP using either control IgG or anti-HuR antibody. The ERBB2, ARTN, and TFF1 mRNA levels in the IgG and HuR IPs were determined by RT-qPCR, with GAPDH as input control. B, levels of total input mRNAs. C (top), schematic depiction of the ERBB2 5′-UTR, CR, and 3′-UTR, as well as the biotinylated RNAs synthesized for use in biotin pulldown analysis. (Bottom), the biotinylated RNAs corresponding to different regions of the ERBB2 transcript were incubated with MCF7/TAMR cell lysates. The interaction of HuR with each biotinylated RNA segment was analyzed by Western blotting, with GAPDH as input control. *, p < 0.05. Error bars, S.E.
HuR and miR-26 regulate ERBB2 expression in a competitive manner
Since the ERBB2 3′-UTR sequence contains the HuR binding site in close proximity to the miR-26a/b seed sequence (Fig. 5A), we hypothesized that miR-26a/b and HuR, which act antagonistically in the post-transcriptional regulation of ERBB2, might compete for association with ERBB2 mRNA. To test this hypothesis, we studied the effect of co-transfection of miR26a/b mimics and HuR expression plasmids in T47D cells. The forced expression of miR-26a/b decreased the ERBB2 protein level (Fig. 5B) and ERBB2 3′-UTR–regulated luciferase reporter activity (Fig. 5C). Although the forced expression of HuR did not significantly increase the ERBB2 protein level and 3′-UTR luciferase reporter activity, it attenuated the miR-26a/b–mediated suppression of ERBB2 expression (Fig. 5, B and C). In addition, the forced expression of miR-26a/b decreased the association of HuR with ERBB2 mRNA in a graded manner (Fig. 5D). Similarly, the forced expression of HuR decreased the association of miR-26a/b with ERBB2 mRNA, whereas the depletion of HuR increased the levels of miR-26a/b–ERBB2 mRNA complexes (Fig. 5E). These data suggest that ERBB2 expression is subject to negative regulation by miR-26a/b and positive regulation by HuR, both acting on ERBB2 3′-UTR in a competitive manner.
Figure 5.

miR-26a/b and HuR regulate ERBB2 expression in a competitive and antagonistic manner. A, sketch of the miR-26 seed region and the HuR binding site in the 3′-UTR of ERBB2 transcript. The ERBB2 protein levels (B) and ERBB2 3′-UTR–driven luciferase reporter activities (C) were measured in cells co-transfected with either scrambled miRNA (NC) or miR26a/b and either pIRESneo3-VEC or pIRESneo3-HuR, as indicated. β-Actin was used as input control in the Western blot analysis. D, the lysates of scrambled miRNA (NC) or miR26a/b-transfected cells were subjected to RNP IP using either control IgG or anti-HuR antibody. The ERBB2 mRNA levels in the IgG and HuR IPs were determined by RT-qPCR, with GAPDH as input control. E, ERBB2 mRNA levels in the pulldown fractions of biotin–miR-26a, biotin–miR-26b, and biotin-scrambled oligomer, from cells transfected with either pIRESneo3-HuR or siHuR, were measured by RT-qPCR analysis with GAPDH as input control. *, p < 0.05; **, p < 0.01. Error bars, S.E.
Decreased expression of miR-26 or increased expression of HuR mediates acquired tamoxifen resistance
As we observed that miR-26a/b expression was reduced, whereas HuR expression was increased in TAMR cells, we went on to investigate whether this dysregulated expression of miR26a/b and HuR is responsible for acquired tamoxifen resistance in ER+ breast cancer cells. We examined the effect of forced expression of miR26a/b or depletion of HuR on acquired tamoxifen resistance in MCF7/TAMR cells. The effect of miR26a/b depletion or forced expression of HuR on the tamoxifen responsiveness of MCF7 cells was also studied. The forced expression of miR26a/b decreased the basal total cell number and cell viability of MCF7/TAMR cells (Fig. 6, A and B). Whereas tamoxifen treatment did not significantly decrease the total cell number and cell viability of MCF7/TAMR cells, the forced expression of miR-26a/b significantly decreased the total cell number and cell viability of tamoxifen-treated MCF7/TAMR cells to the levels of the control cells transfected with scrambled oligomer (Fig. 6, A and B), suggestive of re-sensitization of the MCF7/TAMR cells to tamoxifen. In contrast, the inactivation of miR26a/b by antisense oligonucleotides resulted in significantly increased total cell number and cell viability of MCF7 cells in the presence of tamoxifen, to the levels of the untreated control cells (Fig. 6, C and D), suggestive of decreased sensitivity of MCF7 cells to tamoxifen. Furthermore, although tamoxifen treatment did not significantly decrease the total cell number and cell viability of MCF7/TAMR cells, the depletion of HuR significantly decreased the total cell number and cell viability of tamoxifen-treated MCF7/TAMR cells to the levels of the control cells transfected with scrambled siRNA (Fig. 6, E and F), suggestive of re-sensitization of the MCF7/TAMR cells to tamoxifen. In contrast, the forced expression of HuR resulted in significantly increased total cell number and cell viability of MCF7 cells in the presence of tamoxifen, to the levels of the untreated control cells (Fig. 6, G and H), suggestive of decreased sensitivity of MCF7 cells to tamoxifen. Furthermore, the concomitant forced expression of miR-26a/b and depletion of HuR resulted in a greater decrease in the total cell number and cell viability of MC7/TAMR cells as compared with either the forced expression of miR-26a/b or depletion of HuR alone (Fig. 6, I and J).
Figure 6.

Forced expression of miR-26a/b and/or depletion of HuR re-sensitizes TAMR breast cancer cells to tamoxifen. MCF7/TAMR cells were transfected with either scrambled oligomer (NC) or miR-26a/b mimics (A and B), either scrambled oligomer or miR-26a/b antisense oligonucleotides (C and D), either control siRNA or HuR siRNA (E and F), either empty vector or HuR expression plasmid (G and H), and miR-26a/b mimics and HuR siRNA in combination (I and J). 24 h after transfection, the cells were seeded in 6- or 96-well plates and treated with either the ethanol vehicle or 1 mm 4OH-tamoxifen. A, C, E, G, and I, at the indicated time points after plating, cells were trypsinized, and the total number of cells was counted. B, D, F, H, and J, at 96 h after plating, an MTT assay was performed to determine the viability of the MCF7/TAMR cells. **, p < 0.01. Error bars, S.E.
Elevated levels of HuR mRNA with shortened 3′-UTR are associated with increased HuR expression in TAMR breast cancer cells
Recent studies have revealed widespread mRNA APA in cancer cells (33, 40). The production of mRNA isoforms with shorter 3′-UTR via APA results in increased mRNA stability and increased protein production and thus has been implicated in oncogene activation (33). We observed that the 3′-UTR of the HuR gene contains several APA signals (PASs) (Fig. 7A). To determine the relative abundance of HuR mRNA isoforms terminated at the different PASs in the TAMR cells, we performed RT-qPCR using primers flanking different regions of the HuR 3′-UTR (Fig. 7A). The levels of HuR mRNA isoform terminated at PAS2 were observed to be highly enriched in MCF7/TAMR cells as compared with the MCF7 cells, whereas the levels of the HuR mRNA isoform terminated at the later PASs were observed to be not significantly different between the MCF7 TAMR and control cells (Fig. 7B and supplemental Fig. S2). Additionally, the 3′-rapid amplification of cDNA ends (RACE) assay was carried out to verify the relative abundance of the different HuR mRNA isoforms. The HuR mRNA isoforms terminated at PAS2, PAS3, and PAS8 were designated 3′-UTR-short (3′-UTR-S), 3′-UTR-medium (3′-UTR-M), and 3′-UTR-long (3′-UTR-L), respectively (Fig. 7C). Similarly, in the 3′ RACE assay, the levels of HuR mRNA isoform terminated at PAS2 (3′-UTR-S) were significantly higher in the TAMR cells as compared with the respective control cells, whereas the levels of 3′-UTR-M and 3′-UTR-L HuR mRNA isoforms were not significantly different between the TAMR and control cells (Fig. 7D). In particular, the ratio of HuR mRNA isoforms with short 3′-UTR to HuR mRNA isoforms with long and medium 3′-UTR (3US/3UM + 3UL) in MCF7/TAMR cells and T47D/TAMR cells is 1.85- and 3.26-fold higher as compared with their respective parental cells (Fig. 7D). As we observed that the HuR mRNA isoform with shortened 3′-UTR was increased in TAMR cells, we went on to determine whether this isoform is responsible for the elevated expression of HuR protein. We cloned each of the three 3′-UTR isoforms of HuR downstream of a luciferase reporter gene in the psiCHECK2 vector. To ensure that the reporter mRNA isoforms with long and medium 3′-UTR were expressed, any functional upstream PASs were mutated (ATTAAA or AATAAA was mutated to ACACAC). The luciferase reporter gene with downstream HuR 3′-UTR-S exhibited a higher luciferase reporter activity as compared with the luciferase reporter genes with downstream HuR 3′-UTR-M and 3′-UTR-L (Fig. 7E). Moreover, we determined the levels of HuR protein expression derived from the different HuR mRNA isoforms. We transduced cells with adenovirus vectors expressing the different HuR mRNA isoforms, namely CR, CR plus short 3′-UTR (CR + 3US), CR plus medium 3′-UTR (CR + 3UM), and CR plus long 3′-UTR (CR + 3UL). The HuR protein expression derived from the short 3′-UTR–containing mRNA isoform was higher than that derived from the long or medium 3′-UTR–containing mRNA isoforms (Fig. 7F). These results suggest that the HuR 3′-UTR is shortened in TAMR ER+ breast cancer cells through APA, resulting in increased HuR expression.
Figure 7.

The 3′-UTRs of HuR transcripts were shortened via APA in TAMR breast cancer cells. A, schematic illustration of HuR mRNA with several PASs in the HuR 3′-UTR. Positions of the primer pairs used in mRNA expression analyses are marked by black dashed lines. Positions of some miRNAs targets are marked by black arrows. B, the levels of HuR mRNA isoforms terminated at different PASs (amplified using the indicated primer pairs) in MCF7 control and TAMR cells were assessed by RT-qPCR analysis. C, schematic illustration of different HuR mRNA isoforms mediated through APA. D, cDNA was synthesized from total RNA isolated from MCF7, MCF7/TAMR, T47D, or T47D/TAMR using anchor oligo(dT) from the 3′-RACE kit (Roche Applied Science). The resulting amplification products were analyzed using agarose gel electrophoresis. E, HuR short, medium, and long 3′-UTRs were cloned downstream of the Renilla luciferase gene in psiCHECK2, and the resulting vectors were transfected into MCF7 and T47D cells. Luciferase reporter activities regulated by the different HuR 3′-UTR isoforms were determined using the Dual-Luciferase assay. F, the HuR coding sequences (CDS) with short, medium, or long 3′-UTRs were cloned into pIRESneo3. The plasmids carrying the corresponding sequences were transfected into MCF7 cells, and HuR protein levels were determined using Western blotting with β-actin as an input control. *, p < 0.05. Error bars, S.E.
CSTF2 increases HuR expression in TAMR breast cancer cells
CSTF2 was previously reported to regulate the APA of pre-mRNA 3′-UTR and is involved in cancer progression (34, 35, 40). We observed that the expression of CSTF2 protein was increased in MCF7/TAMR cells as compared with control MCF7 cells (Fig. 8A). To explore whether the increased expression of CSTF2 regulates the production of HuR mRNA with shortened 3′-UTR, which in turn results in increased HuR protein expression, we depleted CSTF2 in MCF7/TAMR cells using siRNA. The depletion of CSTF2 resulted in a decrease in the proportion of HuR mRNA isoforms with short 3′-UTR and an increase in the proportion of HuR mRNA isoforms with long or medium 3′-UTR (Fig. 8, B and C). Consequently, the depletion of CSTF2 decreased the level of HuR protein (Fig. 8D). The depletion of CSTF2 decreased the basal total cell number and cell viability of MCF7/TAMR cells (Fig. 8, E and F). Whereas tamoxifen treatment did not significantly decrease the total cell number and cell viability of MCF7/TAMR cells, the depletion of CSTF2 significantly decreased the total cell number and cell viability of tamoxifen-treated MCF7/TAMR cells to the levels of the control cells transfected with scrambled siRNA (Fig. 8, E and F), suggestive of re-sensitization of the MCF7/TAMR cells to tamoxifen. Hence, the increased expression of CSTF2, which in turn results in increased HuR expression, contributes to tamoxifen resistance in MCF7/TAMR cells.
Figure 8.

CSTF2 overexpression promotes the 3′-UTR shortening of the HuR transcript in TAMR breast cancer cells. A, CSTF2 protein levels in MCF7 control and TAMR cells were determined using Western blotting, with β-actin as an input control. B, the levels of HuR mRNA isoforms terminated at different PASs in MCF7/TAMR transfected with either scrambled siRNA (NC) or siCSTF2 were assessed by RT-qPCR analysis. C, cDNA was synthesized from total RNA isolated from MCF7/TAMR/siNC or MCF7/TAMR/siCSTF2 cells using anchor oligo(dT) from the 3′-RACE kit (Roche Applied Science). The resulting amplification products were analyzed using agarose gel electrophoresis. D, the CSTF2 and HuR protein levels in MCF7 TAMR cells transfected with either siNC or siCSTF2 were determined using Western blotting with β-actin as an input control. E and F, MCF7/TAMR cells transfected with either siNC or siCSTF2 were seeded in 6- or 96-well plates and treated with 1 mm 4OH-tamoxifen or ethanol vehicle. E, at the indicated time points, cells were trypsinized, and total cells were counted. F, at 96 h after plating, an MTT assay was performed to determine the viabilities of the MCF7/TAMR/siNC and MCF7/TAMR/siCSTF2 cells. *, p < 0.05. Error bars, S.E.
Discussion
Although many early and metastatic ER+ breast cancer patients benefit from tamoxifen, a large proportion of patients eventually relapse from the disease, making tamoxifen resistance an important clinical problem (4). However, the underlying mechanisms contributing to clinical endocrine resistance have not been fully elucidated (4). This reinforces the need to identify potential biomarkers to predict tamoxifen response and therapeutic targets to overcome tamoxifen resistance in ER+ breast cancer (4). Elevated expression of ERBB2 has been widely demonstrated to be a critical mediator of tamoxifen resistance (3, 5, 6, 53), whereas the inhibition of ERBB2 has been shown to reverse tamoxifen resistance (54, 55). To date, the increased expression of ERBB2 in TAMR breast cancer cells is generally attributed to either the amplification of the ERBB2 locus (53, 56) or the transcriptional activation of the ERBB2 gene (12). Nevertheless, several mechanisms of ERBB2 post-transcriptional regulation have previously been reported (14–16). Herein, we present the mechanisms of post-transcriptional up-regulation of ERBB2 that contribute to tamoxifen resistance in ER+ breast cancer cells.
In this study, we provided evidence that the reduced levels of miR26a/b are responsible for the post-transcriptional up-regulation of ERBB2 in TAMR ER+ breast cancer cells. Consistently, increased levels of miR-26a have previously been reported to be associated with both higher clinical benefit and prolonged time to progression in metastatic breast cancer patients receiving first-line tamoxifen monotherapy (28, 45). The same study also demonstrated an association between reduced levels of the miR-26a target, EZH2, and a favorable clinical outcome (28). Furthermore, the expression of the cell cycle–regulatory proteins, namely cyclin-dependent kinase 1 (CDK1 or CDC2) and cyclin E1 (CCNE1), which was observed to be negatively correlated with that of miR-26a and positively correlated with that of EZH2, was also shown to be useful in predicting the outcome of tamoxifen therapy (28). Although several mechanisms of ERBB2 post-transcriptional regulation have previously been reported, we have observed for the first time that miR26-a/b binds to ERBB2 3′-UTR and decreases ERBB2 translation. Our observation of miR-26a/b–mediated repression of ERBB2 expression provides a mechanistic link to the association of reduced miR-26a/b levels with tamoxifen resistance in ER+ breast cancer cells.
In addition, we have found that in this study, HuR expression is increased in TAMR ER+ breast cancer cells, contributing to the post-transcriptional up-regulation of ERBB2. This is consistent with previous reports implicating HuR in the regulation of ER and ERBB2 transcript stability, and tamoxifen resistance (29, 57, 58). It was demonstrated that decreasing cytoplasmic HuR levels increases tamoxifen sensitivity of ER+ breast cancer cells, whereas increased HuR expression confers tamoxifen resistance on MCF7 cells (31). Acute tamoxifen treatment has been shown to activate JNKs that can, in turn, increase cytoplasmic accumulation of HuR, potentially resulting in increased stability of mRNA transcripts coding for genes involved in tamoxifen resistance (31). In contrast, treatment with the DNA methyltransferase inhibitor (5-aza-2′-deoxycytidine (AZA)) and histone deacetylase inhibitor (trichostatin A (TSA)) was observed to decrease JNK activation, leading to reduction in the availability of cytoplasmic HuR levels to bind and stabilize the relevant mRNA transcripts (31, 59). A follow-up study by the same group provided further evidence for the critical involvement of HuR in tamoxifen resistance, being attributed to the role of HuR in increasing ER transcript stability (32). It was demonstrated that although the epigenetic inhibitors (AZA and TSA) restore ER expression in ER-negative breast cancer cells and reverse their resistance to tamoxifen, AZA and TSA treatment simultaneously result in reduced cytoplasmic HuR and a corresponding decrease in ER mRNA stability (32). This accounted for the absence of a robust increase in tamoxifen sensitivity in the ER-negative breast cancer cells upon AZA/TSA treatment and necessitates the specific timing of drug administration (i.e. prior administration of AZA to increase ER expression, followed by the administration of TSA with tamoxifen to increase cytoplasmic HuR and enhance ER transcript stability) (32). Besides regulating the stability of ER transcript, HuR has also been reported to be involved in the regulation of ERBB2 transcript stability. HuR has been shown to bind the 3′-UTR of the ERBB2 transcript and de-repress the translational inhibition mediated by the 5′ upstream open reading frames (uORFs) of the ERBB2 transcripts (14). In ERBB2-overexpressing breast cancer cells, HuR was observed to associate with histone deacetylase 6 (HDAC6) to maintain ERBB2 transcript integrity, suggestive of the potential of HuR or HDAC6 inhibition in destabilizing ERBB2 transcripts and thereby promoting the growth arrest of the ERBB2-positive breast cancer cells (15). Similarly, HuR has been shown to increase ERBB2 expression in prostate cancer cells through binding to a U-rich element in the ERBB2 3′-UTR (16). Furthermore, because the HuR binding site is in close proximity to the miR-331-3p target site in the ERBB2 3′-UTR, HuR was demonstrated to antagonize the miR-331-3p–mediated repression of ERBB2 expression in the prostate cancer cells (16). Likewise, our current study has reported an interplay between miR-26a/b and HuR in the post-transcriptional regulation of ERBB2 in ER+ breast cancer cells. We have shown that decreased miR-26a/b and increased HuR levels act synergistically to post-transcriptionally up-regulate ERBB2 expression in tamoxifen-resistant ER+ breast cancer cells.
APA is a pervasive mechanism in the regulation of most human genes, and its implication in diseases, including cancer, is only beginning to be appreciated (33, 60). Most genes processed by APA in cancer possess shorter 3′-UTRs that can avoid microRNA-mediated repression (33). Previous studies have shown that HuR, as a well-known RNA-binding protein, has several alternative polyadenylated isoforms (61, 62). In this study, we observed that the shorter isoform of the HuR gene, which lacks many miRNA binding sites, is predominantly expressed in tamoxifen resistant breast cancer cells. We further demonstrated that the shortening of the HuR 3′-UTR via APA results in increased HuR translation in tamoxifen-resistant breast cancer cells. This is probably due to the absence of repression of the short 3′-UTR-possessing HuR transcript isoform by HuR-targeting miRNAs, including miR-29 (63) and miR-125 (64). Although the mechanism regulating the selection of PAS in the HuR transcript is unclear, one study has reported that CSTF2 is a regulator of the mouse HuR gene APA (65). Consistently, our results suggest that CSTF2, an essential polyadenylation factor, is a master regulator of the 3′-UTR shortening of the HuR transcript. We correspondingly observed that the depletion of CSTF2 resulted in reversal of tamoxifen resistance, similar to the phenotypes induced by HuR depletion. Therefore, the current study carries implications for the development of novel predictive biomarkers based on the detection of 3′-UTR shortening of HuR transcript to identify patients who might show poor response to tamoxifen therapy.
Taken together, our study has identified a novel post-transcriptional mechanism, involving miR-26a/b and HuR, that underlies the elevated expression of ERBB2 in acquired tamoxifen resistance (Fig. 9). The levels of miR-26a/b and/or cytoplasmic HuR levels can potentially serve as biomarkers for determining tamoxifen response or predicting the development of tamoxifen resistance (31). Additionally, therapeutic strategies to restore miR-26a/b expression (66), inhibit HuR, or reduce the cytoplasmic accumulation of HuR, including the use of HuR or JNK inhibitors (31), can potentially overcome tamoxifen resistance in ER+ breast cancer.
Figure 9.

Schematic model of the interaction between CSTF2, HuR, miR-26a/b, and the ERBB2 mRNA 3′-UTR.
Experimental procedures
Cell culture
MCF7 and T47D cells were purchased from American Type Culture Collection and cultured in conditions as recommended. All cells were maintained in a humidified incubator at 37 °C and 5% CO2. MCF7/TAMR and T47D/TAMR cells were derived by long-term exposure to tamoxifen (54) and grown in parallel with the wild-type MCF7 cells.
RNA oligonucleotides and transfection
miRNAs and siRNAs were synthesized by GenePharma (Shanghai, China). miRNA mimics are synthetic duplexes representing mature miRNAs. siRNA and miRNA transfection was performed using Lipofectamine3000 (Qiagen). 20 nmol/liter of siRNA or miRNA was used for transfection. Total RNA and protein were prepared 48–72 h after transfection and were further used in qPCR or Western blot analysis: siRNA-HuR#1, CCAGUUUCAAUGGUCAUAATT; siRNA-HuR#2, CACGCUGAACGGCUUGAGGTT.
Quantitative analysis of miRNAs and mRNAs
Total RNA and miRNA were extracted from the breast cancer cells using the miRVana miRNA isolation kit (Ambion) according to the manufacturer's protocol. The miRNA expression was assessed using the TaqMan stem-loop RT-PCR approach (Applied Biosystems). For quantitative analysis of mRNA expression, 100–200 ng of total RNA was used for synthesis of random-primed single-stranded cDNA using Primescript RT reagent kit (TaKaRa), and cDNA was subjected to qPCR using SYBR Green master mix (Applied Biosystems). The relative amount of gene transcripts was normalized to GAPDH transcript level. Three independent experiments were each performed in triplicate.
Protein extraction and Western blotting
Cells were lysed using cell lysis buffer (Cell Signaling) with protein concentration determined with the BCA protein assay kit (Pierce). Equal amounts of total proteins were resolved in 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Bio-Rad). Membranes were blocked for 1 h with 1% BSA in Tris-buffered saline containing 0.05% Tween 20, incubated overnight with primary antibody, washed and incubated with secondary antibody, and finally visualized by chemiluminescence. The antibodies used were as follows: ERBB2 (Proteintech), EZH2 (Proteintech), HuR (Santa Cruz Biotechnology, Inc.), CSTF2 (Santa Cruz Biotechnology), and β-actin (Santa Cruz Biotechnology).
Luciferase reporter assay
The full-length 3′-UTR of ERBB2 was amplified and cloned downstream of Renilla luciferase in a psiCHECK2 vector (Promega). Cells plated on 24-well plates were transfected with 100 ng of psiCHECK2 plasmids and 200 nmol/liter miR-26a/b mimics, pIRESneo3-HuR, siHuR, or their respective controls. 48 h after transfection, cells were lysed, and the luciferase reporter activities were analyzed with a Dual-Luciferase assay kit (Promega) according to the manufacturer's instructions. The HuR short, medium, and long 3′-UTRs were amplified and cloned downstream of Renilla luciferase in a psiCHECK2 vector (Promega). Cells plated on 24-well plates were transfected with 100 ng of plasmid or empty plasmid. 48 h after transfection, cells were lysed and analyzed with the Dual-Luciferase assay kit (Promega). Renilla/firefly luciferase read-outs from the constructs were normalized to that of empty psiCHECK2, which was set to 1. Three independent experiments were performed in triplicate.
Biotin-labeled miR-26 pulldown assays
Cells were transfected with biotin-labeled miR-26a or miR-26b. 48 h after transfection, whole-cell lysates were collected (51, 67, 68), mixed with Streptavidin-Dynal beads (Invitrogen), and incubated at 4 °C with rotation overnight. After the beads were washed thoroughly, the bead-bound RNA was isolated and subjected to RT-qPCR analysis. Input RNA was extracted and served as control.
RNP IP RT-PCR
RNP IP RT-PCR assays were performed as described previously (16, 51, 52) using extracts prepared from MCF7/TAMR cells. The anti-HuR antibody (sc-5261) used was obtained from Santa Cruz Biotechnology. PCR primers for GAPDH, ERBB2, EZH2, ARTEMIN, and TFF1 are listed below. The forward and reverse primers used were TGCACCACCAACTGCTTAGC and GGCATGGACTGTGGTCATGAG for GAPDH, GGCTCTCACACTGATAGACACC and TCCCCAGCAGCGGGAGCCCTTAC for ERBB2, GATGATGATGGAGACGATCCTG and CTTCTGCTGTGCCCTTATCTG for EZH2, CTGAGCAGCGTCGCAGAG and GCTCTTCCACTGCACCAGC for ARTEMIN, and GGTCCTGGTGTCCATGCTG and GAACGGTGTCGTCGAAACAG for TFF1.
Biotin pulldown analysis
After purification of the template PCR products, biotinylated transcripts were synthesized using the MAXIScript T7 kit (Invitrogen), and whole-cell lysates (200 g/sample) were incubated with 3 g of purified biotinylated transcripts for 30 min at room temperature. The resulting complexes were isolated with streptavidin-coupled Dynabeads (Invitrogen). Proteins present in the pulldown fractions were studied by Western blot analysis.
MTT assay
Cells (103/well) were plated in 96-well plates in a final volume of 100 μl. 24 h after plating, 10 pmol of miRNA mimics, siRNAs, or negative control oligonucleotides were transfected into the cells using Lipofectamine3000 (Qiagen). The MTT assay was performed at 24, 48, 72, and 96 h as described previously (27).
Statistics
All experiments were performed at least three times. All numerical data are expressed as mean ± S.E. from a representative experiment performed in triplicate. Data were analyzed using an unpaired two-tailed t test or analysis of variance by GraphPad Prism version 5. p values < 0.05 were considered as statistically significant.
Author contributions
S. T. and K. D. maintained all of the cell cultures, designed and performed the siRNA experiment, and ran qRT-PCR and Western blots. Q.-Y. C., J. Z., and Y. L. helped S. T. to perform the RNP-IPRT-PCR. Y. S., Y. Z., and Q. Y. helped K. D. to perform the luciferase assays. Z. X., W. Z., and M. Z. helped with data collection and drafted statistical methods. G. L., X. L., X. K., A. A., Z. W., Q. W., and X. Z. helped with data analysis and provided critical review of the manuscript. S. T., P. E. L., and T. Z. conceived of the ideas of the manuscript. S. T., Q.-Y. C., P. E. L., and T. Z. wrote the manuscript. T. Z., P. E. L., and X. Z. provided funding for the experiments performed in the paper. All authors read and approved the final version of the manuscript.
Supplementary Material
This work was supported by the National Key R&D Program of China (Grant 2016 YFC1302305); the National Natural Science Foundation of China Grants (81672609, 81472494, and 81672615); the Specialized Research Fund for the Doctoral Program of Higher Education (20133402120032 and 20133420120006); the CAS Grant XDA01040410, the Cancer Science Institute of Singapore through grants from the National Research Foundation and Ministry of Education of Singapore and grants from the National Medical Research Council of Singapore (R-713-000-163-511 and R-713-000-206-511). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1 and S2.
- ER
- estrogen receptor
- miRNA
- microRNA
- APA
- alternative polyadenylation
- AZA
- 5-aza-2′-deoxycytidine
- TSA
- trichostatin A
- HuR
- human antigen R
- RNP
- ribonucleoprotein
- IP
- immunoprecipitation
- CR
- coding region
- PAS
- polyadenylation signal
- qPCR
- quantitative PCR
- RACE
- rapid amplification of cDNA ends
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
References
- 1. Tyson J. J., Baumann W. T., Chen C., Verdugo A., Tavassoly I., Wang Y., Weiner L. M., and Clarke R. (2011) Dynamic modelling of oestrogen signalling and cell fate in breast cancer cells. Nat. Rev. Cancer 11, 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomas C., and Gustafsson J. Å. (2011) The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 11, 597–608 [DOI] [PubMed] [Google Scholar]
- 3. Ring A., and Dowsett M. (2004) Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 11, 643–658 [DOI] [PubMed] [Google Scholar]
- 4. Musgrove E. A., and Sutherland R. L. (2009) Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 9, 631–643 [DOI] [PubMed] [Google Scholar]
- 5. Osborne C. K., and Schiff R. (2011) Mechanisms of endocrine resistance in breast cancer. Ann. Rev. Med. 62, 233–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shou J., Massarweh S., Osborne C. K., Wakeling A. E., Ali S., Weiss H., and Schiff R. (2004) Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 96, 926–935 [DOI] [PubMed] [Google Scholar]
- 7. Dowsett M. (2001) Overexpression of HER-2 as a resistance mechanism to hormonal therapy for breast cancer. Endocr. Relat. Cancer 8, 191–195 [DOI] [PubMed] [Google Scholar]
- 8. Osborne C. K., Bardou V., Hopp T. A., Chamness G. C., Hilsenbeck S. G., Fuqua S. A., Wong J., Allred D. C., Clark G. M., and Schiff R. (2003) Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 95, 353–361 [DOI] [PubMed] [Google Scholar]
- 9. Benz C. C., Scott G. K., Sarup J. C., Johnson R. M., Tripathy D., Coronado E., Shepard H. M., and Osborne C. K. (1992) Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res. Treat. 24, 85–95 [DOI] [PubMed] [Google Scholar]
- 10. Gutierrez M. C., Detre S., Johnston S., Mohsin S. K., Shou J., Allred D. C., Schiff R., Osborne C. K., and Dowsett M. (2005) Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J. Clin. Oncol. 23, 2469–2476 [DOI] [PubMed] [Google Scholar]
- 11. Lipton A., Leitzel K., Ali S. M., Demers L., Harvey H. A., Chaudri-Ross H. A., Evans D., Lang R., Hackl W., Hamer P., and Carney W. (2005) Serum HER-2/neu conversion to positive at the time of disease progression in patients with breast carcinoma on hormone therapy. Cancer 104, 257–263 [DOI] [PubMed] [Google Scholar]
- 12. Hurtado A., Holmes K. A., Geistlinger T. R., Hutcheson I. R., Nicholson R. I., Brown M., Jiang J., Howat W. J., Ali S., and Carroll J. S. (2008) Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature 456, 663–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zuo T., Wang L., Morrison C., Chang X., Zhang H., Li W., Liu Y., Wang Y., Liu X., Chan M. W., Liu J. Q., Love R., Liu C. G., Godfrey V., Shen R., et al. (2007) FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell 129, 1275–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mehta A., Trotta C. R., and Peltz S. W. (2006) Derepression of the Her-2 uORF is mediated by a novel post-transcriptional control mechanism in cancer cells. Genes Dev. 20, 939–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scott G. K., Marx C., Berger C. E., Saunders L. R., Verdin E., Schäfer S., Jung M., and Benz C. C. (2008) Destabilization of ERBB2 transcripts by targeting 3′ untranslated region messenger RNA associated HuR and histone deacetylase-6. Mol. Cancer Res. 6, 1250–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Epis M. R., Barker A., Giles K. M., Beveridge D. J., and Leedman P. J. (2011) The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331–3p in prostate cancer cells. J. Biol. Chem. 286, 41442–41454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scott G. K., Goga A., Bhaumik D., Berger C. E., Sullivan C. S., and Benz C. C. (2007) Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J. Biol. Chem. 282, 1479–1486 [DOI] [PubMed] [Google Scholar]
- 18. Filipowicz W., Bhattacharyya S. N., and Sonenberg N. (2008) Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 9, 102–114 [DOI] [PubMed] [Google Scholar]
- 19. Esquela-Kerscher A., and Slack F. J. (2006) Oncomirs: microRNAs with a role in cancer. Nat. Rev. Cancer 6, 259–269 [DOI] [PubMed] [Google Scholar]
- 20. Kasinski A. L., and Slack F. J. (2011) Epigenetics and genetics: microRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 11, 849–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu X. X., Li X. J., Zhang B., Liang Y. J., Zhou C. X., Cao D. X., He M., Chen G. Q., He J. R., and Zhao Q. (2011) MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett. 585, 1363–1367 [DOI] [PubMed] [Google Scholar]
- 22. Maillot G., Lacroix-Triki M., Pierredon S., Gratadou L., Schmidt S., Benes V., Roche H., Dalenc F., Auboeuf D., Millevoi S., Bénès V., Roché H., Dalenc F., Auboeuf D., Millevoi S., and Vagner S. (2009) Widespread estrogen-dependent repression of micrornas involved in breast tumor cell growth. Cancer Res. 69, 8332–8340 [DOI] [PubMed] [Google Scholar]
- 23. Ebert M. S., Neilson J. R., and Sharp P. A. (2007) MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 4, 721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bhat-Nakshatri P., Wang G., Collins N. R., Thomson M. J., Geistlinger T. R., Carroll J. S., Brown M., Hammond S., Srour E. F., Liu Y., and Nakshatri H. (2009) Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucleic Acids Res. 37, 4850–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tan S., Ding K., Li R., Zhang W., Li G., Kong X., Qian P., Lobie P. E., and Zhu T. (2014) Identification of miR-26 as a key mediator of estrogen stimulated cell proliferation by targeting CHD1, GREB1 and KPNA2. Breast Cancer Res. 16, R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miller T. E., Ghoshal K., Ramaswamy B., Roy S., Datta J., Shapiro C. L., Jacob S., and Majumder S. (2008) MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J. Biol. Chem. 283, 29897–29903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tan S., Li R., Ding K., Lobie P. E., and Zhu T. (2011) miR-198 inhibits migration and invasion of hepatocellular carcinoma cells by targeting the HGF/c-MET pathway. FEBS Lett. 585, 2229–2234 [DOI] [PubMed] [Google Scholar]
- 28. Jansen M. P., Reijm E. A., Sieuwerts A. M., Ruigrok-Ritstier K., Look M. P., Rodríguez-González F. G., Heine A. A., Martens J. W., Sleijfer S., Foekens J. A., and Berns E. M. (2012) High miR-26a and low CDC2 levels associate with decreased EZH2 expression and with favorable outcome on tamoxifen in metastatic breast cancer. Breast Cancer Res. Treat. 133, 937–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kotta-Loizou I., Vasilopoulos S. N., Coutts R. H., and Theocharis S. (2016) Current evidence and future perspectives on HuR and breast cancer development, prognosis, and treatment. Neoplasia 18, 674–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brennan C. M., and Steitz J. A. (2001) HuR and mRNA stability. Cell. Mol. Life Sci. 58, 266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hostetter C., Licata L. A., Witkiewicz A., Costantino C. L., Yeo C. J., Brody J. R., and Keen J. C. (2008) Cytoplasmic accumulation of the RNA binding protein HuR is central to tamoxifen resistance in estrogen receptor positive breast cancer cells. Cancer Biol. Ther. 7, 1496–1506 [DOI] [PubMed] [Google Scholar]
- 32. Hostetter C. L., Licata L. A., and Keen J. C. (2009) Timing is everything: order of administration of 5-aza 2′ deoxycytidine, trichostatin A and tamoxifen changes estrogen receptor mRNA expression and cell sensitivity. Cancer Lett. 275, 178–184 [DOI] [PubMed] [Google Scholar]
- 33. Mayr C., and Bartel D. P. (2009) Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Di Giammartino D. C., Nishida K., and Manley J. L. (2011) Mechanisms and consequences of alternative polyadenylation. Mol. Cell 43, 853–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Millevoi S., and Vagner S. (2010) Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 38, 2757–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Elkon R., Ugalde A. P., and Agami R. (2013) Alternative cleavage and polyadenylation: extent, regulation and function. Nat. Rev. Genet. 14, 496–506 [DOI] [PubMed] [Google Scholar]
- 37. Proudfoot N. J. (2011) Ending the message: poly(A) signals then and now. Genes Dev. 25, 1770–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jenal M., Elkon R., Loayza-Puch F., van Haaften G., Kühn U., Menzies F. M., Oude Vrielink J. A., Bos A. J., Drost J., Rooijers K., Rubinsztein D. C., and Agami R.(2012) The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 149, 538–553 [DOI] [PubMed] [Google Scholar]
- 39. Masamha C. P., Xia Z., Yang J., Albrecht T. R., Li M., Shyu A. B., Li W., and Wagner E. J. (2014) CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510, 412–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xia Z., Donehower L. A., Cooper T. A., Neilson J. R., Wheeler D. A., Wagner E. J., and Li W. (2014) Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat. Commun. 5, 5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jin K., Park S., Teo W. W., Korangath P., Cho S. S., Yoshida T., Győrffy B., Goswami C. P., Nakshatri H., Cruz L. A., Zhou W., Ji H., Su Y., Ekram M., Wu Z., et al. (2015) HOXB7 Is an ERα cofactor in the activation of HER2 and multiple ER target genes leading to endocrine resistance. Cancer Discov. 5, 944–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lü M., Ding K., Zhang G., Yin M., Yao G., Tian H., Lian J., Liu L., Liang M., Zhu T., and Sun F. (2015) MicroRNA-320a sensitizes tamoxifen-resistant breast cancer cells to tamoxifen by targeting ARPP-19 and ERRgamma. Sci. Rep. 5, 8735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arpino G., Wiechmann L., Osborne C. K., and Schiff R. (2008) Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr. Rev. 29, 217–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schiff R., Massarweh S. A., Shou J., Bharwani L., Mohsin S. K., and Osborne C. K. (2004) Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin. Cancer Res. 10, 331S–336S [DOI] [PubMed] [Google Scholar]
- 45. Egeland N. G., Lunde S., Jonsdottir K., Lende T. H., Cronin-Fenton D., Gilje B., Janssen E. A., and Søiland H. (2015) The role of microRNAs as predictors of response to tamoxifen treatment in breast cancer patients. Int. J. Mol. Sci. 16, 24243–24275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wong C. F., and Tellam R. L. (2008) MicroRNA-26a targets the histone methyltransferase Enhancer of Zeste homolog 2 during myogenesis. J. Biol. Chem. 283, 9836–9843 [DOI] [PubMed] [Google Scholar]
- 47. Krüger J., and Rehmsmeier M. (2006) RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 34, W451–W454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Heinonen M., Bono P., Narko K., Chang S. H., Lundin J., Joensuu H., Furneaux H., Hla T., Haglund C., and Ristimäki A. (2005) Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res. 65, 2157–2161 [DOI] [PubMed] [Google Scholar]
- 49. Denkert C., Weichert W., Winzer K. J., Müller B. M., Noske A., Niesporek S., Kristiansen G., Guski H., Dietel M., and Hauptmann S. (2004) Expression of the ELAV-like protein HuR is associated with higher tumor grade and increased cyclooxygenase-2 expression in human breast carcinoma. Clin. Cancer Res. 10, 5580–5586 [DOI] [PubMed] [Google Scholar]
- 50. Heinonen M., Fagerholm R., Aaltonen K., Kilpivaara O., Aittomäki K., Blomqvist C., Heikkila P., Haglund C., Nevanlinna H., and Ristimäki A. (2007) Prognostic role of HuR in hereditary breast cancer. Clin. Cancer Res. 13, 6959–6963 [DOI] [PubMed] [Google Scholar]
- 51. Zhuang R., Rao J. N., Zou T., Liu L., Xiao L., Cao S., Hansraj N. Z., Gorospe M., and Wang J. Y. (2013) miR-195 competes with HuR to modulate stim1 mRNA stability and regulate cell migration. Nucleic Acids Res. 41, 7905–7919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tominaga K., Srikantan S., Lee E. K., Subaran S. S., Martindale J. L., Abdelmohsen K., and Gorospe M. (2011) Competitive regulation of nucleolin expression by HuR and miR-494. Mol. Cell. Biol. 31, 4219–4231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Borg A., Baldetorp B., Fernö M., Killander D., Olsson H., Rydén S., and Sigurdsson H. (1994) ERBB2 amplification is associated with tamoxifen resistance in steroid-receptor positive breast cancer. Cancer Lett. 81, 137–144 [DOI] [PubMed] [Google Scholar]
- 54. Knowlden J. M., Hutcheson I. R., Jones H. E., Madden T., Gee J. M., Harper M. E., Barrow D., Wakeling A. E., and Nicholson R. I. (2003) Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 144, 1032–1044 [DOI] [PubMed] [Google Scholar]
- 55. Chu I., Blackwell K., Chen S., and Slingerland J. (2005) The dual ErbB1/ErbB2 inhibitor, lapatinib (GW572016), cooperates with tamoxifen to inhibit both cell proliferation- and estrogen-dependent gene expression in antiestrogen-resistant breast cancer. Cancer Res. 65, 18–25 [PubMed] [Google Scholar]
- 56. Dowsett M., Harper-Wynne C., Boeddinghaus I., Salter J., Hills M., Dixon M., Ebbs S., Gui G., Sacks N., and Smith I. (2001) HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res. 61, 8452–8458 [PubMed] [Google Scholar]
- 57. Kang H., Kim C., Lee H., Kim W., and Lee E. K. (2013) Post-transcriptional controls by ribonucleoprotein complexes in the acquisition of drug resistance. Int. J. Mol. Sci. 14, 17204–17220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yuan Z., Sanders A. J., Ye L., and Jiang W. G. (2010) HuR, a key post-transcriptional regulator, and its implication in progression of breast cancer. Histol. Histopathol. 25, 1331–1340 [DOI] [PubMed] [Google Scholar]
- 59. Pryzbylkowski P., Obajimi O., and Keen J. C. (2008) Trichostatin A and 5 aza-2′ deoxycytidine decrease estrogen receptor mRNA stability in ER positive MCF7 cells through modulation of HuR. Breast Cancer Res. Treat. 111, 15–25 [DOI] [PubMed] [Google Scholar]
- 60. Erson-Bensan A. E., and Can T. (2016) Alternative polyadenylation: another foe in cancer. Mol. Cancer Res. 14, 507–517 [DOI] [PubMed] [Google Scholar]
- 61. Mansfield K. D., and Keene J. D. (2012) Neuron-specific ELAV/Hu proteins suppress HuR mRNA during neuronal differentiation by alternative polyadenylation. Nucleic Acids Res. 40, 2734–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Al-Ahmadi W., Al-Ghamdi M., Al-Haj L., Al-Saif M., and Khabar K. S. (2009) Alternative polyadenylation variants of the RNA binding protein, HuR: abundance, role of AU-rich elements and auto-regulation. Nucleic Acids Res. 37, 3612–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Al-Ahmadi W., Al-Ghamdi M., Al-Souhibani N., and Khabar K. S. (2013) miR-29a inhibition normalizes HuR over-expression and aberrant AU-rich mRNA stability in invasive cancer. J. Pathol. 230, 28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guo X., Wu Y., and Hartley R. S. (2009) MicroRNA-125a represses cell growth by targeting HuR in breast cancer. RNA Biol. 6, 575–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dai W., Zhang G., and Makeyev E. V. (2012) RNA-binding protein HuR autoregulates its expression by promoting alternative polyadenylation site usage. Nucleic Acids Res. 40, 787–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li C., Feng Y., Coukos G., and Zhang L. (2009) Therapeutic microRNA strategies in human cancer. AAPS J. 11, 747–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Orom U. A., and Lund A. H. (2007) Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods 43, 162–165 [DOI] [PubMed] [Google Scholar]
- 68. Li S., Zhu J., Fu H., Wan J., Hu Z., Liu S., Li J., Tie Y., Xing R., Zhu J., Sun Z., and Zheng X. (2012) Hepato-specific microRNA-122 facilitates accumulation of newly synthesized miRNA through regulating PRKRA. Nucleic Acids Res. 40, 884–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.