

Summary
Metabolic deregulation is a hallmark of human cancers, and the glycolytic and glutamine metabolism pathways were shown to be deregulated in pancreatic ductal adenocarcinoma (PDAC). To identify new metabolic regulators of PDAC tumor growth and metastasis, we systematically knocked down metabolic genes that were overexpressed in human PDAC tumor samples using short hairpin RNAs. We found that p53 transcriptionally represses paraoxonase 2 (PON2), which regulates GLUT1-mediated glucose transport via stomatin. The loss of PON2 initiates the cellular starvation response and activates AMP-activated protein kinase (AMPK). In turn, AMPK activates FOXO3A and its transcriptional target, PUMA, which induces anoikis to suppress PDAC tumor growth and metastasis. Pharmacological or genetic activation of AMPK, similar to PON2 inhibition, blocks PDAC tumor growth. Collectively, our results identify PON2 as a new modulator of glucose transport that regulates a pharmacologically tractable pathway necessary for PDAC tumor growth and metastasis.
eTOC Blurb
Nagarajan et al., shows that PON2 is overexpressed in pancreatic cancer and is necessary for pancreatic cancer growth and metastasis. PON2 increases glucose uptake to protect pancreatic cancer cells from detachment-induced cell death, which in part occurs through suppression of AMPK→FOXO3A→PUMA signaling pathway.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related deaths in the United States. The 5-year disease-free survival rate for PDAC patients is less than 6%, and all current therapies, including immunotherapies, have failed to provide meaningful clinical benefits to patients (Hidalgo, 2010). Therefore, a better understanding of PDAC is urgently needed to develop new and more effective therapies for PDAC patients.
Genome-scale sequencing identified 12 cellular signaling pathways that were deregulated in 67% to 100% of human PDAC tumor samples (Jones et al., 2008). The same study showed that all PDACs harbored oncogenic mutations in KRAS (Jones et al., 2008). Similarly, mutations in TP53 (commonly known as p53) are found in up to two-thirds of PDACs (Scarpa et al., 1993). Previous studies have shown that oncogenic KRAS mutations are necessary to initiate and maintain PDAC tumor growth (Bryant et al., 2014; Ying et al., 2012), and p53 mutations facilitate the metastatic progression of PDAC (Weissmueller et al., 2014).
Cancer cell-specific hallmarks are used to differentiate cancer cells from normal cells (Hanahan and Weinberg, 2011), and a hallmark of PDAC is its dependency on cellular metabolic pathways for tumor growth and metastasis (Ying et al., 2012). There is some evidence for the deregulation of metabolic pathways in PDAC, such as the glycolytic and glutamine metabolism pathways (Son et al., 2013; Ying et al., 2012). However, the role of metabolic alterations in PDAC tumors and their metastatic growth is not fully understood.
To identify new metabolic regulators of PDAC tumor growth and metastasis, we developed an integrative genomics approach by combining gene expression profiling of PDAC tumor samples with RNA interference-mediated gene knockdown. Using this experimental approach, we identified paraoxonase 2 (PON2) as a previously undocumented regulator of PDAC tumor growth and metastasis that functions by regulating glucose transporter 1 (GLUT1)-mediated glucose transport and consequential activation of the AMP-activated protein kinase (AMPK)→forkhead box O3A (FOXO3A)→p53-upregulated modulator of apoptosis (PUMA) pathway. We also show that the PON2-regulated pathway in PDAC can be targeted by AMP kinase-activating drugs to inhibit tumor growth.
RESULTS
PON2 is Necessary for PDAC Tumor Growth
To identify metabolic genes necessary for PDAC tumor growth, we used an integrative genomics approach, combining gene expression profiling of human PDAC tumor samples with the functional genomics approach of RNA interference screening. We first analyzed four publicly available gene expression datasets (Badea et al., 2008; Grutzmann et al., 2004; Ishikawa et al., 2005; Pei et al., 2009). Collectively, these four studies compared the mRNA expression profiles of 113 human PDACs and 91 normal human pancreatic tissue samples to identify genes that are specifically altered in PDAC tumors. We combined these four datasets to eliminate data bias generated by array platforms and probe efficiencies, to avoid artifacts associated with sample processing, and to minimize the population-based biases of each of these studies. We focused on the top 10% significantly overexpressed genes common to all four datasets (P<0.05), which included 13 metabolic genes (Table S1). We then asked whether any of these 13 genes are necessary to maintain the tumorigenic ability of PDAC cells (Figure 1A; Table S2). To test this, we knocked down the expression of all 13 genes individually using two sequence-independent and knockdown-validated lentiviral short hairpin RNAs (shRNAs) in two PDAC cell lines, PANC1 and AsPC-1 (Figure S1A–S1C). PANC1 and AsPC-1 cells expressing nonspecific shRNA were used as controls. PANC1 and AsPC-1 cells expressing target shRNAs were tested for their ability to grow in an anchorage-independent manner in soft agar. Our approach was based on the idea that knockdown of a metabolic gene necessary for PDAC tumor growth would inhibit its tumorigenic ability. We found that knockdown of the genes encoding hexokinase 2 (HK2), solute carrier family 2, member 1 (SLC2A1), and lactate dehydrogenase A (LDHA) significantly reduced soft-agar colony formation of PANC1 and AsPC-1 cells (Figure S1D and S1E). HK2, SLC2A1, and LDHA have been implicated in PDAC tumor growth (Barretina et al., 2012; Mohammad et al., 2016; Ying et al., 2012). In addition, PON2 knockdown strongly inhibited the soft-agar colony formation of PANC1, AsPC-1, and two additional PDAC cell lines (MIA PaCa-2 and SU.86.86) (Figure 1B; Table S2). We also tested whether PON2 knockdown in PDAC cells inhibits tumor formation in mice. To this end, we used two mouse models of PDAC tumor growth: a subcutaneous tumor xenograft model and an orthotopic pancreatic tumor xenograft model. We found that PON2 knockdown efficiently inhibited the growth of PDAC tumors in both mouse models (Figure 1C and 1D; Figure S1F; Table S3). Collectively, these results demonstrate that PON2 is necessary for tumor development in a wide variety of human PDAC cell lines, both in cell culture and in mice. Because PON2 has not been previously implicated in pancreatic cancer, we decided to study its role in PDAC in greater detail.
Figure 1. Integrative genomics approach identifies metabolic genes necessary for pancreatic ductal adenocarcinoma (PDAC) growth.

A. Schematic of the analysis to identify genes necessary for PDAC tumor growth. B. Representative images show soft-agar colony formation by PDAC cell lines expressing PON2 or nonspecific (NS) shRNAs. C. PANC1 cells expressing PON2 or NS shRNAs were injected subcutaneously and analyzed for tumor formation in athymic nude mice (n=5). Average tumor volumes are shown. D. PANC1 cells expressing PON2 or NS shRNAs were injected orthotopically into the pancreas of athymic nude mice (n=3) and analyzed for tumor formation. Representative bioluminescence images are shown. E. Representative images show soft-agar colony formation in the absence or presence of doxycycline (left) or average tumor volume in mice (n=5) in the presence of doxycycline (right) using iKRAS mouse model-derived pancreatic cancer cells that were engineered to express empty vector or PON2 cDNA. F. Representative images show soft agar-colony formation (top) or average tumor volume in mice (n=5) (bottom) using human HPNE-hTERT E6/E7/st cells that express empty vector or PON2 cDNA. Data are mean ± SEM. **p<0.05. See also Figure S1, Figure S2, Table S2 and Table S3.
PON2 Cooperates with KRASG12D to Promote PDAC Tumor Growth
Mutations of the KRAS gene (typically for KRASG12D) are present in over 90% of PDAC tumors and are necessary for PDAC initiation and tumor maintenance (Collins et al., 2012). Therefore, we tested whether PON2 regulates KRASG12D-induced transformation or cooperates with KRASG12D to accelerate PDAC tumor growth. To this end, we used cells derived from the inducible KRASG12D (iKRAS) PDAC mouse model (Ying et al., 2012) and immortalized human pancreatic ductal epithelial cells (HPNE-hTERT E6/E7/st) (Campbell et al., 2007). In iKRAS-derived cells, KRASG12D expression can be transiently induced by doxycycline, which results in the transformation of these cells (Ying et al., 2012). To determine whether PON2 expression is necessary for KRASG12D to induce transformation, we knocked down PON2 expression using shRNA in iKRAS-derived and HPNE-hTERT E6/E7/st cells (Figure S2A and S2B). We found that PON2 knockdown did not attenuate KRASG12D induction of cellular transformation in either iKRAS-derived or HPNE-hTERT E6/E7/st cells (Figure S2C–S2E). However, we noted that ectopic expression of PON2 enhanced the ability of KRASG12D transformed cells to form colonies in soft agar and accelerated tumor formation in mice using both iKRAS-derived and HPNE-hTERT E6/E7/st cells (Figure 1E and 1F; Table S2). Collectively, these results show that although PON2 is not necessary for KRASG12D-induced transformation but it cooperates with KRASG12D to accelerate PDAC tumor progression.
PON2 is Necessary for Anoikis Resistance and Multi-Organ Metastatic Growth of PDAC
We then asked whether PON2 affects other aspects of PDAC tumor progression. To this end, we analyzed the effects of PON2 knockdown on metastatic PDAC cell migration, invasion, and anoikis. We found that knockdown of PON2 did not significantly affect the migration or invasiveness of PDAC cells (Figure S3A and S3B) but did result in anoikis induction (Figure 2A). Consistent with these results, detached PDAC cells expressing PON2 shRNA showed a significantly higher proportion of annexin V-positive cells (Figure 2B) and increased poly(ADP-ribose) polymerase (PARP) cleavage (Figure 2C) compared with PDAC cells expressing nonspecific shRNA. Treatment with pan-caspase inhibitor Z-VAD-FMK prevented anoikis induction after PON2 knockdown, as shown by the reductions in annexin V-positive cells (Figure 2B) and PARP cleavage (Figure 2C) in cells expressing PON2 shRNA compared with those expressing nonspecific shRNA. Similarly, Z-VAD-FMK rescued the anchorage-independent growth and anoikis induction induced by PON2 knockdown in PDAC cells (Figure S3C and S3D). These findings indicate that anoikis induction after PON2 knockdown is caspase dependent.
Figure 2. Loss of PON2 results in anoikis induction and metastasis inhibition.

A. PDAC cell lines expressing PON2 or nonspecific (NS) shRNAs were seeded on poly(HEMA)-coated plates. Viable cells were counted after staining with trypan blue, and the relative numbers of viable cells are plotted at the indicated times. B. PANC1 cell lines expressing the indicated shRNAs were analyzed for apoptosis under attached and detached conditions by flow cytometry after staining with annexin V-FITC and propidium iodide. Percentage of annexin V-FITC-positive cells (attached and detached) after treatment with Z-VAD-FMK (10 μM) is shown. C. PANC1 cells expressing PON2 or NS shRNAs were grown under attached or detached conditions. Detached cells were either left untreated or treated with Z-VAD-FMK (10 μM). Cell lysates were analyzed for PARP cleavage by immunoblot analysis. ACTINB was used as a loading control. D. Athymic nude mice (n=3) were injected with PANC1 cells expressing PON2 or NS shRNAs via the tail vein. Lung metastasis was monitored by bioluminescence imaging. Representative images are shown at week 1 and week 4. E. Athymic nude mice (n=3) received intrasplenic injections of luciferase-tagged PANC1 cells expressing either PON2 or NS shRNA to cause liver metastasis. Metastatic tumor growth in the livers was visualized after 6 weeks. Bioluminescence images of livers isolated from the mice are shown. F. Luciferase-tagged PANC1 cells carrying PON2 or NS shRNA were injected into the pancreases of athymic nude mice (n=3). Spontaneous metastasis to the livers and lungs was visualized after 6 weeks. Bioluminescence images of livers and lungs isolated from the mice are shown. G. qPCR analysis of circulating tumor cells originating from primary tumors formed by injecting PANC1 cells carrying PON2 or NS shRNA into the pancreas of athymic nude mice (n=3). Data are mean ± SEM. *p<0.05, **p<0.005. See also Figure S3, Table S2 and Table S3.
Anoikis resistance is a key feature of transformed and metastatic cells, promoting cell growth upon detachment from the extracellular matrix and the survival of circulating tumor cells (Paoli et al., 2013). Therefore, we asked whether PON2 expression plays a role in PDAC metastasis. We injected athymic nude mice via the tail vein with PDAC cells expressing PON2 shRNA to mimic lung metastasis or via the spleen to mimic liver metastasis because the lungs and liver are among the most common locations of PDAC metastases (Hishinuma et al., 2006; Teo et al., 2013). We found that PON2 knockdown significantly decreased the metastases of PANC1 and AsPC-1 PDAC cell lines in the lungs (Figure 2D; Figure S3E; Table S3) and the metastasis of PANC1 cells to the liver after splenic injection (Figure 2E; Table S3).
To further evaluate the role of PON2 in PDAC metastatic progression, we used an orthotopic mouse model of spontaneous PDAC metastasis. Specifically, we injected PANC1 or AsPC-1 cells expressing PON2 or nonspecific shRNA into the pancreas and imaged the lungs and liver of the mice after 6 weeks. In agreement with the results of tail vein and splenic injections, we found that PON2 knockdown significantly inhibited lung and liver metastases compared with that in mice injected with PDAC cells expressing nonspecific shRNA (Figure 2F; Figure S3F; Table S3). In this model, we also tested the effect of PON2 knockdown on the abundance of circulating tumor cells. Figure 2G shows that PON2 knockdown reduced the numbers of circulating tumor cells in vivo compared with the numbers of PDAC cells expressing nonspecific shRNA, which explains the reduced metastatic potential of PDAC cells after PON2 knockdown. Collectively, our results demonstrate that PON2 is necessary for anoikis resistance, which facilitates tumor growth and multi-organ metastasis in PDAC.
Inactivation of Tumor Suppressor p53 in PDAC Cells Results in Transcriptional Overexpression of PON2
Our analysis of previously published datasets revealed that PON2 is overexpressed at the mRNA level in patient-derived PDAC tumor samples (Figure 3A; Table S1). We sought to confirm these results by independently analyzing primary samples of patient-derived PDAC and normal pancreatic tissues by immunohistochemistry. Our results showed that PON2 protein levels were higher in PDAC tumor samples than in normal pancreatic tissues (Figure 3B; Table S4). Our results also showed membrane localization of PON2 in the ductal cells of PDAC tumor samples (Figure 3B; Table S4). We next investigated the mechanism by which PON2 is overexpressed in PDAC. The KRASG12D mutation is present in 95% of PDACs, resulting in constitutive activation of the oncogenic KRAS pathway. KRASG12D promotes tumor growth by transcriptionally activating glycolytic enzymes via the transcription factor HIF1α or MYC (Chen et al., 2003; He et al., 2015; Ying et al., 2012). In agreement with results from previous studies, we found that shRNA-induced KRAS knockdown or pharmacological inhibition of the MAP kinase pathway by the MEK inhibitor trametinib significantly decreased HK2 and SLC2A1 mRNA levels (Figure S3G and S3H). However, neither KRAS knockdown nor trametinib treatment affected PON2 expression in PDAC cell lines (Figure S3G and S3H), which suggests that KRASG12D-mediated signaling does not regulate PON2 expression in PDAC cells.
Figure 3. Tumor suppressor p53 transcriptionally represses PON2.

A. Analyses of PON2 mRNA levels of normal pancreatic tissues and PDAC tumor samples from four indicated studies in the Oncomine database. B. Immunohistochemistry analyses of PON2 levels in normal pancreatic tissue and PDAC tumor samples. Corresponding H&E staining is shown in the top panel. C. Transcript levels of p21 and PON2 in AsPC-1 cells infected with adenovirus expressing p53 (Ad-p53) or control adenovirus expressing the β-galactosidase gene (Ad-LacZ) were evaluated by qRT-PCR. D. AsPC-1 cells infected with Ad-p53 or Ad-LacZ were analyzed for the indicated proteins by immunoblotting. E. (left) Schematics showing the p53 binding site on the PON2 promoter. (right) PON2 promoter-firefly luciferase reporters, with intact or mutated p53 binding sites, were transfected into AsPC-1 cells along with adenovirus expressing LacZ or p53-GFP. Luciferase activity was measured after 48 h and is presented as relative luciferase units. F. AsPC-1 cells expressing Ad-p53 or Ad-LacZ were analyzed using the ChIP assay to evaluate the binding of p53 to the PON2 promoter. G. Transcript levels of p53, p21, and PON2 in HPNE cells carrying p53 or nonspecific (NS) shRNA were evaluated by qRT-PCR. H. Immunoblot analyses of the indicated proteins in HPNE cells carrying p53 or NS shRNAs. I. ChIP assay to measure p53 binding on the PON2 promoter using IgG or anti-p53 antibody. The ACTINB, GAPDH, and p21 promoters were used as controls. Data are mean ± SEM. *p<0.05, **p<0.005.
To identify potential transcriptional regulators of PON2 expression in PDAC cells, we analyzed the PON2 promoter sequence and identified a binding site for tumor suppressor p53. Up to two-thirds of PDACs lack functional p53 because of acquired somatic mutations or genetic deletion (Bardeesy and DePinho, 2002). We hypothesized that p53 transcriptionally represses PON2 in normal pancreatic ductal epithelial cells, and its inactivation leads to PON2 overexpression in PDAC cells. To test this hypothesis, we ectopically expressed p53 in the p53-deficient PDAC cell line, AsPC-1, using a recombinant adenovirus (Ad-p53). We found that ectopic expression of p53 in AsPC-1 cells resulted in the downregulation of PON2 mRNA (Figure 3C) and protein (Figure 3D) compared with that in cells infected with control adenovirus expressing the gene encoding β-galactosidase (Ad-LacZ). Consistent with p53 being the direct transcriptional repressor of PON2, ectopic expression of p53 in AsPC-1 cells prevented PON2 promoter-driven luciferase activity, which was restored by mutating the p53 binding site (Figure 3E). To further demonstrate that p53 directly regulates PON2 transcription, we performed chromatin immunoprecipitation (ChIP) to evaluate the association of p53 with the PON2 promoter in AsPC-1 cells expressing Ad-p53 or Ad-LacZ. Our results showed that p53 directly associates with the PON2 promoter in vivo (Figure 3F). In addition, we knocked down p53 using shRNA in immortalized primary HPNE-hTERT cells and analyzed PON2 expression and the association of p53 with the PON2 promoter. We found that p53 knockdown increased PON2 mRNA and protein levels and reduced the enrichment of p53 to the PON2 promoter (Figure 3G–3I). Collectively, these results demonstrate that tumor suppressor p53 functions as a direct transcriptional repressor of PON2 and indicate that p53 inactivation due to mutation or genetic deletion is a key event that results in PON2 overexpression in PDAC.
PON2 Loss Activates the Cellular Starvation Response and AMPK-induced Stimulation of the FOXO3A Tumor Suppressor Pathway
We next examined a mechanism that might explain why PON2 inhibition attenuates PDAC tumor growth and metastasis. Previous studies indicated that PON2 may function as a reactive oxygen species (ROS) scavenger, thereby promoting cell survival (Witte et al., 2011). Therefore, we tested the effect of PON2 knockdown on ROS levels in PDAC cells. Specifically, we measured ROS levels using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) in PDAC cells expressing PON2 or nonspecific shRNA. However, we did not observe significant changes in cellular ROS level (Figure S3I). Because of phenotypic similarity to knockdowns of GLUT1 (SLC2A1), HK2, and LDHA, encoding enzymes involved in glucose metabolism, we asked whether PON2 is also involved in glucose metabolism, which is the major source of cellular energy. Therefore, we measured ATP/ADP and ADP/AMP ratios, which are the best indicators of cellular energy status, in PON2 shRNA-expressing PDAC cells. Our results showed that the loss of PON2 expression significantly decreased ATP/ADP and ADP/AMP ratios (Figure 4A), an indicator of the cellular starvation response. On the basis of our result that PON2 knockdown leads to anoikis induction, we analyzed the expressions of a number of pro- and anti-apoptotic genes (Table S5) and found that PON2 knockdown activated several pro-apoptotic genes. However, only the gene encoding PUMA was significantly and consistently upregulated in all tested PDAC cell lines, particularly under detached conditions (Figure 4B; Figure S4A and S4B). Thus, we focused on PUMA as a mediator of PON2 function in PDAC. Analyses of the PUMA promoter identified FOXO transcription factor binding sites. We systematically knocked down all three FOXO transcription factors (FOXO1, FOXO3A, and FOXO4) using shRNAs (Figure S4C) and measured the effect on PUMA expression. We found that the knockdown of only FOXO3 in detached cells significantly lowered PUMA expression (Figure 4C; Figure S4D). To demonstrate that FOXO3A directly regulates PUMA transcription in PON2-deficient cells, we first evaluated the effect of PON2 loss on PUMA promoter activity using a luciferase reporter. We found that PON2 knockdown resulted in increased PUMA promoter-driven luciferase activity, but this luciferase activity was significantly attenuated upon mutagenesis of the FOXO3A binding site on the PUMA promoter (Figure 4D). We then performed a ChIP assay to evaluate the binding of FOXO3A to the PUMA promoter and found that PON2 shRNA-expressing PDAC cells displayed increased enrichment of FOXO3A to the PUMA promoter compared with that in cells expressing nonspecific shRNA (Figure 4E).
Figure 4. Loss of PON2 activates FOXO3→PUMA-mediated apoptotic pathway to suppress tumor growth.

A. ATP/ADP and ATP/AMP ratios were determined in AsPC-1 and PANC1 cells expressing PON2 or nonspecific (NS) shRNAs by LC-MS/MS. Absolute ATP/ADP and ATP/AMP ratios are plotted. B. Immunoblot analysis of PUMA, PON2, and ACTINB in PANC1 and AsPC-1 cells expressing PON2 or NS shRNAs. C. PUMA expression was assessed by qRT-PCR (left) and immunoblotting (right) in cells carrying the indicated shRNAs grown under attached or detached conditions. D. Luciferase assay using the PUMA promoter-firefly luciferase reporter with wild type or mutated FOXO1 binding site in PANC1 cells expressing PON2 or NS shRNAs in attached and detached conditions. Inset shows the location and sequence of the forkhead response element (FHRE) in the PUMA promoter E. ChIP assay to evaluate FOXO3A binding to the PUMA promoter in PANC1 cells carrying PON2 or NS shRNAs. Relative fold enrichment of FOXO3A on PUMA promoter under indicated conditions is shown. F. Immunoblot analyses of indicated proteins in PANC1 cells expressing PON2 or NS shRNAs under attached and detached conditions. G. Immunoblot analyses of the indicated proteins in PANC1 (left) or AsPC-1 (right) cells expressing PON2 or NS shRNAs with DMSO or AMPK inhibitor (AMPKi), Compound C (10 μM), treatment for 24 h. Data are presented as mean ± SEM. *p<0.05. See also Figure S4.
Previous studies have shown that AMPK can be activated by the cellular starvation response (Faubert et al., 2013; Hardie et al., 2012; Jones et al., 2005). It was also shown that activated AMPK phosphorylates and activates FOXO3A (Greer et al., 2007). Consistent with these reports, we observed an increased phosphorylation of AMPK upon PON2 knockdown and an increased AMPK-mediated phosphorylation of FOXO3A (Figure 4F; Figure S4E), a signal for FOXO3A transcriptional activation, in the same cells. We tested the effect of AMPK on FOXO3A activation using AMPK inhibitors. Our results showed that treating PON2 shRNA-expressing PANC1 and AsPC1 cells with an AMPK inhibitor prevented FOXO3A phosphorylation (Figure 4G). FOXO3A targets include genes that also regulate cell cycle progression (Zhang et al., 2012). Therefore, we performed a cell cycle analysis to determine the effect of PON2 knockdown on cell cycle progression. We found that the cell cycle profiles of PDAC cell lines were not altered by PON2 knockdown (Figure S4F). Finally, we sought to determine the mechanism by which detached conditions increase the activation of the AMPK→FOXO3A→PUMA pathway. We found that under detached conditions, PDAC cells were less efficient in glucose uptake (Figure S4G). Accordingly, the ATP/ADP ratio in PDAC cells was decreased under a detached condition compared with that under an attached condition (Figure S4H). PDAC cells also required more glucose for proliferation under a detached condition than under an attached condition (Figure S4I). Collectively, these results demonstrate that PON2 loss leads to an activation of the cellular starvation response, which in turn results in AMPK-mediated activation of the tumor suppressor FOXO3A pathway and its downstream pro-apoptotic target gene, PUMA. These results also explain the more potent activation of the AMPK→FOXO3A→PUMA pathway under detached conditions.
PON2 Regulates GLUT1-Mediated Glucose Transport
The effects of PON2 loss in PDAC appeared to be driven by reduced ATP/ADP and ADP/AMP ratios and by the activation of the AMPK pathway, which acts as a sensor of energy status. Therefore, we measured key intermediates of bioenergetics pathways in PANC1 and AsPC-1 cells expressing PON2 shRNA. We found that glucose 6-phosphate (G6P) was significantly decreased in both PANC1 and AsPC-1 cells expressing PON2 shRNA compared with that in cells expressing nonspecific shRNA (Figure 5A; Figure S5A). G6P is the first metabolite in the glycolytic and pentose phosphate pathways, both of which are major contributors of cellular ATP. This decrease in G6P may be due to reduced glucose uptake or decreased levels of hexokinase or glucose 6-phosphatase (G6Pase). We did not observe any changes in HK2 expression in cells with PON2 knockdown, and G6Pase was not detectable (Figure S6A). Based on these results, we measured glucose uptake using 14C-labeled 2-deoxyglucose (2-DG) and observed decreased glucose uptake in PANC1 and AsPC1 cells expressing PON2 shRNA (Figure 5B; Figure S5B). In addition, results obtained using the Seahorse extracellular flux analyzer showed lower rates of oxygen consumption (Figure 5C; Figure S5C) and extracellular acidification (Figure 5D; Figure S5D) in PON2 shRNA-expressing PANC1 and AsPC1 cells. We then measured glucose carbon flow through the glycolytic and TCA cycles using U-13C6-labeled glucose. A 3-hour glucose uptake assay was found to be sufficient to label key metabolites of the glycolytic and TCA cycles without saturating the system (Mitsuishi et al., 2012). As expected, we found that PON2-deficient cells had significantly lower amounts of label incorporated into glycolytic and TCA cycle intermediates (Figure S5E–S5G; Table S6).
Figure 5. PON2 regulates the cellular starvation response by regulating GLUT1 activity via STOM.

A. Glucose 6-phosphate/taurine ratio in PANC1 cells expressing PON2 or nonspecific (NS) shRNAs. B. Relative [14C]-2-DG uptake in PANC1 cells expressing PON2 or NS shRNAs. C. Oxygen consumption rate expressed as pmol/min in PANC1 cells expressing PON2 or NS shRNAs. D. Extracellular acidification rate (ECAR) expressed as mpH/min in PANC1 cells expressing PON2 or NS shRNAs. E. Co-immunoprecipitation assay in PANC1 cells using IgG or anti-PON2 antibody. Immunoprecipitates and lysates were analyzed for the indicated proteins. F. Relative [14C]-2-DG uptake in PANC1 cells co-expressing GLUT1 along with vector or PON2. G. Relative [14C]-2-DG uptake in a glucose competition assay in PANC1 cells expressing PON2 or NS shRNAs. H. Relative [14C]-2-DG uptake in PANC1 expressing STOM or PON2 and STOM or NS shRNAs. I. Co-immunoprecipitation assay in PANC1 cells expressing NS or PON2 shRNAs using IgG or anti-STOM antibody. Immunoprecipitates and lysates were analyzed for the indicated proteins. J. Immunoblot analysis to measure indicated proteins in PANC1 cells expressing PON2, PON2 and STOM, or NS shRNAs. K. Soft-agar colony formation by PANC1 cell lines expressing PON2, PON2 and STOM, or NS shRNAs. Data are presented as mean ± SEM. *p<0.05, **p<0.005. See also Figure S5, Figure S6 and Table S2.
Because glucose transport was affected in PON2-deficient cells, we assessed the expression of the ubiquitous glucose transporters, GLUT1 and GLUT3, in PDAC and AsPC1 cells but did not observe any changes in their protein levels despite an increase in SLC2A3 mRNA (GLUT3) levels after PON2 knockdown (Figure S6A and S6B). Next, we performed a cell surface biotinylation assay to examine membrane expression of GLUT1 and GLUT3 in PON2-deficient cells; however, we did not observe any differences in their surface expressions (Figure S6C).
Previous studies in Caenorhabditis elegans showed that the PON2 homolog, MEC-6, interacts with MEC-2 (Chelur et al., 2002), the human stomatin (STOM) homolog. In a co-fractionation assay, PON2 was shown to interact with STOM (Havugimana et al., 2012). STOM interacts with many membrane-bound transporters and modifies their activities (Lapatsina et al., 2012). It specifically binds and inhibits GLUT1 (Zhang et al., 2001). Therefore, we hypothesized that PON2 interacts with GLUT1 to facilitate GLUT1-mediated glucose transport in PDAC cells. Results of a co-immunoprecipitation assay showed that PON2 interacts with GLUT1 in PANC1 cells (Figure 5E), and co-expression of PON2 with GLUT1 enhanced 2-DG uptake (Figure 5F).
In competitive glucose uptake assay, 14C-labeled 2-DG uptake in PON2 shRNA-expressing cells was significantly reduced by much lower levels of unlabelled glucose as compared with cells expressing nonspecific shRNA (Figure 5G).
Neither the PON2-interaction with GLUT1 nor the PON2-mediated increase in GLUT1 activity was affected by the H114Q or H133Q mutation, which abrogates PON2 lactonase activity (Altenhofer et al., 2010; Horke et al., 2015), nor was there an effect of the commonly occurring polymorphism, S311C (Altenhofer et al., 2010) (Figure S6D and S6E). Furthermore, ectopic expression of GLUT1 restored the ability of PANC1 cells expressing PON2 shRNAs to form colonies in soft agar (Figure S6F). However, this restoration was abrogated by treatment with a non-metabolizable glucose analogue, 2-deoxy-D-glucose (Figure S6G). Also, PON2 did not interact with GLUT3 nor did it stimulate GLUT3-mediated glucose uptake, implying that the effects of PON2 are specific to GLUT1 and independent of GLUT3 (Figure S6H and S6I).
To better understand the mechanism by which PON2 regulates GLUT1 function, we asked if the PON2-mediated increase in GLUT1 activity is via its interaction with STOM. STOM has been shown to repress the ability of GLUT1 to transport glucose (Montel-Hagen et al., 2008; Zhang et al., 2001). We found that knockdown of STOM in PON2 shRNA-expressing PANC1 and AsPC1 cells restored glucose uptake (Figure 5H; Figure S5H and S5I). Additionally, co-immunoprecipitation experiments revealed that knockdown of PON2 enhanced the STOM and GLUT1 interaction (Figure 5I). Furthermore, knockdown of STOM in PON2 shRNA-expressing cells inhibited AMPK activation (Figure 5J; Figure S5J) and restored growth in a soft-agar assay (Figure 5K; Table S2). Collectively, these results demonstrate that PON2 regulates glucose metabolism by mediating the interaction of STOM and GLUT1, thereby regulating glucose transport in PDAC cells.
Repression of FOXO3A Tumor Suppressor Pathway is Necessary for PON2-Driven Tumor Growth and Metastasis
After confirming the mechanism behind the cellular starvation response in PON2-deficient cells and the activation of FOXO3A and its downstream target PUMA, we investigated the functional implication of the FOXO3A→PUMA pathway in PON2 inhibition-induced anoikis induction and tumor and metastasis suppression. We first tested whether the knockdown of FOXO3A or PUMA could rescue the PON2 loss-induced suppression of tumor growth. To test this, we generated PANC1 and AsPC-1 cell lines expressing PON2 shRNA alone or simultaneously expressing FOXO3A or PUMA shRNAs. We found that the simultaneous knockdown of PON2 and FOXO3 significantly rescued PDAC tumor growth, as assessed by the soft-agar colony formation and mouse tumorigenesis assays (Figure 6A; Figure S7A–S7C). Similar results were obtained with the simultaneous knockdown of PON2 and PUMA, which partially rescued growth in vitro and in vivo (Figures S7A–S7C; Table S3). We then tested if the simultaneous knockdown of PON2 with FOXO3 or PUMA could prevent anoikis induction and metastasis in PDAC cells. Our results showed that knockdown of PON2 and FOXO3 prevented anoikis induction in PDAC cells and increased PDAC lung metastasis formation in mice, whereas simultaneous knockdown of PON2 and PUMA only partially showed these effects (Figure 6B–6D; Table S3). Finally, we supplemented culture medium with glucose and downstream intermediates of the glycolytic and TCA pathways (pyruvate and succinate, respectively) and found that these decreased AMPK activation in PON2-deficient cells (Figure 6E), and high glucose rescued soft-agar growth in PON2-deficient PANC1 cells (Figure 6F; Table S2). Collectively, these results demonstrate that the loss of PON2 in PDAC cells results in cellular starvation response-mediated activation of FOXO3A and its downstream target PUMA, which uppresses tumor growth and metastasis.
Figure 6. PON2 inhibits FOXO3-mediated induction of PUMA transcription to promote tumor growth and anoikis resistance.

A. Subcutaneous tumor formation in athymic nude mice following injections of PANC1 cells carrying the indicated shRNAs. B. Anoikis assay measuring the proliferation of PANC1 cells carrying the indicated shRNAs in poly(HEMA)-coated plates. Live cell counts on the indicated days are shown. C. Immunoblot assay to measure PARP cleavage in PANC1 cells carrying the indicated shRNAs. D. Lung metastasis imaging following tail veil injection of luciferase-tagged PANC1 cells carrying the indicated shRNAs in athymic nude mice (n=3). Bioluminescence images at week 1 and week 4 under indicated conditions are shown. E. Immunoblot analysis to measure phosphorylated AMPK levels in PANC1 cells expressing PON2 or nonspecific (NS) shRNAs in DMEM supplemented with the indicated concentrations of glucose (top) or 10 mM pyruvate or succinate (Bottom). Total AMPK was used as a control. F. Soft-agar colony formation by PANC1 cell lines expressing PON2 shRNA or NS shRNA (left) grown in regular DMEM with 25 mM or 75 mM glucose. Data are presented as mean ± SEM. *p<0.05, **p<0.005. See also Figure S7 and Table S2.
Inducible Knockdown of PON2 and Pharmacological Activation of the AMPK Pathway Inhibits PDAC Tumor Growth
To test the clinical utility of targeting PON2 expression or the PON2-driven pathway in PDAC, we performed two sets of experiments. First, we asked whether inhibiting PON2 expression in established metastatic PDAC could suppress tumor growth. To this end, we used a mouse model of PDAC lung metastasis and knocked down PON2 expression using doxycycline-inducible shRNA in PDAC cells. Our results showed that the transient knockdown of PON2 inhibited the growth of established metastatic PDAC tumors in the lungs (Figure 7A). Because AMPK activation in PON2-deficient cells suppressed tumor growth and PON2 confers metastatic potential through anoikis resistance, we decided to test whether the AMPK agonist metformin could inhibit PDAC growth. We found that metformin treatment inhibited soft-agar colony formation (Figure 7B; Figure S7D; Table S2) and tumorigenesis in mice (Figure 7C). To further substantiate these results, we also used another AMPK agonist, AICAR (Corton et al., 1995). Similarly, treatment with AICAR inhibited soft-agar colony formation (Figure 7D; Figure S7E; Table S2) and tumorigenesis in mice (Figure 7E). Finally, we validated these findings using a genetic approach in which ectopic expression of constitutively active AMPK inhibited soft-agar colony formation (Figure 7F; Figure S7F; Table S2). Collectively, these results demonstrate that activation of the AMPK pathway mimics the suppression of tumor growth by PON2 inhibition. Thus, the PON2-driven pathway, identified in our study, provides a pharmacological target for the treatment of PDAC.
Figure 7. The PON2→AMPK→FOXO3A→PUMA Pathway is pharmacologically tractable for PDAC Treatment.

A. PANC1 cells expressing doxycycline-inducible PON2 or nonspecific (NS) shRNAs were injected into athymic nude mice (n=3) via the tail vein, and lung metastases were allowed to form. After 1 week, one group of mice was given water-containing doxycycline (20 μg/ml), and the other group was given regular water (without doxycycline). Representative images at week 1 and week 4 are shown. B. Soft-agar colony formation of PANC1 or AsPC-1 cells was evaluated after treatment with 500 μM metformin or PBS (control). Representative wells are shown. C. PANC1 cells (1×106) were injected subcutaneously into the flank of athymic nude mice (n=5). The mice were then treated with either vehicle or the indicated doses of metformin by oral gavage once a day for 2 weeks. Tumor volumes were measured at the indicated time points. D. Soft-agar colony formation of PANC1 or AsPC1 cells was evaluated after treatment with 5 mM AICAR or PBS (control). Representative wells are shown. E. PANC1 cells (1×106) were injected subcutaneously into the flanks of athymic nude mice (n=5). The mice were then treated with either vehicle or the indicated dose of AICAR by intraperitoneal injections once a day for 3 weeks. Tumor volumes were measured at the indicated time points. F. PDAC cells expressing constitutively active AMPK (AMPK-CA) were analyzed for anchorage-independent growth by a soft-agar assay. Representative images under the indicated conditions are shown. G. Model showing the mechanism by which PON2 promotes PDAC tumor growth and metastasis. Data are presented as mean ± SEM. **p<0.005. See also Figure S7, Table S2 and Table S3.
DISCUSSION
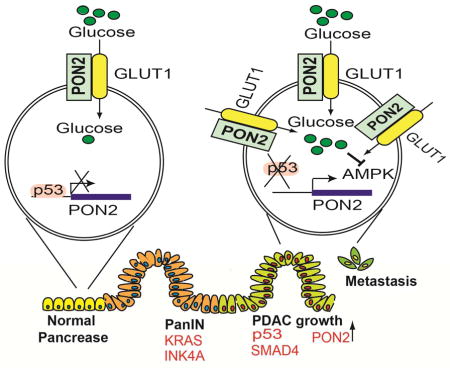
In this study, we identified PON2 as a new regulator of PDAC tumor growth and metastasis that functions by regulating GLUT1-mediated glucose transport. Our results enable us to draw several important conclusions (Figure 7G). First, PON2 modulates glucose transport and function by regulating GLUT1 transport activity. Second, PON2 is transcriptionally repressed by p53, which is consistent with previous reports that the loss of p53 is necessary for PDAC progression and metastasis (Morton et al., 2010; Weissmueller et al., 2014). Finally, PON2 inhibits the AMPK→FOXO3A→PUMA pathway to promote PDAC tumor growth and metastasis, and this pathway can be pharmacologically targeted by AMPK agonists to block PDAC tumor growth.
PON2 as a Regulator of GLUT1-Mediated Glucose Transport
Cancer cells rewire their metabolism to differently utilize glucose for their energy needs. In cancer cells, glucose is converted to energy primarily by aerobic glycolysis (i.e., the Warburg effect) (Cantor and Sabatini, 2012). This faster conversion of glucose to energy is required to meet the needs of rapidly growing cancer cells. In addition, cancer cells undergo other alterations that affect total glucose uptake and utilization, such as the overexpression of glucose transporters (GLUT1, GLUT2, and GLUT3) and glycolytic enzymes (Adekola et al., 2012; Boado et al., 1994; Yamamoto et al., 1990). Consistent with this important role of glucose metabolism in PDAC tumor growth and metastasis, we identified GLUT1 and HK2 as two candidate metabolic genes that were previously implicated in PDAC tumor growth and metastasis (Ogawa et al., 2015; Yu et al., 2015). Here, we describe a new role for PON2 in the regulation of glucose metabolism.
A previous study suggested that PON2 functions as a redox regulator in cells and that this ability might explain the oncogenic function of PON2 (Witte et al., 2011). However, in our study, we did not detect changes in ROS levels upon PON2 knockdown in PDAC cells. There are several possible reasons for this finding. First, the studies that detected the influence of PON2 on redox signaling were performed in different cell types. Second, the previous studies were performed by overexpressing PON2 rather than by modulating the endogenous levels of PON2. Our studies were performed in a physiological setting, in which we tested the effect of PON2 on cellular ROS levels by knocking down the endogenous protein.
p53-Mediated Transcriptional Repression of PON2 in PDAC
The transcription factor p53 has been shown to drive PDAC tumor progression (Weissmueller et al., 2014). In this study, we found that PON2 is overexpressed in PDAC cells, and p53 inactivation is necessary for PON2 overexpression. Tumor suppressor p53 has been shown to activate the transcription of genes that mediate its tumor suppressor function. However, p53 can also repress the transcription of a number of genes (Ginsberg et al., 1991), including SLC2A1 and TERT (Schwartzenberg-Bar-Yoseph et al., 2004; Shats et al., 2004). Our finding that PON2 loss is not involved in PDAC initiation, but cooperates with KRAS to promote tumor growth and metastasis, is consistent with the finding that mutations or deletion of p53 is a late-stage event in PDAC progression. It was previously shown that p53 influences glucose metabolism by regulating both the glycolytic pathway and oxidative phosphorylation (OXPHOS). The ability of p53 to regulate either the glycolytic pathway or OXPHOS is largely dependent upon its transcriptional targets. For example, p53 suppresses glycolysis via activating p53-induced glycolysis and apoptosis regulator (TIGAR) (Bensaad et al., 2006), while it enhances OXPHOS by upregulating the expression of cytochrome C oxidase assembly protein (SCO2) (Matoba et al., 2006). Similarly, p53 inhibits the glycolytic pathway and cellular transformation by downregulating phosphoglycerate mutase (Kondoh et al., 2005). However, in that case, it is believed that p53 stabilizes phosphoglycerate mutase at the protein level rather than affecting its transcription (Kondoh et al., 2005). Our finding that p53 represses the transcription of both SLC2A1 and PON2, which in turn stimulate GLUT1-mediated glucose transport, shows that limiting glucose uptake is a major tumor suppression mechanism. It also demonstrates that p53 is able to activate the AMPK→FOXO3A→PUMA tumor suppressor pathway by inducing the cellular starvation response as a consequence of reduced glucose uptake.
Regulation of PDAC Tumor Growth and Metastasis by PON2
Our studies identify PON2 as a regulator of PDAC tumor growth and metastasis. Loss of PON2 results in AMPK activation due to decreased GLUT1-mediated glucose transport. AMPK in turn activates the tumor suppressor FOXO3A and its downstream target PUMA, thereby inducing anoikis. PUMA is a transcriptional target of both p53 and FOXO3A (Nakano and Vousden, 2001; You et al., 2006). However, p53 is either mutated or deleted in two-thirds of PDACs; thus, PUMA activation through the AMPK→FOXO3A pathway provides a route for anoikis induction. We showed that anoikis resistance is important for PDAC tumor growth and metastasis progression and that anoikis induction, as a result of PON2 inhibition, can inhibit PDAC tumor growth and metastasis.
Targeting the PON2→AMPK→FOXO3A→PUMA Pathway for PDAC Treatment
We found that, similar to the effect of PON2 knockdown, the AMPK agonist metformin can block PDAC tumor growth. This finding supports the idea that the PON2→AMPK→FOXO3A→PUMA pathway is a pharmacologically tractable pathway for treating PDAC. In good agreement with our observations, the National Cancer Institute is currently conducting clinical trials testing metformin in combination with other therapeutic agents in the treatment of metastatic pancreatic cancer (see ClinicalTrials.gov and NCT01210911, NCT02005419, NCT01666730, NCT02336087, NCT01488552, NCT02048384, and NCT01971034).
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following
KEY RESOURCES TABLE
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| p53 | Santa Cruz Biotechnology | Cat#sc-126 |
| PON2 | Santa Cruz Biotechnology | Cat#sc-374158 |
| FOXO3A | Cell Signaling | Cat#2497 |
| β-actin | Cell signaling | Cat#4970 |
| Phospho-FOXO3A | Cell Signaling | Cat#8174 |
| Phospho-AMPK | Cell Signaling | Cat#2535 |
| AMPK | Cell Signaling | Cat#2532 |
| PARP | Cell Signaling | Cat#9542 |
| PUMA | Cell Signaling | Cat#4976 |
| GLUT1 | Cell Signaling | Cat#12939 |
| GLUT1 | Santa Cruz Biotechnology | Cat#Sc-7903, #sc- 377228 |
| GLUT3 | Santa Cruz Biotechnology | Cat #Sc-74497 |
| STOM | Bethyl Laboratories | Cat #A304-425A |
| Bacterial and Virus Strains | ||
| Lenti Virus pLKO.1 | Open Biosystems | Table S7 for clone ID |
| Lenti Virus pGIPZ | Open Biosystems | Table S7 for clone ID |
| Retro virus pBabe-Puro/KRasG12D | Forloni et al., 2014 | N/A |
| Adenovirus-p53 | Vector biolabs | Cat#1260 |
| Adenovirus-LacZ | Vector biolabs | Cat#1080 |
| Biological Samples | ||
| Human Normal Pancreata slides | Yale Pathology Tissue Services (YPTS) | N/A |
| Pancreatic ductal adeno carcinoma slides | Yale Pathology Tissue Services (YPTS) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AMPK inhibitor, Compound C | Calbiochem | Cat# 171261 |
| Metformin | Sigma | Cat#D150959 |
| AICAR | Cayman Chemicals | Cat#CAS 2627-69-2 |
| Trametenib | Selleckchem | Cat#S2673 |
| Z-VAD-FMK | Calbiochem | Cat#CAS 187389-52-2 |
| Critical Commercial Assays | ||
| FITC Annexin V Apoptosis Detection Kit I | BD Pharmingen | Cat#556547 |
| Matrigel Invasion assay | BD Biosciences | Cat#354483 |
| Cell Surface Protein Analysis | Pierce | Cat#PI89881 |
| Metabolite Measurement after Pulse Labeling with Glucose | Metabolon | N/A |
| Deposited Data | ||
| Raw data files (immunoblots, microscopy and bioluminescence imaging) | This paper | Mendeley data: http://dx.doi.org/10.17632/p64m5r9g36.1 |
| Experimental Models: Cell Lines | ||
| PANC1 | ATCC | ATCC-CRL-1469 |
| AsPC-1 | ATCC | ATCC-CRL-1682 |
| MiaPaCa-2 | ATCC | ATCC-CRL-1420 |
| SU.86.86 | ATCC | ATCC-CRL-1837 |
| HPNE-hTERT | ATCC | ATCC-CRL-4036 |
| HPNE-hTERT E6/E7/st | ATCC | ATCC-CRL-4037 |
| IKRAS2 | Ying et al., 2012 | N/A |
| Experimental Models: Organisms/Strains | ||
| mouse: NCr nude | Taconic | CrTac:NCr-Foxn1nu |
| Oligonucleotides | ||
| PON2 Forward: 5′ AATCCACATGGCATCAGCAC 3′ Reverse: 5′ CAAGGTCCAAGGCCAAGAAG 3′ |
This paper | N/A |
| HK2 Forward: 5′ AGCTGCTGGAGGTCAAGAGG 3′ Reverse: 5′ CGGTCCAACAGCTGTGATGT 3′ |
This paper | N/A |
| SLC2A1 Forward: 5′ GATGATGCGGGAGAAGAAGG 3′ Reverse: 5′ GCCTTCTCGAAGATGCTCGT 3′ |
This paper | N/A |
| LDHA Forward: 5′ GTGCCTGTGTGGAGTGGTGT 3′ Reverse: 5′ ATGGCCCAGGATGTGTAACC 3′ |
This paper | N/A |
| For primers see Table S7 and S5 | This paper | N/A |
| Recombinant DNA | ||
| PON2 cDNA | Open Biosystems | Clone ID: 6056277 |
| pEGFP-PON2 | This paper | N/A |
| pBabe-PON2 | This paper | N/A |
| pGL4.14-PON2 promoter | This paper | N/A |
| pGL4.14-PUMA promoter | This Paper | N/A |
| GLUT1-eGFP/pcDNA-DEST47 | Takanaga et al., 2008 | Addgene plasmid # 18729 |
| PMXS-SLC2A3 | Birsoy et al., 2014 | Addgene plasmid # 72877 |
| PEBG-AMPKα1(1-312) | Egan et al., 2010 | Addgene plasmid # 27632 |
| Software and Algorithms | ||
| GraphPad Prism 7 software | Graphpad | https://www.graphpad.com |
| ImageJ software | NIH | https://imagej.nih.gov/ij/ |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Narendra Wajapeyee (Narendra.Wajapeyee@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strain of mouse and details of cell lines used are included in the key resource table and description of the animal model and culture conditions for cell lines are included under relevant sections of method details.
METHOD DETAILS
Bioinformatics Analysis to Identify Overexpressed Metabolic Genes
Datasets that compared gene expression between pancreatic cancer and normal pancreatic tissues were identified by searching the Oncomine cancer profiling database. The Grutzmann, Ishikawa, Pei, and Badea datasets were chosen for data analysis (Badea et al., 2008; Grutzmann et al., 2004; Ishikawa et al., 2005; Pei et al., 2009). We analyzed the top 10% of significantly overexpressed genes from each of the datasets for genes implicated in cellular metabolism. Using a comprehensive list of 2,752 genes encoding human metabolic enzymes (Possemato et al., 2011), we identified 13 metabolic regulator genes that were common in all four datasets (Table S1).
Cell Culture, shRNAs, Transfection, and Retrovirus and Lentivirus Preparation
The PANC1, AsPC-1, MiaPaCa-2, SU.86.86, HPNE-hTERT, and HPNE-hTERT E6/E7/st cell lines were obtained from American Type Culture Collection (ATCC) and maintained as recommended. Mouse iKRAS-derived cells were a kind gift from Dr. Ronald A. DePinho. The cells were maintained in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum, penicillin, and streptomycin.
All lentiviral shRNA plasmids were obtained from Open Biosystems (Table S1). Lentivirus particles were generated by co-transfecting shRNA plasmids and lentiviral packaging plasmids pSPAX2 and pMD2.G into 293T cells using the Effectene transfection reagent (Qiagen) as per the manufacturer’s instructions. pBABE-puro and pBABE-puro/KRASG12D were used to generate retrovirus particles as described previously (Forloni et al., 2014).
Plasmids and Cloning
PON2 cDNA was obtained from Open Biosystems (Clone ID: 6056277) and cloned in the pBABE-hygro retroviral vector between the BamHI and SalI restriction sites and between HindII and BamHI sites in pEGFP-C1 vector. The PON2 and PUMA promoters were PCR-amplified from the BAC vectors RP11-104H16 and RP11-104K20, respectively, and cloned into pGL4.14 upstream of the firefly luciferase gene. Site-directed mutagenesis was performed with QuikChange II XL Site-Directed Mutagenesis Kit (Agilent), as per the manufacturer’s protocol. GLUT1-eGFP/pcDNA-DEST47 was a gift from Wolf Frommer (Addgene plasmid # 18729) (Takanaga et al., 2008). The GLUT3 construct, PMXS-SLC2A3 was a gift from David Sabatini (Addgene plasmid # 72877) (Birsoy et al., 2014). The constitutively active AMPKα1 construct, pEBG-AMPKα1(1-312) was a gift from Reuben Shaw (Addgene plasmid # 27632) (Egan et al., 2011) Primer sequences for the PON2 gene, PON2 and PUMA promoters, and mutagenesis are listed in Table S7.
RNA Isolation, RT-qPCR, and Chromatin Immunoprecipitation (ChIP) Assay
Total RNA was extracted using TRIzol (Invitrogen) and purified using the RNeasy Mini Kit (Qiagen), as per the manufacturer’s instructions. Reverse transcription was performed using ProtoScript First Strand cDNA Synthesis Kit (New England Biolabs). Quantitative PCR was then performed using Power SYBR Green Master Mix (Life Technologies). Chromatin immunoprecipitation (ChIP) was performed as described previously (Wajapeyee et al., 2008). Briefly, AsPC-1 cells were infected with adenovirus expressing p53-GFP or GFP (control). After 48 hours, the cells were paraformaldehyde-fixed and lysed in SDS lysis buffer (1% SDS, 50 mM Tris-HCl [pH 8.0], 10 mM EDTA, and protease inhibitor cocktail [Roche]) and then sonicated at 4°C. The lysates were diluted with ChIP buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl [pH 8.0], 16.7 mM NaCl, and protease inhibitor cocktail [Roche]), and the samples were incubated with antibodies against p53 (Santa Cruz Biotechnology), followed by immobilization on protein A/G agarose beads (Life Technologies). The chromatin was eluted, and DNA was extracted following phenol-chloroform treatment. Quantitative PCR (qPCR) was performed using PON2 promoter-specific primers. The ChIP assay using HPNE-hTERT cells was performed similarly using a control IgG antibody or antibody against p53 (Santa Cruz Biotechnology). The ChIP assay to measure FOXO3A occupancy of the PUMA promoter was performed on PANC1 cells expressing nonspecific or PON2 shRNAs and grown in attached or detached conditions. After paraformaldehyde fixing and lysis in SDS lysis buffer as described above, chromatin immunoprecipitation was performed with control IgG antibody (Santa Cruz Biotechnology) or antibody against FOXO3A (Cell Signaling Technology). Fold-enrichment was calculated as the ratio of immunoprecipitated DNA to input DNA. Primer sequences for the RT-qPCR and ChIP assays are listed in Table S7.
Luciferase Reporter Assay
AsPC-1 cells infected with adenovirus carrying p53-GFP or GFP (control) or HPNE-hTERT cells expressing p53 or nonspecific shRNAs were transfected along with the PON2 promoter-firefly luciferase reporter containing either wild type or mutated p53 DNA binding site. Firefly luciferase activity was normalized to Renilla luciferase activity from the co-transfected control pRL-TK. PANC1 cells expressing shRNAs were transfected with the PUMA promoter-firefly luciferase reporter containing either wild type or mutated forkhead response element (FHRE) (You et al., 2006). After 24 hours, the cells were seeded in regular or poly(HEMA)-coated plates. After another 24 hours, the cells were lysed in passive lysis buffer, and the luciferase reporter assay was performed using the Dual-Luciferase Reporter Assay Kit (Promega). Relative reporter activity was measured as ratio of firefly to Renilla luciferase activity and reported as the average of triplicate measurements.
Immunoblot Analyses
Immunoblot analysis was performed as described previously (Santra et al., 2009). Subcutaneous tumor samples from mice were chopped into small pieces and lysates were prepared by disruption in RIPA buffer using TissueLyzer II (Qiagen) followed by centrifugation. Protein concentrations were estimated using the Bradford Protein Assay Kit (Bio-Rad) or Pierce BCA Protein Assay Kit (Thermo Scientific), as per the manufacturers’ instructions. All antibodies used for immunoblot analyses and their dilutions are listed in Table S7.
Immuno-histochemistry
Paraffin embedded PDAC slides from primary tumor site, metastatic site and normal pancreas were obtained from Yale Pathology Tissue Services (YPTS). H&E staining and PON2 IHC were performed by histology services of the Department of Pathology, Yale school of medicine. PON2 antibody was used at a dilution of 1:500 dilution followed by secondary anti-mouse horseradish peroxidase (HRP)-conjugated antibody. The PDAC and normal pancreata slides stained for PON2 were scored by Dr. Guoping Cai for PON2 staining after the clinical identity of the slide was obscured. Summary of immunohistochemistry staining for PON2 in human pancreata and PDAC samples are provided in Table S4. PON2 antibody used for IHC analyses and its dilution is listed in Table S7.
Measurement of ATP/ADP ratio
PANC1 or AsPC-1 cells (1×105) were seeded in regular or ultra low-attachment 12 well plates and cultured for 48 hrs. Adherent and non-adherent cells were collected and re-suspended at a density of 104 cells/10μl in PBS and transferred to clear bottom, white-walled 96 well plates. ATP/ADP ratio was measured using ADP/ATP Ratio Assay Kit (Sigma-Aldrich) as per manufacturer’s protocol.
Soft Agar Assay and Anoikis Assay
Soft agar assays were performed by seeding 5×103 cells (PANC1, AsPC-1, SU.86.86, or MiaPaCa-2) stably expressing the indicated constructs on 0.35% low melting point agarose (Sigma) and layered on a 0.7% low melting point agarose base. For soft agar assays in high glucose conditions the basal glucose concentration of 25mM in DMEM was increased to 75mM through supplementation with exogenous D-Glucose. 10 mM of 2DG was added where indicated. After 4 weeks of growth, the colonies were stained with 0.005% crystal violet solution. Colony size was measured using microscopy and plotted as percent relative colony size with respect to the control cells. Colony numbers were counted using ImageJ software (https://imagej.nih.gov/ij/) and average number of the replicates are tabulated. Statistical analysis was performed using Student’s t test using GraphPad Prism 7 software. Colony numbers and relative colony size for all soft agar assays are provided in Table S2.
To evaluate anoikis, 6-well tissue culture plates were coated with 200μL poly(2-hydroxyethyl methacrylate) (poly[HEMA]; Sigma) and left for 103hours in a laminar flow hood. PANC1 or AsPC-1 cells (1×105) expressing shRNAs were seeded in the poly(HEMA)-coated plates in regular DMEM or RPMI respectively and incubated for indicated days at 37°C. Live cells identified through trypan-blue dye exclusion were counted using a hemocytometer. For measuring cell proliferation in attached or detached condition under varying concentrations of glucose, PANC1 or AsPC-1 cells (1×105) were seeded in regular or ultra low-attachment 12 well plates in glucose free DMEM or RPMI respectively supplemented with 10% dialyzed fetal bovine serum and indicated concentration of glucose. The experiments were performed in triplicate.
Mouse Tumorigenesis Experiments
PANC1 and AsPC-1 cells stably expressing firefly luciferase under a CMV promoter were generated by co-transfecting the transposon vector piggyBac GFP-Luc and the helper plasmid Act-PBase, as described previously (Ding et al., 2005). Cells with stable transposon integration were selected with blasticidin S (Invitrogen). Athymic nude mice (both male and female) (NCr nu/nu, 8 weeks old) were used for all animal experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Yale University and were performed in accordance with the IACUC guidelines.
a. Subcutaneous Injection
Athymic nude mice were injected subcutaneously with PANC1 or AsPC-1 cells (1×106) expressing shRNAs or iKRAS-derived cells stably expressing PON2 cDNA or carrying an empty vector. Tumors were measured every week, and tumor volumes were calculated using the formula: length × width2 × 0.5. For mice injected with iKRAS-derived cells, doxycycline (Sigma) was given from day 1 of the injection (20 μg/ml in drinking water). Metformin was administered by oral gavage at 250 or 500 mg/kg body weight every day starting 10 days after the injection of cancer cells until the end of the experiment.
b. Orthotopic Injection of Pancreas
Athymic nude mice were anesthetized with isoflurane, and the spleen and underlying pancreas were located through an intra-abdominal incision. Matrigel suspension (20 μl) containing 1×105 PANC1 or AsPC-1 cells carrying shRNAs were injected into the tail of the pancreas. The abdominal wall and skin were then closed with sutures. The mice were imaged every week until the completion of the experiment, at which time they were injected with D-luciferin and euthanized, and the blood, liver, and lungs were collected. The liver and lungs were placed in a petri dish, and images were obtained using the IVIS Xenogen imaging system.
c. Splenic Injection of Cancer Cells
PANC1 cells (5×105) carrying nonspecific or PON2 shRNAs were suspended in 100 μl PBS and injected into the spleen of anesthetized athymic nude mice through an intra-abdominal incision. The abdominal wall and skin were then closed with sutures. The mice were imaged every week until the completion of the experiment.
d. Tail Vein Injection
PANC1 or AsPC-1 cells (5×105) carrying shRNAs were injected into athymic nude mice via the tail vein. The mice were imaged using the IVIS Xenogen imaging system every week until the completion of the experiment. For experiments involving doxycycline-inducible shRNA, doxycycline (20 μg/ml; Sigma) was added to the drinking water starting 1 week after the injection. Total luminescence counts of tumor bearing areas were measured using Living Image® software and statistical analysis was performed using student’s t test. Average total luminescence counts for all in vivo luciferase imaging experiments are listed in Table S3.
Circulating Tumor Cell Measurement
At the completion of the tumorigenesis experiment, the mice were sacrificed, and 1 ml blood was collected from each mouse by cardiac puncture. Genomic DNA was isolated using the DNeasy Blood and Tissue Kit (Qiagen). The abundance of circulating tumor cells of human origin was determined by qPCR using primers specific to the human Alu repeat sequence. Mouse actin was used as a control. Quantitative PCR was performed using the Power SYBR Green Master Mix (Life Technologies). A group of three mice was used to determine the relative circulating tumor cell load, with the mouse bearing tumor cells carrying nonspecific shRNA set as 1. Primers used for this analysis are listed in the Table S7.
Apoptosis Measurement Using Annexin V/Propidium Iodide Staining and PARP Cleavage Immunoblots
PANC1 or AsPC-1 cells carrying shRNAs were grown in attached or detached conditions in poly(HEMA)-coated plates. Apoptosis was analyzed by Annexin V and propidium iodide staining and flow cytometry using the FITC-Annexin Apoptosis Detection Kit I (BD Pharmingen), as per the manufacturer’s protocol. PARP cleavage was detected by immunoblotting using an anti-PARP antibody (Cell Signaling Technology). The pan-caspase inhibitor Z-VAD-FMK (Calbiochem) was added as indicated.
Wound-Healing and Matrigel Invasion Assay
For the wound healing assay, PANC1 cells carrying PON2 or nonspecific shRNAs were grown in 12-well plates until fully confluent. A scratch was created using a sterile 20-μl pipette tip, and the migration of cells into the wound was monitored by light microscopy at 0, 4, and 12 hours. The invasion assay was carried out in a BioCoat Growth Factor Reduced Matrigel Invasion Chamber (BD Biosciences, Cat# 354483) using PANC1 cells carrying nonspecific or PON2 shRNAs. Briefly 5×104 cells/insert were seeded in triplicate in low-serum medium after 6 hours of serum starvation. The cells were allowed to migrate towards serum-rich medium in the bottom well for 20 hours. The number of cells migrating through the Matrigel was quantified by imaging after DAPI staining; 8–12 fields per membrane were counted, and quantification of nuclei was performed using Image J.
Measurement of Reactive Oxygen Species
PANC1 cells (5×104) expressing either PON2 or nonspecific shRNAs were labeled with H2DCFDA (Invitrogen) as per the manufacturer’s protocol. ROS levels were measured by fluorescence-activated cell sorting.
Measurement of Oxygen Consumption and Extracellular Acidification Rates
Oxygen consumption and extracellular acidification rates were measured using the XFe24 analyzer (Seahorse Bioscience). Briefly PANC1 or AsPC-1 cells carrying nonspecific or PON2 shRNAs were seeded at a density of 40,000 cells/well in triplicates. After 24 hours, the culture medium was replaced with assay medium, and oxygen consumption and extracellular acidification rates were measured for 2 hours.
Metabolite Analysis
PANC1 or AsPC-1 cells (5×106) were plated in 100-mm dishes. After 24 hours, the cells were washed with ice-cold calcium- and magnesium-free phosphate-buffered saline (PBS) and the cells were scraped from the dishes. Cells were extracted with ice-cold 50% aqueous acetonitrile with 10 mM spermine (20 μl buffer/mg cells). Nucleotide concentrations in the supernatant were analyzed by liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) in selected ion monitoring mode for ion pairs of ATP (506/79, 506/134, and 506/159), ADP (426/79, 426/134, and 426/159), AMP (346/79 and 346/134), and UDP-glucose (565/323). Concentrations were calculated from the concentration standard curves of absorbance of nucleotides compared with those of the internal standard (d4-taurine: 129/80).
Metabolite Measurement after Pulse Labeling with Glucose
To measure the flux of glucose-derived carbon through the major bioenergetics pathways, we used U-13C6-labeled glucose. PANC1 cells (5×106) carrying nonspecific or PON2 shRNAs were seeded in 100-mm dishes. After 24 hours, the cells were incubated in glucose-free DMEM supplemented with 10 mM [U-13C6]glucose (Cambridge Isotope Laboratories, Inc.) for 3 hours. Cells were washed twice with 5% mannitol and scraped from the dishes. The cell pellets were shipped to Human Metabolome Technologies (Boston, USA) for additional processing. Cells were extracted with methanol and an internal standard solution (10 μM) was added. The extracted metabolites were freeze dried and analyzed using capillary electrophoresis-time of flight mass spectrometry (CE-TOFMS) by Human Metabolome Technologies.
Glucose Uptake Assay
PANC1 or AsPC-1 cells (2×105), infected with lentiviral shRNA particles or transfected with indicated plasmids, were seeded into 12-well plates and incubated in low-glucose medium (5 mM glucose) for 2.5 hours followed by glucose-free medium for 30 minutes. To each well, 0.2 μCi of [1-14C]-2-DG was added alone or with nonradioactive 2-DG, and the cells were incubated for 5 min at room temperature. The reaction was terminated by adding 200 mM 2-DG. The plates were washed three times in ice-cold PBS, and cells were scraped into 500 μl deionized water. To assess glucose uptake, radioactivity was determined by scintillation counting. The relative glucose uptake rate was calculated by setting the radioactivity of cells expressing nonspecific shRNA or the vector control as 1.
Cell Surface Protein Analysis (Biotinylation Assay)
The membrane expression of glucose transporters was assessed in PANC1 cells expressing nonspecific or PON2 shRNA using the Pierce cell surface protein isolation kit according to the manufacturer’s protocol. The eluted protein and input lysate were analyzed for GLUT1 and GLUT3 levels by immunoblotting using anti-GLUT1 and anti-GLUT3 antibodies (Table S5).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification with statistical parameters for soft agar assays is provided in Table S2. Quantification of total luminescence counts of tumor bearing areas was measured using Living Image® software. Quantification for bioluminescence assays with statistical parameters is provided in Table S3. Statistical methodologies are reported in the figure legends.
DATA AND SOFTWARE AVAILABILITY
Data pertaining to immunoblots, bioluminescent imaging and microscopy are deposited at Mendeley data http://dx.doi.org/10.17632/p64m5r9g36.1
Supplementary Material
HIGHLIGHTS.
PON2 is necessary for PDAC tumor growth and metastasis.
Tumor suppressor p53 transcriptionally represses PON2.
PON2 regulates glucose metabolism by modulating GLUT1-mediated glucose transport.
PON2 inhibits FOXO3A and PUMA via AMPK to promote tumor growth and metastasis.
Acknowledgments
This work was in part supported by the grant from the Hirshberg Foundation for Pancreatic Cancer Research. We gratefully acknowledge the following grants from the National Institutes of Health: R21CA197758-01 (N.W.), R21CA191364-01 (N.W.), R21CA195077-01A1 (N.W.), R01CA200919-01 (N.W.), and 1R41CA195908-01A1 (N.W.). N.W. is also supported by a research scholar grant from the American Cancer Society (128347-RSG-15-212-01-TBG).
Footnotes
AUTHOR CONTRIBUTIONS
A.N. and N.W. conceived the idea for and designed the experiments. A.N. performed most of the experiments with the help of L.S., and T.H., and P.K. helped with animal experiments. R.A.C. performed the all survival surgeries with help from N.G for orthotopic injections and splenic injections. Immunohistochemical analysis was performed by G. C. LC-MS/MS analysis was performed by G. Cline. Bioinformatics analyses were performed by S.K.D. A.N. and N.W. analyzed and interpreted the data. A.N. and N.W. co-wrote the manuscript. All authors commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650–654. doi: 10.1097/CCO.0b013e328356da72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenhofer S, Witte I, Teiber JF, Wilgenbus P, Pautz A, Li H, Daiber A, Witan H, Clement AM, Forstermann U, et al. One enzyme, two functions: PON2 prevents mitochondrial superoxide formation and apoptosis independent from its lactonase activity. J Biol Chem. 2010;285:24398–24403. doi: 10.1074/jbc.M110.118604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology. 2008;55:2016–2027. [PubMed] [Google Scholar]
- Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–112. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boado RJ, Black KL, Pardridge WM. Gene expression of GLUT3 and GLUT1 glucose transporters in human brain tumors. Brain Res Mol Brain Res. 1994;27:51–57. doi: 10.1016/0169-328x(94)90183-x. [DOI] [PubMed] [Google Scholar]
- Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PM, Groehler AL, Lee KM, Ouellette MM, Khazak V, Der CJ. K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res. 2007;67:2098–2106. doi: 10.1158/0008-5472.CAN-06-3752. [DOI] [PubMed] [Google Scholar]
- Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2:881–898. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelur DS, Ernstrom GG, Goodman MB, Yao CA, Chen L, ROH, Chalfie M. The mechanosensory protein MEC-6 is a subunit of the C. elegans touch-cell degenerin channel. Nature. 2002;420:669–673. doi: 10.1038/nature01205. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhao S, Nakada K, Kuge Y, Tamaki N, Okada F, Wang J, Shindo M, Higashino F, Takeda K, et al. Dominant-negative hypoxia-inducible factor-1 alpha reduces tumorigenicity of pancreatic cancer cells through the suppression of glucose metabolism. Am J Pathol. 2003;162:1283–1291. doi: 10.1016/s0002-9440(10)63924-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–653. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122:473–483. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forloni M, Dogra SK, Dong Y, Conte D, Jr, Ou J, Zhu LJ, Deng A, Mahalingam M, Green MR, Wajapeyee N. miR-146a promotes the initiation and progression of melanoma by activating Notch signaling. Elife. 2014;3:e01460. doi: 10.7554/eLife.01460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg D, Mechta F, Yaniv M, Oren M. Wild-type p53 can down-modulate the activity of various promoters. Proc Natl Acad Sci U S A. 1991;88:9979–9983. doi: 10.1073/pnas.88.22.9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- Grutzmann R, Pilarsky C, Ammerpohl O, Luttges J, Bohme A, Sipos B, Foerder M, Alldinger I, Jahnke B, Schackert HK, et al. Gene expression profiling of microdissected pancreatic ductal carcinomas using high-density DNA microarrays. Neoplasia. 2004;6:611–622. doi: 10.1593/neo.04295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, et al. A census of human soluble protein complexes. Cell. 2012;150:1068–1081. doi: 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TL, Zhang YJ, Jiang H, Li XH, Zhu H, Zheng KL. The c-Myc-LDHA axis positively regulates aerobic glycolysis and promotes tumor progression in pancreatic cancer. Med Oncol. 2015;32:187. doi: 10.1007/s12032-015-0633-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- Hishinuma S, Ogata Y, Tomikawa M, Ozawa I, Hirabayashi K, Igarashi S. Patterns of recurrence after curative resection of pancreatic cancer, based on autopsy findings. J Gastrointest Surg. 2006;10:511–518. doi: 10.1016/j.gassur.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Horke S, Xiao J, Schutz EM, Kramer GL, Wilgenbus P, Witte I, Selbach M, Teiber JF. Novel Paraoxonase 2-Dependent Mechanism Mediating the Biological Effects of the Pseudomonas aeruginosa Quorum-Sensing Molecule N-(3-Oxo-Dodecanoyl)-L-Homoserine Lactone. Infect Immun. 2015;83:3369–3380. doi: 10.1128/IAI.00141-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M, Yoshida K, Yamashita Y, Ota J, Takada S, Kisanuki H, Koinuma K, Choi YL, Kaneda R, Iwao T, et al. Experimental trial for diagnosis of pancreatic ductal carcinoma based on gene expression profiles of pancreatic ductal cells. Cancer Sci. 2005;96:387–393. doi: 10.1111/j.1349-7006.2005.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–185. [PubMed] [Google Scholar]
- Lapatsina L, Brand J, Poole K, Daumke O, Lewin GR. Stomatin-domain proteins. Eur J Cell Biol. 2012;91:240–245. doi: 10.1016/j.ejcb.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Mohammad GH, Olde Damink SW, Malago M, Dhar DK, Pereira SP. Pyruvate Kinase M2 and Lactate Dehydrogenase A Are Overexpressed in Pancreatic Cancer and Correlate with Poor Outcome. PLoS One. 2016;11:e0151635. doi: 10.1371/journal.pone.0151635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montel-Hagen A, Kinet S, Manel N, Mongellaz C, Prohaska R, Battini JL, Delaunay J, Sitbon M, Taylor N. Erythrocyte Glut1 triggers dehydroascorbic acid uptake in mammals unable to synthesize vitamin C. Cell. 2008;132:1039–1048. doi: 10.1016/j.cell.2008.01.042. [DOI] [PubMed] [Google Scholar]
- Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107:246–251. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- Ogawa H, Nagano H, Konno M, Eguchi H, Koseki J, Kawamoto K, Nishida N, Colvin H, Tomokuni A, Tomimaru Y, et al. The combination of the expression of hexokinase 2 and pyruvate kinase M2 is a prognostic marker in patients with pancreatic cancer. Mol Clin Oncol. 2015;3:563–571. doi: 10.3892/mco.2015.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. 2013;1833:3481–3498. doi: 10.1016/j.bbamcr.2013.06.026. [DOI] [PubMed] [Google Scholar]
- Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–266. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra MK, Wajapeyee N, Green MR. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature. 2009;459:722–725. doi: 10.1038/nature08011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa A, Capelli P, Mukai K, Zamboni G, Oda T, Iacono C, Hirohashi S. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol. 1993;142:1534–1543. [PMC free article] [PubMed] [Google Scholar]
- Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64:2627–2633. doi: 10.1158/0008-5472.can-03-0846. [DOI] [PubMed] [Google Scholar]
- Shats I, Milyavsky M, Tang X, Stambolsky P, Erez N, Brosh R, Kogan I, Braunstein I, Tzukerman M, Ginsberg D, et al. p53-dependent down-regulation of telomerase is mediated by p21waf1. J Biol Chem. 2004;279:50976–50985. doi: 10.1074/jbc.M402502200. [DOI] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanaga H, Chaudhuri B, Frommer WB. GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochim Biophys Acta. 2008;1778:1091–1099. doi: 10.1016/j.bbamem.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo M, Crotty GF, O’Suilleabhain C, Ridgway PF, Conlon KC, Power DG, McDermott RS. Identification of distinct phenotypes of locally advanced pancreatic adenocarcinoma. J Gastrointest Cancer. 2013;44:73–78. doi: 10.1007/s12029-012-9419-9. [DOI] [PubMed] [Google Scholar]
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363– 374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmueller S, Manchado E, Saborowski M, Morris JPt, Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. 2014;157:382–394. doi: 10.1016/j.cell.2014.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte I, Altenhofer S, Wilgenbus P, Amort J, Clement AM, Pautz A, Li H, Forstermann U, Horke S. Beyond reduction of atherosclerosis: PON2 provides apoptosis resistance and stabilizes tumor cells. Cell Death Dis. 2011;2:e112. doi: 10.1038/cddis.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Seino Y, Fukumoto H, Koh G, Yano H, Inagaki N, Yamada Y, Inoue K, Manabe T, Imura H. Over-expression of facilitative glucose transporter genes in human cancer. Biochem Biophys Res Commun. 1990;170:223–230. doi: 10.1016/0006-291x(90)91263-r. [DOI] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A, Mak TW. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203:1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Zhou Q, Zhou Y, Fu Z, Tan L, Ye X, Zeng B, Gao W, Zhou J, Liu Y, et al. Metabolic phenotypes in pancreatic cancer. PLoS One. 2015;10:e0115153. doi: 10.1371/journal.pone.0115153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ, Abbud W, Prohaska R, Ismail-Beigi F. Overexpression of stomatin depresses GLUT-1 glucose transporter activity. Am J Physiol Cell Physiol. 2001;280:C1277–1283. doi: 10.1152/ajpcell.2001.280.5.C1277. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xing Y, Zhang L, Mei Y, Yamamoto K, Mak TW, You H. Regulation of cell cycle progression by forkhead transcription factor FOXO3 through its binding partner DNA replication factor Cdt1. Proc Natl Acad Sci U S A. 2012;109:5717–5722. doi: 10.1073/pnas.1203210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.