


Abstract
Regulator of G Protein Signaling (RGS) 17 is an overexpressed promoter of cancer survival in lung and prostate tumors, the knockdown of which results in decreased tumor cell proliferation in vitro. Identification of drug-like molecules inhibiting this protein could ameliorate the RGS17’s pro-tumorigenic effect. Using high-throughput screening, a chemical library containing natural products was interrogated for inhibition of the RGS17–Gαo interaction. Initial hits were verified in control and counter screens. Leads were characterized via biochemical, mass spectrometric, Western blot, microscopic, and cytotoxicity measures. Four known compounds (1–4) were identified with IC50 values ranging from high nanomolar to low micromolar. Three compounds were extensively characterized biologically, demonstrating cellular activity determined by confocal microscopy, and two compounds were assessed via ITC exhibiting high nanomolar to low micromolar dissociation constants. The compounds were found to have a cysteine-dependent mechanism of binding, verified through site-directed mutagenesis and cysteine reactivity assessment. Two compounds, sanguinarine (1) and celastrol (2), were found to be cytostatic against lung and prostate cancer cell lines and cytotoxic against prostate cancer cell lines in vitro, although the dependence of RGS17 on these phenomena remains elusive, a result that is perhaps not surprising given the multimodal cytostatic and cytotoxic activities of many natural products.
Graphical abstract
Regulator of G protein signaling (RGS) proteins temporally regulate the heterotrimeric G protein signaling cascades elicited by G protein-coupled receptor (GPCR) activation. Agonist binding to GPCRs promotes the exchange of GDP for GTP at the Gα active site, causing dissociation of the heterotrimer Gαβγ into Gα and Gβγ, resulting in initiation of multiple signaling cascades.1,2 RGS proteins, as GTPase accelerating proteins (GAPs), bind the GTP-bound Gα subunit of Gαi/o and Gαq G proteins, stabilizing the transition state and accelerating GTP hydrolysis, returning Gα to its GDP-bound inactive state, promoting association of the heterotrimer and terminating the G protein signaling cascade.3
Normal expression levels of RGS proteins provide critical control of cell signaling events, and dysregulation of RGS expression has been reported in a number of disease pathogeneses.4,5 RGS17 is one such RGS, with transcript and protein enrichment reported for lung and prostate carcinoma and more recently for breast cancer and hepatocellular carcinoma.6–11 For lung and prostate cancer, RGS17 overexpression results in dysregulation of the balance between Gαs and Gαi/o signaling. These two G proteins regulate intercellular cAMP levels through the stimulation or inhibition of adenylyl cyclases. Overexpression of RGS17 shifts the balance due to its function as a negative regulator of Gαi/o, promoting increased levels of cAMP, increased activation of protein kinase A (PKA), and increased activation of CRE response element binding protein (CREB),6 a mechanism that is likely present in breast cancer and hepatocellular carcinoma.7,8 Most importantly, RGS17 knockdown results in decreased cell proliferation in culture for all of these cancer types, and injection of RGS17-depleted H1299 cells into nude mice resulted in decreased tumor load and volume.6–8 These results have established RGS17 as a promising therapeutic target for these malignancies and suggest that identification of a small-molecule inhibitor of RGS17 may act to restore proper cell signaling and inhibit the RGS17-dependent proliferation of these cancers. Importantly, since RGS17 is expressed primarily in the brain,12 a selective RGS17 inhibitor that remains blood brain barrier impermeable would function as an inhibitor of tumor growth while exhibiting minimal off-target side effects, making RGS17 a very attractive anticancer target.
Therefore, a screen interrogating a library of 2320 compounds comprising known drugs, drug-like molecules, and natural products against the RGS17–Gαo protein–protein interaction was performed, the results of which are presented herein. This led to the identification of four natural products (1–4) that inhibit the RGS17–Gα interaction and inhibit the functional activity of the RGS. The mechanism of action for inhibition of the protein–protein interaction was investigated, with characterization focused on potential cysteine reactivity and covalent modification, a mechanism observed with previously reported RGS4 inhibitors.13–16 Additionally, it was established that the lead compounds are capable of disrupting the RGS–G protein interaction in cells. Finally, the cytostatic and cytotoxic potential of the compounds against lung and prostate cancers in cell culture was assessed.
RESULTS AND DISCUSSION
Identification of RGS17 Natural Product Inhibitors
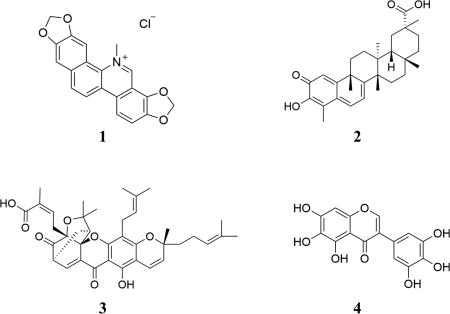
Our previously published HTS paradigm17 was successfully miniaturized from 384- to a 1536-well format with a calculated Z-factor of 0.65,18 and the 2320-compound MicroSource SPECTRUM Diversity library (Discovery Systems, Inc., Gaylordsville, CT, USA) was screened for inhibitors of the RGS17–Gαo interaction (Figure S1A,B, Supporting Information, Table 1). A cutoff of 50% inhibition or greater was established to consider a compound as an initial hit. Initial hits were assessed in a variety of follow-up assays including single-point counter screens, filtration of putative problematic substructures by computational Pan Assay Interference Structure analysis,19–21 and dose–response analysis against the RGS17–G protein interaction as well as a biotinylated-GST counterscreen to identify compounds that directly interfere with the assay. Hits were triaged at each stage, resulting in a reduction from 99 initial hits to four compounds (1–4) considered putative lead molecules, all of which are natural products (Table 1). Compound 1 is the quaternary benzo[c]-phenthridine alkaloid sanguinarine, which can be isolated from several plant sources including the seed oil of Argemone mexicana.22 Compound 2 is the pentacyclic triterpenoid celastrol from Tripterygium wilfordii.23 Compound 3 is the xanthonoid (−)-gambogic acid from Garcinia hanburyi.24 Finally, compound 4 is the hexahydroxy flavonoid irigenol, which has been synthesized from the isolated products of Morus tinctoria.25 Each of these four molecules was subjected to additional characterization to determine the mechanism of RGS inhibition as described below.
Table 1.
Summary of Screening Results
| spectrum library | 2320 compounds |
hit rate |
|---|---|---|
| initial AlphaScreen hits | 99 compounds | 4.26% |
| single point biotin-GST control counterscreenfilter | 41 compounds | 1.76% |
| favorable IC50’s and structures filter | 7 compounds | 0.30% |
| dose−response curves IC50’s PPI vs biotin-GSTfilter | 4 compounds | 0.17% |
| RGS17 specific differential scanning fluorimetry | 3 compounds | 0.13% |
Compounds 1–4 Inhibit RGS17 GAP Activity
The ability of the lead compounds to inhibit the functional activity of RGS17 was examined using a previously described malachite green assay.26 In this assay, compounds 1–4 were determined to inhibit RGS17’s GAP activity with IC50 values of 12, 6, 10, and 35 µM, respectively (Figure S2A, Supporting Information). These were comparable to the IC50 values determined in initial follow-up assessment of hits using the AlphaScreen platform (data not shown). To verify that the compounds bind to the RGS protein rather than the G protein, differential scanning fluorimetry (DSF) was utilized. Treatment with 1, 2, and 4 led to only minor melting temperature (Tm) shifts for RGS17 that were statistically insignificant. However, compound 3 resulted in a drastic negative shift in Tm for the G protein (data not shown). This indicates that 3 is likely binding to Gα protein and would therefore be undesirable toward the goal of this work. A small molecule that inhibits the function of RGS17 would act to restore proper G protein signaling regulation, while an inhibitor of Gα would serve to cause further dysregulation of the system. Therefore, compound 3 was excluded from further evaluation.
Isothermal Titration Calorimetry (ITC)
Isothermal titration calorimetry was utilized to further characterize the binding of the lead compounds to RGS17. Two compounds, 1 and 4, were amenable to ITC and exhibited affinity for RGS17 with Kd’s of 1 µM and 710 nM, respectively (Figure S2B,C, Supporting Information). Compound 2 was not soluble in aqueous buffer at the high concentration needed to conduct the ITC experiment. Compound 1 exhibited a protein:ligand binding stoichiometry of 0.82, while compound 4 exhibited a stoichiometry of 0.33, suggesting 4 may have multiple binding sites. ΔG binding values were −8168 and −8389 cal/mol for 1 and 4, respectively. Decomposing the binding thermodynamics revealed that these two compounds have favorable binding enthalpies. While 1 exhibited a lower change in enthalpy (ΔH = −148 cal/mol) compared to 4, the larger entropic change (TΔS = 8020.2 cal/mol) during the binding reaction of this compound fully compensated for the lower change in enthalpy. Compound 4 was determined to have a significantly higher change in enthalpy (ΔH = −2158 cal/mol) as well as a lower change in entropy (TΔS = 6231.3 cal/mol) compared to 1. This indicates that these two compounds are binding RGS17 through distinct mechanisms. However, the contribution of entropy and enthalpy to hydrophobic, hydrogen bonding, and electrostatic interactions is complex, and it would be inappropriate to draw conclusions on how the compounds are binding RGS17 (i.e., through electrostatic or hydrophobic interactions). Nonetheless, these compounds do bind RGS17 with high nanomolar to low micromolar affinity.
The Newly Identified Inhibitors Exhibit a Dependence on Cysteine Residues
For ease of purification and with the future goal of identifying inhibitor binding sites by protein crystallization and/or NMR, an RGS17 construct that differs slightly from the screening construct was utilized. This construct (Asn72 to Ser206) was used previously to determine the crystal structure of RGS17 by others (PDB 1ZV4)27 and differs from the screening construct (Met61 to Ser210) in that it lacks 11 amino acids on the N-terminus. Using this alternative construct, the compounds were retested to determine if the change in construct affected the compound activity. All three compounds still showed a dose-dependent inhibition of the protein–protein interaction via AlphaScreen (Figure 1A), although the potencies of 1 and 4 were found to be reduced compared to the screening construct assessed using the malachite green assay. This indicates that both these compounds may have binding sites that contribute to inhibition within the 11 residues at the N-terminus. Interestingly, this region contains an additional cysteine residue, and a characteristic of many previously identified small-molecule inhibitors of RGS proteins is the covalent modification of cysteine residues.13–16 With this in mind, the dependence of cysteine for compound activity was assessed using a cysteine-null mutant of the truncated construct RGS17 (C117A). Introduction of this mutation had no effect on the GAP activity of RGS17 in vitro (Figure 1B), which indicated there were no gross structural abnormalities caused by the mutation. The C117A mutant was then assessed in the screening paradigm. All three compounds lost inhibitory activity against the mutant, indicating that the mechanism of inhibition is dependent on this cysteine residue, with 1 and 2 demonstrating the most dramatic reduction in activity (Figure 1C), suggesting that either all three compounds covalently modify the cysteine residue to impart inhibition or they require cysteine for binding coordination.
Figure 1.

Lead compounds inhibit RGS17 in a cysteine-dependent manner. (A) Lead compound dose–response curves against WT RGS17–Gα interaction via AlphaScreen. IC50 values of 72.8, 10.5, and 47.2 µM for compounds 1, 2, and 4, respectively. (B) Malachite Green GAP activity results of WT and C117A RGS17. (C) Lead compound dose–response curves against RGS17 C117A–Gα interaction via AlphaScreen. IC50 values were >100 µM for both 1 and 2 and 81.8 µM for 4. All data are n = 3 in at least duplicate means ± SD.
Mass Spectrometry Revealed Compound 2 to Form Covalent Adducts with RGS17
Having determined that 1, 2, and 4 require the presence of the cysteine residue for inhibition, it was next investigated as to whether the compounds covalently modify cysteine residues or if the interaction is noncovalent. First, Ellman’s reagent, 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), was used to determine the thiol reactivity of the test compounds following published protocols.28–30 Using a 4 h incubation and 1:1 molar ratio of test compound to N-acetylcysteine, both 1 and 4 demonstrated reactivity with N-acetylcysteine, while 2 did not (Figure 2). Next, the ability of the test compounds to covalently modify RGS17 at longer time periods (i.e., 72 h) was assessed via ESITOFMS. At a ratio of 5:1 test compound to RGS17 and an incubation period of 72 h, for 1 (Figure S3B, Supporting Information) and 4 (Figure S3D, Supporting Information) the test-compound-adducted RGS17 could not be detected, while incubation with 2 (Figure S3C, Supporting Information) led to the identification of both singly and doubly adducted species (Table 2). These data demonstrate the thiol reactivity for 1 and 4 at a shorter time (4 h) and that a longer incubation (72 h) may be required for 2 to modify cysteines. It was also assessed whether the binding of the compounds could be detected via NMR spectroscopy; however, no chemical shift perturbations were observed (data not shown). This is likely due to the high concentration of RGS17 needed for these experiments and the corresponding high concentration of compound required. Indeed, the ratio of compound to RGS17 was roughly 3:1 due to solubility limits of the test compounds, and therefore it is likely the fold molar excess of compound to protein was not sufficient for chemical shifts to be observed.
Figure 2.

Thiol reactivity of the lead compounds was assessed using DTNB. Compounds 1, 4, and control N-ethylmaleimide all reduced the absorbance at 412 nm compared to the control N-acetylcysteine (p values, * <0.05, **** <0.0001). Data are n = 3 ± SD.
Table 2.
Deconvoluted Mass Values of Samples in Figure 4
| sample | theoretical sample MW (Da) |
theoretical single adduct MW (Da) |
theoretical dual adduct MW (Da) |
observed MW (Da) |
|---|---|---|---|---|
| RGS17 apo | 16 235 | 16 232 | ||
| RGS17 + 1 | 332 (compound alone) | 16 567 | 16 899 | 16 234 |
| RGS17 + 2 | 451 (compound alone) | 16 686 | 17 137 | 16 233; 16 685 (452); 17 135 (902) |
| RGS17 + 4 | 318 (compound alone) | 16 553 | 16 871 | 16 235 |
For 2, the variation between DTNB and MS results may simply be the difference in molar excess compound and incubation time between the assays (1:1 and 4 h for thiol reactivity, 5:1 and 72 h for MS). However, due to the solubility of 2 and due to increased assay noise when using a reduced concentration of N-acetylcysteine, a greater molar ratio of 2:N-acetylcysteine in the DTNB assay could not be utilized. However, previous reports have demonstrated that 2 is capable of modifying thiols, which supports the data obtained via MS.31,32 Compound 4 contains a pyrogallol functionality, which is known to be sensitive to oxygen and therefore oxidation.33,34 Thus, the biochemical thiol reactivity may be a function of assay interference whereby the compound is promoting the oxidation of N-acetylcysteine and the formation of N-acetylcysteine–N-acetylcysteine disulfide bonds, which then prevents the dimerized N-acetylcysteine from reacting with DTNB. This may also be true for 1, as sanguinarine has been reported as a protein thiol oxidizing agent.35 Nonetheless, the MS data indicate that 1 and 4 may not necessarily form covalent adducts with RGS17.
The presence of multiple adducts is intriguing for 2, since the RGS17 construct employed contains only one cysteine (C117). This implies that 2 is capable of covalently modifying additional amino acids, most likely lysine or histidine. However, since the compound lost all activity against the C117A mutant, this secondary adduct likely does not contribute to RGS17–Gα interaction inhibition.
In summary, all three compounds rely on the presence of C117 to inhibit RGS17, as these compounds lacked activity against the C117A mutant. Compound 2 is capable of forming covalent adducts at two sites as determined via MS, whereas employing a DTNB assay showed that 1 and 4 are capable of either reaction with sulfhydryl groups or promoting a redox mechanism. Although cysteine reactivity may implicate increased promiscuity of these compounds, this does not necessarily exclude the compounds or future analogues from therapeutic tractability. Indeed, the RGS4 small-molecule inhibitor CCG-50014 and close analogue CCG-203769 are cysteine-reactive molecules that have reported functional activity in vivo in mouse models of neuropathic pain and Parkinson’s disease.36–38 Therefore, further validation of these molecules was pursued via evaluation of compound activity in a variety of cell-based systems.
The Lead Compounds Disrupt RGS17 Expression at the Membrane
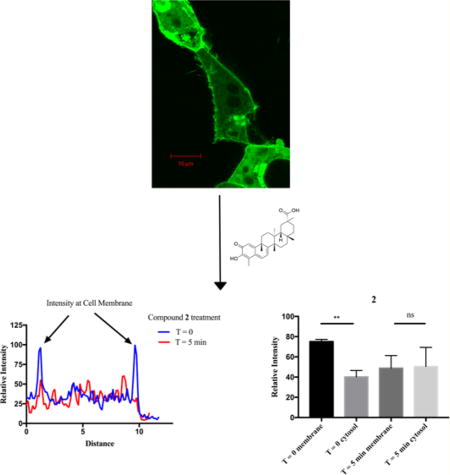
Previous reports have shown that RGS17 has an affinity for Gαo in a biological system,39 and so it was therefore hypothesized that RGS17 is suitable for subcellular localization experiments similar to those employed by Blazer et al. in the assessment of RGS4 lead molecules.14 Indeed, transfection of an N-terminal Green Fluorescent Protein (GFP) full-length RGS17 fusion construct alone exhibited a diffuse expression pattern, while cotransfection with unlabeled Gαo resulted in an enrichment of expression at the cell membrane of HEK293T cells (Figure 3A,B).
Figure 3.

Localization pattern of RGS17 in HEK293T cells. pAcGFPRGS17FL transfected into HEK293T in the absence (A) and presence (B) of hGαo. Treatment with 1 using pAcGFPRGS17FL and Gαo at 0 and 10 min (C and D, respectively). pAcGFPRGS17ΔN transfected into HEK293T in the absence (E) and presence (F) of Gαo. Scale bars represent 10 µm.
It was next assessed whether this localization pattern might be disrupted by treatment with the lead compounds. After treatment with 100 µM 1 for 10 min, RGS17 remained at the membrane (Figure 3C,D). It was hypothesized that palmitoylation of RGS17’s N-terminal cysteine rich domain40 resulted in RGS17 being anchored in the cell membrane once trafficked to the membrane upon cotransfection with Gαo, making it unable to diffuse to the cytoplasm upon treatment with compound, even if its interaction with Gαo was disrupted. Therefore, a truncated RGS17 construct, RGS17ΔN (missing RGS17 residues 1–60), was constructed and utilized. Like the ful-llength construct, RGS17ΔN is diffusely expressed in the cytoplasm in the absence of Gαo, yet upon cotransfection with Gαo RGS17ΔN is recruited to the cell membrane (Figure 3E,F).
It was next assessed whether treatment with the test compounds promoted delocalization of RGS17ΔN from the cell membrane, using the paradigm outlined via the control conditions in Figure 3E and F. Upon treatment with 100 µM of the lead compounds, RGS17ΔN diffused to the cytoplasm, indicating that these compounds disrupt the RGS17–Gα protein–protein interaction (Figure 4). Treatment with 10 µM of each compound also promoted disruption of the interaction, although a longer time course was required (Figure S4, Supporting Information). As a control, a known RGS4 inhibitor (CCG-50014) was tested against RGS17ΔN, which did not result in RGS17ΔN delocalization from the membrane (Figure 4J–L).14 Treatment of cells with vehicle also did not alter RGS localization (data not shown). As a control, pAcGFP vector was cotransfected with hGαo and the expression pattern assessed. GFP was observed as diffuse throughout the cytoplasm, indicating that the localization observed previously was due to the RGS17ΔN–Gαo protein–protein interaction (data not shown). The delocalization observed at 100 and 10 µM compound was quantified using the NIH ImageJ tool. (Figure 4, Figure S4, Supporting Information). For 100 µM lead compound treatment, there was a statistically significant enrichment of GFP signal at the membrane at T = 0, which was then lost when assessed at T = final. Compounds 1 and 2 exhibited this same trend at 10 µM compound treatment, demonstrating a statistically significant enrichment of GFP signal at the membrane at T = 0 and a loss of this enrichment at T = final (Figure S4, Supporting Information). Compound 4 demonstrated the same trend at 10 µM compound treatment; however the membrane enrichment at T = 0 fails to reach statistical significance. Additionally, the control condition of CCG-50014 exhibited no significant change of signal in either the membrane or cytosolic portions.
Figure 4.

Lead compounds disrupt RGS17–Gα interaction in cells. HEK293T cells cotransfected with GFPRGS17ΔN and hGαo were treated with 100 µM 1 (A), 2 (D) and 4 (G) or 100 µM RGS4 inhibitor CCG-50014 (J), and fluorescent protein cellular localization was monitored by inspection. T = 0 and T = final images were quantified with cross-sectional fluorescence intensity analysis (B, E, H, K). Representative intensity analysis is shown. Quantification of intensity analysis from all trials is shown at right (C, F, I, L). Compounds 1, 2, and 4 were performed with n = 3; CCG-50014 was performed with n = 2. Representative images are shown. Scale bars represent 10 µm. * = p ≤ 0.05; ** = p ≤ 0.01.
For 1, 2, and 4, the time course and lack of gross morphological changes that are indicative of cytotoxicity (cell shrinkage, plasma membrane blebbing, etc.) for the 100 µM compound treatments indicate that the observed diffusion from the membrane is indeed a consequence of interruption of the protein–protein interaction and not a consequence of loss of cellular integrity. Additionally, the control compound CCG-50014 demonstrated significant cell shrinkage over 30 min, yet the localization of RGS17 at the membrane remained intact. This is evidence that the diffusion of RGS17 into the cytoplasm for the 10 µM compound conditions is due to compound disruption of the protein–protein interaction and not due to a cytotoxic response despite several imaged cells displaying phenotypes consistent with those listed above indicating cytotoxicity over the longer imaging time course.
Compounds 1 and 2 Cause Growth Arrest of Lung and Prostate Cancer Cells in Vitro
Having demonstrated that the natural product lead compounds could disrupt the RGS17–G-protein interaction in cells, it was investigated whether or not these compounds can inhibit the growth of lung and prostate tumor cell lines. A549 cells were used as the lung tumor cell line, in which the expression of RGS17 has been detected.41,42 Both 1 and 2 demonstrated sub-micromolar inhibition of A549 cell growth compared to DMSO-treated controls, while 4 did not demonstrate any level of growth inhibition up to 1 µM compound (Figure 5). These results correlate with the fluorescence localization described above, as 4 demonstrated a much slower delocalization response, which may be a function of permeability of these compounds. An examination of the physical properties of the lead compounds shows that 1 and 2 have few hydrogen bond donors (0 and 2, respectively), few hydrogen bond acceptors (4 and 4, respectively), and low topical polar surface area (TPSA) values (40.8 and 74.6 Å2, respectively). For reference, compounds that can cross the blood brain barrier tend to have a total number of hydrogen bond donors and acceptors of 5 or less and a TPSA of less than 90 Å2.43 Compounds that have greater hydrogen-bonding potential and a greater TPSA are less blood brain barrier penetrant and by extension are less cell membrane permeable. Compound 4 has six H-bond donors, eight H-bond acceptors, and a TPSA of 148 Å2, which indicated that this substance may be less cell permeable than the other two lead compounds.
Figure 5.

Cytostatic activity of lead compounds against lung tumor cells. Growth response of A549 lung tumor cells with up to 1 µM 1 (A), 2 (B), or 4 (C) over 6 days in culture. Growth is expressed relative to the DMSO control, which is indicated at x = 0, for each compound. Data are n = 3. Error bars represent one standard deviation.
Compounds 1 and 2 were also tested for their ability to inhibit prostate tumor cell growth (Figure S6A,B, Supporting Information). Cell lines PC3E (PC3 cell line enriched for E-cadherin expression), TEM4-18 (PC3 subpopulation selected for mesenchymal phenotype),44 and RWPE-1 (immortalized epithelial, normal) were utilized for this assessment. RGS17 is detected in all three cell lines (Figure S5, Supporting Information)41,42 with TEM4-18 cells demonstrating an enrichment of expression of roughly 8-fold compared to PC3E cells. Both compounds inhibited the growth the PC3E and TEM4-18 tumor cell lines in a dose-dependent manner. However, both compounds also caused growth arrest of RWPE-1 cells at similar potencies. Additionally, overexpression of RGS17 in PC3E cells via transient transfection did not result in a sensitization of these cells to either 1 or 2 (data not shown). These data suggest that 1 and 2 may be affecting tumor cell growth by mechanisms in addition to the inhibition of RGS17 or perhaps in an RGS17-independent manner.
Investigation of Test Compound Mechanism of Action in Cancer Cells
Given that 1 and 2 inhibited lung and prostate cancer cell growth, it was assessed whether these compounds cause growth inhibition of lung and prostate tumors via the proposed mechanism, i.e., modulation of cAMP levels by restoring the negative regulation of adenylyl cyclase by Gαi/o. Previous reports have shown that knockdown of RGS17 in lung cancer cell lines results in a decrease of cAMP generation and CREB phosphorylation upon stimulation with forskolin compared to unaltered controls.6 Therefore, the ability of the compounds to modulate the phosphorylation of CREB upon stimulation with forskolin was assessed. Despite several attempts, a statistically significant reduction of CREB phosphorylation upon compound treatment could not be detected compared to DMSO-treated controls (data not shown). Compounds 1 and 4 trended toward the proposed mechanism, but a high degree of variability between trials prevents significant conclusions from being drawn.
The high degree of variability observed may in fact be a function of compound promiscuity and the compounds’ influence on additional signaling systems. Both 1 and 2 have published cytostatic and cytotoxic activity in other carcinomas targeting separate signaling cascades. Compound 1 (sanguinarine) has been reported to inhibit the growth of several gastric cancer cell lines in culture45 and to be cytotoxic to melanoma cells in culture46 and has reported activity against pancreatic cancer and neuroblastoma cell lines in culture.47,48 Compound 2 (celastrol) has been reported to have activity against osteosarcoma, head and neck cancer, and glioblastoma.49–51 Additionally, both compounds 1 and 2 have been reported to have activity against lung and prostate cancer cell lines through a variety of mechanisms. Compound 1 was reported to inhibit A549 and PC3 cell growth through glutathione depletion mechanisms,52,53 while 2 inhibited A549 cells through activation of mitochondrial and Fas/FasL (Fas receptor/Fas ligand) apoptotic pathways and to have activity against DU145 prostate cancer cells via an hERG-dependent mechanism.54,55 These reports show that these compounds are quite promiscuous. Given that these compounds were identified as inhibitors of RGS17 and that these compounds have reported activities in lung, prostate, hepatocellular carcinoma, and breast cancer, the pathologies in which RGS17 has been reported to play a role, it is possible that inhibition of RGS17 may be one of the many mechanisms by which these compounds alter cancer cell growth.56–58 Extensive pathway analysis is required to determine exactly what signaling events are being altered in the lung and prostate cell lines used here to determine the primary mechanism of action of these compounds.
In conclusion, several natural products were identified that inhibit the activity of RGS17. While these natural products may indeed be promiscuous in the targeting of cancer cells, the information gleaned for the mechanism of RGS17 inhibition will provide the foundation for future discovery and design efforts. The normal expression pattern of RGS17 suggests that a selective RGS17 inhibitor that is incapable of crossing the blood brain barrier would present minimal off-target effects while inhibiting cancer cell growth. This makes RGS17 a very attractive anticancer target, and further investigation of these compounds as well as identification of novel scaffolds through larger screening efforts is essential for the development of selective RGS17 pretherapeutic leads.
EXPERIMENTAL SECTION
General Experimental Procedures
Methods for protein expression and purification, chemical biotinylation on Gαo, Z factor calculation in 1536-well format, malachite green protocol, ITC, quantitative PCR, and prostate cell line dose–response can be found in the Supporting Information, Experimental Section.
Screening of Chemical Library
Altogether, 2320 compounds from the MicroSource SPECTRUM chemical library were screened at a final concentration of 40 µM. RGS17–anti-GST and Gαo–biotin–streptavidin bead conjugations and reagent addition to respective wells were performed as described in the Supporting Information Experimental Section “Z factor calculation in 1536-well format”, with the exception of all Gαo prepared, which was +AMF.
AlphaScreen Dose–Response Analysis
Experiments were carried out similarly to the high-throughput AlphaScreen assay using Corning 384-well white flat-bottomed plates (Corning, NY, USA). Compounds were assessed in a half-log dilution series from 100 µM to 1 nM final. RGS17 (WT or mutant) conjugated to the appropriate bead was added to wells already containing compound at a final protein concentration of 10 nM. After a 30 min incubation, Gαo conjugated to the appropriate bead in the presence of AMF and GDP was added for a final G protein concentration of 10 nM. Negative controls were determined in the absence of AMF.
AlphaScreen Biotinlyated GST Counterscreen
Compounds that exhibited inhibition of the protein–protein interaction in a dose-dependent manner were counterscreened in a biotinylated GST control assay in Corning 384-well white plates. Compounds were assessed in a half-log dilution manner as above. Biotinylated GST (600 pM) was first incubated with anti-GST acceptor beads for 30 min. Streptavidin donor beads were then added and incubated for an additional 30 min. The protein bead mixture was then added to wells already containing compound and incubated for 1 h at RT prior to read. Final protein and bead concentrations were 300 pM protein and 6.25 ng/µL for each bead, respectively. Plates were read on an Envision plate reader (PerkinElmer, Waltham, MA, USA), and data were analyzed using GraphPad Prism 6.
Interrogation of Potential Covalent Interaction by ESI-TOFMS
RGS17ΔN (1ZV4 construct) (20 µM) in 25 mM HEPES, 100 mM NaCl, and 5% DMSO, pH 8, was incubated at 4 °C in either the presence or absence of 100 µM compound for 72 h, prior to dialysis against 25 mM NH4HCO3 and 100 µM NaCl at 4 °C for 24 h to remove noncovalently bound compound and change to MS-compatible buffer. ESITOFMS was conducted using a Waters Q-Tof Premier mass spectrometer using direct infusion and positive ESI ionization. MassLynx was used for data acquisition, while MaxEnt was used for spectra deconvolution. For samples containing 1 and 4 further reduction of NaCl content was required for resolution purposes. These samples were dialyzed in two iterations, resulting in a sub-nanomolar salt concentration. Post dialysis, samples were analyzed identically as above.
Thiol Reactivity
The thiol reactivity was determined for the test compounds using Ellman’s reagent {5,5′[dithiobis(2-nitrobenzoic acid)} following a protocol adapted from the literature.28–30 N-Acetylcysteine at 100 µM was incubated with compound (100 µM) for 4 h at 37 °C. Following incubation, DTNB (1 mM) was added, and absorbance measured at 412 nm using a Molecular Devices SpectraMax plate reader. Data were analyzed using GraphPad Prism 7.
Plasmid Construction and Purification
GFP fusion constructs were cloned using pAcGFP In-Fusion Ready vector (Clontech catalog no. 632500, Mountain View, CA, USA). RGS17 full-length (RGS17FL) and the truncated RGS17 construct (RGS17ΔN) were cloned using the In-Fusion HD cloning system (Clontech, catalog no. 638909). Proper plasmid construction was confirmed by Sanger Sequencing (Iowa Institute of Human Genetics, University of Iowa).
RGS Localization by Confocal Microscopy
HEK293T cells were plated in 96-well tissue culture treated glass-bottom poly-d-lysine-coated view plates (PerkinElmer, Waltham, MA, USA) at a density of 25 000 cells per well and incubated at 37 °C, 5% CO2 overnight. The next day, cells were transfected with hGαo in pcDNA 3.1(+), pAcGFPRGS17 FL, pAcGFPRGS17ΔN, or empty pAcGFP, either singly or in the appropriate combinations using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocols. Then, 24 h post-transfection, the medium was aspirated and replaced with 100 µL of serum-free, phenol-red-free Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies). For compound treatment, 100 µL of DMSO solution was added to wells for final DMSO concentrations ranging from 0.05% to 2%. Compounds were prepared at 2× final desired concentrations in serum-free, phenol-red-free DMEM. Next, 100 µL of compound was added to wells (1× final, 0.07–0.8% DMSO). Cells were imaged using a Zeiss LSM510 confocal microscope (University of Iowa Central Microscopy Core). Images were analyzed using the NIH ImageJ software to perform cross-sectional fluorescence intensity analysis and GraphPad Prism 6. For each analysis, the T = 0 and T = final image pair was normalized to the highest intensity signal in the T = 0 image.
Test Compound Dose–Response in Lung Tumor Cells
Subconfluent A549 lung tumor cells were harvested by trypsinization, counted on a Countess cell counter (Life Technologies), and plated at 1000 cells per well in a 24-well tissue culture plate. After overnight attachment, cells were treated in triplicate with indicated concentrations of test compound or DMSO control in equal concentration, not exceeding 0.2% DMSO final concentration. Cells were placed in an Incucyte FLR live cell imaging system (Essen Bioscience, Ann Arbor, MI, USA) to measure confluence every 2 h over 6 days in culture. Cell images were monitored to exclude morphology differences as a confounding factor affecting confluence measurement. Change in confluence over 6 days relative to DMSO controls was plotted versus drug concentration. IC50 values were calculated using the linear regression equation for each curve using 0.5 relative growth as the value for x and solving for y.
Supplementary Material
Acknowledgments
Special thanks are due to the University of Iowa High-Throughput Screening (UIHTS) Facility and its Director Dr. Meng Wu, the University of Iowa Central Microscopy Research Facility, the University of Iowa High Resolution Mass Spectrometry Facility, and Nadine Bannick for technical assistance. This work was supported by NIH 5R01CA160470 (D.L.R.), NIH R01CA136729 (J.C.D.H.), UIowa IRG-77-004-31 (D.L.R.), UIowa Oberley Award Seed Grant (D.L.R.), NIH T32GM008365-21 (D.I.M.), NIH T32GM067795 (M.P.H.), American Foundation for Pharmaceutical Education Predoctoral Fellowships (D.I.M., C.R.B., M.P.H.), an Institutional Research Grant No. 14-247-29-IRG from the American Cancer Society (Medical College of Wisconsin, M.A.J.), the We Care Fund for Innovation and Discovery (Department of Surgery, Medical College of Wisconsin, M.A.J.), the Donald C. Ausman Family Foundation (Department of Surgery, Medical College of Wisconsin, M.A.J.), and the Theodore W. Batterman Family Foundation (Department of Surgery, Medical College of Wisconsin, M.A.J.). This work also utilized core resources supported by Cancer Center Support Grant (P30 CA086862). Support for the UIHTS facility was provided by NIH S10RR029274-01, while the CMRF is supported by the Vice President for Research and Economic Development, the Holden Comprehensive Cancer Center, and the Carver College of Medicine.
Footnotes
ASSOCIATED CONTENT
- Additional information (ZIP)
The authors declare no competing financial interest.
References
- 1.Gilman AG. Annu. Rev. Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 2.Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. Curr. Opin. Cell Biol. 2014;27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 4.Hurst JH, Hooks SB. Biochem. Pharmacol. 2009;78:1289–1297. doi: 10.1016/j.bcp.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 5.Sjogren B, Neubig RR. Mol. Pharmacol. 2010;78:550–557. doi: 10.1124/mol.110.065219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.James MA, Lu Y, Liu Y, Vikis HG, You M. Cancer Res. 2009;69:2108–2116. doi: 10.1158/0008-5472.CAN-08-3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Li L, Lin J, Hu X, Li B, Xue A, Shen Y, Jiang J, Zhang M, Xie J, Zhao Z. J. Cancer. 2015;6:767–775. doi: 10.7150/jca.11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sokolov E, Iannitti DA, Schrum LW, McKillop IH. Cell. Signalling. 2011;23:1603–1610. doi: 10.1016/j.cellsig.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayes MP, Roman DL. AAPS J. 2016;18:550–559. doi: 10.1208/s12248-016-9894-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bodle CR, Mackie DI, Roman DL. Future Med. Chem. 2013;5:995–1007. doi: 10.4155/fmc.13.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.You M, Wang D, Liu P, Vikis H, James M, Lu Y, Wang Y, Wang M, Chen Q, Jia D, Liu Y, Wen W, Yang P, Sun Z, Pinney SM, Zheng W, Shu XO, Long J, Gao YT, Xiang YB, Chow WH, Rothman N, Petersen GM, de Andrade M, Wu Y, Cunningham JM, Wiest JS, Fain PR, Schwartz AG, Girard L, Gazdar A, Gaba C, Rothschild H, Mandal D, Coons T, Lee J, Kupert E, Seminara D, Minna J, Bailey-Wilson JE, Amos CI, Anderson MW. Clin. Cancer Res. 2009;15:2666–2674. doi: 10.1158/1078-0432.CCR-08-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larminie C, Murdock P, Walhin JP, Duckworth M, Blumer KJ, Scheideler MA, Garnier M. Mol. Brain Res. 2004;122:24–34. doi: 10.1016/j.molbrainres.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 13.Roman DL, Ota S, Neubig RR. J. Biomol. Screening. 2009;14:610–619. doi: 10.1177/1087057109336590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blazer LL, Zhang H, Casey EM, Husbands SM, Neubig RR. Biochemistry. 2011;50:3181–3192. doi: 10.1021/bi1019622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Storaska AJ, Mei JP, Wu M, Li M, Wade SM, Blazer LL, Sjogren B, Hopkins CR, Lindsley CW, Lin Z, Babcock JJ, McManus OB, Neubig RR. Cell. Signalling. 2013;25:2848–2855. doi: 10.1016/j.cellsig.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vashisth H, Storaska AJ, Neubig RR, Brooks CL., 3rd ACS Chem. Biol. 2013;8:2778–2784. doi: 10.1021/cb400568g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mackie DI, Roman DL. J. Biomol. Screening. 2011;16:869–877. doi: 10.1177/1087057111410427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang JH, Chung TD, Oldenburg KR. J. Biomol. Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 19.Lagorce D, Sperandio O, Baell JB, Miteva MA, Villoutreix BO. Nucleic Acids Res. 2015;43:W200–W207. doi: 10.1093/nar/gkv353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baell JB, Holloway GA. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 21.Baell J, Walters MA. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- 22.Sarkar SM. Nature. 1948;162:265. doi: 10.1038/162265a0. [DOI] [PubMed] [Google Scholar]
- 23.Wu S, Sun C, Wang K, Pan Y. J. Chromatogr. A. 2004;1028:171–174. doi: 10.1016/j.chroma.2003.11.072. [DOI] [PubMed] [Google Scholar]
- 24.Lu GB, Yang XX, Huang QS. Yao Xue Xue Bao. 1984;19:636–639. [PubMed] [Google Scholar]
- 25.Haley TJ, Bassin M. J. Am. Pharm. Assoc. Sci. Ed. 1951;40:111–112. [PubMed] [Google Scholar]
- 26.Monroy CA, Mackie DI, Roman DL. PLoS One. 2013;8:e62247. doi: 10.1371/journal.pone.0062247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soundararajan M, Willard FS, Kimple AJ, Turnbull AP, Ball LJ, Schoch GA, Gileadi C, Fedorov OY, Dowler EF, Higman VA, Hutsell SQ, Sundstrom M, Doyle DA, Siderovski DP. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6457–6462. doi: 10.1073/pnas.0801508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellman GL. Arch. Biochem. Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 29.Allen EM, Anderson DG, Florang VR, Khanna M, Hurley TD, Doorn JA. Chem. Res. Toxicol. 2010;23:1843–1850. doi: 10.1021/tx100317q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allen EM, Florang VR, Davenport LL, Jinsmaa Y, Doorn JA. Chem. Res. Toxicol. 2013;26:1043–1054. doi: 10.1021/tx300458b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Gibney PA, West JD, Morano KA. Mol. Biol. Cell. 2012;23:3290–3298. doi: 10.1091/mbc.E12-06-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang H, Chen D, Cui QC, Yuan X, Dou QP. Cancer Res. 2006;66:4758–4765. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- 33.Siegel SM, Porto F, Frost P. Arch. Biochem. Biophys. 1959;82:330–334. doi: 10.1016/0003-9861(59)90128-6. [DOI] [PubMed] [Google Scholar]
- 34.Siegel SM, Siegel BZ. Nature. 1958;181:1153–1154. doi: 10.1038/1811153a0. [DOI] [PubMed] [Google Scholar]
- 35.Choy CS, Cheah KP, Chiou HY, Li JS, Liu YH, Yong SF, Chiu WT, Liao JW, Hu CM. J. Appl. Toxicol. 2008;28:945–956. doi: 10.1002/jat.1360. [DOI] [PubMed] [Google Scholar]
- 36.Blazer LL, Storaska AJ, Jutkiewicz EM, Turner EM, Calcagno M, Wade SM, Wang Q, Huang XP, Traynor JR, Husbands SM, Morari M, Neubig RR. ACS Chem. Neurosci. 2015;6:911–919. doi: 10.1021/acschemneuro.5b00063. [DOI] [PubMed] [Google Scholar]
- 37.Bosier B, Doyen PJ, Brolet A, Muccioli GG, Ahmed E, Desmet N, Hermans E, Deumens R. Br. J. Pharmacol. 2015;172:5333–5346. doi: 10.1111/bph.13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoon SY, Woo J, Park JO, Choi EJ, Shin HS, Roh DH, Kim KS. Anesth. Analg. 2015;120:671–677. doi: 10.1213/ANE.0000000000000607. [DOI] [PubMed] [Google Scholar]
- 39.Mao H, Zhao Q, Daigle M, Ghahremani MH, Chidiac P, Albert PR. J. Biol. Chem. 2004;279:26314–26322. doi: 10.1074/jbc.M401800200. [DOI] [PubMed] [Google Scholar]
- 40.De Vries L, Elenko E, Hubler L, Jones TL, Farquhar MG. Proc. Natl. Acad. Sci. U. S. A. 1996;93:15203–15208. doi: 10.1073/pnas.93.26.15203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Bjorling L, Ponten F. Nat. Biotechnol. 2010;28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 42.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. Science. 2015;347:1260419-1–1260419-9. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 43.Pajouhesh H, Lenz GR. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drake JM, Strohbehn G, Bair TB, Moreland JG, Henry MD. Mol. Biol. Cell. 2009;20:2207–2217. doi: 10.1091/mbc.E08-10-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang R, Wang G, Zhang PF, Zhang J, Huang YX, Lu YM, Da W, Sun Q, Zhu JS. J. Cell. Mol. Med. 2017;21:1117–1127. doi: 10.1111/jcmm.13043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosen J, Landriscina A, Adler BL, Krauz A, Doerner J, Navati M, Musaev T, Gravekamp C, Nosanchuk J, Friedman AJ. J. Drugs Dermatol. 2015;14:453–458. [PubMed] [Google Scholar]
- 47.Singh CK, Kaur S, George J, Nihal M, Pellitteri Hahn MC, Scarlett CO, Ahmad N. Oncotarget. 2015;6:10335–10348. doi: 10.18632/oncotarget.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cecen E, Altun Z, Ercetin P, Aktas S, Olgun N. Asian Pac. J. Cancer Prev. 2014;15:9445–9451. doi: 10.7314/apjcp.2014.15.21.9445. [DOI] [PubMed] [Google Scholar]
- 49.Yu X, Wang Q, Zhou X, Fu C, Cheng M, Guo R, Liu H, Zhang B, Dai M. Oncol. Lett. 2016;12:3423–3428. doi: 10.3892/ol.2016.5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fribley AM, Miller JR, Brownell AL, Garshott DM, Zeng Q, Reist TE, Narula N, Cai P, Xi Y, Callaghan MU, Kodali V, Kaufman RJ. Exp. Cell Res. 2015;330:412–422. doi: 10.1016/j.yexcr.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boridy S, Le PU, Petrecca K, Maysinger D. Cell Death Dis. 2014;5:e1216. doi: 10.1038/cddis.2014.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jang BC, Park JG, Song DK, Baek WK, Yoo SK, Jung KH, Park GY, Lee TY, Suh SI. Toxicol. In Vitro. 2009;23:281–287. doi: 10.1016/j.tiv.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 53.Debiton E, Madelmont JC, Legault J, Barthomeuf C. Cancer Chemother. Pharmacol. 2003;51:474–482. doi: 10.1007/s00280-003-0609-9. [DOI] [PubMed] [Google Scholar]
- 54.Mou H, Zheng Y, Zhao P, Bao H, Fang W, Xu N. Toxicol. In Vitro. 2011;25:1027–1032. doi: 10.1016/j.tiv.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 55.Ji N, Li J, Wei Z, Kong F, Jin H, Chen X, Li Y, Deng Y. BioMed Res. Int. 2015;2015:308475. doi: 10.1155/2015/308475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H, Li Y, Liu D, Sun H, Liu J. Cell. Physiol. Biochem. 2013;32:448–458. doi: 10.1159/000354450. [DOI] [PubMed] [Google Scholar]
- 57.Cao L, Zhang X, Cao F, Wang Y, Shen Y, Yang C, Uzan G, Peng B, Zhang D. BMC Cancer. 2015;15:873. doi: 10.1186/s12885-015-1909-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dong XZ, Zhang M, Wang K, Liu P, Guo DH, Zheng XL, Ge XY. BioMed Res. Int. 2013;2013:517698. doi: 10.1155/2013/517698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.