

Abstract
To investigate the effects of alkylation at 5-OH and 20-OH of 2,3-dehydrosilybin on prostate cancer cell proliferation, the synthetic approaches to 5- or/and 20-O-alkyl-2,3-dehydrosilybins, through a multi-step sequence from commercially available silybin, have been successfully developed. The first three reactions in the syntheses were completed through a one-pot procedure by managing anaerobic and aerobic conditions. With these synthetic methods in hand, twenty-one 2,3-dehydrosilybins, including seven 20-O-alkyl, seven 5,20-O-dialkyl, and seven 5-O-alkyl-2,3-dehydrosilybins, have been achieved for the evaluation of their biological profiles. Our WST-1 cell proliferation assay data indicate that nineteen out of the twenty-one 2,3-dehydrosilybins possess significantly improved antiproliferative potency as compared with silybin toward both androgen-sensitive (LNCaP) and androgen-insensitive prostate cancer cell lines (PC-3 and DU145). 5-O-Alkyl-2,3-dehydrosilybins were identified as the optimal subgroup that can consistently inhibit cell proliferation in three prostate cancer cell models with all IC50 values lower than 8 μM. Our flow cytometry-based assays also demonstrate that 5-O-heptyl-2,3-dehydrosilybin effectively arrests the cell cycle in the G0/G1 phase and activates PC-3 cell apoptosis.
Keywords: 2,3-dehydrosilybin derivatives; synthesis; anti-proliferative activity; prostate cancer; cell cycle regulation; cell apoptosis; Trimethoxyflavonol derivatives; Cell proliferation
Graphical abstract
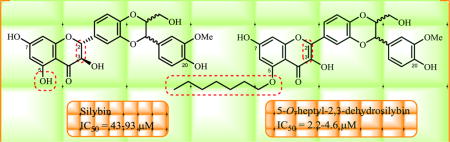
1. Introduction
Over forty naturally occurring flavonolignans comprise a unique and relatively new class of hybrid molecules biogenetically originated from flavonoids and phenylpropanoids (lignans).1,2 Silybin (also named as silibinin, 1, Figure 1) is the key chemical component of milk thistle (Silybum marianum L. Gaertner, Asteraceae) and the first well-studied member of flavonolignans. Silybin is an equivalent mixture of diastereoisomeric silybin A and silybin B (Figure 1) that possess opposite configurations at C-10 and C-11.3 Milk thistle and its crude extract silymarin are well-known traditional European medicines that have originally been used for the prevention and treatment of various hepatotoxicity,4,5 with its earliest written record dating back to 1539 by Hieronymus Bock.6 In the early 1990s, Agarwal and co-workers initiated the investigation on anticancer activities of silymarin.7,8 Their studies, together with the data collected later by other research groups, have identified the potential of several flavonolignans in preventing and treating various cancers, including prostate cancer.9,10 However, the in vitro cell-based studies reveal that silybin, with its IC50 values in the range of 40–106 μM, is barely active as an anti-proliferative agent.2,9,11
Figure 1.
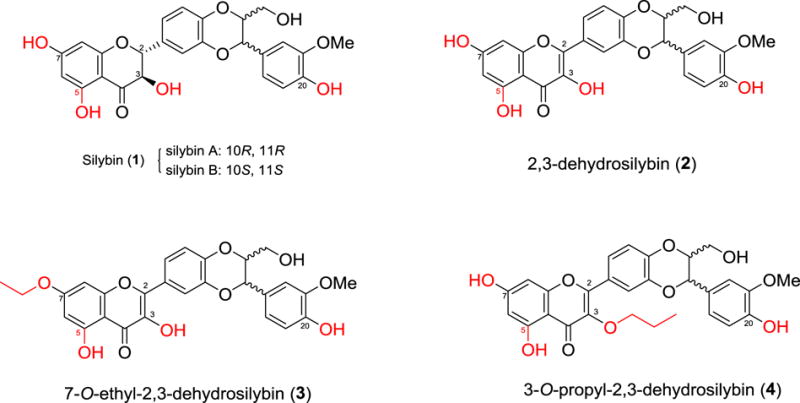
Structures of silybin and 2,3-dehydrosilybins
Structural modifications on silybin have been demonstrated to be a viable strategy to enhance its potency. For example, 2,3-dehydrosilybin (2), first synthesized from aerobic oxidation of silybin (1),6,12 is more potent than silybin against human prostate cancer cells.13,14 Our previous studies indicate that in vitro antiproliferative potency of 2,3-dehydrosilybin against three prostate cancer cell lines can be further improved through appropriate chemical modification on 7-OH and 3-OH, as exemplified by 7-O-ethyl-2,3-dehydrosilybin (3) and 3-O-propyl-2,3-dehydrosilybin (4) (Figure 1).14,15 This encouraged us to further investigate the effects of modifications on 5-OH and/or 20-OH of 2,3-dehydrosilybin on the biological profiles in prostate cancer cell models. Additionally, we aim to explore the synthetic approaches to 5-O-substituted- and 20-O-substituted-2,3-dehydrosilybins. Consequently, this study started with the development of general synthetic approaches to 20-O-substituted- and 5-O-substituted-2,3-dehydrosilybins followed by the syntheses of twenty one 2,3-dehydrosilybins, including seven 20-O-alkyl-2,3-dehydrosilybins, seven 5,20-O-dialkyl-2,3-dehydrosilybins, and seven 5-O-alkyl-2,3-dehydrosilybins. The in vitro anticancer activities of these 2,3-dehydrosilybins have been assessed in three prostate cancer cell models. To date, only two 5- or/and 20-O-alkyl-2,3-dehydrosilybin derivatives, 5-O-methyl-2,3-dehydrosilybin and 20-O-methyl-2,3-dehydrosilybin, have been reported for their antiproliferative and anti-migration activities towards human umbilical vein endothelial cells.16
2. Results and Discussion
2.1 Chemistry
Silybin, as a diastereomeric mixture, was selected as our starting material to synthesize 5-O-substituted-, 5,20-O-disubstituted- and 20-O-substituted-2,3-dehydrosilybins because the optically pure silybin A and silybin B possess similar antiproliferative potency in prostate cancer cell models and two pure enantiomers of 2,3-dehydrosilybin do not exhibit clear difference in suppressing PC-3 prostate cancer cell proliferation.13,17 However, the in vitro antiproliferative potency of the optically pure enantiomers for the specific 2,3-dehydrosilybin derivative should be thoroughly investigated if it were selected for further in vivo animal studies and in-depth mechanism exploration. The reason is because 2,3-dehydrosilybin A has been revealed to be more effective than its enantiomer, 2,3-dehydrosilybin B, in extending lifespan and preventing Aβ aggregation in cell and whole organism models.18 The syntheses of 20-O-alkyl-2,3-dehydrosilybins (14–20) and 5,20-O-dialkyl-2,3-dehdyrosilybins (21–27) were accomplished following the procedures as illustrated in Scheme 1, which were designed by taking advantage of (i) the reactivity of the phenolic hydroxyl groups at different positions in silybin toward etherification is 7-OH > 20-OH > 5-OH;6 (ii) aerobic oxidation of silybin to 2,3-dehydrosilybin only can be achieved in the simultaneous presence of base (e.g. potassium carbonate and potassium acetate) and air;6,14 (iii) the 3-OH becomes the most active functional group toward etherification once silybin is oxidized to the 2,3-dehydrosilybin,6,14,19 which has been rationalized by the electrochemistry measurements and bond dissociation energy calculations,20 and (iv) the 3-OH in 7-0-benzylsilybin is much easier to be aerobically oxidized than that in silybin, which we noticed from our recent experiments. Specifically, we first converted silybin (1) to 7-O-benzylsilybin (5) by using acetone as solvent and 1.1 equivalents of benzyl bromide, as well as 4 equivalents of potassium carbonate, under strictly anaerobic conditions (under argon). After switching the solvent from acetone to DMF and adding additional 1.1 equivalents of benzyl bromide and 2 equivalents of potassium carbonate, the crude 7-O-benzylsilybin was readily oxidized to 7-O-benzyl-2,3-dehydrosilybin under aerobic conditions (open to air), in which the 3-OH was further selectively benzylated to furnish 3,7-O-dibenzyl-2,3-dehydrosilybin (6). Treatment of 3,7-O-dibenzyl-2,3-dehydrosilybin (6) with 2 equivalents of the appropriate alkyl halide using potassium carbonate as the base and DMF as the solvent furnished 20-0-alkyl-3,7-0-dibenzyl-2,3-dehydrosilybins (7–13). Debenzylation of dibenzylsilybins (7–13) catalyzed by Pd/C using ammonium formate as hydrogen source gave the desired 20-O-alkyl-2,3-dehydrosilybins (14–20). The only difference between the synthesis of 5,20-O-dialkyl-2,3-dehydrosilybins (21–27) and that of 20-O-alkyl-2,3-dehydrosilybins (14–20) is the use of 4 equivalents of the appropriate alkyl halide for the former and 2 equivalents for the latter. It was observed that dialkylation of 3,7-O-dibenzyl-2,3-dehydrosilybin with the longer chain alkyl (such as pentyl, hexyl, and heptyl) halide needs more equivalents of alkyl halide and longer reaction time for a comparable yield.
Scheme 1.
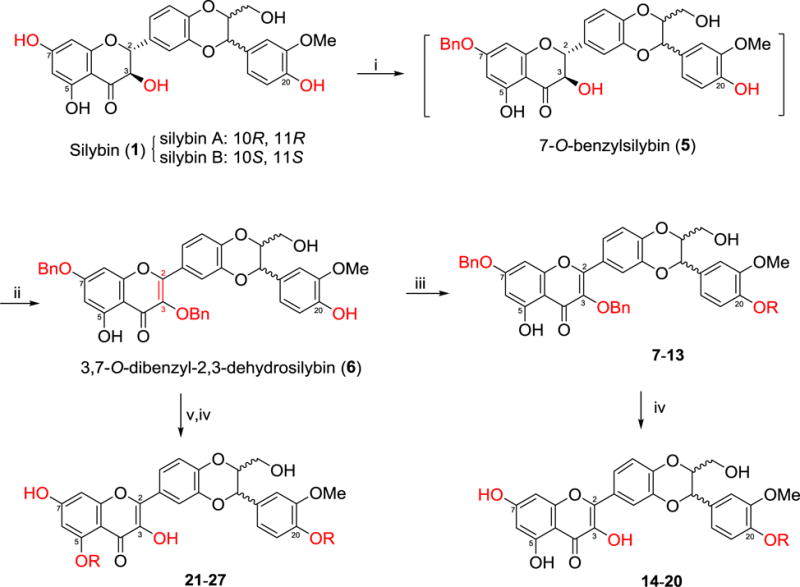
Synthesis of 20-O-alkyl-2,3-dehydrosilybins (14–20) and 5,20-O-dialkyl-2,3-dehydrosilybins (21–27). Reactants and conditions: (i) BnBr (1.1 eq), K2CO3 (4 eq), acetone (0.1 M), argon, reflux for 4 h; (ii) BnBr (1.1 eq), K2CO3 (2 eq), DMF (0.25 M), Air, rt, 6 h; (iii) RI or RBr (2eq), K2CO3 (2 eq), DMF (0.25 M), rt, 24 h; (iv) ammonium formate (10 eq), MeOH/Ethyl Acetate (50/50, v/v), Pd/C, reflux, overnight, argon: (v) RI or RBr (4–10 eq), K2CO3 (10–20 eq), DMF (0.25 M), rt, 24–48 h.
A similar synthetic strategy was applied to the synthesis of 5-O-alkyl-2,3-dehydrosilybins (28–35), as illustrated in Scheme 2. The synthesis started from a one-pot three-step procedure, including benzylation of 7-OH in silybin under strict anaerobic conditions, aerobic oxidation of 7-O-benzylsilybin to 7-O-benzyl-2,3-dehydrosilybin, and dibenzylation at 3-OH and 20-OH in 7-O-benzyl-2,3-dehydrosilybin, to give 3,7,20-O-tribenzyl-2,3-dehydrosilybin (28). Alkylation of 5-OH in 28 followed by global debenzylation in the presence of ammonium formate catalyzed by palladium carbon generated 5-O-alkyl-2,3-dehydrosilybins (29–35). It is worth noting that the synthetic methods described in Schemes 1–2 can also be used for the general syntheses of other 20-O-substituted- and/or 5-O-substituted-2,3-dehydrosilybins.
Scheme 2.
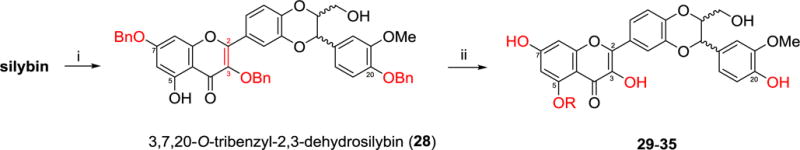
Synthesis of 5-O-alkyl-2,3-dehydrosilybins (29–35). Reactants and conditions: i) (a) BnBr (1.1 eq), K2CO3 (4 eq), acetone (0.1 M), argon, reflux overnight; (b) Remove acetone, DMF (0.25 M), BnBr (2.2 eq), K2CO3 (3 eq), rt, 4 h, air; ii) (a) RI or RBr (8 eq), K2CO3 (8 eq), DMF (0.2 M), rt, 4 h; (b) ammonium formate (10 eq), EtOAc and MeOH, Pd/C (20%), reflux, overnight, argon.
2.2 Anti-proliferative activities towards three prostate cancer cell lines
To evaluate the in vitro anticancer potential of the 5- or/and 20-O-alkyl-2,3-dehydrosilybins (14–27, 29–35), we first measured their antiproliferative activity against androgen-sensitive (LNCaP) and androgen-insensitive (PC-3 and DU145) human prostate cancer cell lines using the WST-1 cell proliferative assay. Silybin was employed as a positive control for comparison in the parallel experiment. The IC50 values in Table 1 show that nineteen out of twenty-one 2,3-dehydrosilybins are more potent than silybin in three prostate cancer cell lines, suggesting that 2,3-dehydrosilybin is a better scaffold of anti-prostate cancer agents. All 5-O-alkyl-2,3-dehydrosilybins (29–35) consistently exhibit promising antiproliferative potency towards three prostate cancer cell lines with IC50 values below 8 μM and 7–29 folds more potent than silybin. In contrast, all IC50 values for 20-O-alkyl-2,3-dehydrosilybins (14–20) toward androgen-insensitive (PC-3 and DU145) prostate cancer cell lines are greater than 8 pM. 5,20-O-Dialkyl-2,3-dehydrosilybins (21–27) displayed very similar potency to those of 20-O-alkyl-2,3-dehydrosilybins (14–20) towards androgen-sensitive (LNCaP) prostate cancer cells, but significantly less potency against androgen-insensitive (PC-3 and DU145) prostate cancer cell lines. It was also observed that 20-O-alkyl-2,3-dehydrosilybins (14–20) and 5,20-O-dialkyl-2,3-dehydrosilybins (21–27) were more effective in suppressing androgen-sensitive (LNCaP) cell proliferation than in inhibiting androgen-insensitive (PC-3 and DU145) cell proliferation. In contrast, both androgen-sensitive and androgen-insensitive prostate cancer cell lines have similar responses to 5-O-alkyl-2,3-dehydrosilybins (29–35). At our present level of understanding, the counterintuitive IC50 values for compounds 15 and 22 are hard to rationalize. At this point, the possibility that the compromised activity was caused by the impurities not detectable by NMR cannot be excluded.
Table 1.
In vitro anti-proliferative activity of the 2,3-dehydrosilybins against prostate cancer cell lines
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Comp. No | R | IC50 (μM)a | IC50 (silybin)/IC50 (derivative) | ||||
|
|
|||||||
| LNCaPb | DU145c | PC-3d | LNCaP | DU145 | PC-3 | ||
|
| |||||||
| silybin | – | 43.73 ± 10.90 | 93.34 ± 13.76 | 72.65 ± 3.15 | 1 | 1 | 1 |
| 14 | methyl | 4.74 ± 1.04 | 27.44 ± 10.15 | 8.89 ± 2.21 | 9 | 3 | 8 |
| 15 | ethyl | >100 | >100 | >100 | <1 | <1 | <1 |
| 16 | propyl | 9.95 ± 1.85 | 13.41 ± 2.91 | 12.94 ± 0.74 | 4 | 7 | 6 |
| 17 | butyl | 8.62 ± 1.11 | 14.01 ± 0.83 | 11.39 ± 0.93 | 5 | 7 | 6 |
| 18 | pentyl | 6.83 ± 2.07 | 17.66 ± 3.04 | 12.69 ± 1.39 | 6 | 5 | 6 |
| 19 | hexyl | 10.78 ± 2.20 | 35.37 ± 10.84 | 20.83 ± 6.20 | 4 | 3 | 3 |
| 20 | heptyl | 6.95 ± 1.28 | 20.53 ± 2.21 | 16.67 ± 0.79 | 6 | 5 | 4 |
|
| |||||||
| 21 | methyl | 4.57 ± 0.42 | 103.76 ± 7.73 | 29.3 ± 3.69 | 10 | 1 | 2 |
| 22 | ethyl | 33.63 ± 0.95 | >100 | >100 | 1 | <1 | <1 |
| 23 | propyl | 3.52 ± 1.02 | 10.00 ± 2.06 | 19.49 ± 2.48 | 12 | 9 | 4 |
| 24 | butyl | 3.26 ± 0.33 | 22.12 ± 3.81 | 49.94 ± 3.89 | 13 | 4 | 1 |
| 25 | pentyl | 6.47 ± 0.67 | 30.66 ± 3.28 | 39.95 ± 5.39 | 7 | 3 | 2 |
| 26 | hexyl | 4.84 ± 0.86 | 20.54 ± 5.06 | 20.21 ± 2.45 | 9 | 5 | 4 |
| 27 | heptyl | 2.35 ± 0.29 | 9.70 ± 1.90 | 5.88 ± 1.19 | 19 | 10 | 12 |
|
| |||||||
| 29 | methyl | 6.55 ± 0.49 | 6.95 ± 0.48 | 5.84 ± 1.78 | 7 | 13 | 12 |
| 30 | ethyl | 2.62 ± 1.35 | 4.61 ± 1.54 | 7.08 ± 1.68 | 17 | 20 | 10 |
| 31 | propyl | 3.17 ± 0.15 | 3.95 ± 0.59 | 7.62 ± 1.45 | 14 | 24 | 10 |
| 32 | butyl | 3.35 ± 0.47 | 4.22 ± 1.07 | 7.46 ± 1.93 | 13 | 22 | 10 |
| 33 | pentyl | 2.34 ± 0.87 | 4.30 ± 0.22 | 7.92 ± 1.46 | 19 | 22 | 9 |
| 34 | hexyl | 2.53 ± 1.34 | 4.31 ± 0.32 | 5.99 ± 0.64 | 17 | 22 | 12 |
| 35 | heptyl | 2.24 ± 0.21 | 3.23 ± 0.77 | 4.61 ± 0.64 | 20 | 29 | 16 |
IC50 is the drug concentration effective in inhibiting 50% of the cell viability measured by the WST-1 cell proliferation Assay after 3 days exposure.
Human androgen-sensitive prostate cancer cell line
Human androgen-insensitive prostate cancer cell line
Human androgen-insensitive prostate cancer cell line
2.3 Effects of 5-O-heptyl-2,3-dehydrosilybin (35) on PC-3 cell cycle progression and apoptosis
Silybin has been demonstrated to arrest rat (H-7 and I-8) and human (LNCaP and PC-3) prostate cancer cell cycle at the G1 phase.21–23 Our previous study revealed that treatment of PC-3 cells with 2,3-dehydrosilybin (2), 7-O-ethyl-2,3-dehydrosilybin (3), and 3-O-propyl-2,3-dehydrosilybin (4) led to appreciable cell cycle arrest at the G0/G1 phase.14,15 To evaluate the effect of 5-O-alkyl group on the PC-3 prostate cancer cell cycle regulation, compound 35, as one of the most potent 5-O-alkyl-2,3-dehydrosilybins, was selected for further assessment using flow cytometry analysis with propidium iodide DNA staining. When PC-3 cells were treated with 5-O-heptyl-2,3-dehydrosilybin (35) at 10 and 20 μM (Fig. 3 and Table 2), a similar cell cycle regulation was observed. Specifically, 35 causes accumulation of PC-3 cells in the G0/G1 phase from 34.7% and 41.9% for control cells at 16 hours and 24 hours, respectively, to 49–59% and 50–59% for the compound-treated cells. The data imply that PC-3 cell proliferation of 5-O-heptyl-2,3-dehydrosilybin is linked to its cell cycle regulation in the G0/G1 phase.
Figure 3.
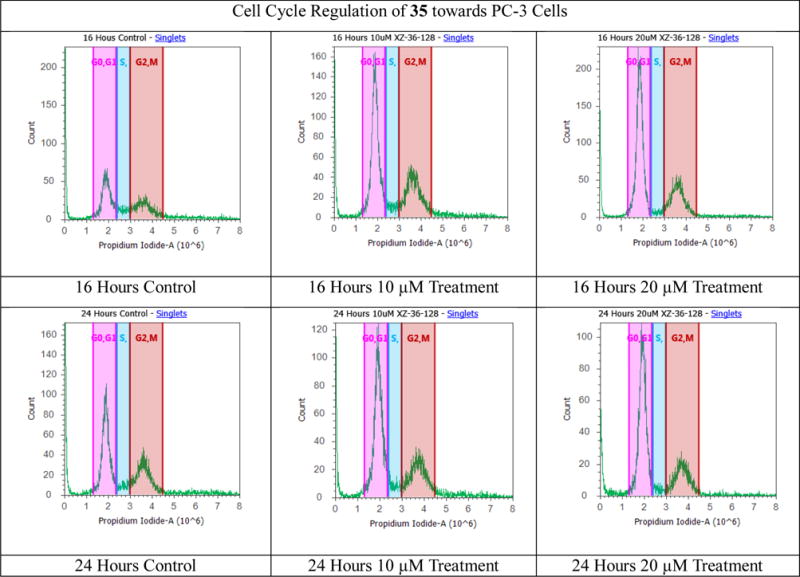
Cell cycle analysis of PC-3 cells. PC-3 cancer cells were untreated or treated with 5-O-heptyl-2,3-dehydrosilybin (35). Cells were harvested after 16 and 24 hours, fixed, stained, and analyzed for DNA content. The distribution and percentage of cells in G0/G1, and G2/M phase of the cell cycle are indicated.
Table 2.
The distribution and percentage of PC-3 cells in G0/G1, S, and G2/M phase of the cell cycle after treatment with 35
| PC-3 cells | G0/G1(%) | S (%) | G2/M (%) |
|---|---|---|---|
| 16 h control | 34.7 | 8.5 | 27.4 |
| 16 h 10 μM Treatment | 49.1 | 5.3 | 29.6 |
| 16 h 20 μM Treatment | 59.0 | 2.8 | 26.9 |
|
| |||
| 24 h control | 41.9 | 5.6 | 28.8 |
| 24 h 10 μM Treatment | 50.1 | 5.4 | 26.6 |
| 24 h 20 μM Treatment | 59.5 | 3.4 | 24.1 |
Cell apoptosis induction by silybin in PC-3 tumor xenografts has been revealed by Agarwal and co-workers.24 Our previous studies indicate that the capability of 2,3-dehydrosilybin in promoting PC-3 cell apoptosis can be altered or reversed by modifying the structure.14,15 For example, incorporation of an ethyl group to 7-OH in 2,3-dehydrosilybin promotes the apoptotic activation; while introduction of a propyl group to 3-OH in 2,3-dehydrosilybin reverses the apoptotic response. To evaluate the effect of 5-O-alkylation on the PC-3 cell apoptosis, one of the most promising derivatives, 35, was chosen for F2N12S and CYTOX AADvanced double staining flow cytometry-based assay at 0–100 μM dosing range. The results (Figure 4) imply that treatment of PC-3 cells with 35 for 16 h led to PC-3 cells dying from apoptosis rather than necrosis in a dose-responsive manner. As shown in Table 3, chemical modification on 3-, 5-, or 7-OH in 2,3-dehydrosilybin can significantly improve the antiproliferative potency toward the human androgen-sensitive prostate cancer cell line. However, PC-3 cell apoptosis can only be greatly promoted by introducing an alkyl group to 7-OH or 5-OH. It is worth noting that 5-O-heptyl-2,3-dehydrosilybin (35) induces PC-3 cell apoptosis more effectively than 7-O-ethyl-2,3-dehydrosilybin (3), suggesting that the heptyl group on 5-OH of 2,3-dehydrosilybin is beneficial to cell apoptosis activation. For example, 30 μM of 7-O-ethyl-2,3-dehydrosilybin (3) can induce 68% apoptotic cells as compared with control cells;15 whereas 30 μM of 5-O-heptyl-2,3-dehydrosilybin (35) can induce 81% apoptotic cells.
Figure 4.
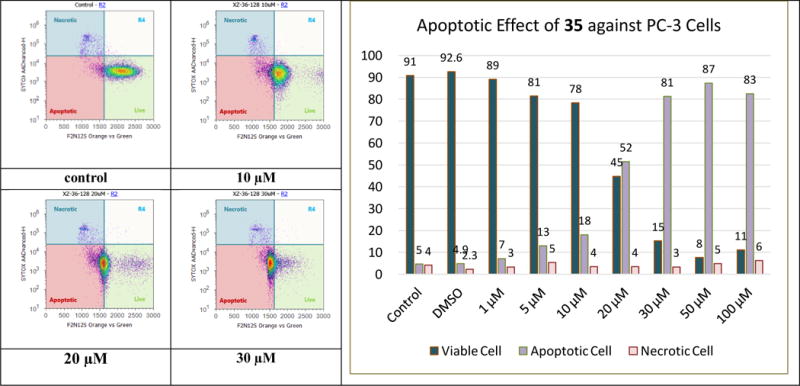
Evolution of viable, apoptotic, and necrotic PC-3 cells populations in response to increasing dosages of 5-O-heptyl-2,3-dehydrosilybin (35).
Table 3.
Comparison of antiproliferative potency and cell apoptosis induction of 2,3-dehydrosilybins in the PC-3 cell model
| Compound | IC50 (μM) | Apoptotic cell population | Ref. |
|---|---|---|---|
| 2,3-dehydrosilybin (2) | 9.45 ± 0.56 | 14% (treated with 60 μM) | 14 |
| 3-O-propyl-2,3-dehydrosilybin (4) | 1.71 ± 0.45 | 8% (treated with 60 μM) | 14 |
| 7-O-ethyl-2,3-dehydrosilybin (3) | 3.25 ± 0.31 | 66% (treated with 50 μM) | 15 |
| 5-O-heptyl-2,3-dehydrosilybin (35) | 4.61 ± 0.64 | 87% (treated with 50 μM) | New data |
3. Conclusion
In summary, the synthetic approaches to 5-O-substituted and/or 20-O-substituted-2,3-dehydrosilybins through a multi-step sequence starting from commercially available silybin have been developed, which include selective benzylation at 7-OH in silybin under strict anaerobic conditions, aerobic oxidation, benzylation, alkylation, and global debenzylation. The fact that the first three reactions were completed by a one-pot procedure makes these synthetic approaches efficient and practical. Employing these synthetic approaches, seven 20-O-alkyl-2,3-dehydrosilybins, seven 5,20-O-dialkyl-2,3-dehydrosilybins, and seven 5-O-alkyl-2,3-dehydrosilybins have been successfully synthesized. The WST-1 cell proliferation assay data indicate that nineteen out of the twenty-one 2,3-dehydrosilybins possess significantly improved antiproliferative potency as compared with silybin toward both androgen-sensitive (LNCaP) and androgen-insensitive prostate cancer cell lines (PC-3 and DU145). 5-O-Alkyl-2,3-dehydrosilybins represent the optimal subgroup consistently suppressing cell proliferation in three human prostate cancer cell models with all IC50 values below 8 μM, as compared with the 3-O-alkyl, 7-O-alkyl, 20-O-alkyl, and 5,20-O-dialkyl-2,3-dehydrosilybins. Our flow cytometry-based assays demonstrate that 5-O-heptyl-2,3-dehydrosilybin effectively inhibits the growth of PC-3 prostate cancer cells by arresting the cell cycle in the G0/G1 phase and activating PC-3 cell apoptosis.
4. Experimental
4.1. General synthetic procedures
NMR spectra were obtained on a Bruker Fourier 300 spectrometer in CDCl3, CD3OD, or DMSO-d6. The chemical shifts are given in δ (ppm) referenced to the respective solvent peak, and coupling constants are reported in Hz. Anhydrous THF and dichloromethane were purified by PureSolv MD 7 Solvent Purification System from Innovative Technologies (MB-SPS-800). All other reagents and solvents were purchased from commercial sources and were used without further purification. Silica gel column chromatography was performed using silica gel (32–63 μM). Preparative thin-layer chromatography (PTLC) separations were carried out on thin layer chromatography plates loaded with silica gel. Silybin (> 98.0%) was purchased from Fisher Scientific (TCI America, Cat # 50-014-46874).
4.2. Synthesis of 3,7-O-dibenzyl-2,3-dehydrosilybin (6)
The solution of silybin (1 equiv.) and potassium carbonate (2 equiv.) in acetone (0.1 M) was stirred under argon atmosphere for 10 min prior to being added benzyl bromide (1.1 equiv.) through syringe. The reaction was allowed to proceed with refluxing under argon for 4 h. After cooling down to room temperature, the acetone was removed by evaporation. To the reaction flask, DMF was added (0.5 M) followed by additional potassium carbonate (2 equiv.) and benzyl bromide (1 equiv.). The reaction flask was stirred at room temperature under aerobic conditions for twelve hours. The reaction was quenched by addition of 1 M HCl followed by ethyl acetate. The subsequent mixture was rinsed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo to give a crude product. PTLC purification of the crude product, using 3% methanol in DCM as eluent, yielded the desired product. The 1H NMR data of 3,7-O-dibenzyl-2,3-dehydrosilybin are consistent with those previously reported by us.15
4.3. General procedure for the synthesis of 20-O-alkyl-2,3-dehydrosilybins (14–20)
The mixture of 3,7-O-dibenzyl-2,3-dehydrosilibin (6, 1 equiv.), alkyl iodide or alkyl bromide (2 equiv.), and potassium carbonate (2 equiv.) in DMF (0.5 M) was stirred under argon at room temperature for 24 h prior to being quenched with 1M HCl. The subsequent mixture was diluted with ethyl acetate and diethyl ether (1:1, v/v), which was rinsed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo to give a crude product. PTLC purification of the crude product, using 3% methanol in DCM as eluent, yielded the desired 20-O-alkyl-3,7-dibenzyl-2,3-dehydrosilibin (7–13). To the solution of 20-O-alkyl-7-O-dibenzyl-2,3-dehydrosilybin (1 eq) in methanol/ethyl acetate (50/50, v/v) (0.2 M) was sequentially added Pd-C (50% wet, 10% w/w) and ammonium formate (10 eq). The reaction mixture was refluxed overnight under argon. After cooling to room temperature, the reaction mixture was filtered through a silica gel pad eluting with ethyl acetate. The filtrate was concentrated under vacuum and purified by PTLC eluting with 3% methanol in DCM to give the desired product (14–20).
4.3.1. 20-O-Methysilybin (14)
This derivative was prepared as a white powder in 16% yield. IR (film) vmax: 3417, 1653, 1557, 1505 cm−1. 1H NMR (300 MHz, CDCl3 + CD3OD) δ 7.83–7.71 (overlapped, 2H), 7.05–6.98 (overlapped, 1H), 6.96 (d, J = 8.7 Hz, 1H), 6.92 (s, 1H), 6.86 (d, J = 7.5 Hz, 1H), 6.32 (s, 1H), 6.18 (s, 1H), 4.95 (d, J = 6.3 Hz, 1H), 4.06 (m, 1H), 3.83 (s, 6H), 3.73–3.61 (overlapped, 1H), 3.46 (d, J = 12.0 Hz, 1H). HR-MS (ESI) m/z: calcd for C26H22O10 [M+H]+: 495.1291; found 495.1302.
4.3.2 20-O-Ethylsilybin (15)
This derivative was prepared as a light yellowish solid in 31%. m.p. 225–227 °C. IR (film) vmax: 3379, 1657, 1597, 1502 cm−1. 1H NMR (300 MHz, CDCl3 and DMSO-d6) δ: 12.16 (s, 1H), 10.32 (s, 1H), 8.71 (s, 1H), 7.79 (d, J = 1.8 Hz, 1H), 7.75 (dd, J = 8.7, 2.0 Hz, 1H), 7.00 (d, J = 8.7 Hz, 1H), 6.97 (s, 2H), 6.89 (d, J = 8.7 Hz, 1H), 6.33 (d, J = 1.9 Hz, 1H), 6.15 (d, J= 1.9 Hz, 1H), 4.97 (d, J = 7.9 Hz, 1H), 4.09 (br.s, 1H), 4.05 (q, J = 6.8 Hz, 3H), 3.83 (s, 3H), 3.69 (dd, J = 12.3, 1.7 Hz, 1H), 3.43 (dd, J = 11.9, 3.7 Hz, 1H), 1.39 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3 and DMSO-d6) δ:176.1, 164.4, 161.2, 156.7, 149.5, 148.8, 145.6, 145.3, 143.6, 136.7, 129.0, 124.4, 121.6, 120.3, 117.0, 116.7, 112.8, 111.0, 103.5, 98.7, 93.8, 79.1, 76.2, 64.3, 60.7, 56.1, 14.9. HR-MS (ESI) m/z: calcd for C27H24O10 [M+H]+: 509.1447; found 509.1458.
4.3.3. 20-O-Propylsilybin (16)
This derivative was made in 21% yield as a light yellow solid. m.p. 169–170 °C. IR (film) vmax: 3311, 2957, 2923, 2871, 1653, 1596, 1569, 1505, 1464, 1420 cm−1. 1H NMR (300 MHz, CDCl3 and DMSO-d6) δ 12.07 (br.s, 1H), 7.77 (s, 1H), 7.75 (d, J = 10.0 Hz, 1H), 7.49 (d, J = 2.9 Hz, 1H), 6.98 (d, J = 8.5 Hz, 1H), 6.94 (d, J = 8.0 Hz, 1H), 6.93 (s, 1H), 6.84 (d, J= 8.0 Hz, 1H), 6.30 (s, 1H), 6.16 (s, 1H), 4.96 (d, J = 8.0 Hz, 1H), 4.06–4.3 (m, 1H), 3.91 (t, J = 6.8 Hz, 2H), 3.81 (s, 3H), 3.69 (d, J = 11.6 Hz, 1H), 3.43 (dd, J = 12.1, 3.4 Hz, 1H), 1.84–1.72 (m, 2H), 0.967 (t, J= 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3 and DMSO-d6): δ 176.5, 164.9, 161.7, 157.3, 150.2, 149.6, 146.0, 145.8, 144.2, 137.1, 129.4, 125.0, 122.3, 120.9, 117.6, 117.2, 113.5, 111.6, 104.1, 99.4, 94.4, 79.6, 76.8, 71.1, 61.4, 56.7, 23.1, 11.1. HR-MS (ESI) m/z: calcd for C28H26O10 [M+H]+: 523.1604; found 523.1588.
4.3.4. 20-O-Butylsilybin (17)
This compound was prepared in 15% yield as a light yellow solid. m.p. 171–173 °C. IR (film) vmax: 3379, 1656, 1620, 1566, 1502 cm−1. 1H NMR (300 MHz, CDCl3 and DMSO-d6) δ: 12.13 (s, 1H), 10.24 (s, 1H), 8.53 (s, 1H), 7.78 (d, J = 2.0 Hz, 1H), 7.75 (dd, J = 8.5, 2.0 Hz, 1H), 7.00 (d, J = 8.6 Hz, 1H), 6.96 (d, J = 6.1 Hz, 1H), 6.95 (s, 1H), 6.86 (d, J = 8.7 Hz, 1H), 6.32 (d, J = 2.0 Hz, 1H), 6.16 (d, J = 2.0 Hz, 1H), 4.98 (d, J = 7.9 Hz, 1H), 4.09–4.05 (m, 1H), 3.97 (t, J = 6.6 Hz, 2H), 3.82 (s, 3H), 3.70 (dd, J = 12.5, 2.4 Hz, 1H), 3.44 (dd, J = 12.3, 4.08 Hz, 1H), 1.80−1.70 (m, 2H), 1.51–1.38 (m, 2H), 0.92 (t, J= 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3 and DMSO-d6) δ 176.0, 164.4, 161.2, 156.7, 149.6, 149.1, 145.5. 145.2, 143.6, 136.6, 128.9, 124.4, 121.7, 120.3, 117.0, 116.6, 113.0, 111.1, 103.5, 98.7, 93.8, 79.1, 76.2, 68.7, 60.8, 56.1, 31.2, 19.2, 14.0. HR-MS (ESI) m/z: calcd for C29H28O10 [M+H]+: 537.1760; found 537.1774.
4.3.5. 20-O-Pentylsilybin (18)
This derivative was prepared in 15% yield as a light yellowish solid. m.p. 170–172 °C. IR (film) vmax: 3364, 1655, 1597, 1503 cm−1. 1H NMR (300 MHz, CDCl3 and DMSO-d6) δ 12.1 (s, 1H), 10.2 (s, 1H), 8.44 (s, 1H), 7.77(d, J = 2.0 Hz, 1H), 7.74 (dd, J = 8.6, 2.1 Hz, 1H), 6.98 (d, J = 8.6 Hz, 1H), 6.94 (d, J = 7.3 Hz, 1H), 6.93 (s, 1H), 6.84 (d, J =8.6 Hz, 1H), 6.30 (d, J = 2.0 Hz, 1H), 6.15 (d, J = 1.8 Hz, 1H), 4.96 (d, J = 8.0 Hz, 1H), 4.07–4.03 (m, 1H), 3.94 (t, J = 6.8 Hz, 2H), 3.80 (s, 3H), 3.68 (dd, J = 12.2, 2.3 Hz, 1H), 3.42 (dd, J = 12.3, 4.0 Hz, 1H), 1.79–1.71 (m, 2H), 1.39–1.30 (m, 4H), 0.85 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3 and DMSO-d6) δ 175.7, 164.0, 160.8, 156.4, 149.3, 148.8, 145.2, 144.9, 143.3, 136.3, 128.5, 124.1, 121.4, 120.0, 116.7, 116.3, 112.6, 110.7, 103.2, 98.4, 93.5, 78.7, 75.9, 68.7, 60.5, 55.8, 28.5, 27.8, 22.1, 13.8. HR-MS (ESI) m/z: calcd for C30H30O10 [M+H]+: 551.1917; found 551.1927.
4.3.6. 20-O-hexylsilybin (19)
This derivative was prepared in 44 % yield as a light yellowish solid. m.p. 199–201°C. IR (neat) vmax: 3379, 2951, 2924, 1654, 1600, 1504 cm−1. 1H NMR (300 MHz, CDCl3 and DMSO-d6) δ 12.11 (s, 1H), 10.21 (s, 1H), 8.50 (s, 1H), 7.77 (d, J = 1.8 Hz, 1H), 7.74 (dd, J = 8.7, 1.9 Hz, 1H), 6.98 (d, J = 8.6 Hz, 1H), 6.94 (d, J = 7.3 Hz, 1H), 6.93 (s, 1H), 6.84 (d, J = 8.6 Hz, 1H), 6.30 (d, J = 1.7 Hz, 1H), 6.14 (d, J = 1.9 Hz, 1H), 4.96 (d, J = 8.0 Hz, 1H), 4.07–4.03 (m, 1H), 3.94 (t, J = 6.7 Hz, 2H), 3.80 (s, 3H), 3.68 (d, J = 13.1 Hz, 1H), 3.42 (d, J = 11.3 Hz, 1H), 1.79–1.70 (m, 2H), 1.41–1.36 (m, 2H), 1.28–1.25 (m, 4H), 0.83 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3 and DMSO-d6) δ 175.7, 164.0, 160.8, 156.4, 149.2, 148.7, 145.2, 144.9, 143.3, 136.3, 128.5, 124.1, 121.3, 120.0, 116.7, 116.3, 112.6, 110.7, 103.2, 98.4, 93.5, 79.7, 75.9, 68.7, 60.4, 55.8, 31.2, 28.8, 25.3, 22.2, 13.8. HR-MS (ESI) m/z: calcd for C31H32O10 [M+H]+: 565.2073; found 565.2092.
4.3.7. 20-O-Heptylsilibin (20)
This derivative was prepared in 46% yield as a light yellow solid. m.p. 175–176 °C. IR (neat) vmax: 3365, 2925, 1631, 1596, 1505, 1467, 1271 cm−1. 1H NMR (300 MHz, CDCl3 + CD3OD) δ 7.83 (s, 1H), 7.78 (d, J = 8.7 Hz, 1H), 7.06 (d, J = 9.6 Hz, 1H), 7.00 (d, J = 8.4 Hz, 1H), 6.97 (s, 1H), 6.92 (d, J = 8.0 Hz, 1H), 6.39 (s, 1H), 6.26 (s, 1H), 4.99 (d, J = 8.2 Hz. 1H), 4.14–4.11 (m, 1H), 4.03 (t, J = 6.8 Hz, 2H), 3.90 (s, 3H), 3.81 (dd, J = 12.2, 1.4 Hz, 1H), 3.55 (dd, J = 12.1, 3.2 Hz, 1H), 1.90–1.80 (m, 2H), 1.48–1.40 (m, 2H), 1.30 (s, 6H), 0.88 (t, J = 6.4 Hz, 3H). 13C NMR (75 MHz, CDCl3 + CD3OD) δ 176.0, 164.7, 161.2, 157.5, 150.3, 149.8, 145.8, 145.7, 144.2, 136.6, 129.0, 124.9, 122.3, 120.8, 117.6, 117.2, 113.5, 111.3, 103.9, 99.3, 94.6, 79.3, 76.8, 69.7, 61.7, 56.6, 32.3, 30.2, 29.6, 23.2, 14.6. HR-MS (ESI) m/z: calcd for C32H34O10 [M+H]+: 579.2230; found 579.2249.
4.4. General procedure for the synthesis of 5,20-O-dialkyl-2,3-dehydrosilybins (21–27)
The mixture of 3,7-O-dibenzyl-2,3-dehydrosilibin (6, 1 equiv.), alkyl iodide or alkyl bromide (4–10 equiv.), and potassium carbonate (10–20 equiv.) in DMF (0.1 M) was stirred under argon at room temperature for 48 h prior to being quenched with 1M HCl. After diluting with diethyl ether and ethyl acetate (1:1, v/v), the subsequent mixture was rinsed with brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude mass obtained was purified by PTLC eluting with 3% methanol in DCM to give the desired 5,20-O-dialkylamino-3,7-dibenzyl-2,3-dehydrosilybin. To the above-made 5,20-O-dialkylamino-3,7-dibenzyl-2,3-dehydrosilybin in methanol/ethyl acetate (50/50, v/v, 0.2 M) was sequentially added Pd/C (50% wet, 10% w/w) and ammonium formate (10 eq). The subsequent mixture was refluxed overnight under argon. After cooling to room temperature, the reaction mixture was filtered through a silica gel pad eluting with ethyl acetate. The filtrate was concentrated under vacuum and purified by PTLC eluting with 3% methanol in DCM to give the desired 5,20-O-dialkyl-2,3-dehydrosilybins (21–27).
4.4.1. 5,20-O-Dimethyl-2,3-dehydrosilybin (21)
This derivative was prepared as a light yellowish solid in 10% yield. m.p. 285–286 °C. IR (neat) vmax: 3356, 2924, 1619, 1599, 1502, 1462 cm−1. 1H NMR (300 MHz, CDCl3/DMSO-d6) δ 7.73 (s, 1H), 7.72 (d, J = 10.4 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 6.93 (s, 1H), 6.85 (d, J = 7.9 Hz, 1H), 6.42 (s, 1H), 6.26 (s, 1H), 4.98 (d, J = 8.0 Hz, 1H), 4.06–4.03 (m, 1H), 3.86 (s, 3H), 3.81 (s, 6H), 3.81–3.68 (overlapped, 1H), 3.43 (dd, J = 12.1, 4.2 Hz, 1H). 13C NMR (75 MHz, CDCl3 + DMSO-d6) δ 171.8, 163.1, 160.7, 158.6, 149.5, 149.2, 144.7, 143.6, 141.3, 137.2, 128.7, 124.5, 121.1, 120.2, 117.0, 115.9, 111.2, 110.3, 105.0, 95.9, 95.3, 78.8, 76.1, 61.0, 56.2, 55.9, 29.6. HR-MS (ESI) m/z: calcd for C27H24O10 [M+H]+: 509.1447; found 509.1461.
4.4.2. 5,20-O-Diethyl-2,3-dehydrosilybin (22)
This derivative was prepared as a light yellowish solid in 26 % yield; m.p. 226–227 °C. IR (neat) vmax: 3054, 2985, 2922, 2852, 1700, 1652, 1605, 1580, 1559, 1506, 1441, 1421 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.76 (s, 1H), 7.74 (d, J = 12.3 Hz, 1H), 7.00 (d, J = 8.7 Hz, 1H), 6.94 (d, J = 9.3 Hz, 1H), 6.92 (s, 1H), 6.84 (d, J = 8.1 Hz, 1H), 6.42 (s, 1H), 6.24 (s, 1H), 4.96 (d, J = 8.1 Hz, 1H), 4.10–3.98 (overlapped, 5H), 3.82 (s, 3H), 3.73 (d, J = 10.4 Hz, 1H), 3.46 (d, J = 9.3 Hz, 1H), 1.45 (t, J = 4.9 Hz, 3H), 1.39 (t, J = 6.5 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 171.9, 163.0, 160.1, 158.7, 149.5, 148.8, 144.6, 143.7, 141.2, 137.3, 128.6, 124.6, 121.1, 120.1, 117.0, 116.0, 112.6, 110.5, 105.2, 96.7, 95.2, 78.8, 76.2, 64.6, 64.3, 61.1, 56.0, 14.7, 14.5. HR-MS (ESI) m/z: calcd for C29H28O10 [M+H]+: 537.1760; found 537.1777.
4.4.3. 5,20-O-Dipropyl-2,3-dehydrosilybin (23)
This derivative was prepared in 35% yield as a yellow solid; m.p. 132–133 °C. IR (neat) vmax: 3355, 2957, 2925, 1657, 1607, 1552, 1467 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.72 (s, 1H), 8.96 (s. 1H), 7.73 (s, 1H), 7.72 (d, J = 8.8 Hz, 1H), 7.10 (d, J = 11.8 Hz, 1H), 7.08 (s, 1H), 6.99 (s, 2H), 6.49 (s, 1H), 6.33 (s, 1H), 5.03–4.98 (overlapped, 1H), 5.01 (d, J = 7.1 Hz, 1H), 4.30–4.26 (m, 1H), 4.00–3.86 (m, 4H), 3.78 (s, 3H), 3.78–3.69 (overlapped, 1H), 3.58 (d, J = 11.8 Hz, 1H), 1.80–1.71 (m, 4H), 1.08 (t, J = 6.4 Hz, 3H), 0.98 (t, J = 7.2 Hz, 3H). 13C NMR (75 MHz, CDCl3 + CD3OD) δ 175.6, 164.2, 160.9, 156.6, 149.5, 149.0, 145.1, 145.0, 143.5, 136.2, 128.5, 124.2, 121.6, 120.1, 116.9, 116.4, 112.8, 110.7, 103.3, 98.7, 93.8, 78.8, 76.1, 68.9, 60.8, 56.0, 31.4, 28.9, 25.5, 22.4, 13.9. HR-MS (ESI) m/z: calcd for C31H32O10 [M+H]+: 565.2073; found 565.2088.
4.4.4. 5,20-O-Dibutyl-2,3-dehydrosilybin (24)
This derivative was prepared in 40% yield as a brown solid; m.p. 195–196 °C. IR (neat) vmax: 3201, 2956, 2924, 2872, 2853, 1708, 1607, 1505, 1465, 1420 cm−1. 1H NMR (300 MHz, CDCl3 and DMSO-d6) δ 7.74 (d, J = 1.6 Hz, 1H), 7.71 (d, J = 8.6 Hz, 1H), 6.99 (d, J= 8.5 Hz, 1H), 6.93 (d, J = 10.1 Hz, 1H), 6.91 (s, 1H), 6.83 (d, J = 7.9 Hz, 1H), 6.40 (s, 1H), 6.23 (s, 1H), 4.96 (d, J = 8.0 Hz, 1H), 4.06–4.03 (m, 1H), 3.97 (t, J = 6.7 Hz, 2H), 3.95 (t, J = 6.7 Hz, 2H), 3.81 (s, 3H), 3.72 (dd, J = 12.4, 1.9 Hz, 1H), 3.46 (dd, J = 12.4, 3.7 Hz, 1H), 1.87–1.70 (m, 4H), 1.55–1.38 (m, 4H), 0.92 (t, J = 6.8 Hz, 3H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3 + DMSO-d6) δ:171.8, 163.0, 160.3, 158.6, 149.6, 149.0, 144.6, 143.6, 141.1, 137.3, 128.6, 124.5, 124.1, 120.1, 117.0, 115.9, 112.8, 110.7, 105.2, 96.6, 95.1, 78.8, 76.2, 68.6, 61.0, 56.0, 53.5, 31.1, 30.9, 29.5, 19.1, 19.0, 13.8. HR-MS (ESI) m/z: calcd for C33H36O10 [M+H]+: 593.2286; found 593.2402.
4.4.5. 5,20-O-Dipentyl-2,3-dehydrosilybin (25)
This derivative was prepared in 7% yield as a light yellowish solid. m.p. 166–167 °C. IR (neat) vmax: 3334, 2954, 2929, 1608, 1505, 1260 cm−1. 1H NMR (300 MHz, CDCl3 + DMSO-d6) δ:7.80 (s, 1H), 7.77 (d, J = 9.0 Hz, 1H), 7.04 (d, J = 8.1 Hz, 1H), 6.98–6.94 (overlapped, 1H), 6.94 (s, 1H), 6.87 (d, J = 8.1 Hz, 1H), 6.45 (s, 1H), 6.27 (s, 1H), 4.98 (d, J = 8.1 Hz, 1H), 4.13–4.06 (m, 1H), 3.99 (t, J = 6.6 Hz, 4H), 3.85 (s, 3H), 3.81–3.75 (overlapped, 1H), 3.52 (d, J = 11.4 Hz, 1H), 1.95–1.74 (m, 4H), 1.54–1.26 (m, 8H), 0.92–0.88 (overlapped, 6H). 13C NMR (75 MHz, CDCl3 + DMSO-d6) δ:171.8, 163.0, 160.3, 158.7, 149.6, 149.1, 144.6, 143.7, 141.1, 137.3, 128.6, 124.6, 121.0, 120.1, 117.0, 115.9, 112.8, 110.7, 105.2, 96.6, 95.1, 78.8, 76.2, 69.1, 69.0, 61.1, 56.1, 29.6, 28.7, 28.5, 28.0, 22.4, 14.0. HR-MS (ESI) m/z: calcd for C35H40O10 [M+H]+: 621.2700; found 621.2715.
4.4.6. 5,20-O-Dihexyl-2,3-dehydrosilybin (26)
This derivative was prepared as a light yellowish solid in 57% yield; m.p. 155–157 °C. IR (neat) vmax: 3303, 2953, 2926, 2870, 2856, 1609, 1537, 1506, 1466 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.71 (s, 1H), 8.96 (s, 1H), 7.73 (s, 1H), 7.71 (d, J = 8.2 Hz, 1H), 7.11 (d, J = 8.4 Hz, 1H), 7.07 (s, 1H), 6.99 (s, 2H), 6.48 (s, 1H), 6.33 (s, 1H), 5.04–4.99 (overlapped, 1H), 5.01 (d, J = 7.3 Hz, 1H), 4.29–4.26 (m, 1H), 3.99 (t, J = 6.2 Hz, 2H), 3.96 (t, J = 3.6 Hz, 2H), 3.78 (s, 3H), 3.57 (dd, J = 11.7, 5.7 Hz, 1H), 3.39–3.35 (overlapped, 1H), 1.79–1.67 (m, 4H), 1.55 (quin, J = 7.3 Hz, 2H), 1.42 (quin, J = 6.3 Hz, 2H), 1.36–1.30 (m, 8H), 0.88 (t, J = 6.4 Hz, 6H). 13C NMR (75 MHz, CDCl3 + DMSO-d6) δ 172.8, 163.4, 160.9, 159.3, 150.2, 149.8, 145.2, 144.2, 142.4, 137.9, 129.1, 125.1, 121.9, 120.8, 117.6, 116.7, 113.5, 111.3, 106.0, 97.1, 95.5, 79.3, 76.8, 69.9, 69.7, 61.8, 56.6, 32.2, 30.3, 29.6, 29.2, 26.2, 26.1, 23.2, 14.6. HR-MS (ESI) m/z: calcd for C37H44O10 [M+H]+: 649.3012; found 649.3035.
4.4.7. 5,20-O-Diheptyl-2,3-dehydrosilybin (27)
This derivative was prepared in 11% yield as a brown solid; m.p. 135.5–136.0 °C. IR (neat) vmax: 3303, 2924, 2955, 2854, 1609, 1507, 1465, 1443 cm−1. 1H NMR (300 MHz, CDCl3) δ:7.84 (s, 1H), 7.80 (d, J = 8.4 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 7.00 (d, J = 10.8 Hz, 1H), 6.98 (s, 1H), 6.90 (d, J = 7.8 Hz, 1H), 6.49 (s, 1H), 6.31 (s, 1H), 5.00 (d, J = 8.1 Hz, 1H), 4.13–4.10 (m, 1H), 4.07–4.00 (m, 4H), 3.89 (s, 3H), 3.81 (d, J = 11.7 Hz, 1H), 3.56 (d, J = 12.0 Hz, 1H), 1.97–1.78 (m, 4H), 1.57–1.22 (m, 16 H), 0.93–0.82 (overlapped, 6H). 13C NMR (75 MHz, CDCl3) δ 171.9, 163.0, 160.4, 158.8, 149.8, 149.3, 144.5, 143.7, 137.4, 132.5, 128.3, 124.9, 121.3, 120.2, 117.1, 116.2, 112.9, 110.6, 105.4, 96.7, 95.3, 78.7, 76.3, 69.3, 69.1, 61.5, 56.1, 31.8, 31.8, 31.7, 29.1,29.1, 29.0, 28.9, 25.9, 22.6, 22.5. HR-MS (ESI) m/z: calcd for C39H48O10 [M+H]+: 677.3325; found 677.3315.
4.5. Synthesis of 3,7,20-O-tribenzyl-2,3-dehydrosilybin (28)
The mixture of silybin (1 equiv.) and potassium carbonate (4 equiv.) in acetone (0.1 M) was stirred under argon atmosphere for 10 min prior to being added benzyl bromide (1.1 equiv.) through syringe. The reaction was allowed to proceed with refluxing under argon for 4 h. After cooling down to room temperature, the acetone solvent was evaporated to dryness. To the reaction flask, DMF was added (0.5 M) followed by additional potassium carbonate (2 equiv.) and benzyl bromide (2.2 equiv.). The reaction flask was stirred at room temperature for 3 hours. The reaction was quenched by addition of 1 M HCl followed by ethyl acetate and diethyl ether (1:1, v/v), which was rinsed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo to give a crude product. The crude product was subjected to column chromatography over silica gel, using ethyl acetate/hexane (1:1, v/v), to furnish the desired product in 51% yield. 1H NMR (300 MHz, CDCl3) δ 12.69 (s, 1H), 7.70 (d, J = 8.7 Hz, 1H), 7.68 (s, 1H), 7.48–7.33 (m, 15H), 7.03 (d, J = 8.7 Hz, 1H), 7.00 (s, 1H), 6.97 (s, 2H), 6.48 (s, 1H), 6.45 (s, 1H), 5.22 (S, 2H), 5.13 (s, 2H), 5.10 (s, 2H), 5.01 (d, J = 8.1 Hz, 1H), 4.15–4.12 (m, 1H), 3.95 (s, 3H), 3.87 (d, J = 12.3 Hz, 1H), 3.60 (dd, J = 12.9, 3.6 Hz, 1H).
4.6. General procedure for the synthesis of 5-O-alkyl-2,3-dehydrosilybins (29–35)
To a solution of 3,7,20-O-tribenzyl-2,3-dehydrosilibin (28, 1 equiv.) in DMF (0.2 M) was sequentially added potassium carbonate (8 equiv.) and alkyl iodide or alkyl bromide (8 equiv.), and the mixture was stirred under argon at room temperature for 24 h prior to being diluted with ethyl acetate and diethyl ether (1:1. v/v). The subsequent mixture was rinsed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo to give a crude product. PTLC purification of the crude product, using 5% methanol in DCM as eluent, gave the desired 5-O-alkyl-3,7,20-O-tribenzyl-2,3-dehydrosilybin. To the solution of above-made 5-O-alkyl-3,7,20-O-tribenzyl-2,3-dehydrosilybin (1 eq) in methanol/ethyl acetate (50/50, v/v, 0.2 M) was sequentially added Pd-C (50% wet, 10% w/w) and ammonium formate (10 eq). The reaction mixture was refluxed overnight under argon atmosphere. After cooling to room temperature, the reaction mixture was filtered through a silica gel pad eluting with ethyl acetate. The filtrate was concentrated under vacuum and purified by PTLC eluting with 8% methanol in DCM to give the desired 5-O-alkyl-2,3-dehydrosilybins (29–35).
4.6.1. 5-O-Methyl-2,3-dehydrosilybin (29)
This derivative was prepared in 41% yield as a yellow solid. m.p. 227–228 °C. IR (neat) vmax: 3363, 2956, 2924, 2870, 1604, 1505, 1462, 1377 cm−1. 1H NMR (300 MHz, DMSO-d6) δ: 7.73 (s, 1H), 7.71 (dd, J = 4.7, 1.8 Hz, 1H), 7.48 (d, J = 4.7 Hz, 1H), 7.00 (d, J = 8.3 Hz, 1H), 6.89 (s, 1H), 6.84 (s, 2H), 6.42 (d, J = 2.0 Hz, 1H), 6.26 (d, J = 1.8 Hz, 1H), 4.93 (d, J = 8.1 Hz, 1H), 4.07–4.02 (m, 1H), 3.86 (s, 3H), 3.82 (s, 3H), 3.69 (dd, J = 12.4, 2.3 Hz, 1H), 3.44 (dd, J = 12.2, 3.8 Hz, 1H). 13C NMR (75 MHz, CDCl3 + DMSO-d6) δ: 172.5, 163.8, 161.4, 159.3, 148.4, 147.7, 145.5, 144.4, 142.0, 137.9, 128.2, 125.0, 121.7, 121.2, 117.7, 116.6, 116.0, 111.1, 105.7, 96.6, 96.0, 79.6, 77.0, 61.6, 56.9, 56.6. HR-MS (ESI) m/z: calcd for C26H22O10 [M+H]+: 495.2859; found 495.2848.
4.6.2. 5-O-Ethyl-2,3-dehydrosilybin (30)
This derivative was prepared in 56 % yield as a yellow solid. m.p. 208–209 °C. IR (neat) vmax: 3315, 2957, 2925, 2870, 1606, 1505, 1463, 1377 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.70 (br.s, 1H), 9.16 (s, 1H), 8.88 (s, 1H), 7.72 (s, 1H), 7.71 (d, J = 6.9 Hz, 1H), 7.10 (d, J = 9.2 Hz, 1H), 7.04 (s, 1H), 6.89 (dd, J = 8.2, 0.96 Hz, 1H), 6.81 (d, J = 8.1 Hz, 1H), 6.49 (d, J = 1.6 Hz, 1H), 6.33 (d, J = 1.3 Hz, 1H), 4.99 (t, J = 5.0 Hz, 1H), 4.96 (d, J = 7.9 Hz, 1H), 4.26–4.23 (m, 1H), 4.06 (q, J = 6.9 Hz, 2H), 3.79 (s, 3H), 3.78–3.74 (overlapped, 1H), 3.56 (br.d, J = 12.3 Hz, 1H), 1.39 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3 + DMSO-d6) δ 172.4, 163.7, 160.7, 159.3, 148.4, 147.7, 145.4, 144.4, 141.8, 137.9, 128.1, 125.0, 121.6, 121.2, 117.7, 116.5, 116.0, 111.2, 105.8, 97.4, 95.8, 79.6, 77.0, 65.3, 61.5, 56.6, 15.1, HR-MS (ESI) m/z: calcd for C27H24O10 [M+H]+: 509.1447; found 509.1440.
4.6.3. 5-O-Propyl-2,3-dehydrosilybin (31)
This derivative was prepared in 48% yield as a yellow solid. m.p. 185–185.5 °C. IR (neat) vmax: 3276, 2957, 2924, 2871, 2853, 1610, 1506, 1464, 1421 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.68 (s, 1H), 9.16 (s, 1H), 8.95 (s, 1H), 7.72 (s, 1H), 7.70 (dd, J = 7.2, 2.0 Hz, 1H), 7.10 (d, J = 9.2 Hz, 1H), 7.03 (d, J = 1.0 Hz, 1H), 6.88 (dd, J = 8.1, 1.3 Hz, 1H), 6.81 (d, J = 8.0 Hz, 1H), 6.48 (d, J = 1.8 Hz, 1H), 6.32 (d, J = 1.7 Hz, 1H), 4.99 (t, J = 5.3 Hz, 1H), 4.96 (d, J = 7.9 Hz, 1H), 4.27–4.23 (m, 1H), 3.96 (t, J = 5.9 Hz, 2H), 3.79 (s, 3H), 3.57 (d, J = 11.6 Hz, 1H), 3.37–3.33 (overlapped, 1H), 1.82–1.75 (m, 2H), 1.08 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3 + DMSO-d6) δ 172.4, 163.7, 161.0, 159.3, 148.5, 147.8, 145.4, 144.4, 141.8, 138.1, 128.2, 125.1, 121.6, 121.2 117.7, 116.6, 116.1, 111.4, 105.9, 97.3, 95.7, 79.7, 77.0, 71.1, 61.5, 56.6, 23.0, 11.3. HR-MS (ESI) m/z: calcd for C28H26O10 [M+H]+: 523.1604; found 523.1595.
4.6.4. 5-O-Butyl-2,3-dehydrosilybin (32)
This derivative was prepared in 54% yield as a yellow solid. m.p. 221–221.5 °C. IR (neat) vmax: 3252, 2956, 2917, 2871, 2849, 1615, 1578, 1540, 1506, 1464 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.68 (br.s, 1H), 9.16 (s, 1H), 8.96 (s, 1H), 7.72 (s, 1H), 7.70 (dd, J = 7.2, 1.9 Hz, 1H), 7.10 (d, J = 9.2 Hz, 1H), 7.03 (s, 1H), 6.88 (dd, J = 8.5, 1.2 Hz, 1H), 6.81 (d, J = 8.1 Hz, 1H), 6.48 (d, J = 1.6 Hz, 1H), 6.32 (d, J = 1.5 Hz, 1H), 4.99 (t, J = 5.3 Hz, 1H), 4.96 (d, J = 7.9 Hz, 1H), 4.26–4.23 (m, 1), 4.00 (t, J = 5.7 Hz, 2H), 3.79 (s, 3H), 3.56 (d, J = 14.8 Hz, 1H), 3.39–3.32 (overlapped, 1H), 1.78–1.71 (m, 2H), 1.62–1.54 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3 + DMSO-d6) 5: 172.3, 163.7, 161.0, 159.2, 148.6, 147.9, 145.4, 144.4, 141.8, 138.3, 128.2, 125.1, 121.5, 121.2, 117.6, 116.6, 116.2, 111.7, 106.0, 97.3, 95.7, 79.7, 76.9, 69.2, 61.4, 56.6, 31.7, 19.8, 14.6. HR-MS (ESI) m/z: calcd for C29H28O10 [M+H]+: 537.1760; found 537.1760.
4.6.5. 5-O-pentyl-2,3-dehydrosilybin (33)
This derivative was prepared in 60% yield as a yellow solid. m.p. 217–218 °C. IR (neat) vmax: 3157, 2955, 2924, 2870, 1606,1579, 1508, 1443, 1416 cm−1. 1H NMR (300 MHz, DMSO-d6) δ: 10.67 (b, 1H), 9.16 (s, 1H), 8.96 (s, 1H), 7.72 (s, 1H), 7.70 (dd, J = 7.5, 1.8 Hz, 1H), 7.10 (d, J = 9.2 Hz, 1H), 7.04 (s, 1H), 6.89 (dd, J = 8.3, 1.0 Hz, 1H), 6.81 (d, J = 8.1 Hz, 1H), 6.48 (d, J = 1.6 Hz, 1H), 6.32 (d, J = 1.2 Hz, 1H), 4.99 (t, J = 5.4 Hz, 1H), 4.96 (d, J = 7.9 Hz, 1H), 4.26–4.23 (m, 1H), 3.99 (t, J = 5.9 Hz, 2H), 3.79 (s, 3H), 3.56 (d, J = 11.6 Hz, 1H), 3.39–3.33 (overlapped, 1H),1.82–1.72 (m, 2H), 1.58–1.48 (m, 2H), 1.42–1.30 (m, 2H), 0.90 (t, J = 7.2 Hz, 3H). 13C NMR (75 MHz, CDCl3 + DMSO-d6) δ: 172.4, 163.7, 160.9, 159.2, 148.5, 147.8, 145.4, 144.4, 141.8, 138.0, 128.1, 125.1, 121.6, 121.2, 117.7, 116.6, 116.2, 111.3, 105.9, 97.3, 95.7, 79.6, 77.0, 69.6, 61.5, 56.6, 29.2, 28.7, 23.0, 14.7. HR-MS (ESI) m/z: calcd for C30H30O10 [M+H]+: 551.1917; found 551.1908.
4.6.6. 5-O-hexyl-2,3-dehydrosilybin (34)
This derivative was prepared in 53% yield as a yellow solid. m.p. 172–173 °C. IR (neat) vmax: 3294, 2957, 2925, 2870, 1610, 1545, 1504, 1460 cm−1. 1H NMR (300 MHz, DMSO-d6) 5: 10.67 (s, 1H), 9.16 (s, 1H), 8.94 (s, 1H), 7.72 (s, 1H), 7.69 (dd, J = 7.5, 2.0 Hz, 1H), 7.10 (d, J = 9.2 Hz, 1H), 7.03 (s, 1H), 6.88 (d, J = 8.3 Hz, 1H), 6.81 (d, J = 8.0 Hz, 1H), 6.48 (d, J = 1.7 Hz, 1H), 6.32 (d, J = 1.4 Hz, 1H), 4.99 (t, J = 5.5 Hz, 1H), 4.96 (d, J = 7.9 Hz, 1H), 4.27–4.22 (m, 1H), 3.99 (t, J = 5.9 Hz, 2H), 3.79 (s, 3H), 3.56 (d, J=14.3 Hz, 1H), 3.33-3.15 (overlapped, 1H), 1.81–1.71 (m, 2H), 1.57–1.53 (m, 2H), 1.32–1.30 (m, 4H), 0.88 (t, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3 + DMSO-d6) δ: 172.4, 163.7, 161.0, 159.3, 148.5, 147.8, 145.4, 144.4, 141.8, 138.1, 128.2, 125.1, 121.6, 121.2, 117.7, 116.6, 116.2, 111.4, 105.9, 97.3, 95.7, 79.7, 77.0, 69.7, 61.5, 56.6, 32.2, 29.5, 26.2, 23.2, 14.8. HR-MS (ESI) m/z: calcd for C31H32O10 [M+H]+: 565.2073; found 565.2064.
4.6.7. 5-O-heptyl-2,3-dehydrosilybm (35)
This derivative was prepared in 49% yield as a yellow solid. m.p. 138–139.5 °C. IR (neat) vmax: 3288, 2954, 2924, 2870, 1608, 1505, 1464 cm−1. 1H NMR (300 MHz, DMSO-d6) δ 10.67 (br.s, 1H), 9.16 (s, 1H), 8.93 (br.s, 1H), 7.72 (s, 1H), 7.69 (dd, J = 7.5, 1.9 Hz, 1H), 7.10 (d, J = 9.1 Hz, 1H), 7.04 (s, 1H), 6.88 (d, J = 8.0 Hz, 1H), 6.81 (d, J = 8.0 Hz, 1H), 6.48 (d, J = 1.5 Hz, 1H), 6.32 (br.s, 1H), 4.99 (t, J = 5.5 Hz, 1H), 4.96 (d, J = 7.9 Hz, 1H), 4.27–4.22 (m, 1H), 3.99 (t, J = 5.9 Hz, 2H), 3.78 (s, 3H), 3.56 (d, J = 14.6 Hz, 1H), 3.53–3.24 (overlapped, 1H), 1.81–1.72 (m, 2H), 1.58–1.49 (m, 2H), 1.35–1.23 (m, 6H), 0.87 (t, J = 6.3 Hz, 3H). 13C NMR (75 MHz, CDCl3 + DMSO-d6) δ 172.5, 163.7, 161.0, 159.3, 148.3, 147.6, 145.3, 144.4, 141.8, 137.9, 128.2, 125.2, 121.7, 121.2, 117.7, 116.6, 115.9, 11 1.0, 105.9, 97.3, 95.8, 79.5, 77.0, 69.7, 61.7, 56.6, 32.3, 29.6, 29.5, 26.5, 23.2, 14.7. HR-MS (ESI) m/z: calcd for C32H34O10 [M+H]+: 579.2230 found 579.2219.
4.3. Cell culture
All cell lines were initially purchased from American Type Culture Collection (ATCC™). The PC-3 and LNCaP prostate cancer cell lines were routinely cultured in RPIM-1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. The DU145 prostate cancer cells were routinely cultured in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% FBS and 1% penicillin/streptomycin. Cultures were maintained in a high humidity environment supplemented with 5% carbon dioxide at a temperature of 37 °C.
4.4. WST-1 cell proliferation assay
PC-3, DU145, and LNCaP cells were plated in 96-well plates at a density of 3,200 each well in 200 μL of culture medium. The cells were then treated with silybin, or synthesized silybin derivatives separately at 5 different doses for 3 days, while equal treatment volumes of DMSO (0.25%) were used as vehicle control. The cells were cultured in a CO2 incubator at 37 °C for three days. 10 μL of the premixed WST-1 cell proliferation reagent (Clontech) was added to each well. After mixing gently for one minute on an orbital shaker, the cells were incubated for additional 3 hours at 37 °C. To ensure homogeneous distribution of color, it is important to mix gently on an orbital shaker for one minute. The absorbance of each well was measured using a microplate reader (Synergy HT, BioTek) at a wavelength of 430 nm. The IC50 value is the concentration of each compound that inhibits cell proliferation by 50% under the experimental conditions and is the average from at least triplicate determinations that reproducible and statistically significant. For calculating the IC50 values, a linear proliferative inhibition was made based on at least five dosages for each compound.
4.5. Cell cycle analysis
PC-3 cells were plated in 24-well plates at a density of 200,000 each well in 400 μL of culture medium. After 3 hours of cell attachment, the cells were then treated with compound 35 at 10 μM and 20 μM for 16 hours and 24 hours, while equal treatment volumes of DMSO were used as vehicle control. The cells were cultured in CO2 incubator at 37 °C for 16 hours and 24 hours, respectively. Both attached and floating cells were collected in a centrifuge tube by centrifugation at rcf value 450 g for 5 minutes. After discarding the supernatant, the collected cells were re-suspended with 500 μL 80% cold ethanol to fix for 30 minutes in 4 °C. The fixed cell could store in −20 °C for one week. After fixation, the ethanol was removed after centrifuging and the cells were washed with PBS. The cells were then re-suspend with 100μL of 100mg/mL ribonuclease and were cultured at 37 °C for 30 minutes to degrade all RNA. The cells were stained with 200 μL of 50 μg/mL propidium iodide stock solution for 30 minutes at −20°C, and then the fluorescence intensity of PI was detected in individual PC-3 cells using an Attune flow cytometer (Life Technologies) within 0.5 to 1 hour after staining.
4.6 F2N12S and CYTOX AADvanced double staining assay
PC-3 cells were plated in 24-well plates at a density of 200,000 each well in 400 μL of culture medium. After 3 hours of cell attachment, the cells were then treated with the test compound at different concentration for 16 hours, while equal treatment volumes of DMSO were used as vehicle control. The cells were cultured in CO2 incubator at 37°C for 15 hours. Both attached and floating cells were collected in a centrifuge tube by centrifugation at rcf value 450 g for 5 to 6 minutes. The collected cells were re-suspended with 500 μL HBSS to remove proteins which may affect flow signal and centrifuged again. After discarding the supernatant, the collected cells were re-suspended with 300 μL HBSS and stained with 0.3 μL of F2N12S for 3–5 minutes followed by 0.3 μL CYTOX AAdvanced for an additional 5 minutes. The fluorescence intensity of the two probes was further measured in individual PC-3 cells using an Attune flow cytometer (Life Technologies) 0.5 to 1 hour after staining.
4.7. Statistical analysis
All data are represented as the mean ± standard deviation (S.D.) for the number of experiments indicated. Other differences between treated and control groups were analyzed using the Student’s t-test. A p-value < 0.05 was considered statistically significant.
Acknowledgments
This work was financially supported by California State University (CSU)-Fresno. The HRMS data were supported by NIH RCMI Program at Xavier University of Louisiana through Grant 2G12MD007595 (G.W.). We are also grateful to (i) the ASI and the Graduate Initiative Net at CSU-Fresno for Graduate Research Fellowships (to X.Z. and S.Z.), (ii) CSUPERB for Presidents’ Commission Scholar Award (to T.L.), and (iii) CSU-LSAMP program funded by NSF under grant # HRD-1302873, CSU Office of the Chancellor, and Fresno State (to T.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pelter A, Haensel R. Tetrahedron Lett. 1968;2911 [Google Scholar]
- 2.Vue B, Chen Q-H. Cur Med Chem. 2016;23:3925. doi: 10.2174/0929867323666160823151833. [DOI] [PubMed] [Google Scholar]
- 3.Kim N-C, Graf TN, Sparacino CM, Wani MC, Wall ME. Org Biomol Chem. 2003;1:1684. doi: 10.1039/b300099k. [DOI] [PubMed] [Google Scholar]
- 4.Flora K, Hahn M, Rosen H, Benner K. Am J Gastroenterol. 1998;93:139. doi: 10.1111/j.1572-0241.1998.00139.x. [DOI] [PubMed] [Google Scholar]
- 5.Abenavoli L, Capasso R, Milic N, Capasso F. Phytother Res. 2010;24:1423. doi: 10.1002/ptr.3207. [DOI] [PubMed] [Google Scholar]
- 6.Biedermann D, Vavrikova E, Cvak L, Kren V. Nat Prod Rep. 2014;31:1138. doi: 10.1039/c3np70122k. [DOI] [PubMed] [Google Scholar]
- 7.Agarwal R, Katiyar SK, Lundgren DW, Mukhtar H. Carcinogenesis. 1994;15:1099. doi: 10.1093/carcin/15.6.1099. [DOI] [PubMed] [Google Scholar]
- 8.Lahiri-Chatterjee M, Katiyar SK, Mohan RR, Agarwal R. Cancer Res. 1999;59:622. [PubMed] [Google Scholar]
- 9.Agarwal R, Agarwal C, Ichikawa H, Singh RP, Aggarwal BB. Anticancer Res. 2006;26:4457. [PubMed] [Google Scholar]
- 10.Kroll DJ, Shaw HS, Oberlies NH. Integr Cancer Ther. 2007;6:110. doi: 10.1177/1534735407301825. [DOI] [PubMed] [Google Scholar]
- 11.Vue B, Zhang S, Chen Q-H. Anticancer Agents Med Chem. 2015;15:1205. doi: 10.2174/1871520615666151008122622. [DOI] [PubMed] [Google Scholar]
- 12.Mericli AH. Planta Medica. 1988;54:44. doi: 10.1055/s-2006-962330. [DOI] [PubMed] [Google Scholar]
- 13.Agarwal C, Wadhwa R, Deep G, Biedermann D, Gazak R, Kren V, Agarwal R. PLoS One. 2013;8:e60074. doi: 10.1371/journal.pone.0060074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S, Vue B, Huang M, Zhang X, Lee T, Chen G, Zhang Q, Zheng S, Wang G, Chen Q-H. Bioorg Med Chem Lett. 2016;26:3226. doi: 10.1016/j.bmcl.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vue B, Zhang S, Zhang X, Parisis K, Zhang Q, Zheng S, Wang G, Chen Q-H. Eur J Med Chem. 2016;109:36. doi: 10.1016/j.ejmech.2015.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karas D, Gazak R, Valentova K, Chambers CS, Pivodova V, Biedermann D, Krenkova A, Oborna I, Kuzma M, Cvacka J, Ulrichova J, Kren V. J Nat Prod. 2016;79:812. doi: 10.1021/acs.jnatprod.5b00905. [DOI] [PubMed] [Google Scholar]
- 17.Davis-Searles PR, Nakanishi Y, Kim N-C, Oberlies TN, Wani MC, Wall ME, Agarwal R, Kroll DJ. Cancer Res. 2005;65:4448. doi: 10.1158/0008-5472.CAN-04-4662. [DOI] [PubMed] [Google Scholar]
- 18.Filippopoulou K, Papaevgeniou N, Lefaki M, Paraskevopoulou A, Biedermann D, Kren V, Chondrogianni N. Free Radic Biol Med. 2017;103:256. doi: 10.1016/j.freeradbiomed.2016.12.042. [DOI] [PubMed] [Google Scholar]
- 19.Dzubak P, Hajduch M, Gazak R, Svobodova A, Psotova J, Walterova D, Sedmera P, Kren V. Bioorg Med Chem. 2006;14:3793. doi: 10.1016/j.bmc.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 20.Psyzkova M, Biler M, Biedermann D, Valentova K, Kuzma M, Vrba J, Ulrichova J, Sokolova R, Mojovic M, Popovic-Bijelic A, Kubala M, Trouillas P, Kren V, Vacek J. Free Radic Biol Med. 2016;90:114. doi: 10.1016/j.freeradbiomed.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 21.Deep G, Singh RP, Agarwal C, Kroll DJ, Agarwal R. Oncogene. 2006;25:1053. doi: 10.1038/sj.onc.1209146. [DOI] [PubMed] [Google Scholar]
- 22.Tyagi A, Bhatia N, Condon MS, Bosland MC, Agarwal C, Agarwal R. Prostate. 2002;53:211. doi: 10.1002/pros.10146. [DOI] [PubMed] [Google Scholar]
- 23.Zi X, Agarwal R. Proc Natl Acad Sci USA. 1999;96:7490. doi: 10.1073/pnas.96.13.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh RP, Deep G, Blouin M-J, Pollak MN, Agarwal R. Carcinogenesis. 2007;28:2567. doi: 10.1093/carcin/bgm218. [DOI] [PubMed] [Google Scholar]