

Abstract
Complement plays an important role in the pathogenesis of rheumatoid arthritis (RA). While the alternative pathway (AP) is known to play a key pathogenic role in models of RA, the importance of the lectin pathway (LP) pattern recognition molecules such as ficolin A (FCN A), ficolin B (FCN B) and collectin-11 (CL-11) as well as the activating enzyme MBL (mannose binding lectin)-associated serine protease-2 (MASP-2) are less well understood. We show here that FCN A−/− and CL-11−/− mice are fully susceptible to collagen antibody-induced arthritis (CAIA). In contrast, FCN B−/− and MASP-2−/−/sMAp−/− mice are substantially protected, with clinical disease activity decreased significantly (p < 0.05) by 47% and 70%, respectively. Histopathology scores, C3, fD, FCN B deposition and infiltration of synovial macrophages and neutrophils were similarly decreased in FCN B−/− and MASP-2−/−/sMAp−/− mice. Our data support that FCN B plays an important role in the development of CAIA, likely through ligand recognition in the joint and MASP activation, and that MASP-2 also contributes to the development of CAIA, likely in a C4-independent manner. Decreased AP activity in the sera from FCN B−/− and MASP-2−/−/sMAp−/− mice with arthritis on adherent anti-collagen antibodies also support the hypothesis that pathogenic antibodies as well as additional inflammation-related ligands are recognized by the LP and operate in vivo to activate complement. Finally, we also speculate that the residual disease seen in our studies is driven by the AP and/or the C2/C4 bypass pathway via the direct cleavage of C3 through a LP-dependent mechanism.
Keywords: complement, arthritis, C4/C2 bypass pathway, inflammation
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease found in ~1% of the adult population (1). The prevalence of RA-related disability and the number of RA patients in the US has been projected to increase by 40% over the next 25 years (2), suggesting that this disease will continue to adversely impact patients and the health care system (3). While the etiology of RA remains unknown, many potential risk factors for the development of RA have been identified (4). Among other factors, it is now appreciated that the complement system plays an important role in the pathogenesis of RA. Complement activation is essential for disease progression in passive transfer mouse models of RA, and complement fragments derived from the activation process have been found in the synovium of RA patients (5–7). Furthermore, complement-directed therapeutics have shown excellent efficacy in mouse models of RA such as collagen antibody-induced arthritis (CAIA) (8, 9). The complement system is activated by three inter-linked pathways: the classical pathway (CP), the lectin pathway (LP) and the alternative pathway (AP). All of these pathways generate two potent pro-inflammatory molecules, C3a and C5a, via C3 and C5 convertases, respectively, as well as the membrane attack complex, each of which plays an important role in CAIA (10).
Our previous studies in the CAIA model have revealed an essential role for the AP in the development of joint damage, with initial studies suggesting that the AP can function fully independently of the CP and LP in this model (6). In contrast, an essential role for the CP has not been observed, as mice deficient in C4 and C1q are fully susceptible to disease (11). The LP has been proposed to be involved in the development of CAIA, but only through the requirement for MASP-1 and/or MASP-3 in cleaving the inactive AP protease pro-Factor D (pro-Df) into active Factor D (Df), thus allowing the AP to function in vivo (12–14). An independent role for the LP recognition and activation mechanisms has not been identified yet in this model, but is evaluated herein.
The LP is activated when specific pattern recognition molecules engage carbohydrate and other ligands either expressed on pathogens or revealed in injured tissue (15, 16). The first pattern recognition molecule described in the LP was mannose-binding lectin (MBL) (17). In addition to MBL, though, ficolins (FCNs) and collectins (CLs) can also recognize unique ligands and activate the LP. With regard to the mechanism of activation of complement, the function of CLs, FCNs and MBLs are similar, as all are soluble pattern recognition molecules which interact with mannose-binding lectin-associated serine proteases (MASPs) -1, 2 and 3 (MASP-1, MASP-2 and MASP-3) and activate these proteins following ligand recognition (18–20).
MBL is a C-type lectin containing a carbohydrate recognition domain (CRD) as well as collagen-like domain (21). MBL binds to mannose-containing molecules as well as N-acetylglucosamine (GlcNAc) (21). Ficolins (FCNs), which also contains a CRD, consist of collagen-like and fibrinogen-like domains and preferentially bind to GlcNAc (18, 22, 23). There are two mouse ficolins; ficolin A (FCN A) (24) and ficolin B (FCN B) (25); in contrast, humans express three ficolins; ficolin M (FCN-M a.k.a. FCN 1), ficolin L (FCN-L a.k.a. FCN 2), and ficolin H (FCN-H a.k.a. FCN 3) (26–30). Mouse FCN A, but not FCN B, exhibit a splice variant known as FCN A variant (31). FCN A is present in the serum and expressed in liver hepatocytes (32). Mouse FCN B was originally found in the lysosomes of macrophages, similar to human FCN-M, which is also found in the secretary granules of monocytes and neutrophils (33, 34). We have reported that FCN-B is also present in the circulation of mice, suggesting that it is secreted from macrophages (35).
The third class, designated Collectins (CLs), are similarly C-type lectins containing carbohydrate-recognition domains (CRDs). Three different human CLs have been identified: Collectin-10 (a.k.a collectin liver 1, CL-L1, or CL-10), Collectin-11 (a.k.a. collectin kidney 1, CL-K1, or CL-11) and Collectin-12 (collectin placenta 1, CL-P1, or CL-12) (36–39). CL-11 is present in the serum, liver, adrenal gland and kidney, and binds L-fucose and D-mannose (36, 40). Recently it has been shown that CL-11 plays an essential role in ischemia reperfusion injury in the kidney (41). The authors demonstrated that CL-11 bound to cell surface L-fucose and required MASP-2 for subsequent complement activation. Although MBL-A/C deficient mice are fully susceptive to CAIA, there are no reports to date regarding the roles of FCNs and CLs in inflammatory arthritis and how these LP pattern recognition molecules might interact with MASPs to cause pathogenic complement activation in the joint.
MASPs are the enzymes of the LP which cleave C2 and C4 to create the C3 convertase C4b2a. They are predominately generated in the liver and consist of five distinct proteins; MASP-1, MASP-2, sMAp, MASP-3, and MAp44. The MASP-1 gene generates three of these proteins: MASP-1, MASP-3 and MAp44 (a.k.a. MAP-1), a truncation of MASP-1/3 containing only the pattern recognition domain (42, 43). MASP-1 triggers the LP and promotes the activation of MASP-2 (13). MASP-3 has been shown to cleave inactive, pro-Df into active Factor D (44). Both MASP-1/3−/− mice as well as WT mice treated with a mixture of MASP-1/3 siRNAs are protected from the development of CAIA (12, 14). The MASP-2 gene generates MASP-2 and sMAp (a.k.a MAP-2 or MAp19), a truncation of MASP-2 (45). Unlike MASP-1 and MASP-2, MASP-3 is not auto-activated when complexed with FCNs or CLs (20, 40, 46). MASP-2 cleaves C4 and C2 to generate the C3 convertase of the LP. MASP-2 is the primary effector protein of the LP, as both mice and humans lacking MASP-2 are defective in this pathway (47, 48). MASP-2 has been shown to play an important role in focal cerebral ischemia as MASP-2−/− mice (in contrast to MASP-1/3−/− mice) were protected from cerebral ischemia (49). Decreased ischemia reperfusion injury of the myocardium, intestine, and kidney has been observed both in mice lacking MASP-2 as well as mice treated with MASP-2 inhibitory antibodies (47). The role of MASP-2/sMAp in experimental arthritis, however, remains to be determined.
We have shown that the AP is necessary for the development of CAIA (6). Given that MASP-1/3−/− mice are resistant to CAIA (12) and that MASPs are activators of the LP, it follows that there must exist a regulatory relationship between the LP and the AP. We and others have shown that MASP-1/3 is an essential activator of fD through the cleavage of pro-Df (13, 14). Another relationship can also be observed in the ability of C2 and C4 deficient patients to activate complement in response to infection. The observation led to the discovery of a C2 independent means of activating either the CP or the LP termed the C4/C2 bypass pathway (50, 51). While the mechanism of the bypass pathway remains to be elucidated, it has been shown that in the presence of the AP, MBL may activate complement in a C2 independent fashion (52). Thus the pathways of complement activation are not necessarily independent, but rather may interact and regulate each other.
In this study we have made two new observations. First, in contrast to other LP ligands, the presence of FCN B is necessary for the full development of CAIA, suggesting that this lectin plays a unique role in recognition of ligand(s) within the inflamed joint. Second, MASP-2 plays an essential role in the development of CAIA, likely through interactions with FCN-B. In addition, the lack of a role for C4 in development of CAIA in conjunction with the residual inflammation in MASP-2 deficient mice suggests the possibility that the alternative pathway activation mechanisms are fully capable of mediating disease, or that MASP-1 and MASP-3 may contribute to the development of disease through the C4/C2 bypass pathway.
Materials and Methods
Mice
All genetically deficient mice i.e. FCN A−/−, FCN B−/−, CL-11−/− and MASP2−/−/sMAp−/− were bred onto the C57BL/6 background. All gene deficient mice used for CAIA studies were age- and sex-matched. In parallel, WT littermates were used for CAIA studies with each cohort. All mice were genotyped before use. Sera from WT, FCN B−/−, and MASP2−/−/sMAp−/− C57BL/6 mice before and after disease induction were also used for various enzyme-linked immunosorbent assay (ELISA) studies. WT C57BL/6, C3−/− and Non-obese diabetic (NOD) mice, purchased from the Jackson Laboratories, were also used as controls in various complement related studies. All mice were kept in a barrier facility and fed breeder’s chow provided by the Center for Laboratory Animal Care, University of Colorado Denver.
Induction of collagen antibody-induced arthritis in mice
CAIA was induced in WT, FCN A−/−, FCN B−/−, CL-11−/− and MASP2−/−/sMAp−/− C57BL/6 mice by using a cocktail of 5 mAb to bovine collagen type II (CII) (Arthrogen-CIA, Chondrex) re-suspended in sterile Dulbecco’s PBS according to our previously published methods (14, 53). All WT, FCN A−/−, FCN B−/−, CL-11−/− and MASP2−/−/sMAp−/− mice received intraperitoneal (ip) injections of 8 mg/mouse of Arthrogen on day 0. The mice then received 50 ug of LPS from E. coli strain 0111B4 on day 3. All mice, after the induction of CAIA, were sacrificed at day 10. The severity of clinical disease activity (CDA) in all groups of WT and gene deficient mice was determined every day by 2 trained laboratory personnel acting independently, and in a blinded fashion, according to our previously published methods (14, 53).
Histopathology, C3 deposition, presence of macrophages and neutrophils in joints
At day 10, both forepaws and the entire right hind limb, including the paw, ankle and knee, were surgically removed from WT, MASP-2−/−/sMAp−/−, FCN A−/−, FCN B−/−, and CL-11−/− mice with CAIA and fixed immediately in 10% buffered formalin (Biochemical Sciences). The preparation of all joint samples and their histological analyses were performed as previously described (6, 14). All sections were read by a trained observer who was also blinded to the genotype and to the CDA of each mouse. The joint sections for WT, MASP-2−/−/sMAp−/−, FCN A−/−, FCN B−/−, and CL-11−/− were scored for the changes in inflammation, pannus, cartilage damage, and bone damage, on a scale of 0–5. C3 deposition in the joints (synovium and cartilage) was localized with polyclonal goat anti-mouse C3 antisera (ICN Pharmaceuticals), and scored as previously described (14, 54). Immunohistochemical (IHC) staining for macrophages and neutrophils was performed using knee joint sections from WT, FCN B−/, and MASP-2−/−/sMAp−/− mice and scored as previously described (14, 54).
IHC analysis for C4d, fD and FCN B deposition in the ankles from mice with CAIA
At day 10, ankles (right and left) from the WT (littermates) and FCN B−/− and WT (littermates) and MASP-2−/−/sMAp−/− mice with CAIA, from the same cohorts, were also processed for the IHC staining analysis for the C4d, fD and FCN B deposition. Knee joints were not used due to the limitation in the thickness of the knee joint and also their prior use for the above mentioned immunopathology studies. An antigen retrieval method, by pressure, was used for C4d detection using 10mM Citrate/1xTBS buffer at 95°C for 20 minutes, for fD detection using 100mM Citrate/1xTBS buffer at 110°C for 10 minutes and for FCN B detection using 100mM Citrate/1xTBS buffer at 110°C for 10 minutes. Immunodetection was performed on the Benchmark stainer (Ventana Medical Systems/Roche Diagnostics, Indianapolis, IN) at an operating temperature of 37°C, with a primary antibody incubation time of 32 minutes. Antibodies were detected with a modified I-VIEW DAB (Ventana) detection kit. The I-VIEW secondary antibody and enzyme were replaced with a species specific rabbit conjugated polymer (Rabbit ImmPress; Vector Labs, Carpinteria, CA; Full Strength in place of the secondary antibody, 50% Strength diluted in 1xPBS pH 7.6 in place of the SA-HRP, 8 minutes each). All sections were counterstained in Acidified Harris hematoxylin (H) for 1.5 minutes, blued in 1% ammonium hydroxide (v/v), dehydrated in graded alcohols, cleared in xylene and cover glass mounted using synthetic resin.
To detect membrane bound C4d deposition in the synovium, cartilage and adipose tissue areas of the ankle, human anti-C4d rabbit polyclonal antibody (American Research Products, Waltham, MA) at 1: 200 dilution was used. Normal mouse liver and sections from rejected human kidneys, after transplantation, were used as controls for C4d deposition assessment. In human kidney, after transplantation rejection, there is substantial C4d deposition and thus the tissue serves as an appropriate positive control. Normal human kidney sections were also used as negative control. No well characterized mouse anti-C4d antibody was available to us at the time for IHC analysis of paraffin-embedded tissues; therefore, we used a human anti-C4d antibody which cross-reacted with mouse C4d as shown by positive control staining in the mouse liver. Overall, we stained sections with two different mouse anti-C4d antibodies, and both of these yielded no detection for mouse C4d using paraffin-embedded ankle sections.
To detect fD deposition on the membrane or in the cytoplasm, in the synovium areas of the ankle, rabbit anti-mouse fD polyclonal antibody (GeneTex, Irvine, CA) at 1:25 dilution was used. Mouse adipose tissue expressing high levels of fD were used as a positive control. Liver expressing no fD was used as a negative control.
To detect FCN B deposition on the membrane or in the cytoplasm, in the synovium areas of the ankle, mouse anti-FCN B (FCN2) rabbit polyclonal antibody (MyBiosource, San Diego, CA) at 1:750 dilution was used. Mouse skin and brain sections which expressed FCN B were used as the positive controls. All antibodies for C4d, fD and FCN B were visualized by using an universal Rabbit modified 1-view detection kit (Abcam, Cambridge, MA) as mentioned above. All stained sections were scored blindly by a trained histology technician using an arbitrary ordinal scale of negative, no staining of cells, low level = few cells (less than 15 positive cells), medium = some cell (from 15–30) and very high level = many cells (more than 30) in the synovium, cartilage and adipose tissue of ankle.
Measurement of C3 deposition and C5a in the serum of mice induced by anti-collagen antibodies
Serum samples from mice with CAIA were analyzed for C3 deposition and C5a levels induced by adherent anti-collagen antibodies. The 96-well costar ELISA plates were pre-coated with a mixture of anti-collagen antibodies (Arthrogen, 25 ug/well) as previously described (14, 54). Sera from mice before and after CAIA were collected by using a standard recommended method as previously described (55). All serum samples from WT, FCN B−/− and MASP-2−/−/sMAp−/− mice before and after disease, were diluted 1:10 in a calcium (Ca++) sufficient (GBV) buffer or in a calcium (Ca++) deficient (Mg++ EGTA) such that all pathways will be active or only the AP will be active under these conditions. Diluted sera were then added to the wells and incubated at 37°C for 1 h. Following washes in PBS/0.5% Tween, HRP-conjugated goat anti-mouse C3 antibody (MP Biomedicals) was added to the wells. The anti-C3 antibody was diluted 1:4000 in freshly made PBS/0.05% Tween to reduce background. Plates were the incubated at room temperature for 1 h. After five more washes, the color reaction was performed and absorbance was determined for C3 deposition adherent to the ELISA plate. C5a generation was examined in the supernatant according to our previously published methods (53). Sera from C3−/− and NOD mice were used as negative controls for C3 deposition and C5a measurements, respectively. Data were expressed as mean OD + SEM value.
Statistics
Data from all WT and gene deficient mice were included in the final analysis of CDA, histology, ELISA and IHC. Analysis of variance ANOVA was used when we compared three cohorts, otherwise, Student’s t-test was used to analyze data. All data were expressed as the mean + SEM, with p < 0.05 considered significant.
Results
Clinical disease activity in FCN, Collectin and MASP-2 deficient mice
CAIA was induced in WT, FCN A−/−, FCN B−/−, CL-11−/− and MASP-2−/−/sMAp−/− mice, and CDA was examined. This CAIA study was performed in four different cohorts comparing WT litter mates to gene targeted mice as shown (Fig. 1). In cohort 1, at day 10 there was no significant difference (p < 0.115) in the CDA between WT and FCN A−/− mice. The CDA was 11.75 ± 0.250 and 10.2 ± 0.734, respectively (Fig. 1A). The prevalence of disease was 100%, at day 10, in WT and FCN A−/− mice. In cohort 2, the CDA in WT and FCN B−/− mice was compared. Interestingly, the CDA of FCN B−/− mice was significantly decreased as compared to WT (p < 0.031 on day 10) (Fig 1B). On day 10, the CDA was 10.75 ± 0.977 and 5.75 ± 1.89 in WT and FCN B−/−mice, respectively (Fig. 1B). Overall there was 47% decrease in the CDA in FCN B−/− mice compared with WT mice (Fig. 1B). The prevalence of disease was 100%, at day 10, in WT and FCN B−/− mice. In cohort 3, there was no significant difference (p < 0.63) in the CDA between WT and CL-11−/− mice (Fig 1C). The CDA was 9.53 ± 0.882 and 10.2 ± 1.04, respectively. The prevalence of disease was 100%, at day 10, in WT and CL-11−/− mice. In cohort 4, on day 10 there was a significant decrease (p < 0.05) in the CDA between WT and MASP-2−/−/sMAp−/− mice. The CDA was 10.8 ± 0.734 and 3.2 ± 0.583, respectively (Fig. 1D). The decrease in the CDA in MASP-2−/−/sMAp−/− compared with the WT mice was ~70%. Notably, significant differences (p < 0.05) in the CDA were seen with MASP-2−/−/sMAp−/− mice compared with the WT mice from day 4 to day 10 (Fig. 1D). The prevalence of disease, at day 10, in WT and MASP-2−/− mice was 100%. These CDA data show that while all mice tested developed disease, FCN B−/− and MASP-2−/−/sMAp−/− mice were significantly protected.
FIGURE 1.
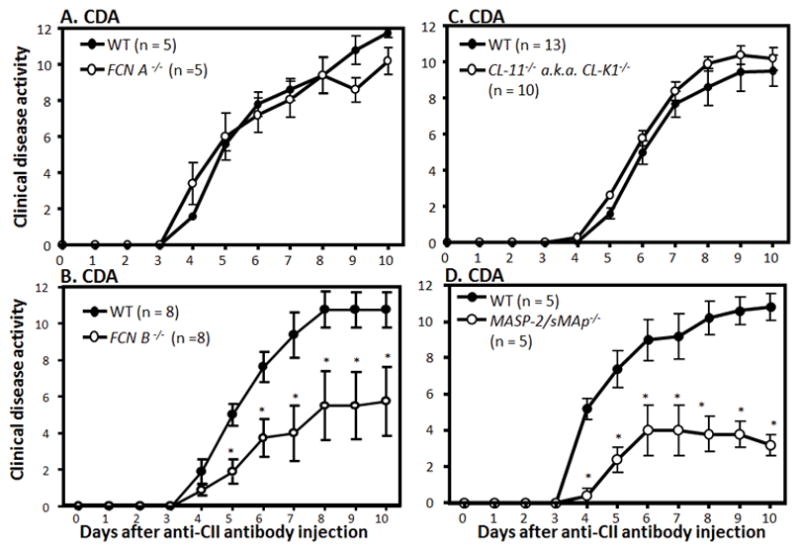
Clinical disease activity (CDA) of CAIA in FCN A−/−, FCN B−/−, CL-11−/− and MASP-2−/−/sMAp−/− mice compared with WT mice. WT, FCN A−/−, FCN B−/−, CL-11−/− and MASP-2−/−/sMAp−/− mice were injected with 8 mg/mouse/i.p. of ArthritoMab at day 0. All mice were also injected on day 3 with 50ug/mouse/i.p. with LPS (E. coli strain, 0111B4). All mice were sacrificed at day 10. CDA over the duration of the experiment is illustrated. A. CDA in WT and FCN A−/− mice (n = 5 each). B. CDA in WT and FCN B−/− mice (n = 8 each). C. CDA in WT and CL-11−/− mice (n = 13 and n = 10, respectively). D. CDA in WT and MASP-2−/−/sMAp−/−mice (n = 5 each). The data are expressed as mean of disease ± SEM. *p < 0.05 in comparison with WT mice
Histopathology of the joints from mice with CAIA
All four cohorts of mice described above were sacrificed on day 10. Both forelimbs and the right hind limb (five joints) were processed for histopathology studies, and for the measurement of C3 deposition in the joints (Fig. 2). Data are shown as an all-joint-mean (AJM) score. No significant differences were found in inflammation, pannus formation, cartilage damage, bone damage, or C3 deposition in the joints of FCN-A−/− and CL-11−/− compared with the WT mice (cohort 1 and cohort 3) (data not shown). These data are consistent with the CDA data shown in Figure 1. Histopathology scores for cohort 2 show a significant decrease (p < 0.04) of 39% in the AJM score in FCN B−/− mice compared with the WT mice (Fig. 2A). There was a significant decrease in FCN B−/− mice compared with WT mice in inflammation 34% (p <0.04), pannus formation 40% (p < 0.042), cartilage damage 41% (p <0.04) and bone damage 40% (p <0.045) (Fig. 2A). Representative pictures of T blue staining related to histopathology from the knee joints of WT and FCN B−/−mice are shown in Fig S1 (A & B). Histopathology AJM scores for MASP-2−/−/sMAp−/− mice (cohort 4) showed a 68% reduction (p<0.001) (Fig 2E). Not only the AJM but also individual scores for inflammation, pannus formation, cartilage and bone damage were significantly (p < 0.05) decreased in MASP-2−/−/sMAp−/− mice compared to WT mice (Fig. 2E). Representative pictures of T blue staining related to histopathology from the knee joints of WT and MASP-2−/−/sMAp−/− /mice are shown in Fig S2 (A & B).
FIGURE 2.

Histopathology, C3 deposition, monocyte/macrophage infiltration, and neutrophil infiltration, at day 10, from the WT, FCN B−/− and MASP-2−/−/sMAp−/− mice. Two separate cohorts of WT and MASP-2−/−/sMAp−/− and of WT and FCN B−/− mice were examined. A. Histopathology from all joints for inflammation, pannus formation, cartilage damage and bone damage comparing WT and FCN B −/− mice. B. C3 deposition from all joints in the synovium, on the surface of cartilage and total scores (synovium plus cartilage). C. Mean score of macrophages from the knee joints of mice in WT and FCN B −/− mice. D. Mean score of neutrophils from the knee joints of mice in WT and FCN B −/− mice. E. Histopathology from all joints for inflammation, pannus formation, cartilage damage and bone damage comparing WT and MASP-2−/−/sMAp−/−. F. C3 deposition from all joints in the synovium, on the surface of cartilage and total scores (synovium plus cartilage). G. Mean score of macrophages from the knee joints of mice in WT and MASP-2−/−/sMAp−/− mice. H. Mean score of neutrophils from the knee joints of mice in WT and MASP-2−/−/sMAp−/−mice. All data represent the mean + SEM in first cohort based on n = 8 for WT, and n = 8 for FCN B−/− mice and in the second cohort based on n = 5 for WT and n = 5 for MASP-2/sMAp −/− mice. *p < 0.05 in comparison to the WT mice.
C3 deposition in the joints from mice with CAIA
C3 deposition was measured as described in Materials and Methods. Consistent with CDA and histology measurements, FCN-A−/− and CL-11−/− mice showed no significant differences as compared to WT mice (data not shown). C3 deposition was significantly (p < 0.033) reduced by 56% in all joints (synovium and cartilage surface) of FCN B−/− comparing with the WT mice (Fig. 2B). Individual scores for C3 deposition in the synovium (p < 0.03) and also on the surface of cartilage (p < 0.03) were also significantly reduced in FCN B−/− mice compared with the WT mice (Fig. 2B). Representative pictures of C3 deposition performed immunohistochemically from the knee joints of WT and FCN B−/− mice are shown in Fig S1 (C & D). C3 deposition was significantly (p < 0.05) reduced to 81% in all joints (synovium and cartilage surface) of MASP-2−/−/sMAp−/− compared to WT mice (Fig. 2F). Individual scores for C3 deposition in the synovium and also on the surface of cartilage were also significantly (p <0.001) reduced in MASP-2−/−/sMAp−/− mice compared to WT mice (Fig. 2F). Representative pictures of C3 deposition performed immunohistochemically from the knee joints of WT and MASP-2−/−/sMAp−/− mice are shown in Fig S2 (C & D).
Assessment of macrophage and neutrophil infiltration into the joints from mice with CAIA
All four cohorts of mice from the CAIA studies were examined via IHC for the infiltration of macrophages and neutrophils using specific cell surface markers according to our previously published studies (9). Again, no significant differences were seen regarding numbers of macrophages and neutrophils in knees from WT and FCN A−/− or from WT and CL-11−/− mice (data not shown). In contrast, the percentage of synovial macrophages and neutrophils decreased 42% (p < 0.03) and 43% (p < 0.04), respectively, in FCN B−/− mice compared with the WT mice with CAIA (Fig. 2C, 2D). Representative IHC pictures of macrophage and neutrophil infiltrates from the knee joints of WT and FCN B−/− mice are shown in Fig S1 (E, F, G & H). In MASP-2−/−/sMAp−/− mice, synovial macrophages and neutrophils were decreased 74% (p <0.002) and 71% (p <0.002), respectively, as compared to WT mice with CAIA (Fig. 2G, 2H). Representative IHC pictures of macrophages and neutrophils from the knee joints of WT and MASP-2−/−/sMAp−/− mice are shown in Fig S2 (E, F, G & H). Overall, the decrease in infiltration of synovial macrophages and neutrophils in the knee joints was consistent with decrease in CDA observed in FCN B−/− and MASP-2−/−/sMAp−/− mice.
IHC detection of C4d, fD and FCN B deposition in the ankles from mice with CAIA
At day 10, ankles from WT and FCN B−/− and WT and MASP-2−/−/sMAp−/− mice with CAIA were processed and tissue sections were subjected to IHC staining for complement C4d deposition, fD deposition and FCN B deposition (Fig 3 & 4). All staining results have been summarized in Table 1. Representative IHC images for C4d, fD and FCN B deposition in the synovium from the ankles of WT comparing with FCN B−/− mice with CAIA are shown in Fig 3 A–K. First, there were low levels of C4d deposition in the synovium of both FCN B−/− mice and WT mice with CAIA (Fig 3A & B). Mouse liver was used as a positive control for the presence of C4d (Fig 3C) along with human kidney negative and positive controls (Fig 3J & K). Second, there was a very low levels of fD deposition in the synovium in FCN B−/− mice compared with the very high levels deposition in WT mice with CAIA (Fig 3D & E). Mouse adipose tissue (fat) was used as a positive control for the presence of fD (Fig 3F). Finally, there was no detection of FCN B deposition in the synovium in FCN B−/− mice as expected, compared with the very high levels deposition in WT mice with CAIA (Fig 3G & H). Mouse skin was used as a positive control for the presence of FCN B (Fig 3I). These IHC staining data, for C4d, fD and FCN B deposition, in the synovium from the ankles of WT and FCN B−/− mice with CAIA suggest that the FCN B ligand recognition molecule of the LP is directly involved in activating the AP through MASP-1 or MASP-3 proteases but possibly independent of C4 because of low levels of C4d in the ankle.
FIGURE 3.
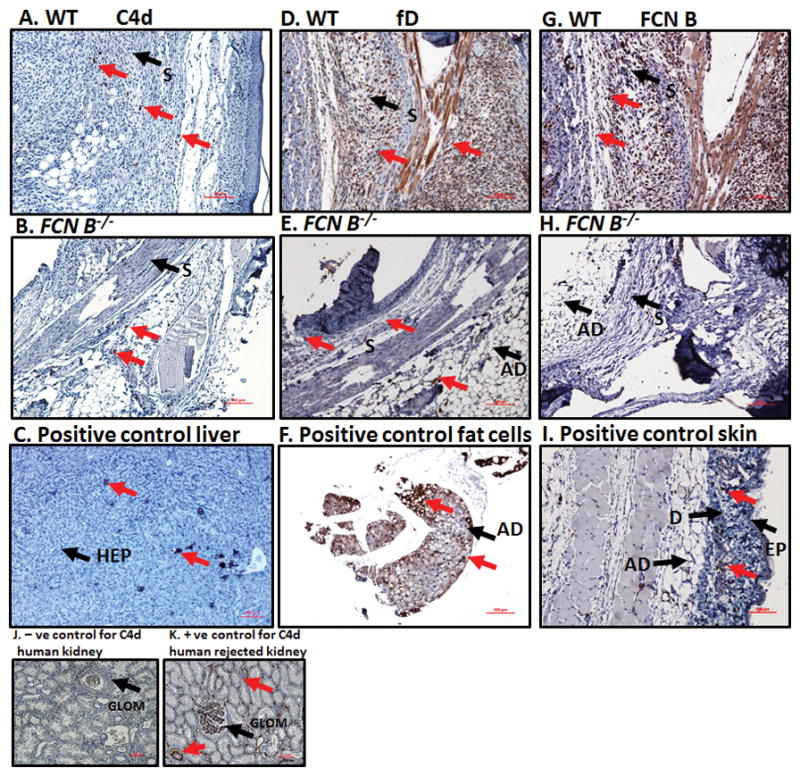
Representative images of the ankle joint synovial immunohistochemistry for C4d, factor D (fD) and ficolin B (FCN B) deposition in WT and FCN B −/− mice with CAIA. Specific area of the ankle synovium with maximum number of positive cell staining has been shown. All ankle joints from mice with CAIA were fixed in a 10% neutral buffered formalin, paraffin-embedded, and sectioned at a thickness of 5 μm followed by specific staining. The first set of three panels from top to bottom (A, B, & C) show staining with anti-C4d Ab (brown color) from the ankle joints of WT mice with CAIA (first left most panel), FCN B −/− mice with CAIA (second left center panel) and a positive liver control for C4d (last left bottom panel). The center set of three panels from top to bottom (D, E & F) show staining with mouse fD antibody (brown color) from the ankle joints of WT with CAIA (first center most panel), FCN B −/− mice with CAIA (second center most panel) and a positive adipose tissue (fat) control for fD (bottom center most panel). The right set of three panels from top to bottom (G, H & I) show staining with mouse FCN B Ab (brown color) from the ankle joints of WT with CAIA (first right most panel), FCN B −/− mice with CAIA (second right most panel) and a positive skin control for FCN B (last right most panel). Two small panels J and K at the bottom left hand show the negative normal human kidney control and positive human kidney control for anti-human C4d staining from rejected transplant. Negative matched-isotype control sections from various tissues were also used with each staining method and there was no non-specific-staining as expected (data not shown). Specific areas of the synovium of ankle has been identified by black arrow (S-synovium), and as well other sites are as labelled (Ad-adipose tissue), (EP-epidermis), (D-dermis), (HEP-hepatocyte) and (GLOM-glomerulus). Specific staining in each panel has been shown by red arrows. The ankle joint sections stained for C4d deposition, fD and FCN B were also counterstained with Hematoxylin (H) only and photographed under the 10× objective using Nikon® Eclipse 80i microscope equipped with Nikon® DS-Qi1MC camera. Red scale bar shown at the bottom right hand corner in images from A-I and J & K equals 0.1 mm (100 μm).
FIGURE 4.
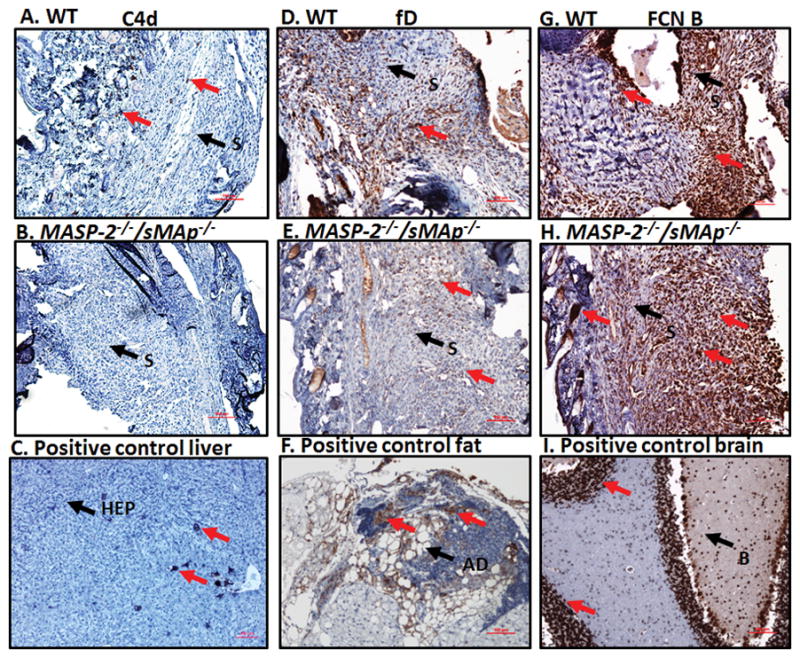
Representative images of ankle joint synovial IHC for C4d, fD and FCN B deposition in WT and MASP-2−/− /sMAp −/− mice with CAIA. Specific area of the ankle synovium with maximum number of positive cell staining has been shown. All ankle joints from mice with CAIA were fixed in a 10% neutral buffered formalin, paraffin-embedded, and sectioned at a thickness of 5 μm followed by specific staining. The first set of three panels from top to bottom (A, B, & C) show staining with anti-C4d Ab (brown color) from the ankle joints of WT mice with CAIA (first left most panel), MASP-2−/− /sMAp −/− mice with CAIA (second left center panel) and a positive mouse liver control for C4d (last left bottom panel). The center set of three panels from top to bottom (D, E & F) show staining with mouse fD antibody (brown color) from the ankle joints of WT with CAIA (first center most panel), MASP-2−/− /sMAp −/− mice with CAIA (second center most panel) and a positive adipose tissue (fat) control for fD (bottom center most panel). The right set of three panels from top to bottom (G, H & I) show staining with mouse FCN B Ab (brown color) from the ankle joints of WT with CAIA (first right most panel), MASP-2−/− /sMAp −/− mice with CAIA (second right most panel) and a positive brain control for FCN B (last right most panel). Negative matched-isotype control sections from various tissues were also used with each staining method and there was no non-specific-staining as expected (data not shown). Specific areas of the synovium of the ankle and other tissues have been identified by black arrow (S-synovium), (Ad-adipose tissue), (HEP-hepatocyte), (B-brain), (GLOM-glomerulus) and (D-dermis). Specific staining (brown color) in each panel has been shown by red arrows. The ankle joint sections stained for C4d deposition, fD and FCN B were also counterstained with Hematoxylin (H) and photographed under the 10× objective using photographed under the 10× objective using Nikon® Eclipse 80i microscope equipped with Nikon® DS-Qi1MC camera. Red scale bar shown at the bottom right hand corner in images from A–I equals 0.1 mm (100 μm).
Table 1.
Comparing the levels of complement C4d, factor D and ficolin B by Immunohistochemical staining (IHC) from the ankles of WT, FCN B−/− and MASP-2/sMAp−/− mice with CAIA
| Complement | 1 Mice | 1 Mice | ||
|---|---|---|---|---|
| WT | FCN B−/− | WT | MASP-2/sMAp−/− | |
|
|
|
|||
| 2 C4d | Low level | Low level | Low level | Negative |
| 3 fD | Very high level | Low level | Very high level | Medium level |
| 4 FCN B | Very high level | Negative | Very High level | Very high level |
Ankle joints formalin fixed, paraffin-embedded sections from the C57BL WT (littermates) and FCN B−/− and WT (littermates) and MASP-2/sMAp−/− mice with collagen antibody induced arthritis (CAIA) were used for the IHC detecting the presence of membrane bound/and or in the cytoplasm of C4d deposition, fD deposition and FCN B deposition. CAIA was induced in these mice as mentioned in the Materials and Methods.
C4d classical pathway C4d component.
fD = factor D alternative pathway component.
FCN B = ficolin B lectin pathway component. An ordinal scoring was method used i.e. Negative = no staining of any cell was seen compared with the positive control at a specific antibody dilution, low level = few cells positive (less than 15) in the synovium, cartilage and adipose cells were positive, medium = some cells positive (from 15–30) in the synovium, cartilage and adipose cells were positive, and very high level = many cells positive (more than 30) in the synovium, on the cartilage surface and adipose cells were clearly visible. An antigen retrieval method was consistently used to detect all three antigens on paraffin-embedded tissue sections. All slides were scored blindly by a trained IHC personal. Specific dilutions used of each primary antibody has been mentioned in the Materials and Methods.
All representative IHC images for C4d, fD and FCN B deposition in the ankles from WT comparing with MASP-2−/−/sMAp−/−mice with CAIA have been shown in Figure 4 A–I. There was a no detection of C4d deposition in the synovium in MASP-2−/−/sMAp−/−mice compared with the low levels of C4d deposition in WT mice with CAIA (Fig. 4 A, B & C) (Table 1). We found fD was present at a very high levels in ankles of WT mice with disease compared with the medium levels of fD in MASP-2−/−/sMAp−/−mice with disease (Fig 4 D, E & F). Again FCN B was present, equally at a high level, both in WT mice and in MASP-2−/−/sMAp−/− mice with CAIA (Fig. 4 G & H). Mouse brain which highly expressed FCN B was used as another positive control (Fig. 4 I). Interestingly these C4d, fD and FCN B deposition IHC data in MASP-2−/−/sMAp−/− mice vs. WT mice with disease showed that MASP-2 appears to contribute to the development of CAIA, likely in a C4-independent manner because of the non-detectable levels of C4d deposition in the synovium from ankles of MASP-2−/−/sMAp−/− mice with CAIA. Nonetheless there was activation of the LP and AP due to the deposition of FCN B and fD in the in the synovium from ankles of MASP-2−/−/sMAp−/− mice with CAIA.
Relationship between in vitro activation of C3, C5a in serum and alterations of in vivo CDA in gene deficient mice with CAIA
ELISAs were performed to find out the relative roles of LP and AP in the C3 activation and generation of C5a in the sera from WT, FCN B−/− and MASP-2−/−/sMAp−/− mice with CAIA (Fig 5 & 6). Sera only from WT, FCN B−/− and MASP-2−/−/sMAp−/− mice, at day 10 with CAIA were analyzed because these mice were partially resistant to disease (Fig 1). Sera from these mice with disease were diluted in a Ca2+ sufficient and Ca2+ deficient buffers separately as mentioned in the Materials and Methods section. C5a generation was examined in parallel in a Ca2+ sufficient buffer or in a Ca2+ deficient buffer from the supernatants of C3 deposition (Fig 5). No statistically significant change in C3 activation was seen comparing sera from WT and FCN B−/− diluted in a Ca2+ sufficient buffer (Fig 5A). However, a highly significant (p < 0.003) decrease of 80% was seen in C3 deposition, in a Ca2+ deficient buffer in the sera from FCN B−/− mice compared with the WT mice with CAIA (Fig 5B). There was a minimal but significant decrease (p < 0.002) of 16% in the levels of C5a, comparing sera diluted in a Ca2+ sufficient buffer from WT and FCN B−/− mice with disease (Fig 5C). Again there was highly significant (p < 0.007) decrease of 60% in the levels of C5a was seen in a Ca2+ deficient buffer the sera from FCN B−/− compared with the WT mice with disease (Fig 5D). Interestingly, although sera from NOD mice with were used as a negative control for C5a ELISA but there was a significant (p < 0.05) decrease in the C3 activation compared with the WT mice in a Ca2+ deficient buffer (Fig 5B). These data regarding C3 activation and C5a levels strongly suggest a direct effect on the levels of potential AP activation due to the deficiency of the LP FCN B ligand.
FIGURE 5.
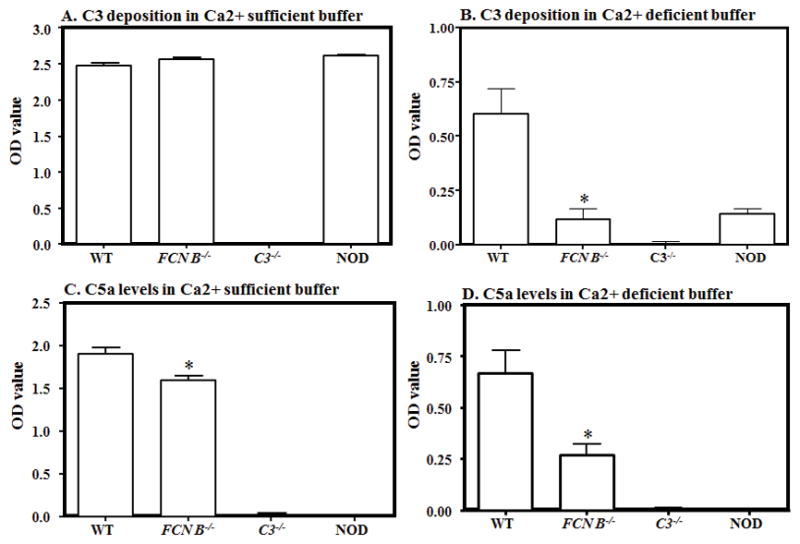
Effect of LP ligand, FCN B deficiency on C3 deposition and C5a levels, at day 10, in WT and FCN B−/− mouse serum with CAIA. All mice were sacrificed after the induction of disease at day 10 and sera were obtained. The ELISA method was used to determine the levels of C3 deposition and C5a generation induced by anti-collagen antibodies as mentioned in Materials and Methods. Sera from WT and FCN B−/− mice were diluted (1:10) in Ca2+ sufficient (GVB+ buffer) (all pathways active) or Ca2+ deficient buffer (Mg2+ EGTA buffer, AP only active and added to the same ELISA plates side by side for an accurate comparison. C3 deposition was determined adherent to the ELISA plate and C5a generation was measured concurrently in the supernatant after the incubation of serum. A. C3 deposition in a Ca2+ sufficient buffer using sera from WT and FCN B−/− mice with disease. B. C3 deposition in a Ca2+ deficient buffer using sera from WT, and FCN B−/− mice with disease. C. C5a generation in a Ca2+ sufficient buffer using sera from WT and FCN B−/− mice with disease. D. C5a generation in a Ca2+ sufficient buffer using sera from WT and FCN B−/− mice with disease. The data were expressed as the optical density (OD) value. Sera from C3−/− and NOD mice (n = 3 each) were used as a negative controls for C3 and C5a respectively for ELISAs. The ELISA data represent the mean ± SEM based on n = 8 for WT and n=8 for FCN B−/− mice. The black asterisks indicate significant differences (p < 0.05) in comparison with WT mouse serum.
FIGURE 6.
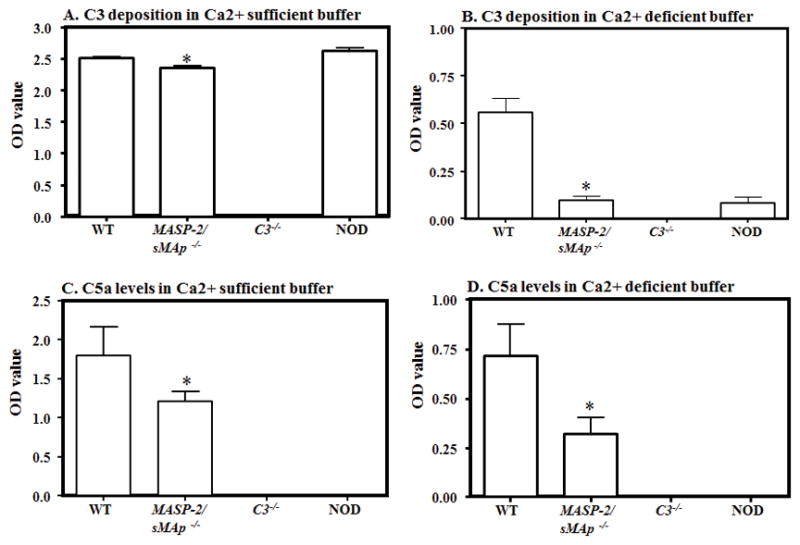
Effect of LP protease, MASP-2 deficiency on C3 deposition and C5a levels, at day 10, in WT and MASP-2−/−/sMAp−/− mouse serum with CAIA. All mice were sacrificed after the induction of disease at day 10 and sera were obtained. The ELISA method was used to determine the levels of C3 deposition and C5a generation induced by anti-collagen antibodies as mentioned in Materials and Methods. Sera from WT and MASP-2−/−/sMAp−/− mice were diluted (1:10) in Ca2+ sufficient (GVB+ buffer) (all pathways active) or Ca2+ deficient buffer (Mg2+ EGTA buffer, AP only active and added to the same ELISA plates side by side for an accurate comparison. C3 deposition was determined adherent to the ELISA plate and C5a generation was measured concurrently in the supernatant after the incubation of serum. A. C3 deposition in a Ca2+ sufficient buffer using sera from WT and MASP-2−/−/sMAp−/− mice with disease. B. C3 deposition in a Ca2+ deficient buffer using sera from WT, and MASP-2−/−/sMAp−/− mice with disease. C. C5a generation in a Ca2+ sufficient buffer using sera from WT and MASP-2−/−/sMAp−/− mice with disease. D. C5a generation in a Ca2+ sufficient buffer using sera from WT and MASP-2−/−/sMAp−/− mice with disease. The data were expressed as the optical density (OD) value. Sera from C3−/− and NOD mice (n = 3 each) were used as a negative controls for C3 and C5a respectively for ELISAs. The ELISA data represent the mean ± SEM based on n = 5 for WT and n = 5 for MASP-2−/−/sMAp−/− mice. The black asterisks indicate significant differences (p < 0.05) in comparison with WT mouse serum.
There was also a significant decrease (p < 0.002) in C3 activation, comparing sera diluted in a Ca2+ sufficient buffer, from WT and MASP-2−/−/sMAp−/− mice with disease but this decrease was a minimal of 6% (Fig 6A). However, a highly significant (p < 0.002) decrease of 82% in the C3 activation was seen, in a Ca2+ deficient buffer, in the sera from MASP-2−/−/sMAp−/− mice compared with the WT mice with disease (Fig 6B). There was a significant decrease (p < 0.002) of 33% in the levels of C5a, comparing sera diluted in a Ca2+ sufficient buffer from WT and MASP-2−/−/sMAp−/− mice with disease (Fig 6C). Again there was a highly significant (p < 0.007) decrease of 55% in the levels of C5a was seen in the sera, in a Ca2+ deficient buffer, from MASP-2−/−/sMAp−/− compared with the WT mice with disease (Fig 6D). Sera from NOD and WT mice were used a negative and a positive controls, respectively, for C5a levels in each ELISA experiment (Fig 6C & 6D). Again, we found that there was a significantly decreased (p < 0.05) level of the C3 activation using sera from NOD mice compared with the WT mice in a Ca2+ deficient buffer (Fig 6B). Sera from C3−/− mice were also used as an additional negative control for C3 activation ELISA (Figs 5 & 6). These results show that there is a relationship between the in vivo CDA and the relative activation of C3 and C5a generation in ex vivo in the presence of the same anti-collagen mAbs that were used to induce disease in vivo in WT, FCB B−/− and MASP-2−/−/sMAp−/− mice with arthritis. The specific decrease in the AP in MASP-2−/−/sMAp−/− mice with disease compared with the WT with disease shows that there is activation of the AP in MASP-2−/−/sMAp−/− might be via C4-independent and MASP-1 or MASP-3-dependent pathway but it is subdued. Nonetheless, this pathway is directly activating C3 and generating C5a through a different C4-indepenent route (Fig 7). However, the decrease in C3 and C5a levels in FCN B−/− mice with CAIA indicate that this ligand might be directly or indirectly activating the AP (Fig. 7).
FIGURE 7.
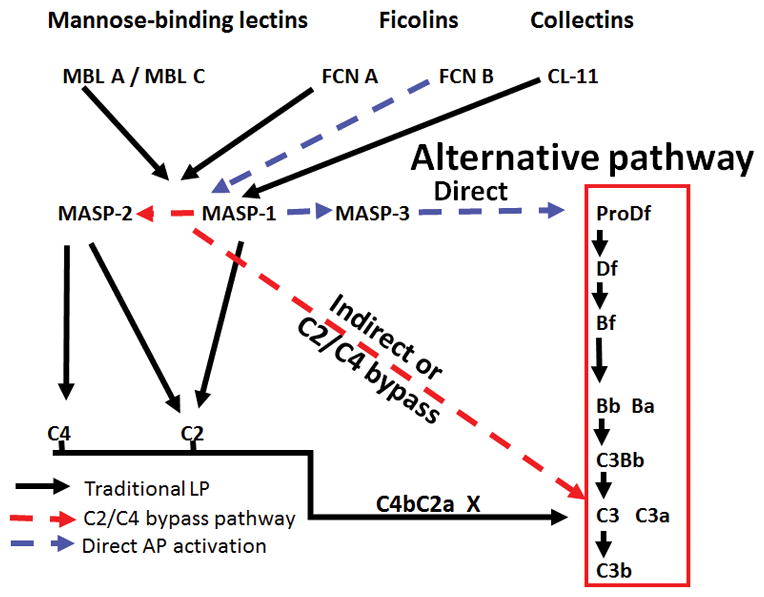
A putative LP-AP pathway model to trigger arthritis using the LP ligand and enzymes to activate the AP. Here we show how FCN B, MASP-1 and MASP-2 may act in concert or independently to trigger AP pathway to induce CAIA. This model regarding the role of FCNs and MASP-2 is based on the finding that both mice lacking FCN B and MASP-2 were partially protected from CAIA. FCN B could be the major ligand in mouse to activate the AP via LP by binding to MASP-1 and activating the C3 directly or directly activating the AP by binding to MASP-3. MASP-2 definitely can cause activation of the AP via C4-independent pathway. Blue and red dotted arrows show possible activation routes to activate the AP and the AP amplification loop.
Discussion
Previously we have shown that mice having only a functional CP plus the FCN and CL pattern recognition molecules of the LP (MBL A/C−/−/Df−/−), or only the intact LP (C1q−/−/Df−/−) are highly resistant to CAIA (56). In contrast, mice having only the AP plus FCNs and CLs (C1q−/−/MBL A/C−/) are highly susceptible to CAIA (6, 35, 54). Furthermore, mice in which the LP is only able to be activated through FCN-B and CLs but with a functional CP and AP (MBL A/C−/−/FCN A−/− or MBL A/C−/−) are fully susceptible to CAIA (35). Thus, in these previous studies, the key role of the AP was clear; however, additional roles for the LP acting through FCN-A, FCN-B and/or CLs were not able to be assessed. In addition, the roles of individual MASPs, proteases which link lectin pattern recognition molecules to downstream complement activation pathways, have not been fully defined in this model. Given that MASP-1 and MASP-3 are alternatively spliced products of a single gene, it has also been difficult to dissect the distinct functions of these two enzymes. We have shown that either disruption or knockdown of the MASP1/3 gene product(s) blocks the development of CAIA (12, 14); however, whether MASP-1or MASP-3 acts solely through cleavage of pro-Df to fD, or alternately through activation of MASP-2 or direct cleavage of C3, or a combination thereof, is uncertain. Meanwhile preliminary data from Takahashi et al. has demonstrated that patients with mutations in a MASP-3 specific exon show no discernable AP in serum samples (44). Numerous studies have shown that MASP-1 activates MASP-2 as an essential step in the activation of the LP (57–59). Additionally, other studies have shown that MASP-1 can activate MASP-3 (60, 61). Together, these data suggest that MASP-3 is the primary activator of the AP via cleavage of pro-Df while MASP-1 is the primary activator of the LP via activation of MASP-2. The cleavage of pro-Df by MASP-3 illustrates an important regulatory relationship between the LP and the AP. While disruption of the MBL A and MBL C genes in conjunction with disruption of the FCN A gene has no effect on disease development in the CAIA model, it is clear that MASP-3 (and thus the LP) plays an important role in the activation of the AP through the generation of active fD.
In the present study, we have uncovered an important dependency of CAIA on FCN B, with a 47% reduction in disease intensity in FCN B−/− mice, while FCN A−/− mice show full susceptibility to CAIA. In mice, FCN A is produced in the liver and spleen, and is readily detectible in the plasma (24, 25). FCN B is expressed primarily in myeloid cells in the bone marrow with very low serum concentrations under normal conditions (32, 33). The location of FCN B protein remains unclear: however, it has been detected in the lysosomes of activated macrophages (34). Lysosomal localization of FCN B is not necessarily surprising. Other pattern recognition receptors such as TLR9 (62) and NOD proteins (63) are also localized in lysosomes where they will interact with phagocytosed pathogens and other molecules. Interestingly, we have found that upon induction of an inflammatory disease state such as CAIA, FCN B can be readily detected in the serum (35), and now its presence in the inflamed ankle suggesting that FCN B is secreted from activated macrophages locally. Comparisons with human FCN genes suggest that mouse FCN A is the ortholog of human FCN 2 while FCN-B is the ortholog of human FCN-M (64, 65). FCN A−/− and FCN B−/−mice both demonstrate decreased survival rates when challenged with intranasal infection of Streptococcus pneumonia, indicating an important role for both FCN-A and FCN-B genes in protection against certain infections (32).
During biosynthesis FCN proteins initially organize into trimeric structures which then combine into oligomers. Given that FCN-A and FCN-B are expressed in different cell types we can assume that they form separate trimers and probably oligomers as well. FCN-A oligomers are found complexed with MASPs in the serum (31). Recombinant FCN-B is capable of interacting with MASP-2 and activating the LP (18). Thus, FCN-B may be contributing to CAIA via its role as a lysosomal sensor of antigen within macrophages or via its ability to interact with specific ligands within the joint, activating MASPs and inducing local complement activation in the joint. This observation regarding local activation of complement further supported by the evidence for now we have confirmed that FCN B and fD are also deposited in the ankle’s synovium of WT mice with CAIA. Previously we have reported the presence of not only of the expression for MASP-1 and MASP-2 but also the deposition of MASP-1 and MASP-2 proteins locally in the knee joint of WT mice with CAIA (8, 12, 66). Macrophage infiltration was decreased in CAIA in FCN B−/− mice along with decreased activation of the AP based on low levels of fD in the ankles of FCN B−/− mice with CAIA. These data support our hypothesis that FCN B compared with MBL, FCN A, CLs of the LP plays a key role in the activation of the AP. However, we cannot yet say whether decreased infiltration caused reduced disease or whether reduced disease led to decreased macrophage infiltration.
In addition, it has been shown that MBL-A and FCN-A are capable of inhibiting LPS-mediated inflammatory processes on mast cells (67) raising the possibility that the presence of FCN-A is somehow inhibiting TLR4 signaling. The CAIA model requires LPS activation of TLR4 for disease development. More work must be performed to further unravel the mechanistic basis and the complex relationship of FCN-A and FCN-B in CAIA.
The second important finding from this study is the dependency of CAIA on the presence of MASP-2. MASP-2 cleaves C4 and C2 to create the C3 convertase of the LP. Previously we showed that disruption of C4 has no effect on CAIA (6). This would then suggest that MASP-2 must have additional functions beyond activation of C4. In this context, it is interesting to note that Asgari and colleagues have shown that MASP-2 is an important mediator of renal ischemia-reperfusion injury and works in a C4-independent fashion that nevertheless is associated with C3 deposition (68). We found C3 and fD deposition to be markedly reduced in joints from MASP-2−/−/sMAp−/− mice after CAIA (Fig 2F, Fig 4) while C3 deposition in C4−/− mice was actually increased (6). Interestingly, when only AP was active, in the sera from arthritic MASP-2−/−/sMAp−/− mice, there was not only a huge decrease in the activation C3 but also a substantial decrease in C5a levels. Furthermore IHC data regarding the negative and medium levels of C4d and fD respectively in ankles of MASP-2−/−/sMAp−/− and FCN B−/− mice with CAIA clearly indicate that MASP-2 appears to contribute to the AP activation and the development of CAIA, likely in a C4-independent manner. Overall, these data are consistent with the hypothesis that MASP-2 is contributing to C3 deposition in CAIA, it could be suboptimal initially and later amplified by the AP, but not through activation of a C4/C2 convertase because no detection of C4d deposition was seen in MASP-2−/−/sMAp−/− mice with CAIA. Given the critical importance of the AP to CAIA disease development it is probable that MASP-2 is contributing in a more direct and independent manner to AP activity, as has been proposed by others (69). Of note, there might be two potential reasons that there is a decrease in C3 activation in the sera from NOD mice, in the absence of AP, compared with the WT mice. The first is that NOD mice exhibit a relative deficiency in the ability of the alternative pathway to be activated. This could be due to relative decreases in factor B or factor D levels. The second possibility is that the “distal positive feedback loop” that we have previously described in vivo (6) is also active in vitro.
Since there was no protection from the CAIA in CL-11−/− mice, we concluded that this ligand plays no essential role in CAIA. We have not examined the expression of CL-11 in the knee joints, and there is a possibility that this site is impenetrable to the circulating CL-11 and thus the molecule has no local function in the knee joint. Recently it has been shown that CL-11−/− mice are protected from renal ischemia reperfusion (I/R) injury (41). Therefore, the requirement of various LP ligands must be target organ and/or context specific.
Our data shows, intriguingly, that disruption of the FCN B gene can partially protect mice from CAIA while disruption of FCN A does not. One possible explanation is that there was a decrease in C3 activation followed by C5a generation in FCN B−/− arthritic mice indicating this LP ligand directly activate the AP though MASP-1/3 or indirectly activate the AP through MASP-2-dependent pathway (Fig 7). Additional work is necessary to define these in vivo. The likely source of FCN-B is a basal infiltrating macrophage population (33, 34). Activation of complement via the LP and amplified by the AP would then result in the generation of C3a and C5a leading to the massive infiltration of macrophages and neutrophils seen in full disease.
In summary, we have shown an important dependence of CAIA on FCN B and MASP-2. It is tempting to speculate that these two dependencies are related as modeled (Fig. 7). This may be tested with the generation of dual FCN B−/− and MASP-2−/−/sMAp−/− mice. In addition, one can ask what is the driver for the partial disease seen in these MASP-2−/−/sMAp−/− mice? It might be wholly independent activity of the AP. However, others have reported the activation of C3 in humans who are missing C2 via a bypass pathway (50). We speculate that MASP-1 may be responsible for this by direct suboptimal cleavage of C3 by C4-independent pathway which then may be augmented by the AP, if then it will be consistent with the promiscuous activities of the MASP-1 (70). Further work will be required to generate data supporting this conjecture. There is also a possibility that blocking FCN-M in humans, which is equivalent to mouse FCN-B, using an inhibitory anti-FCN-M antibody, might be therapeutically beneficial for certain inflammatory diseases including arthritis.
Supplementary Material
Acknowledgments
Supported by National Institutes of Health grant R01AR51749 (VMH-PI and NKB-COI)
The authors are thankful to the Department of Ophthalmology, University of Colorado Anschutz Medical Campus for allowing the use of their Nikon® Eclipse 80i microscope equipped with Nikon® DS-Qi1MC camera and specifically to Dr. David A. Ammar for helping to take excellent images of the IHC sections from the ankles of mice with arthritis.
Abbreviations used in this manuscript
- CDA
clinical disease activity
- AJM
all joint mean
- AP
alternative pathway
- CAIA
collagen antibody-induced arthritis
- CP
classical pathway
- LP
lectin pathway
- MBL
Mannose-binding lectin
- WT
wild type
- C5a
complement 5 anaphylatoxin
- FCNs
ficolins
- FCN A
ficolin A
- FCN B
ficolin B
- CL-11
collectin liver 11
- MASP
mannose-binding lectin-associated serine protease
- mAb
monoclonal antibody
References
- 1.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 2.Hootman JM, Helmick CG. Projections of US prevalence of arthritis and associated activity limitations. Arthritis and rheumatism. 2006;54:226–229. doi: 10.1002/art.21562. [DOI] [PubMed] [Google Scholar]
- 3.Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, Liang MH, Kremers HM, Mayes MD, Merkel PA, Pillemer SR, Reveille JD, Stone JH. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis and rheumatism. 2008;58:15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 4.Arend WP, Firestein GS. Pre-rheumatoid arthritis: predisposition and transition to clinical synovitis. Nature reviews Rheumatology. 2012;8:573–586. doi: 10.1038/nrrheum.2012.134. [DOI] [PubMed] [Google Scholar]
- 5.Ammitzboll CG, Thiel S, Ellingsen T, Deleuran B, Jorgensen A, Jensenius JC, Stengaard-Pedersen K. Levels of lectin pathway proteins in plasma and synovial fluid of rheumatoid arthritis and osteoarthritis. Rheumatology international. 2012;32:1457–1463. doi: 10.1007/s00296-011-1879-x. [DOI] [PubMed] [Google Scholar]
- 6.Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, Holers VM. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J Immunol. 2006;177:1904–1912. doi: 10.4049/jimmunol.177.3.1904. [DOI] [PubMed] [Google Scholar]
- 7.Brodeur JP, Ruddy S, Schwartz LB, Moxley G. Synovial fluid levels of complement SC5b-9 and fragment Bb are elevated in patients with rheumatoid arthritis. Arthritis and rheumatism. 1991;34:1531–1537. doi: 10.1002/art.1780341209. [DOI] [PubMed] [Google Scholar]
- 8.Banda NK, Mehta G, Kjaer TR, Takahashi M, Schaack J, Morrison TE, Thiel S, Arend WP, Holers VM. Essential role for the lectin pathway in collagen antibody-induced arthritis revealed through use of adenovirus programming complement inhibitor MAp44 expression. Journal of Immunology. 2014;193:2455–2468. doi: 10.4049/jimmunol.1400752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehta G, Scheinman RI, Holers VM, Banda NK. A New Approach for the Treatment of Arthritis in Mice with a Novel Conjugate of an Anti-C5aR1 Antibody and C5 Small Interfering RNA. Journal of Immunology. 2015;194:5446–5454. doi: 10.4049/jimmunol.1403012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banda NK, Hyatt S, Antonioli AH, White JT, Glogowska M, Takahashi K, Merkel TJ, Stahl GL, Mueller-Ortiz S, Wetsel R, Arend WP, Holers VM. Role of C3a receptors, C5a receptors, and complement protein C6 deficiency in collagen antibody-induced arthritis in mice. Journal of Immunology. 2012;188:1469–1478. doi: 10.4049/jimmunol.1102310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, Takahashi K, Holers VM, Walport M, Gerard C, Ezekowitz A, Carroll MC, Brenner M, Weissleder R, Verbeek JS, Duchatelle V, Degott C, Benoist C, Mathis D. Arthritis critically dependent on innate immune system players. Immunity. 2002;16:157–168. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- 12.Banda NK, Takahashi M, Levitt B, Glogowska M, Nicholas J, Takahashi K, Stahl GL, Fujita T, Arend WP, Holers VM. Essential role of complement mannose-binding lectin-associated serine proteases-1/3 in the murine collagen antibody-induced model of inflammatory arthritis. Journal of Immunology. 2010;185:5598–5606. doi: 10.4049/jimmunol.1001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi M, Ishida Y, Iwaki D, Kanno K, Suzuki T, Endo Y, Homma Y, Fujita T. Essential role of mannose-binding lectin-associated serine protease-1 in activation of the complement factor D. J Exp Med. 2010;207:29–37. doi: 10.1084/jem.20090633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banda NK, Acharya S, Scheinman RI, Mehta G, Coulombe M, Takahashi M, Sekine H, Thiel S, Fujita T, Holers VM. Mannan-Binding Lectin-Associated Serine Protease 1/3 Cleavage of Pro-Factor D into Factor D In Vivo and Attenuation of Collagen Antibody-Induced Arthritis through Their Targeted Inhibition by RNA Interference-Mediated Gene Silencing. Journal of Immunology. 2016 doi: 10.4049/jimmunol.1600719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holmskov U, Thiel S, Jensenius JC. Collections and ficolins: humoral lectins of the innate immune defense. Annual review of immunology. 2003;21:547–578. doi: 10.1146/annurev.immunol.21.120601.140954. [DOI] [PubMed] [Google Scholar]
- 16.Zhang M, Takahashi K, Alicot EM, Vorup-Jensen T, Kessler B, Thiel S, Jensenius JC, Ezekowitz RA, Moore FD, Carroll MC. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. Journal of Immunology. 2006;177:4727–4734. doi: 10.4049/jimmunol.177.7.4727. [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki T, Etoh R, Yamashina I. Isolation and characterization of a mannan-binding protein from rabbit liver. Biochemical and biophysical research communications. 1978;81:1018–1024. doi: 10.1016/0006-291x(78)91452-3. [DOI] [PubMed] [Google Scholar]
- 18.Endo Y, Iwaki D, Ishida Y, Takahashi M, Matsushita M, Fujita T. Mouse ficolin B has an ability to form complexes with mannose-binding lectin-associated serine proteases and activate complement through the lectin pathway. J Biomed Biotechnol. 2012;2012:105891. doi: 10.1155/2012/105891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garred P, Honore C, Ma YJ, Munthe-Fog L, Hummelshoj T. MBL2, FCN1, FCN2 and FCN3-The genes behind the initiation of the lectin pathway of complement. Molecular Immunology. 2009;46:2737–2744. doi: 10.1016/j.molimm.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Ma YJ, Skjoedt MO, Garred P. Collectin-11/MASP complex formation triggers activation of the lectin complement pathway--the fifth lectin pathway initiation complex. Journal of innate immunity. 2013;5:242–250. doi: 10.1159/000345356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsushita M, Fujita T. Ficolins and the lectin complement pathway. Immunological reviews. 2001;180:78–85. doi: 10.1034/j.1600-065x.2001.1800107.x. [DOI] [PubMed] [Google Scholar]
- 22.Garlatti V, Belloy N, Martin L, Lacroix M, Matsushita M, Endo Y, Fujita T, Fontecilla-Camps JC, Arlaud GJ, Thielens NM, Gaboriaud C. Structural insights into the innate immune recognition specificities of L- and H-ficolins. The EMBO journal. 2007;26:623–633. doi: 10.1038/sj.emboj.7601500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garlatti V, Martin L, Gout E, Reiser JB, Fujita T, Arlaud GJ, Thielens NM, Gaboriaud C. Structural basis for innate immune sensing by M-ficolin and its control by a pH-dependent conformational switch. The Journal of biological chemistry. 2007;282:35814–35820. doi: 10.1074/jbc.M705741200. [DOI] [PubMed] [Google Scholar]
- 24.Fujimori Y, Harumiya S, Fukumoto Y, Miura Y, Yagasaki K, Tachikawa H, Fujimoto D. Molecular cloning and characterization of mouse ficolin-A. Biochemical and biophysical research communications. 1998;244:796–800. doi: 10.1006/bbrc.1998.8344. [DOI] [PubMed] [Google Scholar]
- 25.Ohashi T, Erickson HP. Oligomeric structure and tissue distribution of ficolins from mouse, pig and human. Archives of biochemistry and biophysics. 1998;360:223–232. doi: 10.1006/abbi.1998.0957. [DOI] [PubMed] [Google Scholar]
- 26.Matsushita M, Endo Y, Fujita T. Cutting edge: complement-activating complex of ficolin and mannose-binding lectin-associated serine protease. Journal of Immunology. 2000;164:2281–2284. doi: 10.4049/jimmunol.164.5.2281. [DOI] [PubMed] [Google Scholar]
- 27.Matsushita M, Endo Y, Taira S, Sato Y, Fujita T, Ichikawa N, Nakata M, Mizuochi T. A novel human serum lectin with collagen- and fibrinogen-like domains that functions as an opsonin. The Journal of biological chemistry. 1996;271:2448–2454. doi: 10.1074/jbc.271.5.2448. [DOI] [PubMed] [Google Scholar]
- 28.Endo Y, Sato Y, Matsushita M, Fujita T. Cloning and characterization of the human lectin P35 gene and its related gene. Genomics. 1996;36:515–521. doi: 10.1006/geno.1996.0497. [DOI] [PubMed] [Google Scholar]
- 29.Lu J, Tay PN, Kon OL, Reid KB. Human ficolin: cDNA cloning, demonstration of peripheral blood leucocytes as the major site of synthesis and assignment of the gene to chromosome 9. The Biochemical journal. 1996;313(Pt 2):473–478. doi: 10.1042/bj3130473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugimoto R, Yae Y, Akaiwa M, Kitajima S, Shibata Y, Sato H, Hirata J, Okochi K, Izuhara K, Hamasaki N. Cloning and characterization of the Hakata antigen, a member of the ficolin/opsonin p35 lectin family. The Journal of biological chemistry. 1998;273:20721–20727. doi: 10.1074/jbc.273.33.20721. [DOI] [PubMed] [Google Scholar]
- 31.Endo Y, Nakazawa N, Liu Y, Iwaki D, Takahashi M, Fujita T, Nakata M, Matsushita M. Carbohydrate-binding specificities of mouse ficolin A, a splicing variant of ficolin A and ficolin B and their complex formation with MASP-2 and sMAP. Immunogenetics. 2005;57:837–844. doi: 10.1007/s00251-005-0058-1. [DOI] [PubMed] [Google Scholar]
- 32.Endo Y, Takahashi M, Iwaki D, Ishida Y, Nakazawa N, Kodama T, Matsuzaka T, Kanno K, Liu Y, Tsuchiya K, Kawamura I, Ikawa M, Waguri S, Wada I, Matsushita M, Schwaeble WJ, Fujita T. Mice deficient in ficolin, a lectin complement pathway recognition molecule, are susceptible to Streptococcus pneumoniae infection. Journal of Immunology. 2012;189:5860–5866. doi: 10.4049/jimmunol.1200836. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Endo Y, Iwaki D, Nakata M, Matsushita M, Wada I, Inoue K, Munakata M, Fujita T. Human M-ficolin is a secretory protein that activates the lectin complement pathway. Journal of Immunology. 2005;175:3150–3156. doi: 10.4049/jimmunol.175.5.3150. [DOI] [PubMed] [Google Scholar]
- 34.Runza VL, Hehlgans T, Echtenacher B, Zahringer U, Schwaeble WJ, Mannel DN. Localization of the mouse defense lectin ficolin B in lysosomes of activated macrophages. Journal of endotoxin research. 2006;12:120–126. doi: 10.1179/096805106X102147. [DOI] [PubMed] [Google Scholar]
- 35.Banda NK, Takahashi M, Takahashi K, Stahl GL, Hyatt S, Glogowska M, Wiles TA, Endo Y, Fujita T, Holers VM, Arend WP. Mechanisms of mannose-binding lectin-associated serine proteases-1/3 activation of the alternative pathway of complement. Mol Immunol. 2011;49:281–289. doi: 10.1016/j.molimm.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keshi H, Sakamoto T, Kawai T, Ohtani K, Katoh T, Jang SJ, Motomura W, Yoshizaki T, Fukuda M, Koyama S, Fukuzawa J, Fukuoh A, Yoshida I, Suzuki Y, Wakamiya N. Identification and characterization of a novel human collectin CL-K1. Microbiol Immunol. 2006;50:1001–1013. doi: 10.1111/j.1348-0421.2006.tb03868.x. [DOI] [PubMed] [Google Scholar]
- 37.Ma YJ, Hein E, Munthe-Fog L, Skjoedt MO, Bayarri-Olmos R, Romani L, Garred P. Soluble Collectin-12 (CL-12) Is a Pattern Recognition Molecule Initiating Complement Activation via the Alternative Pathway. Journal of Immunology. 2015;195:3365–3373. doi: 10.4049/jimmunol.1500493. [DOI] [PubMed] [Google Scholar]
- 38.Nakamura K, Funakoshi H, Miyamoto K, Tokunaga F, Nakamura T. Molecular cloning and functional characterization of a human scavenger receptor with C-type lectin (SRCL), a novel member of a scavenger receptor family. Biochemical and biophysical research communications. 2001;280:1028–1035. doi: 10.1006/bbrc.2000.4210. [DOI] [PubMed] [Google Scholar]
- 39.Ohtani K, Suzuki Y, Eda S, Kawai T, Kase T, Yamazaki H, Shimada T, Keshi H, Sakai Y, Fukuoh A, Sakamoto T, Wakamiya N. Molecular cloning of a novel human collectin from liver (CL-L1) The Journal of biological chemistry. 1999;274:13681–13689. doi: 10.1074/jbc.274.19.13681. [DOI] [PubMed] [Google Scholar]
- 40.Hansen S, Selman L, Palaniyar N, Ziegler K, Brandt J, Kliem A, Jonasson M, Skjoedt MO, Nielsen O, Hartshorn K, Jorgensen TJ, Skjodt K, Holmskov U. Collectin 11 (CL-11, CL-K1) is a MASP-1/3-associated plasma collectin with microbial-binding activity. J Immunol. 2010;185:6096–6104. doi: 10.4049/jimmunol.1002185. [DOI] [PubMed] [Google Scholar]
- 41.Farrar CA, Tran D, Li K, Wu W, Peng Q, Schwaeble W, Zhou W, Sacks SH. Collectin-11 detects stress-induced L-fucose pattern to trigger renal epithelial injury. The Journal of clinical investigation. 2016;126:1911–1925. doi: 10.1172/JCI83000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Degn SE, Jensen L, Hansen AG, Duman D, Tekin M, Jensenius JC, Thiel S. Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function. Journal of Immunology. 2012;189:3957–3969. doi: 10.4049/jimmunol.1201736. [DOI] [PubMed] [Google Scholar]
- 43.Skjoedt MO, Hummelshoj T, Palarasah Y, Honore C, Koch C, Skjodt K, Garred P. A novel mannose-binding lectin/ficolin-associated protein is highly expressed in heart and skeletal muscle tissues and inhibits complement activation. The Journal of biological chemistry. 2010;285:8234–8243. doi: 10.1074/jbc.M109.065805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi M, Hideharu S, Endo Y, Schwaeble W, Fujita T. MASP-3 is the main converting enzyme for complement factor D. Molecular Immunology. 2014;61:280–281. [Google Scholar]
- 45.Takahashi M, Endo Y, Fujita T, Matsushita M. A truncated form of mannose-binding lectin-associated serine protease (MASP)-2 expressed by alternative polyadenylation is a component of the lectin complement pathway. International immunology. 1999;11:859–863. doi: 10.1093/intimm/11.5.859. [DOI] [PubMed] [Google Scholar]
- 46.Gulla KC, Gupta K, Krarup A, Gal P, Schwaeble WJ, Sim RB, O’Connor CD, Hajela K. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology. 2010;129:482–495. doi: 10.1111/j.1365-2567.2009.03200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwaeble WJ, Lynch NJ, Clark JE, Marber M, Samani NJ, Ali YM, Dudler T, Parent B, Lhotta K, Wallis R, Farrar CA, Sacks S, Lee H, Zhang M, Iwaki D, Takahashi M, Fujita T, Tedford CE, Stover CM. Targeting of mannan-binding lectin-associated serine protease-2 confers protection from myocardial and gastrointestinal ischemia/reperfusion injury. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7523–7528. doi: 10.1073/pnas.1101748108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olszowski T, Poziomkowska-Gesicka I, Jensenius JC, Adler G. Lectin pathway of complement activation in a Polish woman with MASP-2 deficiency. Immunobiology. 2014;219:261–262. doi: 10.1016/j.imbio.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 49.Orsini F, Chrysanthou E, Dudler T, Cummings WJ, Takahashi M, Fujita T, Demopulos G, De Simoni MG, Schwaeble W. Mannan binding lectin-associated serine protease-2 (MASP-2) critically contributes to post-ischemic brain injury independent of MASP-1. Journal of neuroinflammation. 2016;13:213. doi: 10.1186/s12974-016-0684-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knutzen Steuer KL, Sloan LB, Oglesby TJ, Farries TC, Nickells MW, Densen P, Harley JB, Atkinson JP. Lysis of sensitized sheep erythrocytes in human sera deficient in the second component of complement. Journal of Immunology. 1989;143:2256–2261. [PubMed] [Google Scholar]
- 51.Wagner E, Platt JL, Howell DN, Marsh HC, Jr, Frank MM. IgG and complement-mediated tissue damage in the absence of C2: evidence of a functionally active C2-bypass pathway in a guinea pig model. Journal of Immunology. 1999;163:3549–3558. [PubMed] [Google Scholar]
- 52.Selander B, Martensson U, Weintraub A, Holmstrom E, Matsushita M, Thiel S, Jensenius JC, Truedsson L, Sjoholm AG. Mannan-binding lectin activates C3 and the alternative complement pathway without involvement of C2. The Journal of clinical investigation. 2006;116:1425–1434. doi: 10.1172/JCI25982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banda NK, Levitt B, Glogowska MJ, Thurman JM, Takahashi K, Stahl GL, Tomlinson S, Arend WP, Holers VM. Targeted inhibition of the complement alternative pathway with complement receptor 2 and factor H attenuates collagen antibody-induced arthritis in mice. J Immunol. 2009;183:5928–5937. doi: 10.4049/jimmunol.0901826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banda NK, Takahashi K, Wood AK, Holers VM, Arend WP. Pathogenic complement activation in collagen antibody-induced arthritis in mice requires amplification by the alternative pathway. J Immunol. 2007;179:4101–4109. doi: 10.4049/jimmunol.179.6.4101. [DOI] [PubMed] [Google Scholar]
- 55.Lachmann PJ. Preparing serum for functional complement assays. Journal of immunological methods. 2010;352:195–197. doi: 10.1016/j.jim.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 56.Banda NK, Levitt B, Wood AK, Takahashi K, Stahl GL, Holers VM, Arend WP. Complement activation pathways in murine immune complex-induced arthritis and in C3a and C5a generation in vitro. Clinical and experimental immunology. 2010;159:100–108. doi: 10.1111/j.1365-2249.2009.04035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heja D, Harmat V, Fodor K, Wilmanns M, Dobo J, Kekesi KA, Zavodszky P, Gal P, Pal G. Monospecific inhibitors show that both mannan-binding lectin-associated serine protease-1 (MASP-1) and -2 Are essential for lectin pathway activation and reveal structural plasticity of MASP-2. The Journal of biological chemistry. 2012;287:20290–20300. doi: 10.1074/jbc.M112.354332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heja D, Kocsis A, Dobo J, Szilagyi K, Szasz R, Zavodszky P, Pal G, Gal P. Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10498–10503. doi: 10.1073/pnas.1202588109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kocsis A, Kekesi KA, Szasz R, Vegh BM, Balczer J, Dobo J, Zavodszky P, Gal P, Pal G. Selective inhibition of the lectin pathway of complement with phage display selected peptides against mannose-binding lectin-associated serine protease (MASP)-1 and -2: significant contribution of MASP-1 to lectin pathway activation. Journal of Immunology. 2010;185:4169–4178. doi: 10.4049/jimmunol.1001819. [DOI] [PubMed] [Google Scholar]
- 60.Iwaki D, Kanno K, Takahashi M, Endo Y, Matsushita M, Fujita T. The role of mannose-binding lectin-associated serine protease-3 in activation of the alternative complement pathway. Journal of Immunology. 2011;187:3751–3758. doi: 10.4049/jimmunol.1100280. [DOI] [PubMed] [Google Scholar]
- 61.Oroszlan G, Kortvely E, Szakacs D, Kocsis A, Dammeier S, Zeck A, Ueffing M, Zavodszky P, Pal G, Gal P, Dobo J. MASP-1 and MASP-2 Do Not Activate Pro-Factor D in Resting Human Blood, whereas MASP-3 Is a Potential Activator: Kinetic Analysis Involving Specific MASP-1 and MASP-2 Inhibitors. Journal of Immunology. 2016;196:857–865. doi: 10.4049/jimmunol.1501717. [DOI] [PubMed] [Google Scholar]
- 62.Ahmad-Nejad P, Hacker H, Rutz M, Bauer S, Vabulas RM, Wagner H. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. European journal of immunology. 2002;32:1958–1968. doi: 10.1002/1521-4141(200207)32:7<1958::AID-IMMU1958>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 63.Murray PJ. NOD proteins: an intracellular pathogen-recognition system or signal transduction modifiers? Current opinion in immunology. 2005;17:352–358. doi: 10.1016/j.coi.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 64.Fujita T, Matsushita M, Endo Y. The lectin-complement pathway--its role in innate immunity and evolution. Immunological reviews. 2004;198:185–202. doi: 10.1111/j.0105-2896.2004.0123.x. [DOI] [PubMed] [Google Scholar]
- 65.Garred P, Honore C, Ma YJ, Rorvig S, Cowland J, Borregaard N, Hummelshoj T. The genetics of ficolins. Journal of innate immunity. 2010;2:3–16. doi: 10.1159/000242419. [DOI] [PubMed] [Google Scholar]
- 66.Arend WP, Mehta G, Antonioli AH, Takahashi M, Takahashi K, Stahl GL, Holers VM, Banda NK. Roles of adipocytes and fibroblasts in activation of the alternative pathway of complement in inflammatory arthritis in mice. Journal of Immunology. 2013;190:6423–6433. doi: 10.4049/jimmunol.1300580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ma YJ, Kang HJ, Kim JY, Garred P, Lee MS, Lee BL. Mouse mannose-binding lectin-A and ficolin-A inhibit lipopolysaccharide-mediated pro-inflammatory responses on mast cells. BMB Rep. 2013;46:376–381. doi: 10.5483/BMBRep.2013.46.7.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Asgari E, Farrar CA, Lynch N, Ali YM, Roscher S, Stover C, Zhou W, Schwaeble WJ, Sacks SH. Mannan-binding lectin-associated serine protease 2 is critical for the development of renal ischemia reperfusion injury and mediates tissue injury in the absence of complement C4. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2014;28:3996–4003. doi: 10.1096/fj.13-246306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dobo J, Pal G, Cervenak L, Gal P. The emerging roles of mannose-binding lectin-associated serine proteases (MASPs) in the lectin pathway of complement and beyond. Immunological reviews. 2016;274:98–111. doi: 10.1111/imr.12460. [DOI] [PubMed] [Google Scholar]
- 70.Dobo J, Harmat V, Beinrohr L, Sebestyen E, Zavodszky P, Gal P. MASP-1, a promiscuous complement protease: structure of its catalytic region reveals the basis of its broad specificity. J Immunol. 2009;183:1207–1214. doi: 10.4049/jimmunol.0901141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.