

Abstract
The final step in the biosynthesis of the antibiotic chloramphenicol is the oxidation of an aryl-amine substrate to an aryl-nitro product catalyzed by the N-oxygenase CmlI in three two-electron steps. The CmlI active site contains a diiron cluster ligated by three histidine and four glutamate residues, and activates dioxygen to perform its role in the biosynthetic pathway. It was previously shown that the active oxidant used by CmlI to facilitate this chemistry is a peroxo-diferric intermediate (CmlIP). Spectroscopic characterization demonstrated that the peroxo binding geometry of CmlIP is not consistent with the μ-1,2 mode commonly observed in nonheme diiron systems. Its geometry was tentatively assigned as μ- η2: η1 based on comparison with resonance Raman (rR) features of mixed-metal model complexes in the absence of appropriate diiron models. Here, X-ray absorption spectroscopy (XAS) and rR studies have been used to establish a refined structure for the diferric cluster of CmlIP. The rR experiments carried out with isotopically labeled water identified the symmetric and asymmetric vibrations of an Fe–O–Fe unit in the active site at 485 and 780 cm−1, respectively, which was confirmed by the 1.83-Å Fe–O bond observed by XAS. In addition, a unique Fe•••O scatterer at 2.82 Å observed from XAS analysis is assigned as arising from the distal O atom of a μ-1,1-peroxo ligand that is bound symmetrically between the irons. The (μ-oxo)(μ-1,1-peroxo)diferric core structure associated with CmlIP is unprecedented among diiron cluster-containing enzymes and corresponding biomimetic complexes. Importantly, it allows the peroxo-diferric intermediate to be ambiphilic, acting as an electrophilic oxidant in the initial N-hydroxylation of an arylamine and then becoming a nucleophilic oxidant in the final oxidation of an aryl-nitroso intermediate to the aryl-nitro product.
Keywords: X-ray absorption spectroscopy, Raman spectroscopy, Non-heme iron, Diiron cluster, Oxygen activation, Nonribosomal peptide synthetase
Graphical Abstract
Introduction
Nonheme diiron enzymes are capable of facilitating a variety of difficult chemical transformations via oxygen activation, including C–H bond hydroxylation, C–C bond desaturation, the generation of alkanes from aldehydes, and N-oxygenation of aryl amine substrates, among others.1–8 The diiron cluster-containing N-oxygenase CmlI is a member of this latter class. It serves as a tailoring enzyme on the non-ribosomal peptide synthetase biosynthetic pathway that is responsible for producing the antibiotic chloramphenicol in the soil bacterium Streptomyces venezuelae.9–14 CmlI catalyzes a six-electron oxidation of the aryl-amine precursor to form the aryl-nitro group of the active antibiotic (Figure 1).
Figure 1.
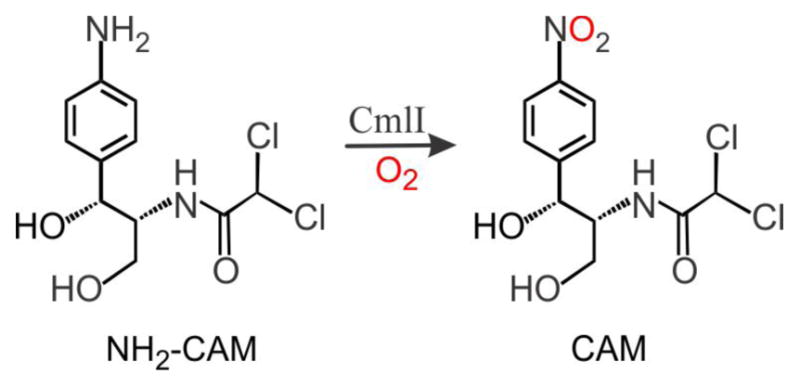
Six-electron oxidation of the aryl-amine precursor to the nitroaryl-containing final product chloramphenicol, catalyzed by the nonheme diiron cluster enzyme CmlI.
Characterization of the aryl-amine conversion by CmlI has provided the strongest evidence to date for the involvement of a new type of oxidant in diiron-cluster-mediated oxygenation reactions.15 The canonical scheme for dioxygen activation by diiron-cluster-containing enzymes is based on the well-studied diiron enzyme soluble methane monooxygenase (sMMOH). Dioxygen binds to the diferrous form of the enzyme to generate a peroxo-diferric intermediate (P), which then undergoes O–O bond cleavage to generate a high-valent bis(μ-oxo)diiron(IV) cluster (Q). Q is the reactive intermediate responsible for C–H bond activation in the sMMOH catalytic cycle. Another canonical diiron enzyme, ribonucleotide reductase (RNR) from E. coli, uses a diiron center in its R2 subunit, to activate O2 and generate a functionally essential tyrosyl radical. This oxidation is carried out by a high-valent oxidant called X with a (μ-oxo)iron(III)iron(IV) core.16–18 sMMOH and RNR are the only enzymes with diiron active sites for which high-valent iron oxidants have been identified, but such oxidants have been postulated to be the reactive intermediate formed in most diiron oxygenases.
Diferric-peroxo (P) species have been trapped and characterized in the reactions of O2 with the diferrous forms of the classical diiron oxygen activating enzymes, sMMOH,19–21 RNR R2,22–24 and stearoyl-CoA Δ9-desaturase (Δ9D),25 but their functions in their respective catalytic cycles have generally been to act as a precursor to the high-valent active oxidant. Analysis of these P intermediates has suggested that diiron-peroxo species generally adopt a cis-μ-1,2-peroxo binding mode (Figure 2) and that P intermediates are not the actual oxidants on the native catalytic cycles, except in rare cases of easily oxidized substrates.26–27 These assumptions are bolstered by the availability of several synthetic (μ-1,2-peroxo)diferric complexes for which the X-ray crystal structures are known.28–32 These complexes, supported by ligands with N- and O-donors to mimic the histidine/carboxylate active sites of the enzymes have been well characterized by various spectroscopic methods to provide a set of parameters to which enzymatic P species can be compared. However, these synthetic peroxo complexes are fairly stable and generally unreactive, and therefore do not provide much insight into the mechanism of oxidation by a P species.
Figure 2.
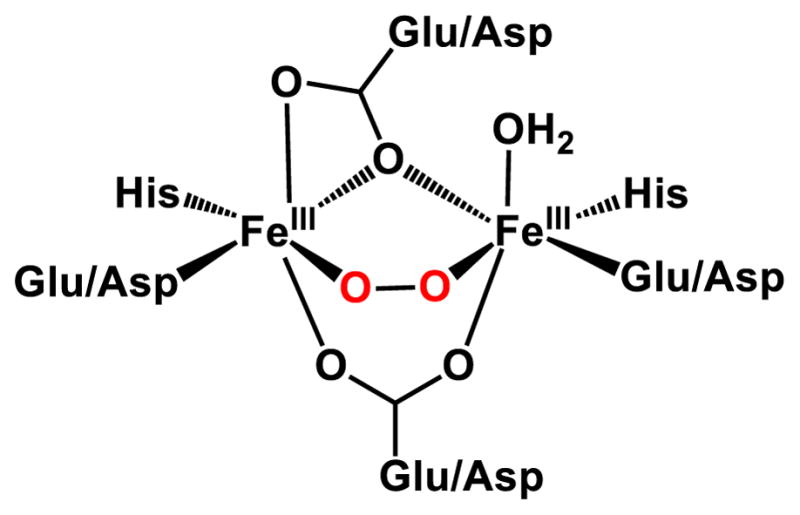
General cis-(μ-1,2-peroxo)diferric intermediate structure in diiron enzymes.
The first reactive P intermediate was discovered in 2005 by Lippard and co-workers,26 when they demonstrated that the P species of sMMOH directly reacts with ether and alcohol substrates. However, definitive structural characterization of this P intermediate is unavailable. In 2009, a hyperstable (t1/2 > 24 hrs at room temperature) (μ-hydroxo)(cis-μ-1,2-peroxo)diferric intermediate was discovered and characterized33 from the human enzyme deoxyhypusine hydroxylase (hDOHH) that decays concomitant with the formation of the native hydroxylated product, demonstrating that stable cis-μ-1,2-peroxo species can be activated.33 In 2013, a peroxo-diferric species was reported for the cyanobacterial aldehyde deformylating oxygenase (cADO) and proposed to function as a nucleophilic oxidant for the aldehyde substrate.34 A final set of examples comes from the related enzymes toluene-4-monooxygenase (T4MO) and toluene/o-xylene monooxygenase (ToMO), which fall into the diiron oxygenase subclass bacterial multicomponent monooxygenases with sMMOH. A recent crystal structure from T4MO was obtained of an arylperoxo species bound in a μ- η2: η2 configuration to the diiron cluster (T4MOPη2),35 which was shown in crystallo to be an intermediate for the hydroxylation of toluene. The putative P intermediate of ToMO (ToMOP) from the wild type enzyme is a short-lived (t1/2 ~ 15 s) diferric species that appears to be active towards arene substrates, but can only be characterized by Mössbauer spectroscopy, and has no accompanying visible chromophore.36 Its Mössbauer parameters significantly differ from those observed for the P from sMMOH and other cis μ-1,2-peroxo-diferric intermediates, and its structure remains unknown. An inactive P intermediate has also been generated from the T201S mutant of ToMO, with spectroscopic signatures that match those of sMMOH.37 These examples provide evidence that diferric-peroxo species can serve as the active oxidant instead of a high-valent intermediate such as Q or X for some types of substrates.
In the past, geometric assignments of enzymatic P intermediates have been largely based on the comparison of their spectroscopic parameters to those of structurally characterized synthetic P analogs. Recently, the X-ray crystal structures of enzymatic P intermediates have been determined for hDOHH,38 a truncated CmlI (CmlIΔ33),39 and T4MO.35, 40 The hDOHH crystal structure confirms the cis-μ-1,2-peroxo binding mode earlier assigned by resonance Raman studies, although the Fe•••Fe distance in the crystallized hDOHH suggests that this enzyme may have suffered from photoreduction by the X-ray beam, which is a well known issue in metalloenzyme crystallography.41–43 Therefore, the specific distances should be viewed with some skepticism. The crystallized CmlIΔ33P also shows a cis-μ-1,2-peroxo binding mode, but the species does not react with substrate, nor does it have the same UV-vis absorption spectrum as the known active P. Consequently, this crystallized CmlI-peroxo species is thought not to represent the active or native peroxo intermediate. Two different reactive peroxo species have been crystallized from the T4MO system, with different peroxo binding modes. A cis-μ-1,2-peroxo intermediate (T4MOP) is prepared by soaking crystals of diferric T4MO with H2O2. Accordingly, T4MO is active via the peroxide shunt as first described for sMMOH,40, 44 though the reaction is ~600 times slower than that carried out with the native biological reductants and O2. The more recent crystal structure reveals a μ-η2: η2-arylperoxo intermediate (T4MOPη2), prepared by soaking crystals of diferrous T4MO with toluene and then exposing them to O2. T4MO Pη2 is proposed to be part of the native reaction cycle. Of the three crystallized enzymatic μ-1,2-peroxo P species, only hDOHHP is shown to be active in the native cycle. The high stability and low reactivity of many of the cis-μ-1,2 P species (synthetic and enzymatic) suggest that this geometry may not be the active species. However, the new evidence implicating enzymatic P species, like T4MOPη2, as the active oxidant in some enzyme cycles emphasizes the notion that alternative types of peroxo intermediates with much higher reactivity can exist.
In 2015, we published the first evidence for a distinct P species from CmlI.8 Kinetic studies showed that CmlIP reacts rapidly with substrates to yield oxygenated products. However, it is unusually stable in the absence of substrate, exhibiting a half-life of ~3 h at 4 ºC. This long lifetime allowed some of the spectroscopic features of CmlIP to be determined in detail, revealing that it is distinct from all but one of the previously reported peroxo intermediates. The N-oxygenase AurF was found to have a P species (AurFP) with similar electronic absorption maximum and Mössbauer parameters as CmlIP, but the half-life of AurFP is much shorter (~7 min at 4 ºC), and structural characterization is unavailable.45 In contrast to the typical cis-μ-1,2-peroxo species with absorbance maxima near 700 nm, CmlIP and AurFP instead have significant absorption at 500 nm. Resonance Raman (rR) experiments of CmlIP showed a ν(O–O) at 791 cm−1 that was definitively assigned using 18O2 and 16O18O labeling experiments.8 The ν (O–O) of CmlIP is much lower in frequency than that of any known biological or synthetic diiron-cluster P species. The commonly observed (μ-1,2-peroxo)diferric species generally exhibit a range of 840 – 925 cm−1 for the ν (O–O) feature. Mössbauer analysis showed that CmlIP has two inequivalent Fe centers, while (μ-1,2-peroxo)-diferric species generally have two similar iron(III) sites.8 Unfortunately, there were no diiron peroxo model complexes available to allow a definitive structural assignment based on this spectroscopic data. Consequently, the CmlIP peroxo structure was suggested to be μ-η1: η2 based on the similarity of its rR parameters to those of mixed-metal peroxo complexes that have comparably low ν (O–O) frequencies.46–49
We have recently proposed that chloramphenicol is synthesized in three 2-electron oxidation steps from its arylamine precursor as illustrated in Scheme 1.15 CmlIP carries out both the arylamine-to-arylhydroxylamine and the arylnitroso-to-arylnitro steps in which the oxygen transfer reactions occur. The intervening arylhydroxylamine-to-arylnitroso step is thought to be an internal redox process in the active site of CmlI that results in diiron cluster reduction to allow re-formation of CmlIP to be used in the final step of chloramphenicol synthesis. The substrates for each of the three oxidation steps in this biosynthetic pathway differ substantially in their electronic nature, requiring that CmlIP be able to react with both electron-rich and electron-poor substrates, a quality we have termed ambiphilic reactivity.
Scheme 1.
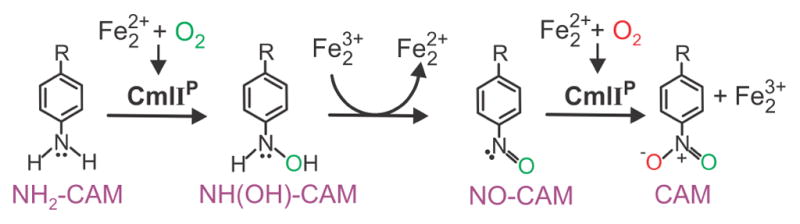
Proposed mechanism for the biosynthesis of chloramphenicol by CmlI.
In the current study, we have used X-ray absorption spectroscopy (XAS) and rR spectroscopy to structurally characterize three different states of CmlI, including CmlIP. A new view of the structure is now presented based on this spectroscopic investigation. Our studies further refine the active site picture for CmlIP, identifying the diiron core as having both an oxo and a μ-1,1-peroxo bridge. This novel diiron peroxo core structure sheds some light on the ambiphilic reactivity of the diiron cluster of CmlI and supports the notion that these are reactive peroxo intermediates that function within the canonical diiron framework.
EXPERIMENTAL PROCEDURES
Overexpression and Purification of CmlI
Expression of CmlI was performed in E. coli BL21(DE3) in M9 minimal medium in the presence of 100 μg/ml ampicillin. A His-tagged construct described previously was used.8 Cells were grown to an OD ~ 1.0 and induced with 150 μM isopropyl-β-D-1-thiogalactopyranoside (IPTG) and 50 μM FeCl3 at which point the temperature was lowered to 20 °C and grown for an additional 15 h. Cells were harvested by centrifugation and stored at −80 °C until further use. Cells were resuspended in 50 mM potassium phosphate pH 7.4, 300 mM NaCl, 10 mM imidazole, lysed via sonication, and centrifuged. The resulting supernatant was loaded onto a Ni nitrilotriacetic acid column (Qiagen) equilibrated in the same buffer. After loading, the column was washed with the above buffer containing 20 mM imidazole, and protein was eluted in the same buffer containing 300 mM imidazole. Protein-containing fractions were pooled and dialyzed against 50 mM Bicine pH 9 and stored at −80 °C until further use. CmlI concentrations were determined by the calculated extinction coefficient (ε280 = 50 mM−1 cm−1).
CmlIOx Sample
As-isolated CmlI (CmlIOx) was thawed and concentrated to 3–4 mM by centrifugal filtration (Amicon Ultra-0.5 mL centrifugal filters, kDa 10 cutoff), and then mixed with glycerol to a final concentration of ~20% v/v. After loading into an XAS cup, the sample was flash frozen with LN2. For the generation of D2O samples (CmlIOx-D2O), a minimum of 5 concentration and re-dilution cycles was used to exchange into 50 mM Bicine pD 9 prepared with D2O. No glycerol was used in D2O-containing samples.
Generation of CmlIR and CmlIP Samples for XAS
An aliquot of CmlI was degassed on an Ar Schlenk line and then brought into an anaerobic chamber where the reductant sodium hydrosulfite (10x molar excess) and the reduction mediator methyl viologen (20 μM final concentration) were added to the protein. The protein was stirred for 35 min to ensure the blue color from the methyl viologen remained, indicating complete reduction. The sodium hydrosulfite and methyl viologen were then removed by passing the CmlI through a PD-10 size exclusion column equilibrated with 50 mM Bicine, pH 9. Several fractions were collected and concentrated to 3–4 mM enzyme anaerobically using a centrifuge located within the anaerobic chamber and spin filters as described above.
Reduced
While in the anaerobic chamber, 400 μl of reduced enzyme was mixed with 100 μl glycerol and then loaded into an XAS cup (CmlIR). The cup was then sealed in a large Reacti-Vial (Thermo Scientific). The vial was brought out of the chamber and submerged in LN2. Only after the sample was frozen was the vial opened and the sample stored under LN2.
Peroxo
A reduced sample was removed from the anaerobic chamber in a Reacti-Vial and then the peroxo intermediate was generated by blowing pure O2 over the surface of the sample while stirring vigorously for 3 min. Glycerol was added to final concentration of ~20% by volume, and then the solution was placed in an XAS cup and frozen in LN2. All work outside of the anaerobic chamber was done on ice and/or in a 4 °C cold room.
Generation of CmlIOx and CmlIP Samples for rR. CmlIOx and CmlIP samples for rR were prepared in the same manner as the corresponding XAS samples, except that samples were loaded into flat-bottomed NMR tubes and were kept frozen until they were analyzed.
D2O
CmlIP in D2O (CmlIP-D2O) was generated as described above, except that after the reduction step the enzyme was passed through a PD-10 size exclusion column equilibrated with buffer prepared with D2O (50 mM Bicine, pD 9). Concentration and re-dilution with D2O buffer a minimum of two times, interspersed with 30 minute incubation periods, ensured nearly complete isotope exchange. Samples prepared with D2O did not contain glycerol.
18OH2
CmlIP and CmlIOx in 18OH2 were prepared in the same manner as their corresponding standard water samples, with the addition of several concentration and dilution cycles and 30 minute incubation periods to exchange the 16OH2-containing buffer with the 18OH2-containing buffer. In the CmlIP preparation, the exchange was performed on the diferrous enzyme in an anaerobic chamber. To prepare 18OH2-containing buffer, 2 mL of standard buffer (50 mM Bicine, pH 9) was lyophilized for 24 h. The resulting salts were then dissolved in 2 ml 18OH2 water, either anaerobically or on the bench top, as required by the sample.
X-ray Absorption Spectroscopy
Iron K-edge X-ray absorption spectra were collected on SSRL beam line 7–3 and 9-3 using a 30 element and 100 element (respectively) solid state Ge detector (Canberra) with a SPEAR storage ring current of ~500 mA at a power of 3.0 GeV. The incoming X-rays were unfocused using a Si(220) double crystal monochromator, which was detuned to 40% of the maximal flux to attenuate harmonic X-rays. For CmlIR and CmlIOx, 16 and 12 scans, respectively, were collected. For CmlIP, two samples with 9 and 10 scans were collected and averaged. All scans were between 6882 eV and 8000 eV at ~10 K using an Oxford Instruments CF1208 continuous flow liquid helium cryostat. An iron foil was placed in the beam pathway prior to the ionization chamber I0 and scanned concomitantly for an energy calibration, with the first inflection point of the edge assigned to 7112.0 eV. A 3 μm Mn filter was used for the collection of CmlIR and CmlIOx, and a “9” μm Mn filter (3 μm + 6 μm) and a Soller slit were used to increase the signal to noise ratio of the spectra for CmlIP. Photoreduction was monitored by scanning the same spot on the sample twice and comparing the first derivative peaks associated with the edge energy during collection.
The detector channels from the scans were examined, calibrated, averaged, and processed for EXAFS analysis using EXAFSPAK50 to extract Χ(k). Theoretical phase and amplitude parameters for a given absorber-scatterer pair were calculated using FEFF 8.4051 and were utilized by the “opt” program of the EXAFSPAK package during curve fitting. Parameters for each species were calculated using a model derived from the crystal structure of analogous enzyme AurF (PDB code 3CHH). In all analyses, the coordination number of a given shell was a fixed parameter and was varied iteratively in integer steps, while the bond lengths (R) and mean-square deviation (σ2) were allowed to freely float. The amplitude reduction factor S0 was fixed at 0.9, while the edge-shift parameters E0 was allowed to float as a single value for all shells. Thus, in any given fit, the number of floating parameters was typically equal to (2 x num shells) + 1. CmlIR has a k range of 2 – 14 Å−1 and CmlIOx and CmlIP have a range of 2 – 15 Å−1.
Pre-edge analysis was performed on data normalized in the “process” program of the EXAFSPAK package, and pre-edge features were fit as described elsewhere52 between 7108 eV to 7118 eV using the Fityk53 program with pseudo-Voigt functions composed of 50:50 Gaussian/Lorentzian functions.
Resonance Raman Spectroscopy
Resonance Raman spectra were obtained at ~4 ºC with excitation at 561 nm (100 mW at source, Cobolt Jive 04-01 series) through the sample in a flat bottom NMR tube using a 90° backscattering arrangement (parallel to the slit direction). The collimated Raman scattering was collected using two Plano convex lenses (f = 12 cm, placed at an appropriate distance) through the appropriate long pass edge filter (Semrock) into an Acton AM-506M3 monochromator equipped with a Princeton Instruments ACTON PyLON LN/CCD-1340x400 detector. The detector was cooled to −120 °C prior to the experiments. Spectral calibration was performed using the Raman spectrum of acetonitrile/toluene 50:50 (v:v). The spectra of CmlIOx were collected with 180, 450 and 225 scans with acquisition times of 5s, 2s and 4s for the 16O, 18O and D2O samples, respectively, for a total of 15 min collections. The spectra of CmlIP were collected with 180 scans with an acquisition time of 10s for the 18 O and D2O samples for a total of 30 min collections, and 225 scans with an acquisition time of 4s for the 16O samples for a total of 15 min. The collected data was processed using Spekwin32,54 and a multipoint baseline correction was performed for all spectra.
RESULTS
X-ray absorption near edge structure (XANES) analysis was utilized to gain structural insight into the active sites of three CmlI species: CmlIR, CmlIOx, and CmlIP (Figure 3). This analysis provides information about the oxidation state (from the Fe K-edge energy) and the symmetry (from the pre-edge peak area) of the target metal centers. The K-edge energy for CmlIR is 7122.1 eV, which falls in the range typical for diferrous species (7121 eV – 7123 eV).55–58 CmlIOx has a K-edge energy of 7124.1 eV, which is two electron volts higher than that of the reduced species. This difference in K-edge energy is consistent with the difference observed between diferrous and diferric states in other diiron enzyme systems (Table 1).57–59 The oxygenated CmlIP has a K-edge of 7124.9 eV, which falls lower than found for hDOHHP (7125.6 eV)57 but within the range found for synthetic (μ-1,2-peroxo)diferric complexes having an additional single-atom bridge (7123 – 7126 eV).60–63
Figure 3.
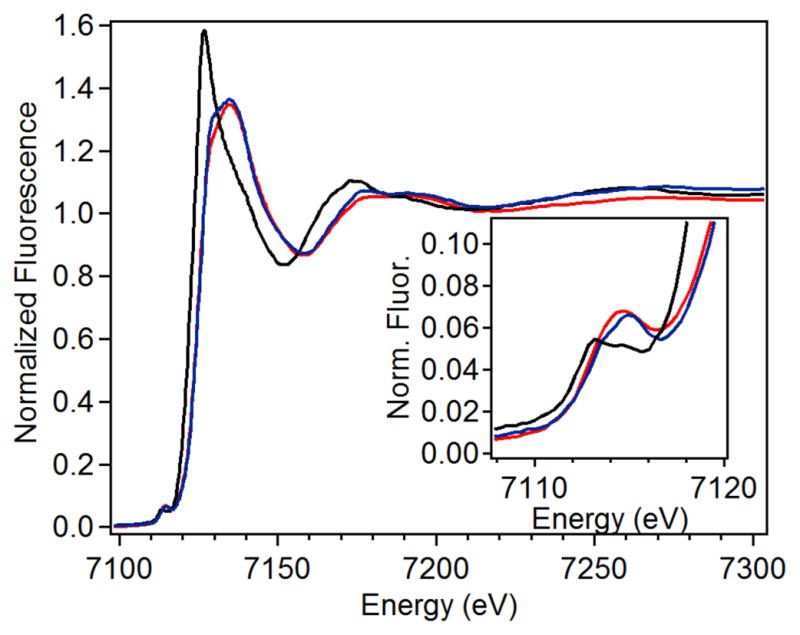
XANES region for CmlIR (solid black), CmlIOx (solid blue) and CmlIP (solid red). Inset: Magnification of the pre-edge region.
Table 1.
XANES analysis of CmlI Complexes and Related Species.
The pre-edge peak in the XANES region corresponds to a formally forbidden 1s → 3d transition in first row transition metal complexes.64 The intensity of the pre-edge peak increases as the metal center is distorted away from centrosymmetry, which is reflected in a larger pre-edge peak area.65. The fits of the pre-edge features are summarized in Table 1 and individual fits of the pre-edge region are shown in Figures S1 – S3. The pre-edge feature of CmlIR is fit with pseudo-Voigt functions that yield an area of 8.4 units, which falls between values for five-coordinate (~11 units) and six-coordinate (~5 units) diferrous centers,66 and is similar to the recently reported value for six-coordinate diferrous CmlA.58 The pre-edge area of CmlIOx is 14.5 units, consistent with a six-coordinate (μ-oxo)diferric center (ave,14.5 units).52, 67 Interestingly, CmlIP has a pre-edge area of 19.2 units, which is significantly higher than that of hDOHHP (12.4 units)57 and those found for synthetic six-coordinate diferric-peroxo complexes (13 – 16 units).60–61, 63 As synthetic (μ-oxo)diferric complexes with two five-coordinate centers have been reported to have pre-edge areas of ~24 units,67 averaging the contributions of one six-coordinate Fe center and one five-coordinate Fe center would give rise a pre-edge area of 19.3 units, so the value of 19.2 units obtained for CmlIP would be consistent with the presence of such a diferric complex.
Extended X-ray absorption fine structure (EXAFS) analysis provides information about the nature of the ligands to the iron centers and the Fe•••Fe distance. Key features of the best fits for each species are listed in Table 2, and the fitting protocols that led to the best fits for individual species can be found in the supplementary information (Figures S5 – S6, Tables S1 – S3). The Fourier transformed (FT) EXAFS data of the various CmlI samples all show an intense feature at R + Δ < 2 Å that is assigned to the scattering atoms of the primary coordination sphere, while the weaker features observed at R + Δ > 2 Å are generally associated with outer sphere interactions such as the other Fe atom in the dinuclear active site as well as the C/N atoms from His residues and carboxylates, as observed in the EXAFS analysis of other nonheme diiron enzymes.18, 57–58, 68–69
Table 2.
Key distances for CmlI species obtained from EXAFS fits.a
| Fe–O | Fe-O/N | Fe••C/O | Fe•••Fe | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fit | N | R(Å) | σ2(10−3) | N | R(Å) | σ2(10−3) | N | R(Å) | σ2(10−3) | N | R(Å) | σ2(10−3) |
| CmlIR | 1 | 1.94 | 5.35 | 5 | 2.10 | 5.02 | 1 | 2.58 | 1.35 | 1 | 3.35 | 9.90 |
| CmlIOx | 1 | 1.83 | 2.71 | 2 | 2.00 | 0.97 | 1 | 3.32 | 3.38 | |||
| 3 | 2.15 | 1.73 | ||||||||||
| CmlIP | 1 | 1.83 | 4.05 | 2 | 1.98 | 2.09 | 1 | 2.82 | 1.50 | 1 | 3.35 | 4.47 |
| 3 | 2.13 | 2.50 | ||||||||||
For complete fitting tables, see Tables S1 – S3.
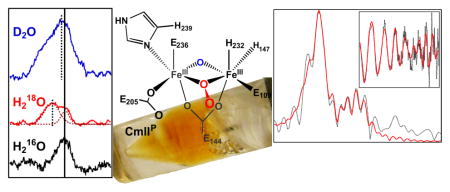
The first coordination sphere of CmlIR consists of 1 Fe–O/N at 1.94 Å and 5 Fe–N/O scatterers at 2.10 Å (Table 2). The scatterers at 2.10 Å arise from protein-derived carboxylate and histidine ligands, as we have established previously for hDOHH and CmlA.57–58 These assignments are based on metal-ligand distances found in the crystal structures of nonheme diiron proteins,7, 70–77 including diferrous CmlI.39 The shorter Fe–N/O distance at 1.94 Å is assigned to a μ-hydroxo bridge based on comparison to the Fe–O distances associated with the μ-hydroxo ligand in the reduced clusters of CmlA58 and synthetic (μ-hydroxo)diferrous complexes.78–80
Beyond the primary sphere, the fits provide evidence for a low-Z scatterer at 2.58 Å and an Fe scatterer at 3.35 Å. The distance associated with the former and its small Debye-Waller factor (σ2) are consistent with the carbon atom from a bidentately bound carboxylate ligand.58–59, 70 The fact that N = 1 for this scatterer indicates that there must be an average of one bidentate carboxylate per Fe in CmlIR. Also observed is an Fe scatterer at 3.35 Å with a σ2 of 9.90 × 10−3 Å2. This relatively high value suggests a greater variance in the Fe•••Fe distance that likely reflects fairly flexible Fe(II)–μ-O(H/R) bonds. Indeed, similarly large σ2 values have been reported for other reduced diiron enzymes such as Fe(II)Fe(III)-uteroferrin (12 × 10−3 Å2) and Fe(II)Fe(II)-sMMOH (13.3 × 10−3 Å2).68, 81 The active site picture thus obtained for CmlIR in frozen solution from EXAFS analysis differs significantly from that derived from the crystal structure of CmlI 33R.39 For the latter, the hydroxo bridge observed in the EXAFS analysis is absent. Instead, there is a bidentate carboxylate bridge (E326) that gives rise to a longer Fe•••Fe distance of 3.6 Å. For reasons that are described in detail in the Discussion, we believe that the EXAFS results better model the active conformation of the enzyme.
The best fit to the EXAFS data for diferric CmlIOx yields three shells of scatterers of the first coordination sphere, with one Fe–O/N at 1.83 Å, two Fe–O/N at 2.00 Å, and three Fe–N/O at 2.15 Å (Table 2). The much shorter Fe–O distance at 1.83 Å corresponds to a μ-oxo bridge that forms by loss of the proton from the μ-hydroxo bridge of CmlIR upon oxidation to CmlIOx. The assignment of an oxo bridge at 1.83 Å would also be consistent with the large pre-edge area associated with CmlIOx presented earlier. The main component of the first coordination sphere associated with 5 N/O scatterers in CmlIR becomes resolved in CmlIOx into two subshells at 2.00 and 2.15 Å with small σ2 values. Unlike for CmlIR, the fit for CmlIOx does not require a carbon scatterer at ~2.6 Å, suggesting that the bidentate carboxylate ligands found in CmlIR have become monodentate. Notably, the Fe•••Fe distance at 3.32 Å for CmlIOx remains essentially the same as that found for the reduced enzyme, a phenomenon that has also been observed for CmlA and hDOHH.57–58
The best fit for CmlIP is similar to that of CmlIOx except for the inclusion of a scatterer at 2.82 Å (Table 2). The major FT peak at R + Δ ~ 1.7 Å (Figure 4, top left) corresponds to one Fe–O/N at 1.83 Å, two Fe–O/N at 1.98 Å, and three Fe–N/O at 2.13 Å. The assignments of these distances correspond to those for CmlIOx, including the 1.83-Å oxygen scatterer as a μ-oxo bridge. The only difference is that one of the scatterers at 1.98 Å for CmlIP is now assigned to the proximal oxygen atom of the peroxo moiety. The Fe•••Fe distance is 3.35 Å, essentially unchanged within error relative to those observed for CmlIOx and CmlIR.
Figure 4.
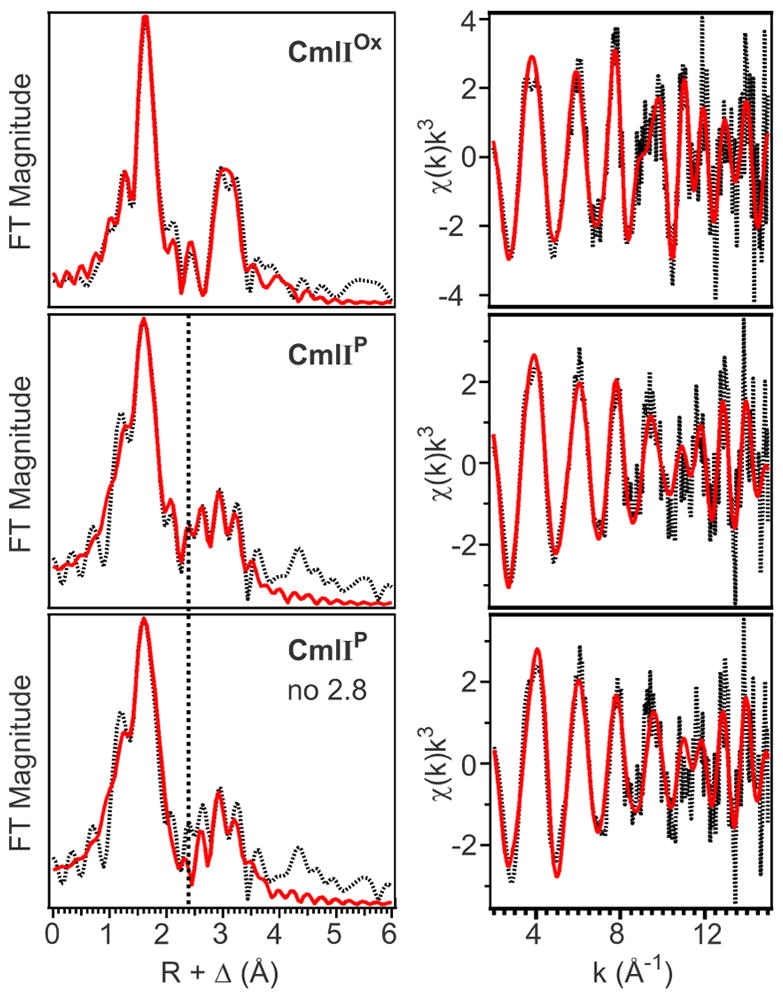
Left Column: Fourier transforms of the EXAFS data (black dotted) with best fit (solid red) for CmlIOx (top; Table S2, fit 15), CmlIP (middle; Table S3, fit 23) and CmlIP without an Fe•••O scatterer at 2.82 Å (bottom; Table S3, fit 26), k range = 2 – 15 Å−1. Vertical black dashed line shows the position of the FT feature associated with the 2.82-Å scatterer for CmlIP. Right column: unfiltered EXAFS data (black dotted) with best fit (red solid) for CmlIOx (top; Table S2, fit 15), CmlIP (middle; Table S3, fit 23) and CmlIP without an Fe–O scatterer at 2.82 Å (bottom; Table S3, fit 26).
The most notable feature of the CmlIP fit is the requirement for a Fe•••O scatterer at 2.82 Å, as demonstrated by the better fit of the FT peaks centered at ~ 2.5 Å (Figure 4, compare middle to bottom) as well as the increase in the goodness-of-fit value upon deletion of this scatterer (see Table S3, fit 23 versus fit 26). This scatterer is not required in the fits for CmlIOx, CmlIR or decayed CmlIP samples and is therefore unique to, and diagnostic of, CmlIP. The 2.82-Å scatterer has a σ2 value of 1.50 × 10−3 Å2, which is remarkably low for a low-Z scatterer beyond the first coordination sphere, implying that this scatterer is held relatively well fixed in space. In addition, it must also be positioned equidistant to the two Fe centers; otherwise, a higher σ2 value should be observed to reflect the different Fe1–O and Fe2–O distances. Reducing the N value for this scatterer to 0.5 to reflect a model in which this scatterer is only 2.82 Å away from one of the two Fe’s in the active site results in an unacceptable negative σ2 value of −1.50 × 10−3 Å2 (Table S3, fit 29). This fit further supports the assignment of a 2.82-Å scatterer that is equidistant from both Fe centers. As such a scatterer has not been found thus far in the EXAFS analysis of any other diferric-peroxo species, its presence suggests that the peroxo ligand of CmlIP must not be bound in the usual μ-1,2 mode, in line with its unusually low ν(O–O) frequency of 791 cm−1.8
Resonance Raman Characterization of CmlIOx and CmlIP
Resonance Raman experiments performed on CmlIOx and CmlIP confirm the presence of the μ-oxo ligand identified in the EXAFS analysis (see Figure S7 for full spectra). The rR spectrum of CmlIOx obtained in 16OH2 shows a peak at 487 cm−1 (Figure 5, left panel), which downshifts 18 cm−1 to 469 cm−1 in a sample of CmlIOx prepared in 18OH2 (Figure 5, middle panel). Both the location of the peak and the magnitude of the shift are consistent with its assignment to νs(Fe-O-Fe), the symmetric Fe–O–Fe stretching mode of a (μ-oxo)diferric center. For comparison, the νs(Fe–O–Fe) vibrational mode for the (μ-oxo)diferric center in CmlA was observed at a similar value of 481 cm−1 with a 18O downshift of 17 cm−1.59
Figure 5.
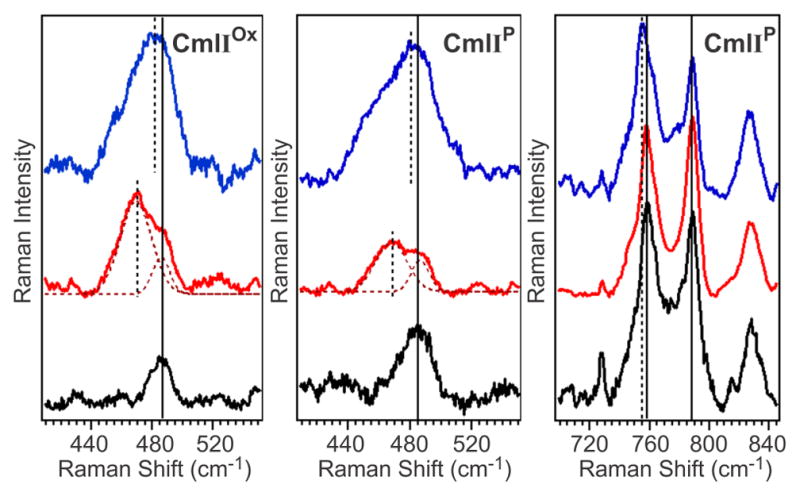
Resonance Raman spectra of CmlIOx (left), and CmlIP (middle, right) in 16OH2 (black), 18OH2 (red) and 16OD2 (blue). The isotopic composition of water affects the Fe–O–Fe unit but not the peroxo moiety. λex = 561 nm, Power = ~100 mW. All spectra were collected in solution at ~4 ºC. Protein concentration ~1 mM for each sample, 50 mM Bicine pH/pD = 9. 18OH2 enrichment of the samples was ~60%.
To put this vibration into a broader context, we compared its frequency with data originally collected by Sanders-Loehr and co-workers on a large number of (μ-oxo)diferric complexes, which established a correlation between ν(Fe–O–Fe) and ∠Fe–O–Fe values.82 Since the original 1989 study, examples of oxo-bridged diiron complexes with an additional μ-1,2-peroxo bridge (green squares) have been characterized,32, 60–62, 83 as well as complexes with bis(μ-oxo)diiron diamond cores.84–86 These new data have been included to the plot shown in Figure 6 and extend the correlation to entries with Fe–O–Fe angles approaching 90°. Based on Figure 6, the observed νs(Fe–O–Fe) frequency of 487 cm−1 would then correspond to an ∠Fe–O–Fe of ~134° for the Fe–O–Fe unit in CmlIOx, which is in good agreement with an angle of 130° that is calculated assuming Fe1–O and Fe2–O distances of 1.83 Å and an Fe•••Fe distance of 3.32 Å.
Figure 6.
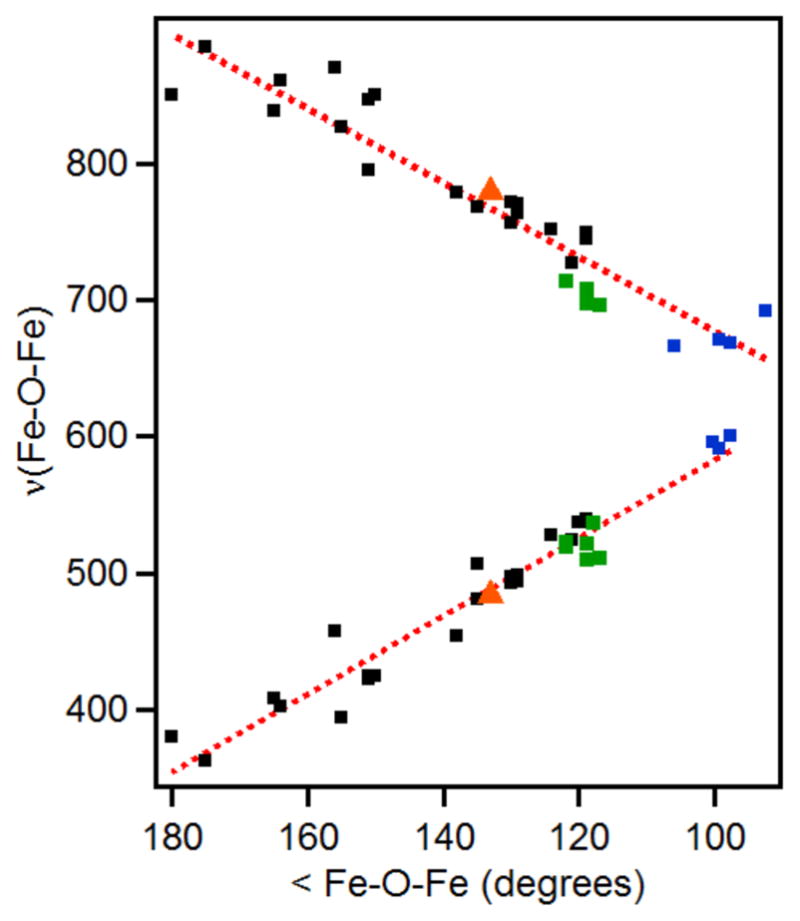
Extended data set for the Sanders-Loehr correlation of ∠Fe–O–Fe and ν(Fe–O–Fe) in oxo-bridged diiron complexes. Data from the original study (black),82 augmented by data for species with (μ-oxo)(μ-1,2-peroxo)diiron cores (green)32, 60–62, 83 and species with bis(μ-oxo)diiron diamond cores (blue).84–85 The data points corresponding to CmlIP are shown as orange triangles. Red dotted lines represent the best linear fits of the data.
Close inspection of the 740–800-cm−1 region in the 16OH2 and 18OH2 samples of CmlIOx shows the loss of signal intensity at ~780 cm−1 when the 16OH2 buffer solution is replaced by 18OH2 , unmasking a peak at ~790 cm−1 that is assigned to protein-related vibration (Figure 7, left top and left middle). Fitting the protein-related features with Gaussian functions did not sufficiently fit the peaks in the Raman spectrum. In the 16OH2 sample an additional function centered at 780 cm−1 was required, while functions at 780 cm−1 and 750 cm−1 had to be included in the 18OH2 sample, which are consistent with their respective assignments to νas(Fe–O–Fe) modes of the 16O and 18O isotopomers. Based on the correlation shown in Figure 6, they correspond to an ∠Fe–O–Fe of 138º, which is in agreement with the EXAFS-derived angle of 135º for CmlIOx.
Figure 7.

Resonance Raman spectra of CmlIOx (left) and CmlIP (right) in the 740–800-cm−1 region in 16 OH2 (top), 18 OH2 (middle) and 16 OD2 buffer (bottom). λex = 561 nm, Power = ~100 mW. The experimental data (solid black, red lines) are fit with Gaussian functions to model the protein feature at ~760 cm−1 and the peroxo/protein peak at 789 cm−1. These peaks are then subtracted from the experimental data (resulting green solid line). All spectra were collected in solution at ~4 ºC. Protein concentration ~1 mM for each sample, pH/pD = 9. 18OH2 enrichment of the samples was ~60%.
Raman data also confirm the retention of the oxo bridge in CmlIP. Examination of the 500-cm−1 region of CmlIP reveals a peak at 485 cm−1, which downshifts to 467 cm−1 in 18OH2 buffer (Figure 5, center) and can be assigned to νs(Fe–O–Fe). The similar frequencies of this feature in both CmlIOx and CmlIP suggest little difference between the Fe–O–Fe angles of CmlIOx and CmlIP, in agreement with the EXAFS analysis. The corresponding νas(Fe–O–Fe) mode is found to be in the same region as a protein-derived mode at 758 cm−1 and the previously identified νs(O–O) vibration at 791 cm−1 (789 cm−1 in the present study). Fitting the 740–800 cm−1 region of the 16OH2 sample only with Gaussian functions at 758 cm−1 and 789 cm−1 is insufficient to match the observed peak shape and requires inclusion of another peak at 780 cm−1 (Figure 7, top right). Fitting the signals in this region for the 60% 18OH2 sample (Figure 7, middle right) requires the inclusion of peaks at 780 cm−1 and 749 cm−1, which can be assigned respectively to the νas(Fe–O–Fe) modes of the 16O and 18O isotopomers and correspond to an ∠Fe-O-Fe of 138º on the Sanders-Loehr correlation (Figure 6), in reasonable agreement with the EXAFS-derived angle of 133º. CmlIP thus represents the only enzymatic diiron-peroxo intermediate to have both an oxo and a peroxo bridge, a combination that presumably has implications on its biological function.
The previously reported Raman spectrum of CmlIP identified the ν(O–O) vibration at 791 cm−1, which downshifted 43 cm−1 when CmlIP was formed with 18O2. In the experiments reported here, the ν(O–O) is observed at 789 cm−1 and does not shift in 18OH2 buffer. The latter result is consistent with recent mechanistic data demonstrating that water is not incorporated into the products of N-oxygenation by CmlI and therefore does not exchange with the bound peroxo moiety.15
Effect of D2O on the Active Site
To interrogate the effect of water on the CmlI active site, we conducted rR experiments with CmlIOx-D2O and CmlIP-D2O. D2O-sensitive vibrations were observed for both CmlIOx and CmlIP (Table 3). For CmlIOx, the νs(Fe–O–Fe) peak downshifts by 7 cm−1 in CmlIOx-D2O (Figure 5, left panel, blue trace), while the νas(Fe–O–Fe) peak could not be deconvoluted from the experimental spectrum (Figure 5, left bottom). For CmlIP, the νs(Fe–O–Fe) peak downshifts by 4 cm−1, while the νas(Fe–O–Fe) peak downshifts by 2 cm−1. These results indicate some interaction between the Fe–O–Fe unit and solvent water, but are different from those reported for metHr-X complexes,87 the only other nonheme protein with a (μ-oxo)diferric active site that has been investigated in the same manner. Interestingly, D2O substitution did not affect the νs(Fe–O–Fe) frequency observed for metHr-X complexes with X = azide, thiocyanate, cyanate, cyanide, chloride or formate. These anions occupy the only coordination site available for exogenous ligands on the diiron center of hemerythrin and preclude the possibility of water binding to this center. However respective upshifts of +4 and +26 cm−1 were found for X = hydroperoxide and hydroxide, anions with O–H functionalities that can hydrogen bond to the oxo bridge of the Fe–O–Fe center.87 Clearly, D2O interacts with the μ-oxo moiety in CmlI in a different manner from the hemerythrin complexes,87 which we will explore in the Discussion.
Table 3.
Select data points from the extended Sanders-Loehr correlation in Figure 6.
| Speciesa | ∠Fe-O-Fe (º)b | νs(Fe-O-Fe) (cm−1)c | νas(Fe-O-Fe) (cm−1)c | Refs |
|---|---|---|---|---|
| CmlIP | 133 | 485 (−18) [−4] | 780 (−31) [−2] | This work |
| CmlIOx | 130 | 487 (−18) [−7] | 780 (−30) [−] | This work |
| CmlA | 135 | 481 (−17) | - | 59 |
| oxyHr | 134 | 486 (−14) [+4] | 753 (−37) | 87 |
| metHrCN | 137 | 512 (−14) [0] | 782 (−28) | 88 |
| 1 | 122 | 523 (−16) | 714 (−14) | 60 |
| 2 | 117 | 511 (−12) | 696 (−30) | 83 |
| 3 | 119 | 522 (−13) | 708 (−32) | 62 |
| 4 | 130 | 497 (−17) | 772 (−37) | 89 |
| 5 | 124 | 528 (−17) | 751 (−30) | 90 |
| 6 | 99 | 591 (−27) | 671 (−31) | 85 |
| 7 | 93 | - | 692 (−32) | 84 |
| 8 | 106 | - | 666 (−32) | 84 |
Synthetic complexes in this column: 1 = [(BnBQA)FeIII2 (O)(O2)(CH3CN)2]2+, BnBQA = N-benzyl-N,N-bis(2-quinolinylmethyl)amine; 2 = [(6-Me3TPA)2FeIII 2(O)(O2)]2+, 6-Me3TPA = tris(6-methyl-2-pyridylmethyl)amine; 3 = [(BQPA)FeIII2 (O)(O2)]2+, BQPA = bis(2-quinolylmethyl)-2-pyridylmethylamine; 4 = [(TPA)2FeIII 2(O)(OBz)]3+, TPA = tris(2-pyridylmethyl)amine, OBz = benzoate; 5 = [(Tp)2FeIII 2(O)(OAc)2], Tp = tris(1–pyrazolyl)borate; 6 = [(6-Me3TPA)2FeIII 2(O)(OH)] 3+; 7 = [(6-Me3TPA)2FeIII 2(O)2]2+; 8 = [(5-Me3TPA)2FeIIIFeIV(O)2]3+, 5-Me3TPA = tris(5-methyl-2-pyridylmethyl)amine.
Angles in italics come from X-ray diffraction studies, while the other angles are calculated from EXAFS data.
Values in parenthesis indicate 18O isotope shifts, while those in brackets are isotope shifts observed in D2O buffer.
The effect of D2O on the Raman properties of the iron(III)-peroxo unit of CmlIP (Figure 5) is also different from that on oxyHr. OxyHr exhibits a ν(O–O) mode at 844 cm−1 that upshifts by 4 cm−1 in the presence of D2O and a ν(Fe–OOH) mode at 503 cm−1 that downshifts by 3 cm−1.88 Parallel behavior is reported for oxymyohemerythrin (oxyMHr).91 The unusual upshift of the ν (O–O) in these two complexes reflects the hydrogen bonding interaction between the hydroperoxo ligand and the Fe–O–Fe unit that is crucial for the reversible O2 binding function of hemerythrin.92 In contrast for CmlIP, the ν(O–O) mode is unaffected by D2O.
DISCUSSION
The reactions of the N-oxygenase CmlI and its analog AurF present the clearest examples to date of a diiron-peroxo intermediate as the major reactive species in a catalytic cycle. The peroxo intermediate involved in the 6-e–-oxidation of an arylamine substrate to a nitroaryl product exhibits unique spectroscopic features that distinguish it from the commonly encountered cis-(μ-1,2-peroxo)diferric intermediates of other well characterized diiron cluster-containing enzymes.8, 24, 33, 93–94 These features include its orange color and a ν (O–O) frequency that is the lowest by far found for a diferric-peroxo complex. Here, we have taken advantage of the exceptionally long lifetime of the CmlIP to prepare nearly homogeneous, high concentration samples for detailed XAS and rR analysis. The results indicate that CmlIP has a geometry that has not been previously described in biological or chemical systems. The basis for this assignment and the implications for the reactivity of peroxo intermediates are discussed here.
Comparison of CmlIP to Synthetic Diferric cis-μ-1,2-Peroxo Intermediates
Besides its orange color and ν(O–O) frequency, perhaps the most distinguishing spectral feature of CmlIP is the 2.82-Å scatterer seen in its EXAFS spectrum. Such a scatterer has not been previously reported for any diiron enzyme. To better address the assignment of this feature, available structural information from synthetic models was used to parse the possibilities. As detailed in the introduction, O2 binds in a cis-μ-1,2 mode (Figure 8, A) in all crystallographically characterized synthetic diferric-peroxo species, and all except one of these have an additional single-atom bridge; examples of these complexes are listed in Table 4 and include [FeIII2(N-Et-HPTB)(O2)(Ph3PO)2]3+ (9) (N-Et-HPTB = N,N,N’,N’-tetrakis(2-benzimidazolylmethyl)-2-hydroxy-1,3-diaminopropane)30 and the acid/base pair of [FeIII2(6Me2-BPP)2(OH)(O2)]+ (10) and [FeIII2(6Me2-BPP)2(O)(O2)] (11) (6Me2-BPP = N,N-bis(6-methyl-2-pyridylmethyl)-3-amino-propionate).95 In these species, the Fe–O–O–Fe torsion angle is ~0º, corresponding to a nearly flat Fe1–O1–O2–Fe2 unit that fixes the distal O (Fe1•••O2 and Fe2•••O1) of the peroxo moiety to a distance of ~2.9 Å in 9 and 10 and ~3 Å in 11 (Fe•••Fe: 9 = 3.46 Å, 10 = 3.40 Å, 11 = 3.17 Å). These distances are longer than the 2.82-Å distance observed in CmlIP. The metal-metal separation for 9 and 10 is ~0.1 Å longer than found for CmlIP, while the oxo-bridged 11 has a metal-metal separation that is ~0.2 Å shorter than that of CmlIP. Importantly, an ~2.8 Å O scatterer has not been reported in the EXAFS analyses for these synthetic peroxo species or related complexes60–63, 83 or for the enzymatic μ-1,2-peroxo species hDOHHP.57 Collectively, these spectroscopic results implicate a peroxo binding geometry for CmlIP that is distinct from the cis-μ-1,2-peroxo configuration commonly found in diiron chemistry.8
Figure 8.
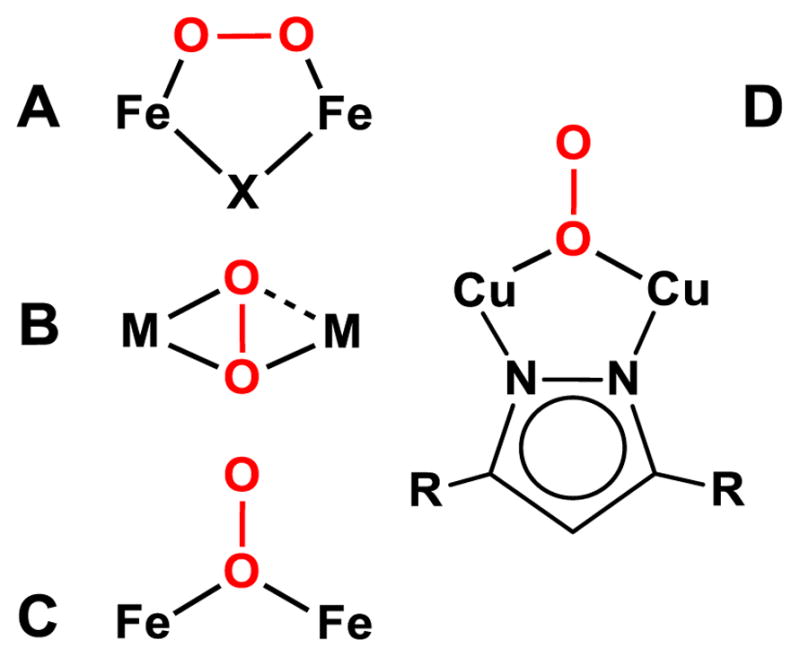
O2 binding modes for dimetal peroxide species relevant to the analysis of CmlIP. X represents an additional bridging ligand, M represents a transition metal center, and R represents the remaining sections of the Let ligand from 15.
Table 4.
Select structural metrics for CmlIP and relevant synthetic peroxo complexes.
| Species | Metal | Modea | M•••M (Å) | M–Op (Å)b | ν(O–O) (cm−1) |
|---|---|---|---|---|---|
| CmlIP | Fe2 | C | 3.35 | 1.98; 2.82 | 789 |
| 9 | Fe2 | A | 3.46 | 1.88; 2.91 | 900 |
| 10 | Fe2 | A | 3.40 | 1.88, 1.89; | 908 |
| 2.86, 2.88 | |||||
| 11 | Fe2 | A | 3.17 | 1.73; 2.97c | 847 |
| 12 | Fe, Cu | B | 3.92 | 1.89, 1.92; | 790 |
| 2.03, 2.66 | |||||
| 13 | Co2 | B | 3.34 | 1.85, 1.92; | 839 |
| 1.93, 2.76 | |||||
| 14 | Co2 | A | 3.77 | 1.86; 2.70 | 866 |
| 15 | Cu2 | D | 3.53 | 1.99; | 860 |
| 2.90, 3.00 |
The mode listed in this column refers to the O2 binding mode shown in Figure 8.
In this column, the M–Oproximal distance is listed first, followed by the M–Odistal distance.
The μ-oxo and μ-1,2-peroxo atoms are disordered over two positions, each with 0.5 occupancy, so these values are not very reliable and represent the average from the crystal structure.
Comparison of CmlIP to Known (μ-η2: η1-Peroxo)dimetal Complexes
In our previous study,8 we suggested that CmlIP might have a η2: η1-peroxo geometry because it exhibits a ν(O–O) frequency nearly identical to the 790-cm−1 value reported for [(TMP-5Me-TPA)FeIII(O2)CuII]+ (12) (TMP-5Me-TPA = 10,15,20-tris(2,4,6-trimethylphenyl)-5-(2’-bis((5’’-methyl-2’’-pyridylmethyl)aminomethyl)pyridine-5’-carboxyamidophenyl)-porphyrin) (Table 4).96 This orthogonal comparison was necessary, because no synthetic (μ- η2: η1-peroxo)diferric complex had yet been reported. In the crystal structure of the Fe/Cu complex 12 reported by Naruta,96 O2 is bound η2 to Fe and η1 to Cu (Figure 8, B), Its structure shows a flat Fe–O–O–Cu plane, with Fe–Operoxo distances of 1.89 and 2.03 Å, Cu–Operoxo distances of 1.92 and 2.66 Å, and an Fe•••Cu separation of 3.92 Å, which is much longer than observed for CmlIP. Moreover, there would be no way to accommodate a 2.82 Å scatterer with an η2: η1 configuration. Although there is a Cu-Odistal distance in 12 of 2.66 Å, the corresponding Fe-Odistal distance at 2.03 Å, so this (μ- η2: η1-peroxo)dimetal core would not be a suitable model for the core deduced for CmlIP by EXAFS analysis.
Another crystallized complex with a μ- η2: η1-peroxo ligand is [(oxapyme)CoIII2(μ- η2: η1-O2)]2+ (13) (oxapyme = 2-(bis-pyridin-2-ylmethyl-amino)-N-[2-(5-{2-[2-(methyl-pyridin-2-ylmethyl-amino)-acetylamino]-phenyl}-[1,3,4]oxadiazol-2-yl)-phenyl]-acetamide) (Figure 8, B).97 This complex has a Co•••Co separation of 3.34 Å, with Co1– η2 -Operoxo bond lengths of 1.85 and 1.93 Å and Co2–η1-Operoxo distances of 1.92 and 2.76 Å. Although the metal-metal separation matches that of CmlIP, only one of the two Co centers has a Co-Odistal distance that approaches the 2.8-Å distance found in CmlIP, as with the Fe/Cu complex discussed above. Furthermore, neither of the two (μ- η2: η1-peroxo)dimetal complexes discussed has the additional oxo bridge found in CmlIP by rRaman and EXAFS analysis.
Complex 13 provides an additional point of comparison because a related complex 14, [(oxapyme)CoIII2(μ-1,2-O2)(NO2)]1+, has been described, and both complexes have been analyzed by FT-Raman. Complex 13 has a ν(O–O) = 839 cm−1, with no ν (Co–O–Co) vibration reported, and 14 has a ν (O–O) of 866 cm−1. A change in the O–O stretching frequency of -27 cm−1 is observed on going from a μ-1,2-peroxo binding mode in 14 to a μ- η2: η1 configuration in 13,. In contrast, the ν(O–O) for CmlIP is at least 60 cm−1 lower in energy than found for any (cis-μ-1,2-peroxo)diferric intermediates.8 This structural and vibrational analysis argues against the η2: η1-O2 assignment for CmlIP.
The μ-1,1-Peroxo Model for CmlIP
An alternative to the cis-μ-1,2-peroxo and μ- η2: η1 peroxo configurations is the μ-1,1-peroxo mode illustrated in Figure 8C. The analysis of the XAS data shows that CmlIOx and CmlIP have very similar diiron(III) core structures, including short Fe– μ-O distances of 1.83 Å indicative of an oxo bridge and identical Fe•••Fe distances (Figure 9 and Table 2). A μ-1,1-peroxo ligand would provide an explanation for the unique 2.82 Å scatterer found in CmlIP. To rationalize the low σ2 associated with this scatterer, the distal oxygen of the peroxo unit must be at approximately this distance from each iron. The only other scatterer with a similar distance and comparably small σ2 is the carbon atom of the bidentate carboxylates at ~2.6 Å seen in the CmlIR sample. It could be argued that bidentate carboxylate ligand(s) of CmlIR could become monodentate upon formation of CmlIP, which would increase the Fe–C distance to 2.82 Å as observed in CmlIP. However, such a monodentate ligand would not be expected to have the rigidity to give rise to the observed 2.82-Å scatterer; indeed scattering from such a ligand has not been reported previously in EXAFS studies of diiron enzymes. The model for CmlIP as a μ-1,1-peroxo species based on the XAS and rR data presented here is compared with those for CmlIOx and CmlIR in Figure 9.
Figure 9.

Structural models of CmlIR (top left), CmlIOx (top right) and CmlIP (bottom) as determined by EXAFS analysis. Numbers in italics represent the best fit scattering distances in angstroms. Fe1 atoms are colored green, and Fe2 atoms are colored black. The CmlIR structure has a Fe•••Fe distance of 3.35 Å and a calculated Fe– μ-OH–Fe angle of 119°, while CmlIOx has an Fe•••Fe distance of 3.32 Å, and a calculated Fe– μ-O–Fe angle of 130°. CmlIP has an Fe•••Fe distance of 3.35 Å, and calculated Fe–μ-O–Fe and Fe–Operoxo–Fe angles of 133° and 116°, respectively. The 2.82 Å values found for the Fe–Odistal distances require the O–O bond to be tilted out of the Fe–μ-Operoxo–Fe plane.
Support for the Assignment of the 2.8-Å Scatterer from the Structure of Product-Bound AurF
Further support for the assignment of the 2.82-Å scatterer as the terminal oxygen of a μ-1,1-peroxo unit comes from the crystal structure of AurF with product bound (PDB code 3CHT).7 The product of the AurF-catalyzed reaction is para-nitrobenzoic acid (pNBA), which is bound in the active site substrate channel with one of the oxygen atoms of the nitro group oriented between the two Fe centers (Figure 10). If the peroxo intermediate of AurF also adopts the proposed μ-1,1-peroxo binding configuration, it would place the distal peroxo oxygen at approximately the position of one of the O atoms of the nitro group that is formed during the reaction. This juxtaposition should facilitate oxygen atom transfer to the precursor aryl-nitroso intermediate during the reaction. The product-bound AurF crystal structure also suggests a means by which the terminal oxygen of the μ-1,1-peroxo can be rigidly positioned, as reflected in the small σ2 value observed for the 2.82-Å scatterer (Table 2). Three of the monodentate carboxylate ligands of the AurF diiron center (Figure 10, labeled 1 – 3) have their distal oxygen atoms pointing into the substrate pocket within 2.8 to 3.1 Å of the oxygen atom of pNBA. These residues are all conserved in the active site pocket of CmlI. Steric interactions may lock the peroxo moiety in the optimal position for reaction. The constrained orientation of the peroxo ligand may also distort the iron coordination geometry sufficiently to account for the unusually large XAS pre-edge feature of CmlIP.
Figure 10.

Crystal structure of product-bound AurF (PDB code: 3CHT). Distances shown are in angstroms. Panel B is a 90º rotation of panel A. Carboxylate ligands that have oxygen atoms pointed toward the substrate channel are labeled 1 to 3. Carbon atoms from AurF are shown in green, oxygen atoms are in red, nitrogen atoms are in dark blue, iron atoms are represented as brown spheres and the carbon atoms of para-nitrobenzoic acid are shown as gray.
Comparison of CmlIP to Known μ-1,1-Peroxo Complexes
Examples of dinuclear complexes with a μ-1,1-(hydro)peroxo ligand are quite scarce. [CuII2(Let)(OOH)](OTf)(BPh4) (15) (Let = 3,5-bis(1-ethyl-4,7-di-isopropyl-1,4,7-triazacyclononane)pyrazole) is a crystallographically characterized example of a (μ-1,1-hydroperoxo)dicopper complex reported by Meyer and co-workers.98 This complex has a nearly planar Cu–O2–Cu core with a metal-metal distance of 3.53 Å, which is ~0.2 Å longer than that in CmlIP due to the pyrazolate bridge in 15. Interestingly, both Cu atoms are approximately 2.95 Å from the Odistal-atom, which is longer than the 2.82 Å distance found for CmlIP; however this distance can be shortened by tilting the O–O bond out of the Cu–Oproximal–Cu plane, as we propose in the case of CmlIP. The rR spectrum of 15 shows a higher ν(O–O) frequency than what is observed for CmlIP (860 cm−1 vs 789 cm−1),98 but the authors mention that there is significant mechanical coupling between the ν (O–O) and ν (Cu–O), which makes direct comparison to the ν (O–O) of CmlIP challenging.
Among the recently reported crystallographic results for T4MO is the crystal structure of the unreactive O2 adduct of the Q228A variant of reduced T4MOH in complex with its effector protein D.35 This structure shows a diiron center with an Fe•••Fe distance of 3.3 Å and μ-1,1-dioxygen bridge, the 1.5-Å O•••O distance of which is indicative of a peroxide ligand. The observed Fe–Odistal distances of 3.2 – 3.7 Å are quite a bit longer than those found for CmlIP, suggesting that the (μ-1,1-peroxo)diiron(III) unit in this T4MO adduct is much closer to a plane than that found in CmlIP. As the distal O-atom of the μ-1,1-peroxo ligand in the Q228A T4MOH adduct occupies the same space as C-3 of the toluene substrate, it seems unlikely to be a catalytically viable intermediate in the T4MOHD cycle.
Structural Effects of Protonation or Hydrogen Bonding in the CmlIP Active Site
The rR analysis of CmlIP and CmlIOx demonstrate that the ν(Fe–O–Fe) vibrations are affected by substitution with D2O. The only other diiron protein for which D2O isotope shifts have been reported is oxyHr. Sanders-Loehr and co-workers showed that the isotope shift from D2O in oxyHr was the result of hydrogen bonding interactions between the end-on hydroperoxo ligand and the μ-oxo bridge.87 Along similar lines, the D2O isotope shift in CmlI presumably results from hydrogen bonding as well. However for CmlIP, the hydrogen bonding interaction cannot arise from a hydroperoxo ligand, as the ν (O–O) is not affected by D2O substitution, unlike in the case of oxyHr, where the ν (O–O) of the OOH ligand is in fact upshifted upon deuterium substitution.88 Protonation of the μ-oxo ligand can also be ruled out based on precedent from synthetic complexes. Direct protonation of an oxo bridge substantially lengthens the Fe-μ-O bond and decreases the νs(Fe–O–Fe) frequency. This effect is demonstrated in the case of the (μ-oxo)(μ-1,2-peroxo)diferric complex 1. When 1 is protonated to its μ-hydroxo conjugate acid, the Fe-μ-O bond lengthens by 0.1 Å from 1.81 to 1.91 Å, and the νs(Fe–O–Fe) vibration decreases by ~100 cm−1, from 523 cm−1 in 1 to 424 cm−1.60 Neither of these changes is observed in the comparison of CmlIP with CmlIOx, so the oxo bridge does not become protonated.
One possible source of hydrogen bonding interactions with the Fe–O–Fe unit is a protonated protein ligand. E236 in the crystal structure of CmlI is able to move to the proposed position of the μ-oxo ligand, so if protonated, E236 could feasibly interact with the Fe–O–Fe unit in both CmlIOx and CmlIP.39 However, at pH = 9 for the CmlI samples, it is not likely for the carboxylate residues to be protonated. A likely alternative is that water hydrogen bonds to the oxo bridge, accounting for its observed rR shift and the lower νs(Fe–O–Fe) relative to values for metHr-X and related synthetic complexes (Table 3). This hydrogen bond would also serve to distribute the formal -2 charge on the CmlIP cluster, which is the largest known among biological diiron cluster complexes.
Disparity Between XAS and XRD of the CmlI Peroxo Intermediate
The structural model for CmlIP that has been developed here differs substantially from that of the peroxo intermediate seen in the XRD structure of CmlIΔ33, revealing a (cis-μ-1,2-peroxo)diiron center without an oxo bridge.39 Similarly, the structural details of the CmlIR state determined here differ in several aspects from what is found in the crystal structure of CmlIΔ33R. Differences may arise from the inherent differences in crystallized versus solution state intermediates and/or from the different pH values at which the samples were prepared, pH 6.3 for the crystallization and pH 9 for our spectroscopic studies. It is likely that the structures derived from XAS and rR data more accurately reflect the active enzyme, because these samples are made directly from solution. In contrast, the peroxo intermediate found in the crystal is thought to be generated by diffusion into the crystal of H2O2 formed as PEG in the mother liquor breaks down.39 Similarly, the reduced state in the crystal is generated by diffusion of a reductant into the pre-formed crystal. In each case, the active site structure in the crystal forces an amino acid side chain into the substrate binding channel, so the enzyme is unlikely to be active. The same inactive conformation is seen in the crystal structure for the dimanganese form of AurF,99 but not the native diiron form, which exhibits an open substrate binding channel.7 AurF also does not spontaneously form a cis-μ-1,2-peroxo intermediate in the crystal of the resting diferric enzyme.
Comparison with Proposed Structure of AurFP
Recently, a model for AurFP has been advanced based on NRVS (nuclear resonance vibrational spectroscopy) and associated computational studies.100 This model of AurFP contains a protonated cis-μ-1,2-peroxo bridge. Like the (μ-oxo)(μ-1,1-peroxo)diferric species proposed for the homologous CmlIP, the protonated cis-μ-1,2-peroxo bridging structure proposed for AurFP is without synthetic precedent, so the interpretation of the NRVS data has been facilitated by computational simulation. The rR spectra presented here for CmlIP show no evidence for protonation of the peroxo unit. The protonation state of the bound peroxo unit can be best ascertained by the observation of a 2H-isotope shift in the ν(Fe–OOH) mode, as seen for oxyHr87 and two synthetic Fe–OOH complexes,101–102 but this mode has not been identified in the rR spectrum of CmlIP. In the computational study of AurFP, the authors tested a (μ-1,1-peroxo)diferric model and found that it could not reproduce the experimental NRVS spectra. However, the authors did not test a (μ-oxo)(μ-1,1-peroxo)diferric model similar to the model proposed here for CmlIP, evidence for the oxo bridge having just been obtained by XAS and rR in the present study. In fact, in all of the models computed in the AurFP study, the carboxylate equivalent to E236 of CmlI acts as a bidentate bridge, as found in the crystal structure of dimanganese(II) AurF.7 This differs from the reported X-ray crystal structures for diiron(III) AurF7 and, as related above, is unlikely to be active because it forces the substrate binding channel to close. It is interesting to note that the AurFP study finds that the protonated cis-μ-1,2-peroxo intermediate is unlikely to undergo O–O bond cleavage, as the frontier molecular orbitals are unfavorably oriented for a reaction with the pNBA substrate. Indeed, reorganization to a (μ-1,1-peroxo)diferric species is predicted to dramatically lower the energy barrier of the native reaction, relative to the protonated cis-μ-1,2-peroxo model. Thus, a peroxo-diferric species with a structure similar to what we propose for CmlIP is also likely to be implicated in the N-oxygenation reaction of the AurF system.
Possible Mechanism for O–O Bond Cleavage in CmlIP
In light of the updated structure of CmlIP as well as mechanistic studies,15 we can better assess how CmlIP carries out reactions with the native substrate. During the course of the native reaction, CmlIP is capable of oxidizing the more electron-rich aryl-amine substrate (Figure 1, left), as well as a less electron-rich aryl-nitroso substrate later in the reaction pathway.15 These mechanistic traits require CmlIP to be ambiphilic, being able to switch from being an electrophilic oxidant in the oxidation of the aryl-amine to acting as a nucleophilic oxidant in the oxidation of the aryl-nitroso substrate. The available kinetic data suggest a direct reaction between substrate and CmlIP without an intervening a high-valent intermediate typically encountered in diiron oxygenases that activate C–H bonds. This ambiphilic reactivity is unprecedented for a peroxo intermediate, but does not extend to the activation of C–H bonds.8, 15 There are two major differences between the proposed CmlIP structure and the canonical cis-μ-1,2 peroxo species. In the latter, both oxygen atoms are tethered to an iron atom, thereby decreasing its accessibility to substrate as well as limiting its reactivity. Subsequent protonation is a common strategy for enhancing its reactivity, by conversion of the cis-μ-1,2 peroxo species to a high-valent oxidant. On the other hand, the postulated terminally bound and unprotonated peroxo unit of CmlIP is more versatile and can react directly with both its amino- and nitroso-substituted substrates
As indicated in Figure 11, the placement of an unprotonated terminal oxygen of a peroxo intermediate immediately next to the position for oxygen transfer to the substrate would allow either nucleophilic or electrophilic reactions. In the first step of the native reaction (Figure 11, A), the substrate amine group would likely be protonated under the reaction conditions and provide the proton required to break the O–O bond in the hydroxylation of the amine function, thereby linking O–O bond cleavage with substrate hydroxylation. In the later step of the biosynthetic pathway, the nitroso substrate would be susceptible to direct nucleophilic attack by the peroxo ligand (Figure 11, B), resulting in heterolytic cleavage of the peroxo bond to generate the chloramphenicol product and a diferric cluster.
Figure 11.
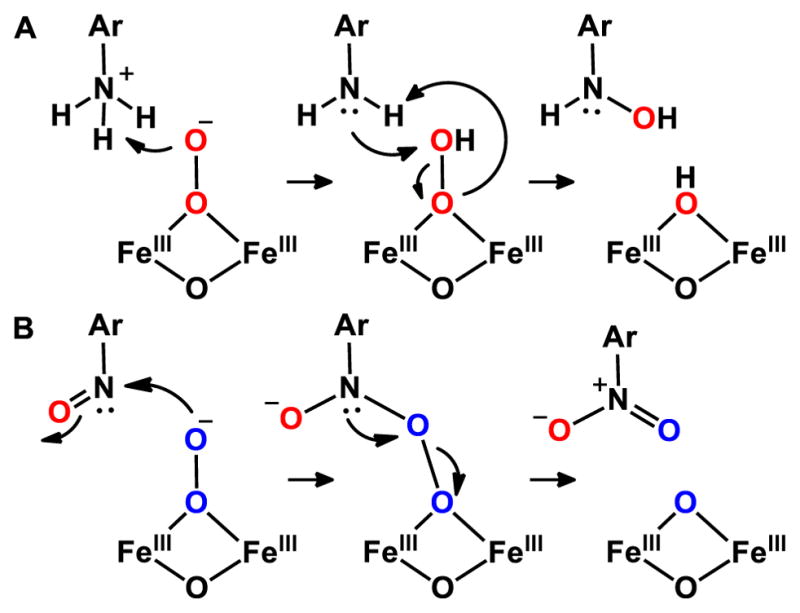
Possible mechanisms for electrophilic (A) and nucleophilic (B) oxidation by CmlIP along the native reaction pathway.
CONCLUSION
Enzymatic peroxo-diferric intermediates have been known for several decades, but every structurally characterized example, until very recently, has contained a μ-1,2-peroxo binding mode.103 CmlIP is proposed here to have a μ-oxo bridge as well as a μ-1,1-peroxo ligand that interacts with protein-derived ligands in the CmlI active site. It is the only example of a μ-1,1-peroxo species in diiron chemistry. Despite having the same 4α-helix fold and a similar active site as C–H activating enzymes like sMMOH, CmlI is unable to facilitate C–H bond oxidation. It seems likely that the novel peroxo structure increases its reactivity over the canonical peroxo intermediate. This boost in oxidizing potency then allows it to carry out both electrophilic and nucleophilic N-oxygenations without creating a species that can also carry out adventitious C–H bond activation reactions.
Supplementary Material
Acknowledgments
The authors acknowledge financial support of this work from grants NIH GM118030 (to J.D.L.), NIH GM38767 (to L.Q.) and graduate traineeship NIH GM08700 (to A.J.K.). XAS data were collected on Beamlines 7-3 and 9-3 at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory. SLAC is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. Use of Beamlines 7-3 and 9-3 is supported by the DOE Office of Biological and Environmental Research and the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). We thank Brent Rivard for assistance with protein growth and helpful discussions.
ABBREVIATIONS USED
- AurF
arylamine oxygenase of the aureothin biosynthesis pathway
- cADO
cyanobacterial aldehyde deformylating oxygenase
- CmlA
β-hydroxylase of the chloramphenicol biosynthetic pathway
- CmlI
arylamine oxygenase of the chloramphenicol biosynthetic pathway
- CmlIΔ33
33-amino-acid-truncated form of CmlI
- CmlIOx
as-isolated diferric CmlI
- CmlIOx-D2O
as-isolated diferric CmlI in D2O buffer
- CmlIP
peroxo-diferric intermediate of CmlI
- CmlIP-D2O
peroxo-diferric intermediate of CmlI prepared in D2O buffer
- CmlIR
chemically reduced CmlI with a diferrous cluster
- Δ9D
stearoyl-CoA Δ9-desaturase
- EXAFS
extended X-ray absorption fine structure
- FT
Fourier transform
- hDOHH
human deoxyhypusine hydroxylase
- LN2
liquid nitrogen
- sMMOH
the hydroxylase subunit of the soluble form of methane monooxygenase
- oxyHr
the oxygenated form of hemerythrin
- pNBA
p-nitrobenzoic acid
- RNR R2
the diiron-cluster-containing subunit of ribonucleotide reductase
- T4MO
toluene-4-monooxygenase
- ToMO
toluene/o-xylene monooxygenase
- rR
resonance Raman spectroscopy
- XAS
X-ray absorption spectroscopy
- XANES
X-ray absorption near-edge structure
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: XANES and EXAFS analysis with fit parameters and full resonance Raman spectra for CmlIR, CmlIOx, and CmlIP
References
- 1.Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 2.Pikus JD, Studts JM, Achim C, Kauffmann KE, Münck E, Steffan RJ, McClay K, Fox BG. Biochemistry. 1996;35:9106–9119. doi: 10.1021/bi960456m. [DOI] [PubMed] [Google Scholar]
- 3.Bochevarov AD, Li J, Song WJ, Friesner RA, Lippard SJ. J Am Chem Soc. 2011;133:7384–7397. doi: 10.1021/ja110287y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fox BG, Lyle KS, Rogge CE. Acc Chem Res. 2004;37:421–429. doi: 10.1021/ar030186h. [DOI] [PubMed] [Google Scholar]
- 5.Krebs C, Bollinger JM, Jr, Booker SJ. Curr Opin Chem Biol. 2011;15:291–303. doi: 10.1016/j.cbpa.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das D, Eser BE, Han J, Sciore A, Marsh ENG. Angew Chem Int Ed. 2011;50:7148–7152. doi: 10.1002/anie.201101552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. Proc Natl Acad Sci USA. 2008;105:6858–6863. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makris TM, Vu VV, Meier KK, Komor AJ, Rivard BS, Münck E, Que L, Jr, Lipscomb JD. J Am Chem Soc. 2015;137:1608–1617. doi: 10.1021/ja511649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu HG, Chanco E, Zhao HM. Tetrahedron. 2012;68:7651–7654. doi: 10.1016/j.tet.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makris TM, Chakrabarti M, Münck E, Lipscomb JD. Proc Natl Acad Sci USA. 2010;107:15391–15396. doi: 10.1073/pnas.1007953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 12.Walsh CT, Chen H, Keating TA, Hubbard BK, Losey HC, Luo L, Marshall CG, Miller DA, Patel HM. Curr Opin Chem Biol. 2001;5:525–534. doi: 10.1016/s1367-5931(00)00235-0. [DOI] [PubMed] [Google Scholar]
- 13.Pacholec M, Sello JK, Walsh CT, Thomas MG. Org Biomol Chem. 2007;5:1692–1694. doi: 10.1039/b703356g. [DOI] [PubMed] [Google Scholar]
- 14.Piraee M, White RL, Vining LC. Microbiology. 2004;150:85–94. doi: 10.1099/mic.0.26319-0. [DOI] [PubMed] [Google Scholar]
- 15.Komor AJ, Rivard BS, Fan R, Guo Y, Que L, Jr, Lipscomb JD. J Am Chem Soc. 2016;138:7411–7421. doi: 10.1021/jacs.6b03341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sturgeon BE, Burdi D, Chen S, Huynh BH, Edmondson DE, Stubbe J, Hoffman BM. J Am Chem Soc. 1996;118:7551–7557. [Google Scholar]
- 17.Doan PE, Shanmugam M, Stubbe J, Hoffman BM. J Am Chem Soc. 2015;137:15558–15566. doi: 10.1021/jacs.5b10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dassama LMK, Silakov A, Krest CM, Calixto JC, Krebs C, Bollinger JM, Jr, Green MT. J Am Chem Soc. 2013;135:16758–16761. doi: 10.1021/ja407438p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SK, Lipscomb JD. Biochemistry. 1999;38:4423–4432. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- 20.Liu KE, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J Am Chem Soc. 1994;116:7465–7466. [Google Scholar]
- 21.Han WG, Noodleman L. Inorg Chem. 2008;47:2975–86. doi: 10.1021/ic701194b. [DOI] [PubMed] [Google Scholar]
- 22.Skulan AJ, Brunold TC, Baldwin J, Saleh L, Bollinger JM, Jr, Solomon EI. J Am Chem Soc. 2004;126:8842–8855. doi: 10.1021/ja049106a. [DOI] [PubMed] [Google Scholar]
- 23.Bollinger JM, Jr, Krebs C, Vicol A, Chen S, Ley BA, Edmondson DE, Huynh BH. J Am Chem Soc. 1998;120:1094–1095. [Google Scholar]
- 24.Moenne-Loccoz P, Baldwin J, Ley BA, Loehr TM, Bollinger JM., Jr Biochemistry. 1998;37:14659–14663. doi: 10.1021/bi981838q. [DOI] [PubMed] [Google Scholar]
- 25.Broadwater JA, Achim C, Münck E, Fox BG. Biochemistry. 1999;38:12197–12204. doi: 10.1021/bi9914199. [DOI] [PubMed] [Google Scholar]
- 26.Beauvais LG, Lippard SJ. J Am Chem Soc. 2005;127:7370–7378. doi: 10.1021/ja050865i. [DOI] [PubMed] [Google Scholar]
- 27.Tinberg CE, Lippard SJ. Biochemistry. 2010;49:7902–7912. doi: 10.1021/bi1009375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ookubo T, Sugimoto H, Nagayama T, Masuda H, Sato T, Tanaka K, Maeda Y, Okawa H, Hayashi Y, Uehara A, Suzuki M. J Am Chem Soc. 1996;118:701–702. [Google Scholar]
- 29.Menage S, Brennan BA, Juarez-Garcia C, Münck E, Que L., Jr J Am Chem Soc. 1990;112:6423–6425. [Google Scholar]
- 30.Dong Y, Yan S, Young VG, Jr, Que L., Jr Angew Chem Int Ed. 1996;35:618–620. [Google Scholar]
- 31.Kim K, Lippard SJ. J Am Chem Soc. 1996;118:4914–4915. [Google Scholar]
- 32.Zhang X, Furutachi H, Fujinami S, Nagatomo S, Maeda Y, Watanabe Y, Kitagawa T, Suzuki M. J Am Chem Soc. 2005;127:826–827. doi: 10.1021/ja045594a. [DOI] [PubMed] [Google Scholar]
- 33.Vu VV, Emerson JP, Martinho M, Kim YS, Münck E, Park MH, Que L., Jr Proc Natl Acad Sci USA. 2009;106:14814–14819. doi: 10.1073/pnas.0904553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pandelia ME, Li N, Noergaard H, Warui DM, Rajakovich LJ, Chang W-c, Booker SJ, Krebs C, Bollinger JM., Jr J Am Chem Soc. 2013;135:15801–15812. doi: 10.1021/ja405047b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Acheson JF, Bailey LJ, Brunold TC, Fox BG. Nature. 2017;544:191–195. doi: 10.1038/nature21681. [DOI] [PubMed] [Google Scholar]
- 36.Murray LJ, Naik SG, Ortillo DO, Garcia-Serres R, Lee JK, Huynh BH, Lippard SJ. J Am Chem Soc. 2007;129:14500–14510. doi: 10.1021/ja076121h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song WJ, Behan RK, Naik SG, Huynh BH, Lippard SJ. J Am Chem Soc. 2009;131:6074–6075. doi: 10.1021/ja9011782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han Z, Sakai N, Boettger LH, Klinke S, Hauber J, Trautwein AX, Hilgenfeld R. Structure. 2015;23:882–892. doi: 10.1016/j.str.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 39.Knoot CJ, Kovaleva EG, Lipscomb JD. J Biol Inorg Chem. 2016;21:589–603. doi: 10.1007/s00775-016-1363-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bailey LJ, Fox BG. Biochemistry. 2009;48:8932–8939. doi: 10.1021/bi901150a. [DOI] [PubMed] [Google Scholar]
- 41.Sousa CM, Carpentier P, Matias PM, Testa F, Pinho F, Sarti P, Giuffrè A, Bandeiras TM, Romão CV. Acta Crystallogr D Biol Crystallogr. 2015;71:2236–2247. doi: 10.1107/S1399004715015825. [DOI] [PubMed] [Google Scholar]
- 42.Gudmundsson M, Kim S, Wu M, Ishida T, Momeni MH, Vaaje-Kolstad G, Lundberg D, Royant A, Ståhlberg J, Eijsink VGH, Beckham GT, Sandgren M. J Biol Chem. 2014;289:18782–18792. doi: 10.1074/jbc.M114.563494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sigfridsson KGV, Chernev P, Leidel N, Popovic-Bijelic A, Gräslund A, Haumann M. J Biol Chem. 2013;288:9648–9661. doi: 10.1074/jbc.M112.438796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersson KK, Froland WA, Lee SK, Lipscomb JD. New J Chem. 1991;15:411–415. [Google Scholar]
- 45.Korboukh VK, Li N, Barr EW, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2009;131:13608–13609. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chufan EE, Puiu SC, Karlin KD. Acc Chem Res. 2007;40:563–572. doi: 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]
- 47.Naruta Y, Sasaki T, Tani F, Tachi Y, Kawato N, Nakamura N. J Inorg Biochem. 2001;83:239–246. doi: 10.1016/s0162-0134(00)00170-7. [DOI] [PubMed] [Google Scholar]
- 48.Li F, Van HKM, Meier KK, Münck E, Que L., Jr J Am Chem Soc. 2013;135:10198–10201. doi: 10.1021/ja402645y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee YM, Bang S, Kim YM, Cho J, Hong S, Nomura T, Ogura T, Troeppner O, Ivanovic-Burmazovic I, Sarangi R, Fukuzumi S, Nam W. Chem Sci. 2013;4:3917–3923. doi: 10.1039/C3SC51864G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.George GN. A suite of computer programs for analysis of X-ray absorption spectra. 1990 [Google Scholar]
- 51.Ankudinov AL, Ravel B, Rehr JJ, Conradson SD. Phys Rev B. 1998;58:7565–7576. [Google Scholar]
- 52.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1997;119:6297–6314. [Google Scholar]
- 53.Wojdyr M. J Appl Crystallogr. 2010;43:1126–1128. [Google Scholar]
- 54.Menges F. Spekwin32 - optical spectroscopy software, 1.72.0. 2015. [Google Scholar]
- 55.Griese JJ, Kositzki R, Schrapers P, Branca RMM, Nordström A, Lehtiö J, Haumann M, Högbom M. J Biol Chem. 2015;290:25254–25272. doi: 10.1074/jbc.M115.675223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hwang J, Krebs C, Huynh BH, Edmondson DE, Theil EC, Penner-Hahn JE. Science. 2000;287:122–125. doi: 10.1126/science.287.5450.122. [DOI] [PubMed] [Google Scholar]
- 57.Jasniewski AJ, Engstrom LM, Vu VV, Park MH, Que L. J Biol Inorg Chem. 2016;21:605–618. doi: 10.1007/s00775-016-1373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jasniewski AJ, Knoot CJ, Lipscomb JD, Que L., Jr Biochemistry. 2016;55:5818–5831. doi: 10.1021/acs.biochem.6b00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vu VV, Makris TM, Lipscomb JD, Que L., Jr J Am Chem Soc. 2011;133:6938–6941. doi: 10.1021/ja201822v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cranswick MA, Meier KK, Shan X, Stubna A, Kaizer J, Mehn MP, Münck E, Que L., Jr Inorg Chem. 2012;51:10417–10426. doi: 10.1021/ic301642w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pap JS, Cranswick MA, Balogh-Hergovich E, Barath G, Giorgi M, Rohde GT, Kaizer J, Speier G, Que L., Jr Eur J Inorg Chem. 2013;2013:3858–3866. doi: 10.1002/ejic.201300162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fiedler AT, Shan X, Mehn MP, Kaizer J, Torelli S, Frisch JR, Kodera M, Que L., Jr J Phys Chem A. 2008;112:13037–13044. doi: 10.1021/jp8038225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frisch JR, Vu VV, Martinho M, Münck E, Que L., Jr Inorg Chem. 2009;48:8325–8336. doi: 10.1021/ic900961k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Groot F. Chem Rev. 2001;101:1779–1808. doi: 10.1021/cr9900681. [DOI] [PubMed] [Google Scholar]
- 65.Sarangi R. Coord Chem Rev. 2013;257:459–472. doi: 10.1016/j.ccr.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Randall CR, Shu L, Chiou YM, Hagen KS, Ito M, Kitajima N, Lachicotte RJ, Zang Y, Que L., Jr Inorg Chem. 1995;34:1036–1039. [Google Scholar]
- 67.Roe AL, Schneider DJ, Mayer RJ, Pyrz JW, Widom J, Que L., Jr J Am Chem Soc. 1984;106:1676–1681. [Google Scholar]
- 68.Rudd DJ, Sazinsky MH, Lippard SJ, Hedman B, Hodgson KO. Inorg Chem. 2005;44:4546–4554. doi: 10.1021/ic048794w. [DOI] [PubMed] [Google Scholar]
- 69.Shu L, Broadwater JA, Achim C, Fox BG, Münck E, Que L., Jr J Biol Inorg Chem. 1998;3:392–400. [Google Scholar]
- 70.Makris TM, Knoot CJ, Wilmot CM, Lipscomb JD. Biochemistry. 2013;52:6662–6671. doi: 10.1021/bi400845b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Whittington DA, Lippard SJ. J Am Chem Soc. 2001;123:827–838. doi: 10.1021/ja003240n. [DOI] [PubMed] [Google Scholar]
- 72.Elango N, Radhakrishnan R, Froland WA, Wallar BJ, Earhart CA, Lipscomb JD, Ohlendorf DH. Protein Sci. 1997;6:556–568. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Logan DT, Su XD, Aberg A, Regnstrom K, Hajdu J, Eklund H, Nordlund P. Structure. 1996;4:1053–1064. doi: 10.1016/s0969-2126(96)00112-8. [DOI] [PubMed] [Google Scholar]
- 74.Eriksson M, Jordan A, Eklund H. Biochemistry. 1998;37:13359–13369. doi: 10.1021/bi981380s. [DOI] [PubMed] [Google Scholar]
- 75.Lindqvist Y, Huang WJ, Schneider G, Shanklin J. EMBO. 1996;15:4081–4092. [PMC free article] [PubMed] [Google Scholar]
- 76.Holmes MA, Le Trong I, Turley S, Sieker LC, Stenkamp RE. J Mol Biol. 1991;218:583–593. doi: 10.1016/0022-2836(91)90703-9. [DOI] [PubMed] [Google Scholar]
- 77.Holmes MA, Stenkamp RE. J Mol Biol. 1991;220:723–737. doi: 10.1016/0022-2836(91)90113-k. [DOI] [PubMed] [Google Scholar]
- 78.Korendovych IV, Kryatov SV, Reiff WM, Rybak-Akimova EV. Inorg Chem. 2005;44:8656–8658. doi: 10.1021/ic051739i. [DOI] [PubMed] [Google Scholar]
- 79.Kryatov SV, Taktak S, Korendovych IV, Rybak-Akimova EV, Kaizer J, Torelli S, Shan X, Mandal S, MacMurdo VL, Mairata i Payeras A, Que L., Jr Inorg Chem. 2005;44:85–99. doi: 10.1021/ic0485312. [DOI] [PubMed] [Google Scholar]
- 80.Chaudhury P, Wieghardt K, Nuber B, Weiss J. Angew Chem Int Ed. 1985;24:778–779. [Google Scholar]
- 81.True AE, Scarrow RC, Randall CR, Holz RC, Que L., Jr J Am Chem Soc. 1993;115:4246–4255. [Google Scholar]
- 82.Sanders-Loehr J, Wheeler WD, Shiemke AK, Averill BA, Loehr TM. J Am Chem Soc. 1989;111:8084–8093. [Google Scholar]
- 83.Dong Y, Zang Y, Kauffmann K, Shu L, Wilkinson EC, Münck E, Que L., Jr J Am Chem Soc. 1997;119:12683–12684. [Google Scholar]
- 84.Wilkinson EC, Dong YH, Zang Y, Fujii H, Fraczkiewicz R, Fraczkiewicz G, Czernuszewicz RS, Que L., Jr J Am Chem Soc. 1998;120:955–962. [Google Scholar]
- 85.Zheng H, Zang Y, Dong Y, Young VG, Jr, Que L., Jr J Am Chem Soc. 1999;121:2226–2235. [Google Scholar]
- 86.Xue G, Wang D, De Hont R, Fiedler AT, Shan X, Münck E, Que L., Jr Proc Natl Acad Sci USA. 2007;104:20713–20718. doi: 10.1073/pnas.0708516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shiemke AK, Loehr TM, Sanders-Loehr J. J Am Chem Soc. 1986;108:2437–2443. doi: 10.1021/ja00269a050. [DOI] [PubMed] [Google Scholar]
- 88.Shiemke AK, Loehr TM, Sanders-Loehr J. J Am Chem Soc. 1984;106:4951–4956. [Google Scholar]
- 89.Norman RE, Yan S, Que L, Backes G, Ling J, Sanders-Loehr J, Zhang JH, O'Connor CJ. J Am Chem Soc. 1990;112:1554–1562. [Google Scholar]
- 90.Armstrong WH, Lippard SJ. J Am Chem Soc. 1984;106:4632–4633. [Google Scholar]
- 91.Xiong J, Phillips RS, Kurtz DM, Jr, Jin S, Ai J, Sanders-Loehr J. Biochemistry. 2000;39:8526–8536. doi: 10.1021/bi9929397. [DOI] [PubMed] [Google Scholar]
- 92.Brunold TC, Solomon EI. J Am Chem Soc. 1999;121:8288–8295. [Google Scholar]
- 93.Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Biochemistry. 1998;37:14664–14671. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- 94.Moenne-Loccoz P, Krebs C, Herlihy K, Edmondson DE, Theil EC, Huynh BH, Loehr T. Biochemistry. 1999;38:5290–5295. doi: 10.1021/bi990095l. [DOI] [PubMed] [Google Scholar]
- 95.Zhang X, Furutachi H, Fujinami S, Nagatomo S, Maeda Y, Watanabe Y, Kitagawa T, Suzuki M. J Am Chem Soc. 2005;127:826–827. doi: 10.1021/ja045594a. [DOI] [PubMed] [Google Scholar]
- 96.Chishiro T, Shimazaki Y, Tani F, Tachi Y, Naruta Y, Karasawa S, Hayami S, Maeda Y. Angew Chem Int Ed. 2003;42:2788–2791. doi: 10.1002/anie.200351415. [DOI] [PubMed] [Google Scholar]
- 97.Gavrilova AL, Qin CJ, Sommer RD, Rheingold AL, Bosnich B. J Am Chem Soc. 2002;124:1714–1722. doi: 10.1021/ja012386z. [DOI] [PubMed] [Google Scholar]
- 98.Kindermann N, Dechert S, Demeshko S, Meyer F. J Am Chem Soc. 2015;137:8002–8005. doi: 10.1021/jacs.5b04361. [DOI] [PubMed] [Google Scholar]
- 99.Zocher G, Winkler R, Hertweck C, Schulz GE. J Mol Biol. 2007;373:65–74. doi: 10.1016/j.jmb.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 100.Park K, Li N, Kwak Y, Srnec M, Bell CB, Liu LV, Wong SD, Yoda Y, Kitao S, Seto M, Hu M, Zhao J, Krebs C, Bollinger JM, Jr, Solomon EI. J Am Chem Soc. 2017;139:7062–7070. doi: 10.1021/jacs.7b02997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ho RYN, Roelfes G, Feringa BL, Que L., Jr J Am Chem Soc. 1999;121:264–265. [Google Scholar]
- 102.Wada A, Ogo S, Nagatomo S, Kitagawa T, Watanabe Y, Jitsukawa K, Masuda H. Inorg Chem. 2002;41:616–618. doi: 10.1021/ic001058h. [DOI] [PubMed] [Google Scholar]
- 103.Trehoux A, Mahy J-P, Avenier F. Coord Chem Rev. 2016;322:142–158. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.