

Abstract
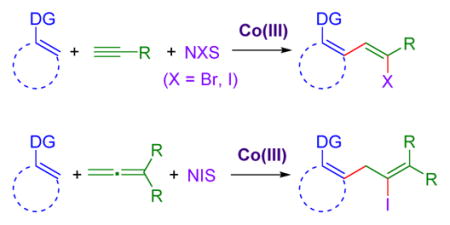
A Co(III)-catalyzed three-component coupling of C(sp2)–H bonds, alkynes, and halogenating agents to give alkenyl halides is reported. This transformation proceeds with high regio- and diastereoselectivity, and is effective for a broad range of aryl and alkyl terminal alkynes. Diverse C–H bond partners also exhibit good reactivity for a range of heteroaryl and aryl systems as well as synthetically useful secondary and tertiary amide, urea, and pyrazole directing groups. This multi-component transformation is also compatible with allenes in place of alkynes to furnish tetra-substituted alkenyl halides, showcasing the first halo-arylation of allenes.
Keywords: C–H activation, homogeneous catalysis, multicomponent reactions, alkynes, allenes
Triple Play
A convergent assembly of functionalized alkenyl halides is reported via Co(III)-catalyzed three-component C–H bond addition across alkynes and halogenating agents. The reaction proceeds with high regio- and diastereoselectivity and is effective for a wide range of alkynes and C–H bond substrates. Allenes are also suitable substrates for the preparation of tetra-substituted alkenyl halide products.
Transition-metal-catalyzed C–H functionalization has become a powerful synthetic approach that relies on simple and readily available starting inputs. In directed C–H bond functionalization, a C–H bond substrate and a transition-metal catalyst typically form a nucleophilic metallocycle intermediate that adds across a single coupling partner to create a functionalized product (Figure 1a).1,2 Three-component C–H functionalization using two different coupling partners theoretically would provide efficient entry to an enormous array of diverse and complex products in a single reaction, especially when one considers the very large number of potential coupling partner combinations (Figure 1b).3
Figure 1.
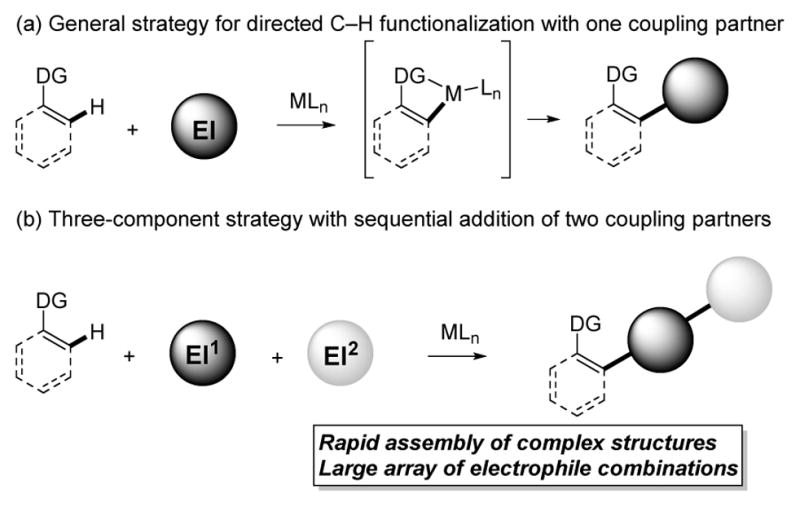
Overview of three-component C–H functionalization approach.
Recently our lab published on the Co(III)-catalyzed three-component diastereoselective synthesis of β-hydroxy ketones, utilizing enones and aldehydes (Figure 2a).3a To expand upon this first example of intermolecular multi-component C–H functionalization,4 we sought to extend this approach to different C–H functionalization substrates and coupling partners. Herein we describe a new Co(III)-catalyzed three-component coupling wherein the C–H bond sequentially adds to terminal alkynes or allenes and halogenating agents to provide functionalized alkenyl halides with high regio- and diastereoselectivity (Figure 2b). Notably, alkenyl halides are versatile synthetic handles for many important transformations.5,6
Figure 2.
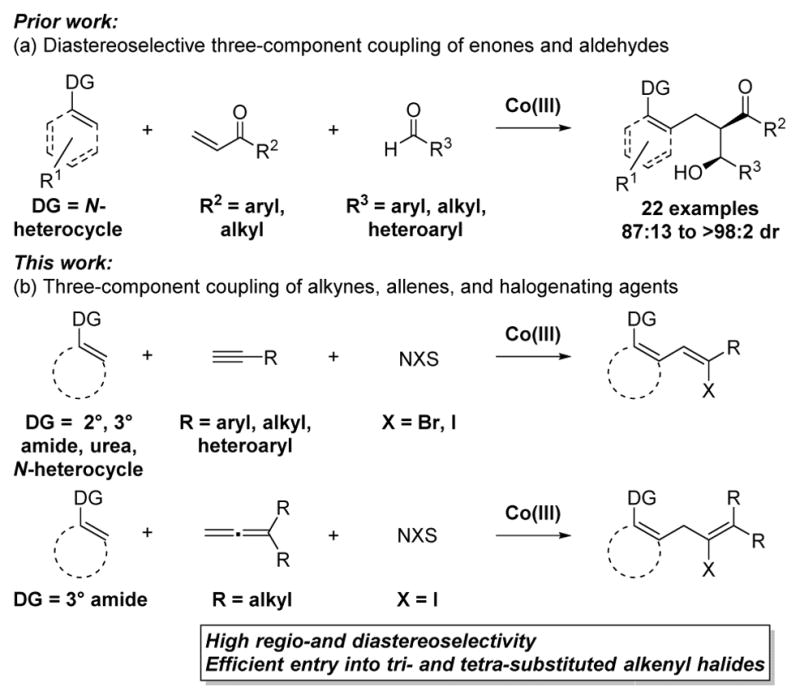
Co(III)-catalyzed three-component transformations.
We began by exploring terminal alkynes7,8,9 and halogenating agents as the two coupling partners. Significantly, neither of these inputs had previously been explored in an intermolecular three-component C–H bond functionalization. Successful coupling required initial addition to the alkyne instead of the more electrophilic halogenating agent.10 More challenging was the identification of suitable conditions for halogenation of the alkenyl-metal intermediate while minimizing well-established and efficient proto-demetallation to give the undesired alkene.7 Additionally, terminal alkynes are susceptible to C–H bond activation, and this mode of activation needed to be avoided to prevent side reactions such as homocoupling or oligomerization.11
A thorough exploration of reaction conditions established that the preformed air-stable Co(III)-catalyst [Cp*Co(MeCN)3][SbF6]2 in conjunction with sparing amounts of acetic acid (2.5 mol %) at 40 °C provided the desired alkenyl iodide 4a in 71% yield from thiophene-amide 1a, phenylacetylene (2a), and N-iodosuccinimide (NIS) (Table 1) (see Table S1 in SI). Direct C–H iodination was not observed, suggesting that under the reaction conditions, alkenylation completely outcompetes iodination.12–14 Acetic acid has previously been found to be an effective additive for facilitating Co(III)-catalyzed C–H bond additions to π-bonds and was crucial to achieving effective alkyne coupling. However, only sub-stoichiometric amounts of acetic acid relative to catalyst loading could be used to minimize undesired proto-demetallation of the alkenyl cobalt intermediate. It is also noteworthy that Rh(III) catalysts, which often show parallel reactivity to Co(III), were ineffective for this transformation.
Table 1.

|
Conditions: 1a (1.0 equiv at 0.1 M), 2 (1.2 equiv), and 3 (1.5 equiv).
Isolated yields.
Using 10 mol % of AcOH.
Using 5 mol % of AcOH.
Once optimal conditions were determined, different halogenating agents and a wide range of terminal alkynes 2 were evaluated (Table 1). In addition to NIS to give 4a, N-bromosuccinimide (NBS) provided alkenyl bromide 4b in good yield (see also 4x in Table 2, vide infra). However, chlorination with N-chlorosuccinimide was unsuccessful.
Table 2.
|
Conditions: 1 (1.0 equiv at 0.1 M), 2 (1.2 equiv), and 3 (1.5 equiv).
Isolated yields.
Using 5 mol % of AcOH.
Using 10 mol % of AcOH.
1 (1.5 equiv), 2 (1.0 equiv), and 3a (1.1 equiv) using 20 mol % of LiOAc at 60 °C.
Next, a range of substituents was examined on the aryl alkyne (Table 1). Electron donating substituents furnished alkene products 4c and 4d in good yields. Alkynes with electron withdrawing chloro and carboxyester groups also coupling efficiently to generate alkenyl iodides 4e and 4f in 68% and 58% yields, respectively. Additionally, a heteroaryl terminal alkyne coupled to give 4g in 56% yield. A broad range of alkyl alkynes were also effective substrates. Linear and β-branched alkyl alkynes gave alkenyl iodides 4h and 4i in high yields, and 4-phenyl-1-butyne led to product 4j in 74% yield. In contrast, α-alkyl branching in ethynylcyclohexane provided alkenyl iodide 4k in lower yield. A variety of heteroatom functionality could be incorporated on the alkyne coupling partner, including a Boc-protected amine (4l), a propargyl ether (4m), a silyl ether (4n) and an ester (4o), all in ≥80% yields. Notably, alkenyl iodides 4 were obtained with >95% regio- and diastereoselectivity for every terminal alkyne examined. However, internal alkynes did not give three-component products with 1a primarily recovered.15
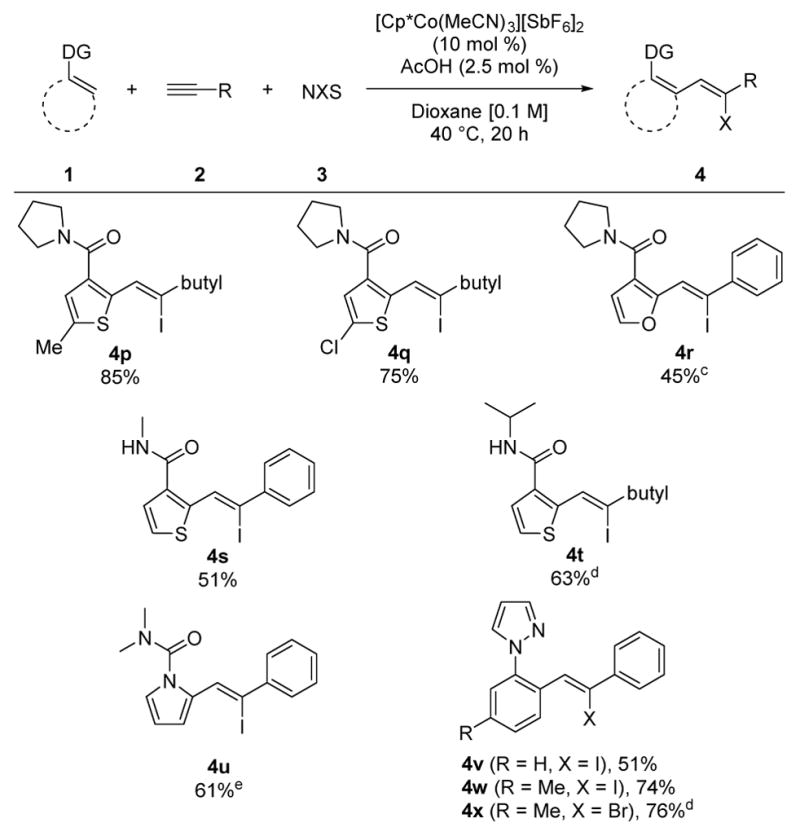
A variety of different C–H bond partners were also effective for coupling with both aryl and alkyl alkynes (Table 2). Thiophenes with alkyl- or halo-substituents at the 5-position underwent three-component coupling to give products 4p and 4q in 85% and 75% yields, respectively. Moreover, the thiophene could be replaced with a furan to provide product 4r in 45% yield. In addition to the tertiary pyrrolidine amide directing group, secondary amides with varying steric character were also effective. For example, N-Me and N-iPr amides furnished products 4s and 4t in good yields. For 4s, the regio- and stereochemistry was rigorously confirmed by X-ray crystallography.16 Furthermore, pyrrole protected as the N, N-dimethyl urea also coupled to give 4u. Finally, aromatic C–H bond functionalization was demonstrated using the pyrazole directing group as exemplified by alkenyl halides 4v – 4x.
A number of experiments were conducted to probe the mechanistic features of this three-component transformation (Schemes 1–2). The potential participation in catalysis of alkene 5a, an undesired two-component coupling byproduct produced in minor amounts under the reaction conditions, was initially investigated. First, alkene product 5a and NIS were subjected to the standard reaction conditions (Scheme 1a). Excellent recovery of alkene 5a was observed and no alkenyl iodide 4a could be detected. Second, substrate 1b, 1-hexyne, and NIS were submitted to the standard reaction conditions with one equiv of 5a as an additive (Scheme 1b). The presence of 5a did not affect the yield of the three component coupling product 4p, and again, 5a was not iodinated. Together, these experiments clearly establish that two-component byproduct 5a is not an intermediate and does not interfere with the catalytic cycle.
Scheme 1.
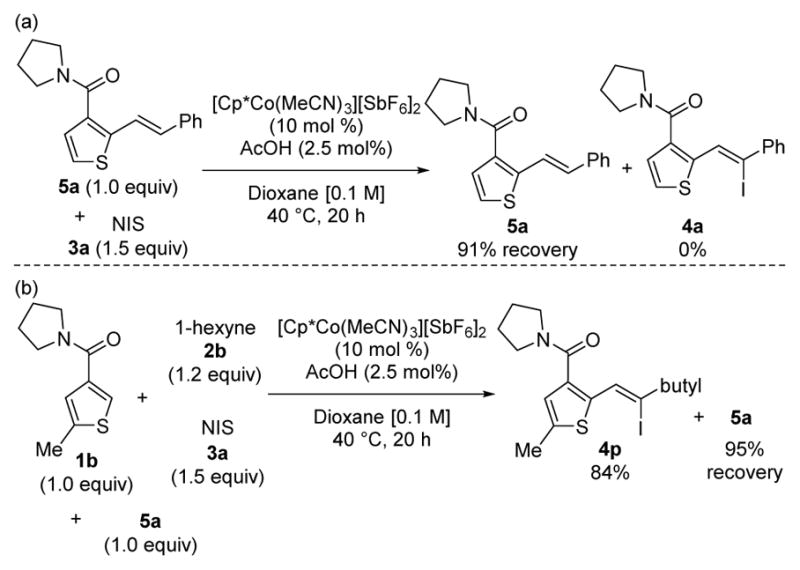
Mechanistic experiments with two-component product 5a
Scheme 2.
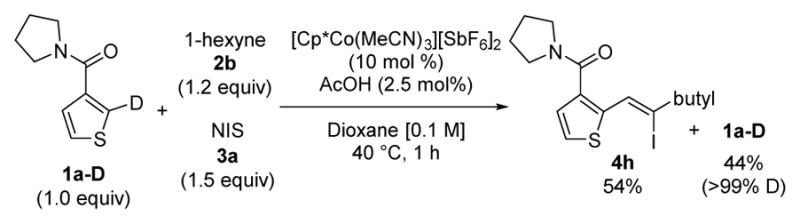
No observed H/D exchange for 1a–D under reaction conditions
Experiments with deuterated substrate 1a–D also defined key features of the reaction (Scheme 2). When deuterated product 1a–D was subjected to the optimal reaction conditions for only 1 h, no deuterium exchange was observed in the recovered starting material. The lack of scrambling at the site of C–H functionalization is unexpected because extensive deuterium exchange by reversible orthometallation is almost always observed for Cp*Co(III)-systems.17 Initial rates were next independently determined for protio 1a and deuterio 1a–D under the standard catalyst and acetic acid loadings (See Section 7 of SI). A primary kinetic isotope effect (KIE) of 2.15 ± 0.16 was observed, which is consistent with orthometallation as a rate-determining step.
A possible mechanism for the reaction consistent with the high regio- and diastereoselectivity and the aforementioned mechanistic studies is depicted in Scheme 3. First, irreversible orthometallation of 1 with the cationic Co(III)-catalyst furnishes cobaltacycle 6. Regioselective syn-insertion of 6 into the terminal alkyne then generates alkenyl-cobalt 7. Finally, intermediate 7 reacts with the halogenating agent 3 to provide the desired three-component product 4 with release of the cationic Co(III)-catalyst. An undesired minor pathway for intermediate 7 is proto-demetallation to give the alkene product 5, also with release of the active catalyst.
Scheme 3.
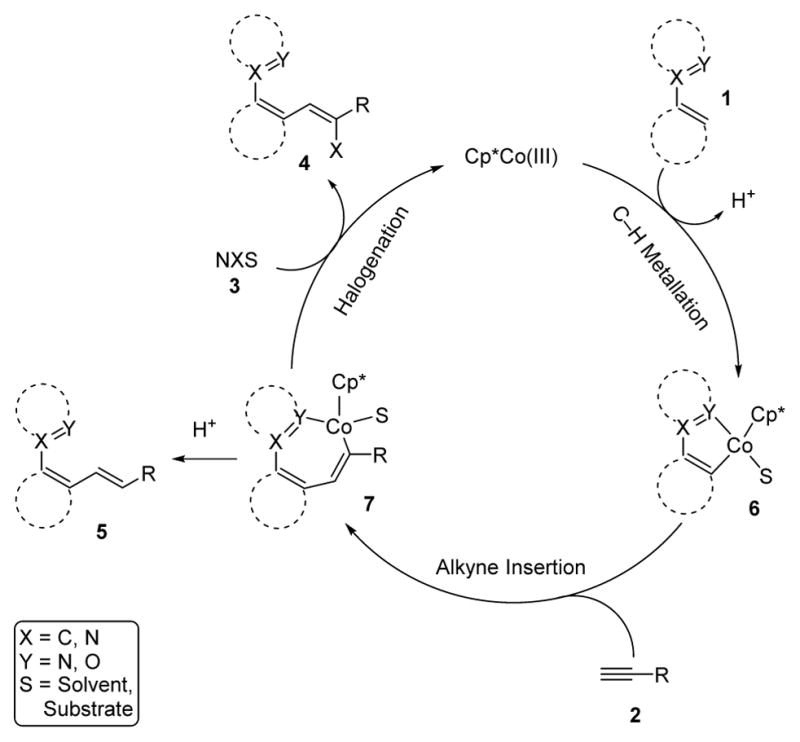
Proposed mechanism for the Co(III)-catalyzed three-component transformation.
Allenes were also investigated as possible three-component coupling partners with NIS to generate tetra-substituted alkenyl iodides (Table 3).18 Few examples of Co(III)-catalyzed C–H bond additions to allenes have been reported,19,20 with the first hydroarylation of allenes appearing only very recently.19 In particular, 1,1-disubstituted allenes 8 were effective three-component coupling electrophiles to give 9a–c in good yields. These allylated thiophene amides nicely complement the alkenylated derivatives obtained with the aforementioned three-component reactions of terminal alkynes. To the best of our knowledge, halo-arylation of allenes has not previously been reported.
Table 3.
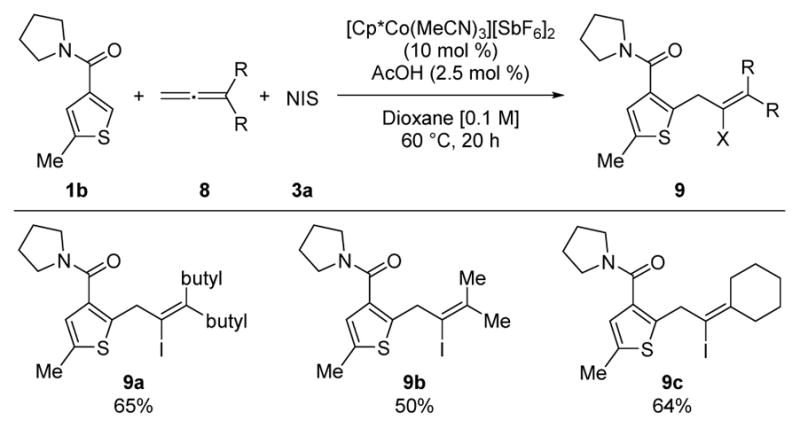
|
Conditions: 1b (1.0 equiv at 0.1 M), 8 (1.2 equiv), and 3a (1.5 equiv).
Isolated yields.
In summary, a novel multi-component transformation has been developed using Co(III) catalysis to form functionalized alkenyl halides with high regio- and stereoselectivity. This coupling proceeds with broad alkyne scope, and is applicable to a variety of C–H bond substrates and directing groups. Additionally, we have showcased that allenes are suitable reaction partners for the synthesis of allylated products incorporating tetra-substituted alkenyl iodides. Significantly, the alkenyl iodide functionality present in products 4 and 9, are well documented to be capable of undergoing a range of metal-catalyzed transformations. We are currently investigating the C–H bond three-component coupling of numerous additional coupling partner combinations for the synthesis of interesting and useful structures in a straightforward and convergent manner.
Supplementary Material
Acknowledgments
This work was supported by the NIH (R35GM122473). We gratefully acknowledge Dr Brandon Mercado (Yale University) for solving the crystal structure of 4s, and Joshua Hummel for helpful input.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.For select relevant reviews on C–H functionalization, see: Hummel JR, Boerth JA, Ellman JA. Chem Rev. doi: 10.1021/acs.chemrev.6b00661. Article ASAP.Gensch T, Hopkinson MN, Glorius F, Wencel-Delord J. Chem Soc Rev. 2016;45:2900. doi: 10.1039/c6cs00075d.Yang L, Huang H. Chem Rev. 2015;115:3468. doi: 10.1021/cr500610p.Wencel-Delord J, Glorius F. Nat Chem. 2013;5:369. doi: 10.1038/nchem.1607.Arockiam PB, Bruneau C, Dixneuf PH. Chem Rev. 2012;112:5879. doi: 10.1021/cr300153j.Li BJ, Shi ZJ. Chem Soc Rev. 2012;41:5588. doi: 10.1039/c2cs35096c.Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem Int Ed. 2012;51:8960. doi: 10.1002/anie.201201666.Angew Chem. 2012;124:9092.
- 2.For recent reviews on Co(III)-catalyzed C–H bond functionalization, see: Yoshino T, Matsunaga S. Adv Synth Catal. 2017;359:1245.Moselage M, Li J, Ackermann L. ACS Catal. 2016;6:498.Wei D, Zhu X, Niu JL, Song MP. ChemCatChem. 2016;8:1242.
- 3.(a) Boerth JA, Hummel JR, Ellman JA. Angew Chem Int Ed. 2016;55:12650. doi: 10.1002/anie.201603831. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2016;128:12840. [Google Scholar]; (b) Boerth JA, Ellman JA. Chem Sci. 2016;7:1474. doi: 10.1039/c5sc04138d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For a three-component approach initiated by Pd-catalyzed C–I oxidative addition followed by C–H functionalization, see: Ye J, Lautens M. Nat Chem. 2015;7:863. doi: 10.1038/nchem.2372.
- 5.For leading references on the synthesis and application of alkenyl halides, see: Petrone DA, Ye J, Lautens M. Chem Rev. 2016;116:8003. doi: 10.1021/acs.chemrev.6b00089.Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417. doi: 10.1021/cr100327p.Evano G, Blanchard N, Toumi M. Chem Rev. 2008;108:3054. doi: 10.1021/cr8002505.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4442. doi: 10.1002/anie.200500368.Angew Chem. 2005;117:4516.
- 6.For examples of transition-metal catalysed syntheses of alkenyl iodides and bromides from alkynes, see: Derosa J, Cantu AL, Boulous MN, O’Duill ML, Turnbull JL, Liu Z, De La Torre DM, Engle KM. J Am Chem Soc. 2017;139:5183. doi: 10.1021/jacs.7b00892.Trost BM, Kalnmals CA. Org Lett. 2017;19:2346. doi: 10.1021/acs.orglett.7b00879.Uehling MR, Rucker RP, Lalic G. J Am Chem Soc. 2014;136:8799. doi: 10.1021/ja503944n.Gao F, Hoveyda AH. J Am Chem Soc. 2010;132:10961. doi: 10.1021/ja104896b.Ye L, Zhang L. Org Lett. 2009;11:3646. doi: 10.1021/ol901346k.Lipshutz BH, Butler T, Lower A. J Am Chem Soc. 2006;128:15396. doi: 10.1021/ja065769b.
- 7.For alkenylation/protonation via Co(III)-catalyzed C–H bond additions to terminal alkynes, see: Wang S, Hou JT, Feng ML, Zhang XZ, Chen SY, Yu XQ. Chem Commun. 2016;52:2709. doi: 10.1039/c5cc09707j.Sen M, Emayavaramban B, Barsu N, Premkumar JR, Sundararaju B. ACS Catal. 2016;6:2792.Tanaka R, Ikemoto H, Kanai M, Yoshino T, Matsunaga S. Org Lett. 2016;18:5732. doi: 10.1021/acs.orglett.6b02997.Ikemoto H, Yoshino T, Sakata K, Matsunaga S, Kanai M. J Am Chem Soc. 2014;136:5424. doi: 10.1021/ja5008432.
- 8.For alkenylation/cyclization via Co(III)-catalyzed C–H bond additions to terminal alkynes, see: Zhou S, Wang J, Wang L, Chen K, Song C, Zhu J. Org Lett. 2016;18:3806. doi: 10.1021/acs.orglett.6b01805.Zhang J, Chen H, Lin C, Liu Z, Wang C, Zhang Y. J Am Chem Soc. 2015;137:12990. doi: 10.1021/jacs.5b07424.Wang H, Koeller J, Liu W, Ackermann L. Chem–Eur J. 2015;21:15525. doi: 10.1002/chem.201503624.Sun B, Yoshino T, Kanai M, Matsunaga S. Angew Chem Int Ed. 2015;54:12968. doi: 10.1002/anie.201507744.Angew Chem. 2015;127:13160.(e) ref 7d.
- 9.For select other examples of metal-catalyzed C–H bond additions to terminal alkynes, see: Lin C, Chen Z, Liu Z, Zhang Y. Org Lett. 2017;19:850. doi: 10.1021/acs.orglett.6b03856.Yang X, Jin X, Wang C. Adv Synth Catal. 2016;358:2436.Jia J, Shi J, Zhou J, Liu X, Song Y, Xu HE, Yi W. Chem Commun. 2015;51:2925. doi: 10.1039/c4cc09823d.Zhou B, Chen H, Wang C. J Am Chem Soc. 2013;135:1264. doi: 10.1021/ja311689k.
- 10.For examples of Co(III)-catalyzed halogenation via C–H activation, see: Pawar AB, Lade DM. Org Biomol Chem. 2016;14:3275. doi: 10.1039/c5ob02640g.Yu DG, Gensch T, de Azambuja F, Vasquez-Cespedes S, Glorius F. J Am Chem Soc. 2014;136:17722. doi: 10.1021/ja511011m.
- 11.Trost BM, Masters JT. Chem Soc Rev. 2016;45:2212. doi: 10.1039/c5cs00892a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For an example of halogenation on a thiophene-3-carboxamide using Rh(III), see: Daniels MH, Armand JR, Tan KL. Org Lett. 2016;18:3310. doi: 10.1021/acs.orglett.6b01205.
- 13.For an example of halogenation on a thiophene-2-carboxamide using Rh(III), see: Schröder N, Lied F, Glorius F. J Am Chem Soc. 2015;137:1448. doi: 10.1021/jacs.5b00283.
- 14.Separate reaction of 1a with alkyne 2a or NIS gave alkenylation or iodination, respectively (see Table S1).
- 15.Bera SS, Debbarma S, Ghosh AK, Chand S, Maji MS. J Org Chem. 2017;82:420. doi: 10.1021/acs.joc.6b02516. [DOI] [PubMed] [Google Scholar]
- 16.Synthetic details, characterization, and molecular structures obtained by X-ray structural analysis are described in the Supporting Information. CCDC 1548918 (4s) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 17.For examples of H/D exchange with Cp*Co(III) catalysts, see: Li J, Zhang Z, Ma W, Tang M, Wang D, Zou LH. Adv Synth Catal. 2017;359:1717.Sauermann N, González MJ, Ackermann L. Org Lett. 2015;17:5316. doi: 10.1021/acs.orglett.5b02678.Hummel JR, Ellman JA. J Am Chem Soc. 2015;137:490. doi: 10.1021/ja5116452.Gensch T, Vásquez-Céspedes S, Yu DG, Glorius F. Org Lett. 2015;17:3714. doi: 10.1021/acs.orglett.5b01701.Yoshino T, Ikemoto H, Matsunaga S, Kanai M. Chem–Eur J. 2013;19:9142. doi: 10.1002/chem.201301505.(f) ref 7e (g) ref 7f (h) ref 7g (i) ref 9a (i) ref 13.
- 18.For stereoselective Co(III)-catalyzed coupling of alkynes to give tetra-substituted alkenes, see; Ikemoto H, Tanaka R, Sakata K, Kanai M, Yoshino T, Matsunaga S. Angew Chem Int Ed. 2017;56:7156. doi: 10.1002/anie.201703193.Angew Chem. 2017;129:7262.
- 19.For Cp*Co(III)-catalyzed hydroarylation of allenes, see: Nakanowatari S, Mei R, Feldt M, Ackermann L. ACS Catal. 2017;7:2511.
- 20.For Co-catalyzed allene additions with stoichiometric oxidants, see: Li T, Zhang C, Tan Y, Pan W, Rao Y. Org Chem Front. 2017;4:204.Boobalan R, Kuppusamy R, Santhoshkumar R, Gandeepan P, Cheng CH. ChemCatChem. 2017;9:273.Kuppusamy R, Muralirajan K, Cheng C-H. ACS Catal. 2016;6:3909.Thrimurtulu N, Dey A, Maiti D, Volla CMR. Angew Chem, Int Ed. 2016;55:12361. doi: 10.1002/anie.201604956.Angew Chem. 2016;128:12549.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.