


Abstract
Vertebrate internal exons are usually between 50 and 400 nt long; exons outside this size range may require additional exonic and/or intronic sequences to be spliced into the mature mRNA. The mouse polymeric immunoglobulin receptor gene has a 654 nt exon that is efficiently spliced into the mRNA. We have examined this exon to identify features that contribute to its efficient splicing despite its large size; a large constitutive exon has not been studied previously. We found that a strong 5′ splice site is necessary for this exon to be spliced intact, but the splice sites alone were not sufficient to efficiently splice a large exon. At least two exonic sequences and one evolutionarily conserved intronic sequence also contribute to recognition of this exon. However, these elements have redundant activities as they could only be detected in conjunction with other mutations that reduced splicing efficiency. Several mutations activated cryptic 5′ splice sites that created smaller exons. Thus, the balance between use of these potential sites and the authentic 5′ splice site must be modulated by sequences that repress or enhance use of these sites, respectively. Also, sequences that enhance cryptic splice site use must be absent from this large exon.
INTRODUCTION
How vertebrate exons are properly recognized from amongst the sequence complexity of a pre-mRNA has been the subject of much attention over the past 20 years. It is clear that multiple parameters can contribute to exon recognition, including the sequence of the splice junctions bordering the exon, internal exon sequences, sequences in the surrounding introns and exon size. Most vertebrate exons are between 50 and 400 nt long, the average being 137 nt; <1% of internal exons are >400 nt (1,2). This observation, along with functional studies of small and large exons, has suggested that a functional exon size limit exists when an exon is surrounded by large (greater than ∼500 nt) introns (see for example 3–6). Interactions among factors bound at the splice sites bordering an exon is an important first step in exon recognition and these interactions are optimal when the exon is at least 50 nt and less than ∼500 nt (2,6,7). Therefore, exons that fall outside these size constraints are more likely to be alternative exons or to have additional features that enhance their recognition by the splice machinery. Indeed, naturally occurring small exons are often alternatively processed such that the exon is included or excluded in different cell types and sequences within the exon and surrounding introns contribute to this regulation (8–11). In several cases, when the splice sites were improved to better match the consensus sequences the small exons became constitutively spliced (4,11,12). Only a few abnormally large internal exons have been examined to date, all of which are alternatively processed; these include the 3500, 1090 and 800 nt exons of the breast cancer 1 (13), caldesmon (14) and neural cell adhesion molecule (NCAM) genes (15), respectively. Improving the 5′ splice site of the NCAM alternative exon resulted in constitutive splicing of this exon (16) and sequences in the upstream exon were shown to contribute to its regulated splicing (17). However, other sequences in and surrounding these or other large exons have not been examined for splice enhancing activity. One might presume that exons >400–500 nt, whether they are alternatively or constitutively spliced, would also require specific cis-acting sequences in order to be efficiently recognized. These sequences may be similar to those found in small or alternatively processed exons. Alternatively, large exons may require multiple sequences, some of which may be similar to those described for other exons while others may be unique to large exons. In order to understand all the intricacies of the splicing machinery and exon recognition it will be important to understand how all types of exons are efficiently recognized. Therefore, we have examined sequences within and surrounding a large constitutively spliced exon and found that while some previously identified sequences may be important for recognition, other features, such as strong splice sites, that are sufficient for small exon inclusion may not be sufficient for large exons.
We have begun to study the fourth exon of the polymeric immunoglobulin receptor (pIgR) gene which is 654 nt long yet is efficiently spliced into the mRNA in all vertebrates examined (18–22), except for the rabbit, where it is either excluded from or included in the mRNA (23). The pIgR protein transports polymeric immunoglobulins (Ig), primarily IgA, from the basolateral to the apical surface of epithelial cells (reviewed in 24). This protein is a member of the Ig superfamily of structurally related cell surface proteins and the large exon 4 encodes two Ig-like domains. Such domains are usually encoded by single exons; pIgR exon 4 is the only example in which two domains are encoded by a single exon (25).
To study pIgR exon 4 splicing we have constructed a chimeric Dd–pIgR gene by placing the large exon 4 from the mouse pIgR gene with part of its surrounding introns into the third intron of the mouse major histocompatibility complex class I Dd gene. We have shown that the pIgR exon is efficiently spliced into the Dd mRNA and have described an exon deletion that creates a cryptic 3′ splice site (26). This site activated an upstream cryptic 5′ splice site, resulting in creation of a cryptic intron. Interestingly, the location of the cryptic splice sites are between protein coding domains where introns exist in other Ig superfamily genes (26). Here we describe our further examination of sequences within the exon, the downstream intron and the normal and cryptic 5′ splice sites to better understand the interactions among these sequences that contribute to efficient recognition of this exon. We found that the strength of the 5′ splice site is a crucial feature of this large exon. However, although the splice sites are strong when compared to consensus sequences, they were not sufficient to confer efficient splicing of a large exon. Other sequences, including at least two exonic sequences and one evolutionarily conserved intronic sequence, are redundant features that contribute to pIgR exon 4 recognition. Also, since cryptic 5′ splice sites were activated by several different mutations, the balance between use of these potential splice sites, which create much smaller exons, and the normal 5′ splice site must be modulated by the presence of sequences that enhance use of the normal 5′ splice site or repress potential cryptic sites. In addition, sequences that enhance use of the cryptic splice sites must be absent from this large exon; when such sequences were introduced, cryptic splice reactions were activated and the large exon was no longer spliced intact.
MATERIALS AND METHODS
Plasmid construction
The construction of Dd–pIgR was described previously (26). All mutations within the pIgR sequences were constructed in a pGEM7 subclone of the 1.4 kb EcoRI–HindIII pIgR genomic fragment and then cloned into the KpnI site of the DdBsm(H) plasmid (Fig. 1). Dd–PpuMI, Dd–Bsu36I and Dd–EcoNI were constructed by removing a 76 bp PpuMI, a 192 bp Bsu36I and a 54 bp EcoNI fragment, respectively, from pIgR exon 4. To construct Dd–Xcm a 116 bp XcmI fragment was removed from the intron downstream of pIgR exon 4.
Figure 1.

Chimeric Dd–pIgR construct. (A) The 654 bp pIgR exon 4 with 262 bp of upstream intron and 420 bp of downstream intron was placed into the KpnI (K) site of the Dd gene as an EcoRI (R)–HindIII (H) fragment. The open box represents the pIgR exon, the black boxes are Dd exons and thin lines are introns. (B) Several deletions between restriction sites within pIgR exon 4 and the downstream intron were constructed as shown. The 5′ splice site sequence at the end of exon 4 is shown; the A at the +4 position was mutated to C in the construct 5′SS. The lengths of the deletions are indicated; (5′) indicates the cryptic 5′ splice site identified in Figure 2; * indicates locations of 7–12 nt long purine-rich sequences; X indicates locations of 7–9 nt long A/C-rich sequences.
For the exon replacement constructs (Fig. 4) a 580 bp AccI–EcoNI fragment, a 325 bp HincII–Bsu36I fragment and an 84 bp HincII fragment were individually removed from pIgR exon 4 and the DNA ends were made blunt with Klenow fragment. Fragments from the Ig µ constant region (Cµ) cDNA, a 582 bp BamHI–PstI fragment or a 326 bp RsaI–RsaI fragment were ligated into the AccI–EcoNI deletion to produce AE/BP, AE/RR and AE/RR(–). The 326 bp Cµ fragment was also ligated into the HincII–Bsu36I and HincII deletions in both orientations to construct HB/RR, HB/RR(–), HH/RR and HH/RR(–).
Figure 4.

pIgR exon 4 replacement constructs. The EcoRI–HindIII pIgR fragment is shown with the restriction sites used to delete internal exon sequence: Ac, AccI; Hc, HincII; Bsu, Bsu36I; EcN, EcoNI. pIgR exon sequence was replaced with cDNA fragments from the constant region of the immunoglobulin M gene (Cµ). Shown at the bottom of the figure is the Cµ region of the gene and cDNA fragment cloned into pIgR; Cµ exons are filled boxes and the hatched box denotes the µs-specific portion of exon Cµ4. The restriction sites are: B, BamHI; R, RsaI; P, PstI. The size of each deletion and replacement fragment and the final chimeric exon are indicated; construct names are shown, with (–) indicating fragment insertion in the antisense orientation.
All the site-directed mutations except pIgR NonGs were created using the megaprimer mutagenesis protocol (27) in the appropriate pGEM7 subclone; all mutations were confirmed by DNA sequencing. The oligonucleotides used are listed in Table 1. To construct the 5′ splice site mutation (5′SS, Fig. 1), the oligonucleotides 5′SS Mut and MSC-4C were used to synthesize the megaprimer which was then used with the AS-STY oligonucleotide. The PCR product was digested with StyI and cloned into the StyI sites of pIgR. To construct the pIgR Gs mutation (Fig. 7) the oligonucleotides CRYG and USAcc were used to synthesize the megaprimer which was then used with the DSNhe oligonucleotide. The PCR product was digested with AccI and NheI and cloned into the AccI and NheI sites of pIgR. To construct the pt1 and pt2 mutations (Fig. 9) the oligonucleotides 370E1 and 370E2 were used with the USAcc oligonucleotide to synthesize the megaprimers which were then used with the DSNhe oligonucleotide. The PCR products were cloned into pIgR and pIgR–Xcm as described above. The pIgR NonGs mutation was created using the NonG-T and NonG-B mutagenic oligonucleotides to amplify around the Dd–pIgR plasmid, followed by DpnI digestion and direct bacterial transformation (28).
Table 1. Oligonucleotides used in mutagenesis and PCR reactions.
| Oligo |
Sequence |
| pIgR oligos | |
| 5′SS Mut | 5′-TGGAGGTTCGCACCTTCATTGAC-3′ |
| AS-STY | 5′-GGCCTATCATTGGTACTGAATCCC-3′ |
| MSC-4C | 5′-GTCCGGACTCCTTCCTCCTACG-3′ |
| CRYG | 5′-GTCCCTTTCATACCCCCCCCTGCTCTGCCTAT-3′ |
| USAcc | 5′-CCACAGTTCCTGAGTTGCC-3′ |
| DSNhe | 5′-TCGTTTGCCACCTCACGG-3′ |
| 370E1 | 5′-GTCTTCTTACCCAGGGATTTC-3′ |
| 370E2 | 5′-GTCTTCTTACCTAGGGATTTCTT-3′ |
| NonG-T | 5′-GCTATATAGGCAGAGCATCGAGATCTATGAAAGGGACC-3′ |
| NonG-B | 5′-GGTCCCTTTCATAGATCTCGATGCTCTGCCTATATAGC-3′ |
| Dd oligos | |
| Dd Ex3 | 5′-AGGCTGGTGCTGCAGAGA-3′ |
| Dd Ex4 | 5′-CCAGGTCAGGGTGATGTC-3′ |
Figure 7.
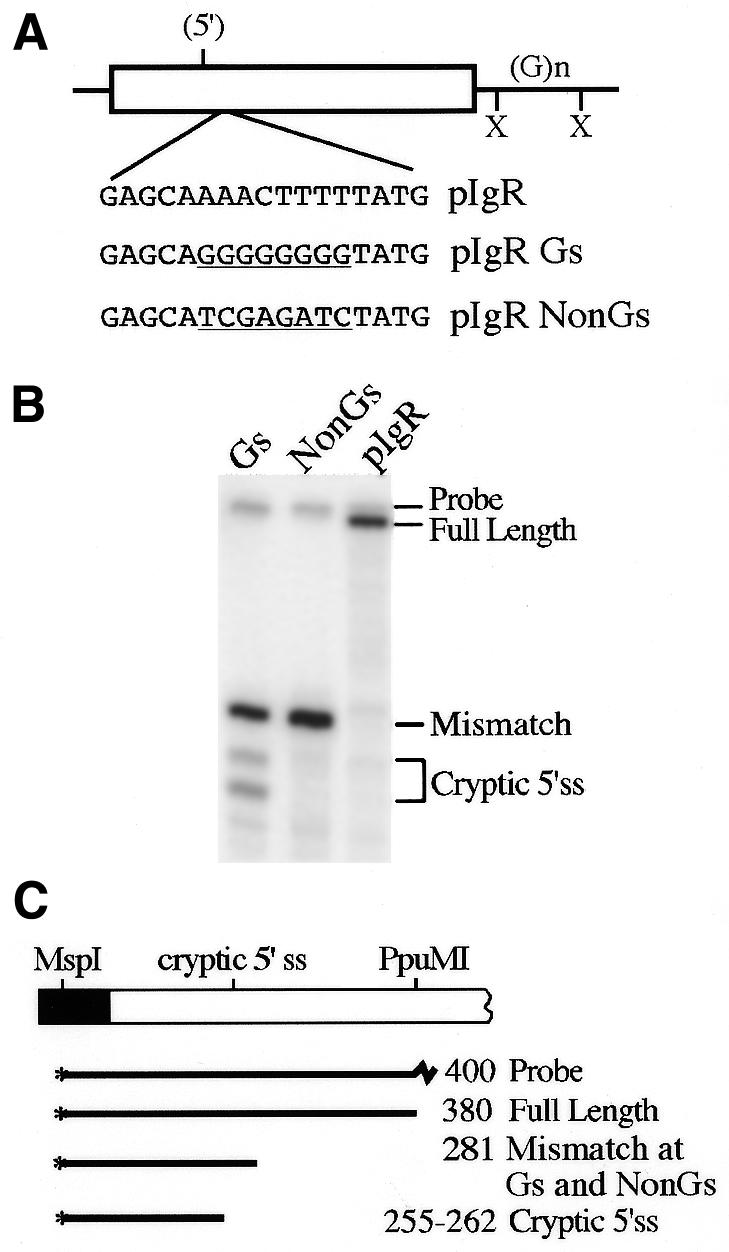
G-rich sequences activate the cryptic 5′ splice site. (A) Sequences beginning 27 nt downstream of the cryptic 5′ splice site were mutated to create a run of eight G residues or changed to a non-G-rich sequence, shown underlined below the wild-type sequence in this region. (B) S1 nuclease analysis of RNA from HepG2 cells transfected with the constructs in (A). Protected bands are identified on the right. (C) Diagram of the S1 nuclease probe and protected fragments; the intact spliced IgR Gs and pIgR NonGs exons protect the probe only to the site of these mutations.
Figure 9.
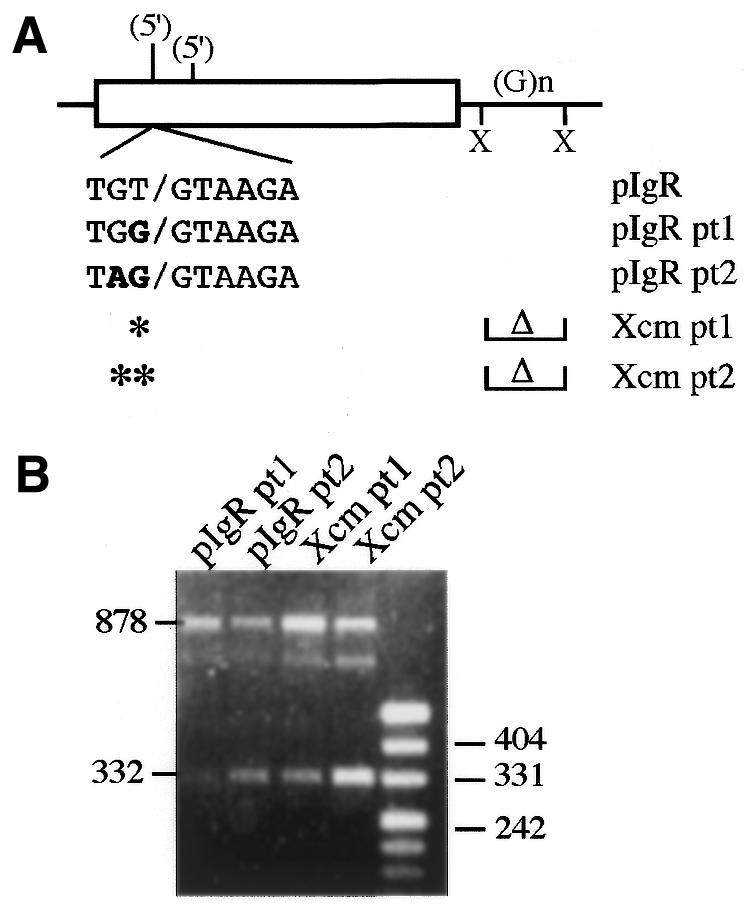
Cryptic 5′ splice site enhancement mutations. (A) A potential cryptic 5′ splice site 50 nt upstream from the identified cryptic site was mutated at one or two nucleotides to more closely resemble the consensus 5′ splice site sequence; the mutations are indicated in bold. The point mutations were analyzed on their own or in combination with the intronic XcmI deletion. (B) RT–PCR analysis of the constructs shown in (A). Activation of the new cryptic splice site is indicated by the 332 bp RT–PCR product; full-length pIgR exon 4 inclusion is also seen as the 878 bp product. The ∼800 bp band likely represents a hybrid product between the full-length product and the cryptic 5′ splice site product as seen before (32).
Transfection and RNA preparation
HepG2 human hepatoma cells (29) were grown in Dulbecco’s modified Eagle’s medium/F12 medium (1:1) supplemented with 10% heat-inactivated fetal bovine serum, 10 µg/ml insulin and 50 U/ml penicillin/streptomycin. Cells were transfected using a calcium phosphate procedure (29). The RNA was harvested 48 h after transfection using a hot phenol method (29).
RT–PCR analysis
One microgram of total cellular RNA was reverse transcribed using oligo(dT) and BRL Superscript Reverse Transcriptase as per the manufacturer’s instructions. Five microliters of this reaction was amplified by PCR using 20 pmol Dd Ex3 and Dd Ex4 primers (Table 1) and BRL Taq polymerase for 25 cycles of 94°C for 1 min, 58°C for 1 min and 72°C for 2 min.
Southern blot analysis was performed by standard methods after separating 1–10 µl of RT–PCR products on a 1.2% agarose gel. Filters were probed with a labeled PstI–PpuMI Dd–pIgR cDNA fragment and were visualized with a phosphorimager.
S1 nuclease analysis
The S1 probe used to differentiate the Dd–pIgR transcripts was derived from a Dd–pIgR cDNA clone; the sequence between the Dd Ex3 PCR primer and the PpuMI site in pIgR exon 4 was subcloned into pGEM4. The probe was 3′-end-labeled at an MspI site using Klenow and [α-32P]dCTP and extends to the EcoRI site in the vector (Fig. 3). A total of 100 µg RNA, a combination of 50–100 µg specific RNA and carrier RNA, was hyridized overnight at 50°C with the labeled probe. Digestion was at 37°C for 30 min with 60 U S1 nuclease (Pharmacia) per reaction. The protected fragments were separated on a 6% acrylamide, 7 M urea gel and quantitated by phosphorimager analysis. Multiple protected fragments are observed with the cryptically spliced RNAs due to fortuitous partial homology between the probe and the Dd exon 4 sequences to which the cryptic site is spliced; by changing the S1 digestion temperature all bands could be combined into one.
Figure 3.

S1 nuclease quantitation of Dd–pIgR spliced products. (A) S1 nuclease protection assays of RNA from HepG2 cells transfected with the constructs shown above each lane. Protected bands are identified on the right. (B) S1 nuclease reactions were quantitated on a phosphorimager and graphed as the ratio of full-length to cryptically spliced RNA. These numbers represent at least two independent transfections, each analyzed two or more times. (C) Diagram of the S1 nuclease probe. RNA that has spliced the full-length pIgR exon into Dd protects the probe to the PpuMI site and cryptically spliced RNA protects the probe to the cryptic 5′ splice site; the expected sizes of each product are indicated. Multiple protected bands are observed with the cryptic 5′ splice site in 5′SS and Xcm 5′SS RNA due to fortuitous partial homology between the probe and sequences in Dd exon 4 to which the cryptic splice site is joined; the fragments were combined for quantitation. There is a single cryptic splice band in Bsu36I since it is not spliced directly to Dd exon 4.
RESULTS
We have previously described a chimeric Dd–pIgR gene constructed to study sequences within the 654 nt pIgR exon 4 that are necessary for its efficient full-length inclusion into the mRNA (26). We placed the 654 bp exon with 262 bp of upstream intron and 420 bp of downstream intron sequence into the large intron 3 of the Dd gene to produce Dd–pIgR; the intron upstream of the pIgR exon is 814 nt and the intron downstream is 1537 nt (Fig. 1A). This placement is important since the normal location of exon pIgR 4 is also between two large introns (2592 and 1237 nt) (30) and introns less than ∼500 nt can affect efficient splicing of large exons by intron definition (6). When we transiently expressed this chimeric construct in the human HepG2 liver cell line, full inclusion of the exon was observed by RT–PCR using primers to the upstream Dd exon 3 and the downstream Dd exon 4 (Fig. 2A, lane 2; 26). This indicates that all sequences necessary for full inclusion of this large exon have been transferred to the heterologous context.
Figure 2.

RT–PCR analysis of the Dd–pIgR construct and its derivatives. (A) RNA from HepG2 cells transiently transfected with the construct shown above each lane were analyzed by RT–PCR. M, marker lane; sizes, in bp, of some of the bands are shown. (B) Diagram of Dd–pIgR splicing reactions. The primers used in the RT–PCR analysis, Dd Ex3 and Dd Ex4, are indicated by the arrows. The 878 bp RT–PCR product is from RNA containing the complete pIgR exon 4, the 224 bp product is from RNA that has omitted the exon (see Dd control in lane 1) and any products migrating between 224 and 878 bp are from RNA that had spliced exon 4 using cryptic splice sites. RT–PCR products from complete exon 4 inclusion from the exon deletion constructs are 878 bp minus the size of the deletion. The cryptic splice reaction activated by the 5′SS mutation is shown on the diagram of the gene. The structure of the 382 bp RT–PCR product, as determined by DNA sequence analysis, is also shown.
5′ splice site mutations activate a cryptic splice reaction
A strong 5′ splice site has been shown to enhance the inclusion of both small and alternatively processed exons (see for example 3,12,16) and the normal 5′ splice site at the 3′-end of pIgR exon 4 has a 7 out of 9 nt match to the 5′ splice site consensus sequence (Fig. 1B; 31). To determine if the 5′ splice site strength was required for recognition of pIgR exon 4 we made a single A→C point mutation at nt +4 that would decrease the match to the consensus sequence of the 5′ splice site (5′SS, Fig. 1B). RNA from 5′SS transiently expressed in HepG2 cells was analyzed by RT–PCR using primers for Dd exons 3 and 4 (Fig. 2B). We detected the full-length 878 bp product plus two smaller products of ∼830 and 380 bp (Fig. 2A, lane 7), which likely result from cryptic splicing within the pIgR exon (Fig. 2B). We cloned and sequenced these smaller RT–PCR products and found that the smallest one corresponds to use of a cryptic 5′ splice site 158 nt into pIgR exon 4 that is being spliced directly to Dd exon 4 (Fig. 2B). Interestingly, this is the same cryptic 5′ splice site that was activated in pIgR–ΔHc when an improved 3′ splice site was created by the deletional junction (26). However, the sequence of the larger potential cryptic splice product matched that of the full exon. Also, this 830 bp PCR product was not present consistently in our PCR reactions. We conclude that it is likely a hybrid between the full-length RT–PCR product and the cryptic 5′ splice site RT–PCR product and not a unique cryptically spliced RNA. Such a hybrid PCR artifact has been described before (32).
To quantitate the relative amounts of cryptically spliced RNA compared to normally spliced RNA in 5′SS, S1 nuclease protection assays were performed (Fig. 3). A cDNA probe that spans the Dd exon 3–pIgR splice junction and extends beyond the cryptic 5′ splice site sequence was used to quantitate these two RNAs (Fig. 3C). This probe would also identify RNAs from which the entire pIgR exon was omitted and those that use any other cryptic splice sites in this region. As seen by RT–PCR, Dd–pIgR RNA was mostly fully spliced (Fig. 3A, lane 1); because of a variable lane background we have determined the ratio of full-length to cryptically spliced RNA for this construct to be ≥30 (Fig. 3B, Table 2). The cryptic 5′ splice site was activated at least 6-fold when the normal 5′ splice site was mutated in 5′SS (Fig. 3A, lane 5 and 3B and Table 2).
Table 2. Expression of Dd–pIgR constructs in HepG2 cells.
| Construct |
Full-length:cryptic splice ratio |
| pIgR | ≥30 |
| 5′SS | 4.7 ± 0.8 |
| pIgR Gs | 2.0 ± 0.2 |
| pIgR NonGs | ≥30 |
| pIgR pt1 | 12 ± 2 |
| pIgR pt2 | 1.8 ± 0.06 |
| Xcm pt1 | 6.2 ± 0.6 |
| Xcm pt2 | 1.3 ± 0.05 |
S1 nuclease protection assays were used to quantitate the full-length:cryptic splice products made by each construct. Each expression ratio value is the result of at least two transfections, each analyzed at least twice.
Deletion of pIgR exon sequences
Alternatively processed and small exons often require sequences within the exon or surrounding intron for full exon inclusion; in some cases these exonic splice enhancer (ESE) sequences are purine-rich or A/C-rich (see for example 33–36). It is likely that large exons also contain splice enhancing sequences to enable efficient splicing. To begin to explore pIgR exon 4 sequences for such activities we made several deletions within the pIgR exon that ranged in size from 54 to 192 bp and that removed some of the 7–12 nt long purine-rich and 7–9 nt long A/C-rich sequences (Fig. 1B). We have previously shown that while the 84 bp HincII deletion resulted in striking activation of a cryptic splice reaction, this fragment did not contain ESE activity in a heterologous context (26). We also constructed a 76 bp deletion that spanned the two PpuMI sites (Dd–PpuMI), a 192 bp deletion that spanned the two Bsu36I sites (Dd–Bsu36I) and a 54 bp deletion that spanned the two EcoNI sites (Dd–EcoNI). These were transiently expressed in HepG2 cells and the RNA was analyzed by RT–PCR. None of the deletion mutations noticeably affected pIgR exon 4 splicing; the full-length RT–PCR product for each construct was predictably shortened by the size of the exon deletion (Fig. 2A, lanes 2–5). RNA from these exon deletions was also examined by S1 nuclease protection analysis. Predominantly full inclusion of the exon was seen with the EcoNI and PpuMI constructs, (Fig. 3A, lanes 2 and 4, and Fig. 3B). However, the Bsu36I construct showed an ∼2-fold increase in use of the same cryptic 5′ splice site that was strongly activated in the 5′SS construct (Fig. 3A, lane 3, and Fig. 3B), suggesting that this 192 nt region may contain sequences with ESE-like activity. By Southern blotting the RT–PCR reaction from the Bsu36I construct we were able to detect a faint band smaller than the full-length product but larger than the 382 bp product of the cryptic 5′ splice site spliced to Dd exon 4, suggesting that a different cryptic splice reaction had been activated (data not shown). However, this low abundance product was not characterized further. This exon deletional data suggests there may be some ESE activity within the Bsu36I region, but there was no evidence for similar activity in the other two fragments. However, it is not clear whether these regions truly lack ESE activity or whether it cannot be detected because redundant elements are present elsewhere in the pIgR sequence.
The 3′ and 5′ splice sites are not sufficient for pIgR exon inclusion
Since most of the pIgR exon 4 deletions had a minimal effect on splicing, but weakening the 5′ splice site activated a cryptic splice site, it was possible that the strength of the splice sites alone was sufficient for recognition of this large exon. Consistent with this idea, the normal 3′ splice site at the 5′-end of exon 4 appears to be fairly strong; it contains a 6 of 7 nt match to the consensus branch point sequence followed by a 17 of 20 nt polypyrimidine tract (30,31). To test whether the pIgR exon 4 splice sites were sufficient for full exon recognition we replaced internal pIgR exon sequence with heterologous sequence and analyzed the splicing patterns of the chimeric exons. We removed a 580 bp AccI–EcoNI fragment from pIgR exon 4, leaving 30 bp at the 5′-end and 44 bp at the 3′-end of the exon. Two cDNA fragments from the Ig µ constant region (Cµ), 326 and 565 bp, were inserted into the pIgR exon; the 326 bp cDNA fragment was also inserted in the opposite orientation (Fig. 4). Thus, the exon sizes of 639 nt in AE/BP and 400 nt in AE/RR and AE/RR(–) were similar to or smaller than the 654 nt pIgR exon. RNA from these constructs was analyzed by S1 nuclease protection; the RNA will protect the probe only to the AccI site in pIgR exon 4 but will detect any RNA that has skipped the modified exon (Fig. 5B). Both AE/BP and AE/RR RNA protected the probe to the AccI site, indicating that the RNA was correctly spliced across the Dd exon 3–pIgR exon 4 junction (Fig. 5A, lanes 4 and 7). However, the chimeric exon is frequently skipped in the AE/RR(–) RNA and only a small amount of protection to the AccI site is seen (Fig. 5, lane 1). These results indicate that the pIgR exon 4 splice site sequences alone are not capable of directing efficient inclusion of the chimeric AE/RR(–) exon, even though it is only 400 nt long, which is within the size range that should be more efficiently recognized by exon definition. They also suggest that there are ESE sequences within the Cµ cDNA BP and RR fragments in the sense orientation that are not present in the antisense orientation. While it is also possible that the RR(–) fragment may contain sequences that inhibit splicing, exons containing this fragment are spliced when additional pIgR exon 4 is included (see below). Therefore, if such negative sequences exist in this fragment, they can be overcome by positive acting elements in the pIgR exon sequence.
Figure 5.

Expression from exon replacement constructs. (A) S1 nuclease protection assays of RNA from HepG2 cells transfected with the constructs shown above each lane. The protected fragments are identified on the right. (B) Diagram of the probe used in this assay. With the AE/RR and AE/BP constructs the S1 probe will only be protected to the AccI site, the site of fragment insertion; the HH/RR and HB/RR constructs will protect the probe to the HincII site. Skipped exon represents those RNAs in which the chimeric pIgR exon is excluded from the Dd gene. (C) Southern blot analysis of RT–PCR reactions from the RNA in (A) using the Dd exon primers (Fig. 2). The sizes in bp of the RT–PCR products are shown on the left; identities of the products are shown on the right. The blot was probed with a cDNA fragment that spans the Dd exon 3–pIgR splice junction.
To delineate the regions within pIgR exon 4 that could enhance exon inclusion we altered the portions of exon 4 remaining in the chimeric exon. We removed a 325 bp HincII–Bsu36I fragment from pIgR exon 4, leaving 227 bp of sequence at the 5′-end and 102 bp of sequence at the 3′-end of the exon and inserted the 326 bp Cµ cDNA sequence in both orientations to make HB/RR and HB/RR(–); this chimeric exon is 655 nt long (Fig. 4). We also removed the 84 bp HincII fragment from the pIgR exon and inserted the 326 bp Cµ cDNA, making the exon 896 nt long in HH/RR and HH/RR(–) (Fig. 4). Correctly spliced RNA from all of these constructs will protect the S1 probe to the HincII site and any exon skipping or cryptic splice site use up to this sequence will also be detected. Both constructs with the Cµ RR fragment in the sense orientation mainly protected the probe to the HincII site (Fig. 5A, lanes 5 and 6). However, RNA from HB/RR(–) used the cryptic 5′ splice site predominantly; very little RNA protecting the probe to the HincII site was seen (Fig. 5A, lane 2). This suggests that the pIgR exon 4 sequences that had been added back to this construct prevent exon skipping but are not sufficient to confer full-length exon inclusion; when the cryptic splice site was added back, in the AccI and HincII fragment, this became the preferred 5′ splice site. Surprisingly, RNA from HH/RR(–) mainly protected the probe to the HincII site, suggesting efficient exon inclusion, even though the exon is now 896 nt long (Fig. 5A, lane 3). Thus, replacing the downstream HincII–Bsu36I region of pIgR exon 4 restored intact exon splicing and prevented use of the cryptic 5′ splice site. These results identify two regions within pIgR exon 4, one upstream and one downstream from the HincII sites, that are likely to contain ESE elements that aid in efficient exon inclusion.
The S1 probe used with these exon replacement constructs only provides information about the splice patterns in the 5′ portion of the exon; it would not detect other cryptic splice reactions within the substituted parts of the exon or further downstream. Thus, we examined the RNA from these constructs by RT–PCR to determine the sizes of the pIgR exons spliced into the Dd RNA (Fig. 5C). The chimeric exon in AE/RR is fully spliced, as indicated by the 624 bp RT–PCR product and AE/RR(–) shows a significant amount of exon skipping, seen as the 224 bp product (Fig. 5C, lanes 1 and 2). The chimeric exon in AE/BP was also fully spliced (data not shown). The RNA from HB/RR mainly included the full chimeric exon, but also skipped the exon and used the cryptic 5′ splice site at detectable levels (Fig. 5C, lane 3); these other minor RNAs were not clearly seen in the S1 analysis. RNA from HB/RR(–) was predominantly spliced using the cryptic 5′ splice site (Fig. 5C, lane 4), consistent with the S1 analysis data. Both HH/RR and HH/RR(–) RNA predominantly included the entire 896 nt chimeric exon, as indicated by the 1120 bp RT–PCR product, although a low level of other smaller products can also be seen (Fig. 5C, lanes 5 and 6). These results obtained by RT–PCR analysis complement and confirm the information from the S1 nuclease assays.
An evolutionarily conserved intronic splice enhancer sequence
Intronic sequences have also been shown to contribute to splice site recognition and regulation (see for example 9,37). When we sequenced the intronic regions of the EcoRI–HindIII mouse pIgR fragment included in our chimeric construct we found a strikingly purine-rich region 13 nt into the intron downstream of pIgR exon 4. In this region 21 of 22 nt are purines, including a run of 11 G residues (Fig. 6). We sequenced this portion of intron 4 from the human gene and also found a purine-rich sequence (23 of 25 nt are purines) in a similar location with respect to the 5′ splice site (Fig. 6). The conservation of this purine-rich sequence led us to investigate whether this region may contribute to exon recognition. It has been shown that intronic G-rich sequences enhance use of a small exon (37) and G-triplets enhance use of an upstream 5′ splice site in small introns (38). We deleted a 116 bp XcmI fragment from the intron which removed the purine-rich region and some additional downstream sequence (Figs 1B and 6); this reduced the intron length minimally, from 1537 to 1421 nt. However, RT–PCR analysis of this construct transiently expressed in HepG2 cells showed that there was no activation of cryptic splice sites or exon skipping; it was expressed identically to the wild-type construct (Fig. 2A, lane 6, Fig. 3A, lane 6 and Fig. 3B).
Figure 6.

A purine-rich sequence within pIgR intron 4 is conserved between the mouse and human genes. Purine-rich sequences just downstream from the 5′ splice junction in the mouse and human pIgR genes are underlined; additional sequence homology extends beyond this stretch of purines and is underlined with a dotted line. Upper case letters are the 3′-end of exon 4, lower case letters are intron sequence. The XcmI sites that were used in the Dd–Xcm construct are shown. The 144 nt of human sequence that is available is shown compared to the same sequence of the mouse intron. The sequence we obtained for the mouse intron matches the recently published sequence, GenBank accession no. AB001489 (30). The sequences shown here were submitted to GenBank, accession nos AF261083 (human) and AF261084 (mouse).
It is likely that both the 5′ splice site strength and additional sequence elements contribute to exon 4 inclusion. If so, the combination of the 5′ splice site mutation with the intron deletion may have a cumulative effect on pIgR exon 4 splicing. To test this idea we combined 5′SS with the XcmI deletion, to make Xcm 5′SS. By RT–PCR we observed activation of the same cryptic splice reaction seen in the 5′SS mutation alone (data not shown). By S1 nuclease assay (Fig. 3A, lane 7) there was a 3-fold increase in cryptic 5′ splice site use in Xcm 5′SS as compared to 5′SS (Fig. 3B). This suggests that the XcmI intronic element does aid recognition of the normal 5′ splice site but this can only be detected experimentally when other features that also contribute to exon recognition are weakened.
G-rich sequences activate the cryptic 5′ splice site
The active sequences of the evolutionarily conserved intronic enhancer element within the XcmI fragment are likely to be the long purine-rich stretch, more specifically, the multiple G residues. To determine whether a G-rich sequence could activate the cryptic 5′ splice site in competition with the normal wild-type 5′ splice site we created a G8 sequence 27 nt downstream from the cryptic 5′ splice site in pIgR, to make pIgR Gs (Fig. 7A). This sequence should not act as an ESE since poly(G) does not have this activity (39). As a control to ensure that the sequences we disrupted did not have regulatory activity, such as suppressing cryptic splice site use or enhancing full exon inclusion, we also changed this sequence to a non-G-rich sequence to make pIgR NonGs. By S1 nuclease analysis (Fig. 7B) we observed significant activation of the cryptic 5′ splice site from the pIgR Gs construct, but not from the pIgR NonGs construct. RT–PCR analysis showed that the cryptic 5′ splice site was being spliced to Dd exon 4 (data not shown). Surprisingly, the G8 mutation activated the cryptic 5′ splice site to a greater extent than did the 5′SS mutation (Table 2). The G-rich sequences are clearly strong activators of the weaker cryptic 5′ splice site even though the normal 5′ splice site remains wild-type and is followed by the downstream purine-rich sequence. These results are consistent with the activities of other G-rich intronic splice enhancing sequences (37,38) and with our proposal that the G-rich sequence within the XcmI fragment also has splice enhancing activity.
Interactions among cryptic splice sites within pIgR exon 4
The frequency with which each nucleotide occurs at each position of a 5′ splice site has been determined and used to calculate a match to consensus score for any real or potential 5′ splice site (31). We have calculated a score for the natural pIgR 5′ splice site of 89 and for the cryptic 5′ splice site of 76 (Fig. 8); by this measure, this cryptic site is the closest match to the consensus sequence within pIgR exon 4. A sequence 50 nt upstream is the next best match to consensus with a score of 69; this site was used to a small extent only when the cryptic 5′ splice site was mutated away from consensus (to a score of 63) in ΔHc547D (26). To further examine the interactions among natural and cryptic splice sites in this exon and the features that contribute to their recognition we mutated the upstream potential cryptic 5′ splice site to enhance its match to the consensus sequence (Fig. 9A). We made a 1 nt change in pt1 and a 2 nt change in pt2 so that these sites now had scores of 81 and 90, respectively (Fig. 8). These mutations were made in pIgR and in pIgR–Xcm to determine whether the intronic element in the XcmI fragment would affect use of the natural 5′ splice site under these conditions. By RT–PCR analysis we observed both full-length pIgR exon 4 inclusion and activation of the new cryptic 5′ splice site in all constructs (Fig. 9B, 878 and 332 bp products, respectively). S1 nuclease protection assays confirmed the location of the cryptic splice site and quantitated the amount of cryptic splice site activation (data not shown and Table 2). The pt1 mutation was able to detectably compete with the normal 5′ splice site, as seen by an ∼3-fold activation of the cryptic site in pIgR pt1 (Fig. 9B and Table 2). Use of the new cryptic site was greatly enhanced by the pt2 mutation; there was a nearly 17-fold increase in the use of this site in pIgR pt2 relative to pIgR (Table 2). In both pt1 and pt2 the amount of new cryptic site activation was ∼2-fold higher when the XcmI fragment was deleted, consistent with the presence of a splice enhancing sequence within this fragment (Table 2).
Figure 8.

Relative use of 5′ splice sites in pIgR constructs compared to their match to consensus scores. The frequency with which each nucleotide occurs at each position of a 5′ splice site has been determined and used to calculate a match to consensus score for individual 5′ splice sites (see for example ref. 31). The scores for the natural pIgR exon 4 5′ splice site, the upstream cryptic 5′ splice sites and the 5′SS, pt1 and pt2 mutations are shown below each splice site. The consensus 5′ splice site sequence is shown below. Also included is relative use of the 5′ splice sites in each construct, taken from the data in Figures 3 and 9 and Table 2.
DISCUSSION
In this paper we have examined the 654 nt mouse pIgR exon 4 to identify features that contribute to its efficient splicing, even though it exceeds the size of >99% of internal exons. This is the first constitutively spliced large exon that has been studied in this way. The strong 5′ splice site of this exon was required for full intact exon splicing as mutating it away from the consensus sequence partially activated a far upstream cryptic site. However, the splice junctions of the pIgR exon were not sufficient to confer efficient splicing on a chimeric large exon, indicating that strong splice sites are not always sufficient for exon recognition. Thus, pIgR exonic sequences must also contribute to overcoming the negative impact of a large exon size. However, rather than finding a single feature or sequence element that was responsible for efficient intact splicing, we found evidence for multiple redundant elements, including at least two exonic and one intronic sequence elements that each contribute to the exon recognition process (summarized in Fig. 10). These elements were only revealed when another positive acting element had been weakened or eliminated. For example, an effect of the intronic element was detected only when the normal 5′ splice site was weakened or was in competition with an improved internal cryptic splice site. Also, a clear effect of the exonic sequences within the Bsu36I fragment were seen when they were added back to an exon that had been expanded by 242 nt; when this region was deleted from the intact exon only a 2-fold effect was observed. However, by deleting the Bsu36I fragment we also shortened the exon to 462 nt, which may have reduced the burden of large exon size and made it less dependent on ESE-like sequences. Indeed, if multiple cis-acting elements are involved in recognizing this large exon the removal of some, with the concomitant decrease in exon size, would make it difficult to identify these elements by this approach. Another feature of the pIgR exon that appears to be important for its intact full-length splicing is that potential internal cryptic 5′ splice sites must be sufficiently weak and lack nearby sequences that would enhance their use (e.g. G-triplets) so as to not compete with recognition of the normal 5′ splice site. It is also possible that the pIgR exon contains sequences that normally suppress the use of potential internal cryptic sites. When we improved potential splice sites and/or introduced nearby regulatory elements into the pIgR exon the cryptic sites were recognized by the splicing apparatus and the exon became alternatively spliced.
Figure 10.

Summary of sequences in and surrounding pIgR exon 4 shown to contribute to the intact splicing of this large exon. The regions identified by each experiment are denoted by the hatched ovals; the experiments are identified on the right. Symbols and restriction sites are as identified in Figures 1 and 4. * indicates locations of the 7–12 nt long purine-rich sequences and X indicates locations of 7–9 nt long A/C-rich sequences that could potentially contribute to exon recognition.
The exon replacement experiments clearly established that multiple exonic sequences contribute to the splicing efficiency of pIgR exon 4. The pIgR splice junctions were not sufficient for recognition of a large chimeric exon; the AE/RR(–) exon was spliced out of the mRNA more frequently than it was spliced in. However, when the 197 nt AccI–HincII fragment near the 5′-end of the exon was added to the chimeric exon the exon was no longer skipped but was spliced nearly exclusively using the cryptic 5′ splice site contained within this fragment. Therefore, this region must contain sequences that contribute to exon recognition, but they are not sufficient for recognition of the entire intact exon. When the 241 nt HincII–Bsu36I fragment was also included in the HH/RR(–) chimeric exon, recognition of the entire exon was restored, even though it was expanded by 242 nt to 896 nt long. Therefore, this region must contain ESE sequences that assist this large exon to be recognized intact and somehow suppress use of the cryptic 5′ splice site. The positive acting element(s) within the AccI–HincII fragment could be one or more of the purine-rich or A/C-rich sequences within this fragment (Fig. 10). Alternatively, it could be the cryptic 5′ splice site sequence itself that eliminates exon skipping since 5′ splice site-like sequences have been shown to act as splice enhancers (40,41). There are also a couple of purine-rich and A/C-rich sequences within the HincII–Bsu36I fragment which may be active. We have tested several subfragments of this exon for ESE activity in the heterologous µAV transcript whose splicing requires an ESE element (10). However, the purine-rich sequences in the Hc fragment (26) and several A/C-rich and purine-rich sequences we have so far tested do not contain ESE activity in this assay (data not shown). As these are not the only types of sequences known to have ESE activity (36,42) it is very likely that other sequences also participate in pIgR exon 4 splicing.
While several of the exon deletions and exon replacements are predicted to change the reading frame of the chimeric Dd–pIgR protein and thus potentially cause the RNA to be less stable, we do not believe this has a major impact on the conclusions we have obtained from our data. We have been able to detect the predicted RNAs whether they are from out-of-frame or in-frame deletions or insertions. All of the exon replacements with the RR fragment are predicted to be out-of-frame, yet full-length splicing of the chimeric exon, exon skipping and cryptic 5′ splice site use are all observed in different constructs, depending on the amount of pIgR exon sequence included. The data fit well with the interpretation of gradual restoration of ESE sequences to the chimeric pIgR Cµ exon.
We found a strikingly purine-rich intronic sequence directly downstream from exon 4 in both the mouse and human pIgR genes. Deleting this sequence from the pIgR intron had no effect on pIgR exon 4 splicing until the 5′ splice site was weakened by a point mutation. Then, use of the normal 5′ splice site was decreased 3-fold in the absence of the XcmI fragment. Also, the wild-type 5′ splice site was less able to compete with use of the improved cryptic splice sites in pt1 and pt2 when this purine-rich sequence was deleted. G-rich sequences in other splice enhancing intronic elements have been suggested to aid in defining exon–intron boundaries (37,38,43). Consistent with this idea, when we mutated the sequence 27 nt downstream from the cryptic 5′ splice site to G8 the cryptic splice site was greatly activated. This site, which is a poorer match to the consensus than the authentic site, with a score of 76 compared to 89 (Fig. 8), competes very effectively with the authentic site when the G-rich sequence is placed downstream. Thus, this element clearly helps to define the cryptic splice site as an authentic site.
When the normal 5′ splice site of pIgR exon 4 was weakened by the 5′SS point mutation, from a score of 89 to 78, a cryptic 5′ splice site located 496 nt upstream from the authentic splice site, which has a score of 76, was activated (Fig. 8). While activation of cryptic splice sites has commonly been seen in the presence of 5′ splice site mutations, the activated site is usually located within 100 nt of the authentic site (44). Thus, it is surprising that a site so far from the authentic site is used in the pIgR exon; this likely reflects the negative impact that large exon size has on the efficiency of splicing. The exon created by the cryptic splice site is 158 nt, close to the median internal exon size and likely to be more easily recognized by the spliceosomal apparatus. Also, when another potential cryptic 5′ splice site further upstream was mutated to better match the consensus site, from a score of 69 to 90, this site created a 108 nt exon and was also used as an authentic splice site. When the cryptic and authentic sites were similarly matched to the consensus sequence, in pIgR pt2 and 5′SS (Fig. 8), the authentic site was still used more frequently than the cryptic site, despite the differences in exon size. Clearly, other features besides the match to the consensus sequence, such as exonic and intronic sequences acting positively and/or negatively, as well as the size of the exon, contribute to the relative use of these two sites.
It is interesting that pIgR exon 4 is the only example of an exon in an Ig superfamily member that encodes two Ig-like domains; such domains are usually encoded by single exons (25). This suggests that during evolution of the pIgR gene two separate exons may have fused to create one large exon. Since large exons are rare, exon fusion events to create exons >400 nt must either be rare or not well tolerated, perhaps due to constraints on exon size. Our results suggest that multiple criteria, such as strong splice sites, the presence of ESE sequences and the absence of internal potential cryptic splice sites, would determine whether two fused exons could be successfully maintained as a large exon.
Despite having an abnormally large size, pIgR exon 4 is efficiently spliced in most mammals. The one exception is in the rabbit pIgR gene, where exon 4 is an alternative exon, being omitted from the mRNA ∼40% of the time (23). The sequence of the rabbit gene is the most divergent of the known pIgR cDNA sequences (mouse, human, rat and bovine; 21); it is intriguing that at least some of the sequence differences would be expected to affect splicing when judged by the data presented here. For example, the rabbit exon 4 does not contain the cryptic 5′ splice site used in the mouse exon, five of the seven purine-rich sequences are interrupted by pyrimidines, only one of the five A/C-rich sequences have been conserved and the 5′ splice site sequence matches that of the 5′SS mutation that we created in the mouse pIgR exon. These observations are consistent with our proposal that efficient splicing of this large exon is due to multiple features of the mouse pIgR pre-mRNA and, likely, the other organisms that constitutively splice exon 4. In addition, the rabbit contains several potential cryptic 5′ splice site sequences that have scores close to or better than the authentic splice site. It is likely that there are also exonic sequences that act to keep these potential splice sites silent, although a detailed RT–PCR analysis has not been performed with the rabbit RNA to determine to what extent they are suppressed. Further comparison of the pIgR exon between rabbit and other species could serve as a model to elucidate how large exons are constitutively spliced, identify potential ESE activity in the mouse exon and examine the role of these ESE sequences in splicing unusually large exons.
The sequences reported in this paper have been submitted to GenBank, accession numbers AF261083 (human pIgR exon4/intron 4) and AF261084 (mouse pIgR fragment used in this study containing exon 4 and portions of introns 3 and 4).
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Tim Boze for expert technical assistance with early parts of this work and Charlotte Kaetzel, Brett Spear, Brian Rymond, Miles Wilkinson, Tom Cooper and Cam Dingle for helpful comments on the manuscript. This work was supported by grants MCB-9507513 and MCB-9808637 from the National Science Foundation.
DDBJ/EMBL/GenBank accession nos AF261083 and AF261084
References
- 1.Hawkins J.D. (1988) A survey of intron and exon lengths. Nucleic Acids Res., 16, 9893–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berget S.M. (1995) Exon recognition in vertebrate splicing. J. Biol. Chem., 270, 2411–2414. [DOI] [PubMed] [Google Scholar]
- 3.Cooper T.A. and Ordahl,C.P. (1989) Nucleotide substitutions within the cardiac troponin T alternative exon disrupt pre-mRNA alternative splicing. Nucleic Acids Res., 17, 7905–7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dominski Z. and Kole,R. (1991) Selection of splice sites in pre-mRNAs with short internal exons. Mol. Cell. Biol., 11, 6075–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tian H. and Kole,R. (1995) Selection of novel exon recognition elements from a pool of random sequences. Mol. Cell. Biol., 15, 6291–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sterner D.A., Carlo,T. and Berget,S.M. (1996) Architectural limits on split genes. Proc. Natl Acad. Sci. USA, 93, 15081–15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robberson B.L., Cote,G.J. and Berget,S.M. (1990) Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell. Biol., 10, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Black D.L. (1992) Activation of C-src neuron-specific splicing by an unusual RNA element in vivo and in vitro. Cell, 69, 795–807. [DOI] [PubMed] [Google Scholar]
- 9.Ryan K.J. and Cooper,T.A. (1996) Muscle-specific splicing enhancers regulate inclusion of the cardiac troponin T alternative exon in embryonic skeletal muscle. Mol. Cell. Biol., 16, 4014–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu R., Teng,J. and Cooper,T.A. (1993) The cardiac troponin T alternative exon contains a novel purine-rich positive splicing element. Mol. Cell. Biol., 13, 3660–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L., Ashiya,M., Sherman,T.G. and Grabowski,P.J. (1996) Essential nucleotides direct neuron-specific splicing of γ2 pre-mRNA. RNA, 2, 682–698. [PMC free article] [PubMed] [Google Scholar]
- 12.Black D.L. (1991) Does steric interference between splice sites block the splicing of a short c-src neuron-specific exon in non-neuronal cells. Genes Dev., 5, 389–402. [DOI] [PubMed] [Google Scholar]
- 13.Miki Y., Swensen,J., Shattuck-Eidens,D., Futreal,P.A., Harshman,K., Tavtigian,S. et al. (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science, 266, 66–71. [DOI] [PubMed] [Google Scholar]
- 14.Humphrey M.B., Bryan,J., Cooper,T.A. and Berget,S.M. (1995) A 32-nucleotide exon-splicing enhancer regulates usage of competing 5′ splice sites in a differential internal exon. Mol. Cell. Biol., 15, 3979–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barthels D., Vopper,G. and Wille,W. (1988) NCAM-180, the large isoform of the neural cell adhesion molecule of the mouse, is encoded by an alternatively spliced transcript. Nucleic Acids Res., 16, 4217–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tacke R. and Gordis,C. (1991) Alternative splicing in the neural cell adhesion molecule pre-mRNA: regulation of exon 19 skipping depends on the 5′-splice site. Genes Dev., 5, 1416–1429. [DOI] [PubMed] [Google Scholar]
- 17.Côté J., Simard,M.J. and Chabot,B. (1999) An element in the 5′ common exon of the NCAM alternative splicing unit interacts with SR proteins and modulates 5′ splice site selection. Nucleic Acids Res., 27, 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kulseth M.A., Krajci,P., Myklebost,O. and Rogne,S. (1995) Cloning and characterization of two forms of bovine polymeric immunoglobulin receptor. DNA Cell Biol., 14, 251–256. [DOI] [PubMed] [Google Scholar]
- 19.Krajci P., Solberg,R., Sandberg,M., Oyen,O., Jahnsen,T. and Brandtzaeg,P. (1989) Molecular cloning of the human transmembrane secretory component (poly-Ig receptor) and its mRNA expression in human tissues. Biochem. Biophys. Res. Commun., 158, 783–789. [DOI] [PubMed] [Google Scholar]
- 20.Banting G., Brake,B., Braghetta,P., Luzio,J.P. and Stanley,K.K. (1989) Intracellular targeting signals of polymeric immunoglobulin receptors are highly conserved between species. FEBS Lett., 254, 177–183. [DOI] [PubMed] [Google Scholar]
- 21.Piskurich J.F., Blanchard,M.H., Youngman,K.R., France,J.A. and Kaetzel,C.S. (1995) Molecular cloning of the mouse polymeric Ig receptor: functional regions of the molecule are conserved among five mammalian species. J. Immunol ., 154, 1735–1747. [PubMed] [Google Scholar]
- 22.Adamski F.M. and Demmer,J. (1999) Two stages of increased IgA transfer during lactation in the marsupial, Trichosurus vulpecula (Brushtail Possum). J. Immunol ., 162, 6009–6015. [PubMed] [Google Scholar]
- 23.Deitcher D.L. and Mostov,K.E. (1986) Alternate splicing of rabbit polymeric immunoglobulin receptor. Mol. Cell. Biol., 6, 2712–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mostov K. and Kaetzel,C.S. (1998) Chapter 12. In Ogra,P.L., Mestecky,J., Lamm,M.E., Strober,W., McGhee,J.R. and Bienstock,J. (eds) Handbook of Mucosal Immunology. Academic Press, San Diego, CA.
- 25.Williams A.F. and Barclay,A.N. (1988) The immunoglobulin superfamily—domains for cell surface recognition. Annu. Rev. Immunol., 6, 381–405. [DOI] [PubMed] [Google Scholar]
- 26.Bruce S.R., Kaetzel,C.S. and Peterson,M.L. (1999) Cryptic intron activation within the large exon of the mouse polymeric immunoglobulin gene: cryptic splice sites correspond to protein domain boundaries. Nucleic Acids Res., 27, 3446–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarkar G. and Sommer,S.S. (1990) The ‘megaprimer’ method of site-directed mutagenesis. Biotechniques, 8, 404–407. [PubMed] [Google Scholar]
- 28.Wang J. and Wilkinson,M.F. (2000) Site-directed mutagenesis of large (13-kb) plasmids in a single-PCR procedure. Biotechniques, 29, 976–978. [DOI] [PubMed] [Google Scholar]
- 29.Spear B.T. and Tilghman,S.M. (1990) Role of α-fetoprotein regulatory elements in transcriptional activation in transient heterokaryons. Mol. Cell. Biol., 10, 5047–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kushiro A. and Sato,T. (1997) Polymeric immunoglobulin receptor gene of mouse: sequence, structure and chromosomal location. Gene, 204, 277–282. [DOI] [PubMed] [Google Scholar]
- 31.Senapathy P., Shapiro,M.B. and Harris,N.L. (1990) Splice junctions, branch point sites and exons: sequence statistics, identification and applications to genome project. Methods Enzymol., 183, 252–278. [DOI] [PubMed] [Google Scholar]
- 32.Eckhart L., Ban,J., Ballaun,C., Weninger,W. and Tschachler,E. (1999) Reverse transcription-polymerase chain reaction products of alternatively spliced mRNAs form DNA heteroduplexes and heteroduplex complexes. J. Biol. Chem., 274, 2613–2615. [DOI] [PubMed] [Google Scholar]
- 33.Fu X.-D. (1995) The superfamily of arginine/serine-rich splicing factors. RNA, 1, 663–680. [PMC free article] [PubMed] [Google Scholar]
- 34.Manley J.L. and Tacke,R. (1996) SR proteins and splicing control. Genes Dev., 10, 1569–1579. [DOI] [PubMed] [Google Scholar]
- 35.Coulter L.R., Landree,M.A. and Cooper,T.A. (1997) Identification of a new class of exonic splicing enhancers by in vivo selection. Mol. Cell. Biol., 17, 2143–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaal T.D. and Maniatis,T. (1999) Selection and characterization of pre-mRNA splicing enhancers: identification of novel SR protein-specific enhancer sequences. Mol. Cell. Biol., 19, 1705–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlo T., Sterner,D.A. and Berget,S.M. (1996) An intron splicing enhancer containing a G-rich repeat facilitates inclusion of a vertebrate micro-exon. RNA, 2, 342–353. [PMC free article] [PubMed] [Google Scholar]
- 38.McCullough A.J. and Berget,S.M. (1997) G triplets located throughout a class of small vertebrate introns enforce intron borders and regulate splice site selection. Mol. Cell. Biol., 17, 4562–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka K., Watakabe,A. and Shimura,Y. (1994) Polypurine sequences within a downstream exon function as a splicing enhancer. Mol. Cell. Biol., 14, 1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiara M.D. and Reed,R. (1995) A two-step mechanism for 5′ and 3′ splice-site pairing. Nature, 375, 510–513. [DOI] [PubMed] [Google Scholar]
- 41.Watakabe A., Tanaka,K. and Shimura,Y. (1993) The role of exon sequences in splice site selection. Genes Dev., 7, 407–418. [DOI] [PubMed] [Google Scholar]
- 42.Liu H.-X., Zhang,M. and Krainer,A.R. (1998) Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev., 12, 1998–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Engelbrecht J., Knudsen,S. and Brunak,S. (1992) G + C-rich tract in 5′ end of human introns. J. Mol. Biol., 227, 108–113. [DOI] [PubMed] [Google Scholar]
- 44.Nakai K. and Sakamoto,H. (1994) Construction of a novel database containing aberrant splicing mutations of mammalian genes. Gene, 141, 171–177. [DOI] [PubMed] [Google Scholar]