

Abstract
Ecological genomics aims to understand the functional association between environmental gradients and the genes underlying adaptive traits. Many genes that are identified by genome-wide screening in ecologically relevant species lack functional annotations. Although gene functions can be inferred from sequence homology, such approaches have limited power. Here, we introduce ecological regulatory genomics by presenting an ontology-free gene prioritization method. Specifically, our method combines transcriptome profiling with high-throughput cis-regulatory sequence analysis in the water fleas Daphnia pulex and Daphnia magna. It screens coexpressed genes for overrepresented DNA motifs that serve as transcription factor binding sites, thereby providing insight into conserved transcription factors and gene regulatory networks shaping the expression profile. We first validated our method, called Daphnia-cisTarget, on a D. pulex heat shock data set, which revealed a network driven by the heat shock factor. Next, we performed RNA-Seq in D. magna exposed to the cyanobacterium Microcystis aeruginosa. Daphnia-cisTarget identified coregulated gene networks that associate with the moulting cycle and potentially regulate life history changes in growth rate and age at maturity. These networks are predicted to be regulated by evolutionary conserved transcription factors such as the homologues of Drosophila Shavenbaby and Grainyhead, nuclear receptors, and a GATA family member. In conclusion, our approach allows prioritising candidate genes in Daphnia without bias towards prior knowledge about functional gene annotation and represents an important step towards exploring the molecular mechanisms of ecological responses in organisms with poorly annotated genomes.
Keywords: cis-regulation, omics data integration, prioritization, crustacea endocrine signaling, functional enrichment, motif discovery
Introduction
Understanding how organisms respond and adapt to their natural environments is a key objective of biology (Feder and Mitchell-Olds 2003). Evolutionary and ecological functional genomics is a discipline of biology that arose from the application of modern sequencing technologies to uncover genetic variation under natural selection (Feder and Mitchell-Olds 2003; Andrew et al. 2013; Pardo-Diaz et al. 2015). In its search for genes and other elements of the genome that underpin adaptive traits, an expanding suite of organisms is emerging as ecological model species, which are accessible to both laboratory and field studies along defined environmental gradients. Discoveries made from ecological genomics include adaptive variation arising from both de novo (Karasov et al. 2010) and standing genetic variation (Colosimo et al. 2005); phenotypes that evolve by selection on multiple small effect loci (Burke et al. 2010) or on a few loci of major effect (Lamichhaney et al. 2016); and new phenotypes resulting from major effect mutations that alter protein coding sequences (Protas et al. 2006) or gene regulation (Manceau et al. 2011). Moreover, tools and approaches that were once reserved to traditional biomedical model species, such as genome-wide forward or reverse genetics, are increasingly being applied to ecological model species (Ekblom and Galindo 2010; Stapley et al. 2010; Alvarez et al. 2015; Pardo-Diaz et al. 2015).
Genome-wide gene expression profiling is a particularly powerful method to identify regulated genes under different ecological conditions and linked to phenotypic change (Purdy et al. 2010). The set of all genes that are expressed in a given condition and in a particular cell or tissue type (the transcriptome) is itself a “molecular phenotype” (Hughes et al. 2000). This molecular phenotype is under the control of transcription regulation that genetically varies, based on the ecological and evolutionary history of populations (Dalziel et al. 2009). Hence, the transcriptional responses of natural isolates to environmental perturbation are a rich source of both phenotypic and genotypic information about the mechanisms of adaptation (Todd et al. 2016). The use of next generation sequencing for data-driven genome-wide investigations of transcriptomes (RNA-Seq, Wang et al. 2009) is increasingly used in studies on ecological model species (Todd et al. 2016), because this method is comprehensive and does not assume prior knowledge of the functional elements of the genome, nor of the gene annotations. However, the biological interpretation of RNA-Seq data obtained from ecological model species remains challenging, because these investigations often provide the first evidence of the biological functions of genes within an ecological context (Colbourne et al. 2011; Alvarez et al. 2015). The challenge is particularly acute for lineage-specific genes having no identified sequence homology to the functionally annotated genes of related model species (Colbourne et al. 2011). Otherwise, the genes that have known homologues are often analyzed using Gene Ontology or pathway enrichment tools to test for the statistical significance of their enrichment within a coexpressed gene set (Primmer et al. 2013; Kanehisa et al. 2012). Although this homology-based approach makes use of the vast knowledge obtained from traditional model species, it makes many assumptions on the reliability of ascribing functional orthology to gene family members across large evolutionary distances (Primmer et al. 2013; Pavey et al. 2012).
Another powerful method at identifying ecologically important elements of genomes is the analysis of cis-regulatory sequences, also referred to as motif discovery (reviewed by Hardison and Taylor 2012; Yáñez-Cuna et al. 2013; Aerts 2012). Motif discovery finds enriched motifs in the promoters or upstream sequences of coexpressed genes, which may represent shared binding sites for a particular transcription factor (TF). If such a TF is known, direct TF-target interactions can be inferred leading to the prediction of gene regulatory networks (GRN). This concept is proven for genetic model species and human, by producing experimentally validated predictions (Van Loo et al. 2008; Warner et al. 2008; Aerts et al. 2010). For ecological model species, however, little is known about gene regulation and its mechanisms. On one hand, rapid turnover of regulatory sequences (Schmidt et al. 2010; Weirauch and Hughes 2010; Sayou et al. 2014; Baker et al. 2011) and complete rewiring of GRNs (Tsong et al. 2006) across evolutionary time scales have been documented. On the other hand, evidence shows that TFs, their binding specificities and even complete GRNs can be highly conserved between distantly related species (Del Bene et al. 2007; Nitta et al. 2015; Weirauch et al. 2014; Villar et al. 2015; Guertin et al. 2010; Green et al. 2015). Therefore, it remains unknown if and to what extend knowledge about the regulatory code (e.g., TF binding specificities) obtained from genetic model species can be transferred to ecological model species.
Here, we propose a motif discovery method for ecological model species, namely for the water fleas Daphnia pulex and Daphnia magna. Water fleas (Crustacea: Cladocera) are keystone species in lake and pond ecosystems and are intensively used as model organisms in ecology, evolutionary biology, and ecotoxicology (Lampert and Kinne 2011; Miner et al. 2012). They are well-studied for their phenotypic responses to various environmental conditions (see e.g., Miner et al. [2012] and a draft genome sequence is available for both D. pulex and D. magna (Colbourne et al. 2011; Wfleabase). However, the GRNs underlying ecological responses in Daphnia are unknown.
To discover GRNs in D. magna and D. pulex, we developed the method Daphnia-cisTarget that identifies overrepresented cis-regulatory motifs and candidate regulators in a set of coexpressed genes. The approach and algorithm are based on cisTargetX. cisTargetX and its follow-up methods i-cisTarget and evo-cisTarget are successfully applied for motif discovery in fruit fly, human, and mouse (Aerts et al. 2010; Herrmann et al. 2012; Janky et al. 2014; Imrichová et al. 2015; Naval Sanchez 2014). We use Daphnia-cisTarget to computationally predict transcription factors and gene regulatory networks in Daphnia, using RNA-Seq data and TF binding specificities from other species. We validate our method by linking the inferred factors and networks to morphological and life history traits.
Materials and Methods
Daphnia-cisTarget
Daphnia-cisTarget is based on the Drosophila melanogaster-specific method cisTargetX (Aerts et al. 2010). To adapt this method to Daphnia, we used an established protocol and default thresholds that are empirically determined. Our approach includes two major steps: 1) Definition of a motif search space and subsequent genome-wide motif cluster prediction, and 2) motif enrichment analysis on a set of coexpressed genes and target gene prediction. Daphnia-cisTarget is available through a web interface at http://daphniacistarget.aertslab.org/ (last accessed July 17, 2017). To delineate candidate cis-regulatory regions, we used the genome sequences of D. magna and D. pulex together with their gene catalogues. First, we assigned potential regulatory regions to each gene. Those regions include all exons and introns, 3′- and 5′-UTRs, and a region upstream of the transcription start site. The upstream region comprises either 300 bp or 5 kb. If another gene at the same strand falls within this region, the region is terminated at the margin of that upstream gene. Next, we scored the potential cis-regulatory sequences generated in the previous step for homotypic motif clusters with the program Cluster-Buster (Frith et al. 2003). Cluster-Buster uses a Hidden Markov Model and allows regions with clusters of multiple motif matches to receive higher scores than regions with single motif instances. The Cluster-Buster cluster score threshold was set to zero to receive a score for every region, resulting in several scores for each gene. Only the highest score was retained and assigned to the gene. The above procedure results in one score for each gene for a given motif. This score was used to rank all genes. We repeated this scoring and ranking step for each motif in a library of 9713 transcription factor motifs collected from various species (Janky et al. 2014) and compiled the resulting 9713 rankings into a database. The search space delineation and motif scoring steps outlined above were done separately for each of the two Daphnia species (pulex and magna) and the two search space delineations (300 bp and 5 kb), resulting in four databases in total. In the motif enrichment step, the database is queried with a set of coexpressed genes to determine which motifs are enriched in this set as compared with the whole-genome background. For each of the 9713 motif rankings within a queried database, Daphnia-cisTarget calculates a cumulative recovery curve of the input gene set along the ranking (blue curve fig. 1C). It also calculates the area under the recovery curve (AUC) as a measure of enrichment. Because we are mainly interested in highly ranked genes, the AUC is calculated for a fraction of the top ranked genes only. This fraction is delimited by the AUC threshold, which defaults to 3% of the total number of ranked genes (927 for D. pulex and 990 for D. magna). Lowering the threshold increases specificity by favoring motifs with steeper enrichment curves, i.e., where the input gene set is highly enriched at the top of the ranking of all genes. A threshold of 3% has proven sensible for cisTargetX (Potier et al. 2012). We have shown that motif and TF recovery are robust across a wide range of AUC thresholds in a version of cisTargetX for human (Janky et al. 2014). Note that the user can adjust the AUC threshold in Daphnia-cisTarget and compare the impact of different settings. To normalize the AUC, Daphnia-cisTarget calculates first an average recovery curve by taking the mean of all AUC scores across all motifs in the database (red curve fig. 1C), and second the normalized enrichment score as NES = (AUC-AUCmean)/AUCstd, where AUCstd is the standard deviation of the mean AUC. Only motifs with a NES above a specified threshold (default 2.5) are considered enriched. To simplify the output, the enriched motifs are grouped into clusters of similar motifs using STAMP (Mahony and Benos 2007). Motifs assigned to the same cluster are given the same color in the results table. To retrieve an optimal subset of the input gene set as putative target genes, a “leading edge” is determined as the rank position (below the AUC threshold) where the difference between the signal (recovery curve: blue curve fig. 1C) and the background (mean recovery curve across all motifs plus two standard deviations: green curve) is largest. The input genes within this leading edge are predicted as target genes for the given motif. To enable the identification of TFs that putatively bind to the enriched motifs and regulate the predicted target genes, candidate transcription factors are mapped to the motifs. Specifically, these candidate TFs comprise Daphnia genes that are homologous to D. melanogaster genes that have been mapped to the same motifs in the motif2TF database developed by Janky et al. (2014).
Fig. 1.—

Daphnia-cisTarget. (A) Transcription factors (TFs) bind to specific motifs in cis-regulatory regions to control gene expression. (B) This in vivo process can be inferred in silico by combining motif discovery and gene expression data, since genes that are coexpressed and share the same motif are likely to be regulated by the same transcription factor. (C) For a given input gene set, Daphnia-cisTarget generates a cumulative recovery curve for each motif ranking in the database (blue line left panel). The area under this curve (AUC) is calculated as a measure of enrichment (grey area), and those motifs that surpass the AUC cut-off (dotted line right panel) are listed in the output. The red line in the left panel represents the average recovery over the entire motif ranking database. This average plus two standard deviations yields the green line.
Heat Shock Data Set
The D. pulex heat shock signature used for the validation of Daphnia-cisTarget was retrieved from an experiment by Becker et al. (unpublished data; GSE91031). Briefly, 20 °C-acclimated animals (adult females with a body length of 2–2.5 mm, carrying parthenogenetic eggs and embryos), were exposed for 2, 4, or 8 h to either 30 ± 0.2 °C (test condition) or 20 ± 0.3 °C (control condition). After exposure, animals were shock-frozen in liquid nitrogen and short-term stored at −80 °C. For each experimental condition, four independent replicates (50 animals each) were analyzed. Whole-body RNA-levels were measured using a 12-plex 60 nt-oligonucleotide microarray platform, which is described elsewhere (Colbourne et al. 2011). For each treatment, four independent RNA samples were processed and the labeled cDNA were competitively hybridized (test vs. control condition) on four replicate microarrays including dye swaps. To obtain differential gene expression profiles, data were analyzed as described elsewhere (Colbourne et al. 2011). We selected upregulated genes with an FDR ≤ 0.05 as input for Daphnia-cisTarget.
Reanalysis of the Chronic Cyanobacteria Data Set
To recover GRNs that are affected by chronic cyanobacteria exposure, we reanalyzed the RNA-Seq data produced by Schwarzenberger et al. (2014) (SRA177938). Briefly, the authors exposed D. magna to two different cyanobacteria strains, one expressing Microcystin-LR (“toxic;” here named “BX”), and a microcystin-free strain (“less-toxic;” here named “BN”). We mapped the reads with TopHat v2.0.12 (Trapnell et al. 2009) (option –max-multhits 1) against the D. magna reference genome (v2.4) and used HTSeq v0.6.1p1 (Anders et al. 2014) (–stranded = no) for read counting. We calculated differential gene expression with the Bioconductor (Gentleman et al. 2004) R-package DESeq2 v1.6.1 (Love et al. 2014) and applied a cut-off of |log2F C|≥2 and FDR ≤ 0.05 to obtain the lists of differentially expressed genes. We ran Daphnia-cisTarget on the four sets of differentially expressed genes (i.e., up-/downregulated genes of BN & BX treatment), and selected all predicted target genes of the top scoring motif clusters (containing NR, ovo/Svb and GATA motifs). To reduce the number of false positives, we used only the motif-gene mappings of the 300 bp version to construct Cytoscape (Shannon et al. 2003) networks.
Acute Exposure of D. magna to Cyanobacteria
To compare the responses to chronic and acute cyanobacteria exposure in D. magna, we designed a follow-up experiment. Experimental conditions and RNA-Seq data generation are described by Orsini et al. (2016) (SRA272145). Briefly, we exposed 5-days-old juvenile females for 4 h to a unicellular, Microcystin-LR producing strain of M. aeruginosa (strain T4, characterized by van Gremberghe et al. (2009)), and the mutant strain CCAP 1450/1, which lacks the gene to produce the toxic cyanobacterial Microcystin-LR. We used the two D. magna Straus, 1820 genotypes described by Routtu et al. (2014), originating from different habitats (Iinb1: Munich, Germany; Xinb3: Tvärminnen, Finland). Replicates were obtained from three maternal lines (“cohorts”) cultured separately for two generations. For full-body RNA extraction, library preparation and Illumina RNA-Seq sequencing details we refer to Orsini et al. (2016). We processed the reads similarly to the chronic data set. To assess the treatment-specific and cohort effects, respectively, we performed two different differential expression analyses. First, for each clone and cohort, we contrasted the control and each of the two treatment samples separately, resulting in 12 comparisons. Expressed genes that had a mean of <10 reads per exon base position across two samples were excluded from the analysis. This filtering step resulted in about 11,300–14,000 genes having sufficient coverage. We calculated the log2-fold change (log2FC) between each treatment and its control using the Bioconductor (Gentleman et al. 2004) R package DESeq v1.18.0 (Anders and Huber 2010) and ranked the genes according to this value. For subsequent analysis with Daphnia-cisTarget, we used 24 gene sets containing the 1000 most differentially expressed genes (i.e., the 500 most up- and downregulated genes of each comparison, respectively), corresponding to 7–8.8% of the whole ranking. The resulting 24 gene sets were objected to a Daphnia-cisTarget analysis. Predicted target genes for each cluster of GATA, NR, ovo/Svb, Blimp-1, and Grh motifs were pooled, and genes that contained a motif in at least three Daphnia-cisTarget runs (300 bp version) were displayed as a network with Cytoscape (Shannon et al. 2003). Second, we considered the three samples (control and two cyanobacteria treatments) of each cohort as replicates and contrasted the cohorts, which resulted in six comparisons (2 clones × 3 cohorts). We filtered for read coverage of <10 reads per exon base position across the six samples that were to be compared (11,700–13,800 genes retained), and calculated differential expression with DESeq2 v1.6.3 (Love et al. 2014). It is important to note that the three cohorts of the I-clone (1–3) are independent from the three X-clone cohorts (1′–3′), as the experiments were carried out on different days. Sample details including SRA accession numbers are available as supplementary table S1, Supplementary Material online.
Expression Data and Gene Network Visualization
The heat maps in figures 3A and 4B and D and supplementary figures S2C and S3A, Supplementary Material online were generated with the MultiExperiment Viewer of the TM4 software suite (Saeed et al. 2003). We normalized the read count matrix obtained by HTSeq by removing all genes having an expression across all samples in the lowest 40% quantile. The quantile filtered data were imported into the MultiExperiment Viewer, where they were library size corrected, log2-transformed, and median-centered per row (i.e., gene). The read mapping in supplementary figure S3C, Supplementary Material online was displayed with the integrative genomics viewer IGV v2.3.70 (Robinson et al. 2011). Gene regulatory networks were generated with the software Cytoscape v3.3.0 (Shannon et al. 2003).
Fig. 3.—

Chronic cyanobacteria treatment in Daphnia magna. The upregulated genes (top, red network) contain nuclear receptor (NR) and ovo/Shavenbaby (ovo/Svb) motifs, along with transcription factors that potentially target those motifs (red boxes). The downregulated gene sets (bottom, blue network) are enriched for GATA motifs. (A) The heatmap shows median-centered expression levels (red: upregulation, blue: downregulation). For illustration purpose, only a selection of differentially expressed genes is displayed. The gene names refer to D. melanogaster homologies (“-”: no homology) or manual annotation (Dam*). (B) Sequence logos of Daphnia-cisTarget top scoring motifs, enrichment curve and normalized enrichment score (NES) of those motifs. The curly brackets indicate which genes in panel A are enriched for the respective motif in panel B. (C) Gene regulatory network including all putative target genes of one or several nuclear receptors, ovo/Svb and GATA factors. The node color intensity corresponds to the log2-fold change in the BX treatment (node center) and BN treatment (node border). The labels at gene subsets indicate gene ontology enrichment results. In subsets without gene ontology labels, genes arranged in left semi-circles do not have annotations based on homologies to D. melanogaster.
Fig. 4.—

Motif recovery, sequence homology and literature curation uncover links to moulting and growth regulation. Grey edges connect Daphnia-cisTarget motifs with their predicted targets, dotted purple edges indicate literature-curated interactions and solid purple edges literature connections that were confirmed by Daphnia-cisTarget. (A) Daphnia-cisTarget establishes a connection between ecdysone-signaling and the formation of cuticle and cuticular structures in D. magna. The curated network contains genes involved in trichome formation, developmental timing and cuticle formation. References to fruit fly literature: 1) Yoshiyama et al. (2006); 2) Gauhar et al. (2009); 3) Agawa et al. (2007); 4) White et al. (1997); 5) Chanut-Delalande et al. (2014); 6) Kondo et al. (2010); 7) Menoret et al. (2013); 8) Lee and Adler (2004); 9) Gangishetti et al. (2012); 10) Jang et al. (2009); 11) Ono et al. (2006). (B) The heatmap demonstrates coexpression of the genes in the network. Note the anticorrelation of ftz-f1 and its transcriptional repressor Blimp-1 (red box). (C) D. magna might react to poor food quality of cyanobacteria by downregulation of Insulin/IGF-signaling (IIS). Among the downregulated genes are many neuropeptides, including the insulin-related peptide homologue DamIrp2, a neuropeptide receptor, and proteases that activate neuropeptides through cleavage. In insects and crayfish, those genes regulate feeding motivation, nutrient storage and starvation resistance. Homologues of targets of the D. melanogaster midgut differentiation factor GATAe (Okumura et al. 2007) (arrows labeled “13”) are enriched. DamJHE and DamJHBP hint at the action of juvenile hormone (JH). (D) The heatmap demonstrates coexpression of DamGATAe (red box) with its putative target genes, although DamGATAe itself is not significantly differentially expressed. References to known interactions: 1) Honegger et al. (2008) (fruit fly); 2) Huang et al. (2015) (fruit fly); 3) Hwang et al. (2000) (fruit fly); 4) Buch et al. (2008) (fruit fly); 5) Ida et al. (2011) (fruit fly); Maeda et al. (2015) (blowfly); 6) Reiher et al. (2011) (fruit fly); 7) Britton et al. (2002) (fruit fly); 8) Veenstra (2015) (crayfish); 9) Veenstra et al. (2008) (fruit fly); Fu et al. (2007) (crab); 10) Chen et al. (2014) (crab); 11) Lorenz et al. (1995) (cricket); 12) Hua et al. (1999); Davis (2003); Yamanaka et al. (2010) (moths); 13) Okumura et al. (2007) (fruit fly); 14) Kataoka et al. (1989) (moth); 15) Kaneko and Hiruma (2015) (moth); 16) Verlinden et al. (2015) (insects); 17) Natzle et al. (1988) (fruit fly); 18) Nijhout et al. (2014) (insects); 19) Mirth et al. (2014) (insects); 20) Kethidi et al. (2005) (fruit fly); 21) Zhao and Campos (2012) (fruit fly).
Functional Annotation and Gene Homology between Species
Protein homologies between Daphnia species were determined by blasting all D. pulex predicted proteins against the D. magna protein catalogue using the NCBI program blastp v2.2.25+ (Altschul et al. 1997). Only the best hit in terms of E-score and total bit score was retained. The gene models of D. magna were functionally annotated by blasting the translated genes against the D. melanogaster protein catalogue v6.02 (blastp, E value cut-off 1 × 10−20, best hit only). D. melanogaster orthologues for the D. pulex gene catalogue were retrieved from Ensembl BioMart (Kinsella et al. 2011). This resulted in 8268 D. magna and 8524 D. pulex genes mapping to 5824 and 7270 D. melanogaster genes, respectively. As gene name, we kept the gene symbol of the closest D. melanogaster homologue. If a closer inspection of the gene revealed a different function or if the gene model had to be modified, the genes were given names preceded by “Dam” for D. magna. We italicize gene names (DamSvb) and use roman type for gene products (DamSvb) and motif names (ovo/Svb).
Sequence Alignment and Phylogenetic Tree
We determined homology between protein sequences through sequence alignment with the online program Clustal Omega v1.2.1 (Sievers et al. 2011). For the transcription factors discussed in this paper, we retrieved the D. melanogaster DNA-binding domain sequences from the CIS-BP database v1.02 (Weirauch et al. 2014), aligned them to the D. magna predicted protein sequence with Clustal Omega, and calculated amino acid identities.
Gene Ontology (GO)
GO-term enrichment analysis was carried out using the Cytoscape plug-in BiNGOv3.0.3 (Maere et al. 2005). Blasting D. magna against D. melanogaster protein sequences frequently yielded several water flea genes that map to the same fruit fly gene and thus share the same annotation. Therefore, we created a custom background for this analysis. To this end, we assigned the GO-terms attributed to D. melanogaster genes to the respective homologous D. magna genes (8268 D. magna genes had sufficient homology) and used those as background set (option “select organism/annotation”). Gene ontologies for D. melanogaster were obtained from the Gene Ontology Consortium (Blake et al. 2015). BiNGO was used with the settings “ontology file”: go.obo; “statistical test”: hypergeometric; “multiple testing correction”: Benjamini and Hochberg; False Discovery Rate (FDR) correction with a significance level of 0.05. Otherwise, default settings were used.
Gene Set Enrichment Analysis (GSEA)
To determine whether the differentially expressed genes from the chronic cyanobacteria experiment are also affected by the acute cyanobacteria treatment, we used the software GSEA v2.1.0 (Subramanian et al. 2005) with the default command-line options, except for “xtools.gsea.GseaPreranked”, “scoring_scheme classic” and “nperm 10,000”. We considered enrichments with FWER < 0.01 and |NES|>2 significant. Enrichments were visualized in supplementary figure S5B, Supplementary Material online as density trace with the R-package beanplot v1.2 (Kampstra 2008).
Overrepresentation Analysis
To test whether D. magna homologues of known D. melanogaster GATAe and Svb targets were enriched among the differentially expressed genes from the chronic cyanobacteria data, we calculated the probability of overlap with the hypergeometric distribution function phyper in R. As background we used the number of genes that have D. melanogaster homology (i.e., 8268).
Explained Variation
To visualize the variation that can be explained by the GRNs predicted with Daphnia-cisTarget, we generated cumulative recovery curves. To this end, we sorted the STAMP-predicted motif clusters by the NES score of their highest-ranking members, and generated a cumulative recovery curve by counting the number of unique predicted target genes (supplementary table S5, Supplementary Material online). We also calculated the percentage of input gene sets recovered by 1) the highest scoring motif cluster, 2) by motif clusters containing GATA, NR, ovo/Svb, Grh, and Blimp-1 motifs, and 3) by all motif clusters recovered with Daphnia-cisTarget, representing the maximal variation that can be explained with motif discovery.
Data Availability
The genomic resources and databases used in this study are listed in supplementary table S4, Supplementary Material online.
Results
Design and Implementation of Daphnia-cisTarget
The motif discovery method Daphnia-cisTarget is comprised of two steps: a computationally intensive motif scoring and ranking step, and a fast motif recovery step (see Materials and Methods). For the motif scoring step, we first define the search space for potential cis-regulatory sequences. We tested our method by delineating two different intervals: a small search space of 300 bp, and a larger search space of 5 kb sequence upstream of the TSS. The average intergenic space is 1,800 bp in D. magna, with 44% and 88% of the genes having an upstream sequence smaller than 300 bp and 5 kb, respectively. For D. pulex, those values are 3,200 bp, 21% and 83%. The defined sequence search spaces capture thus a large proportion of the intergenic space. Next, we scored those sequences for the occurrence of specific DNA sequence motifs. To this end, we used motifs derived from different species including yeast, fruit fly and human (Janky et al. 2014). In the subsequent motif recovery step, a gene set is used to query the database in order to find the motifs that are enriched in this set over the background set of all genes. This gene set can be for, example, a signature of differentially expressed genes, a coexpression cluster, or genes associated with a certain functional annotation. For each motif ranking in the database, a cumulative recovery curve is generated (fig. 1). The motifs with the strongest representation of input genes at the top of their corresponding gene ranking receive the strongest enrichment score (NES). Since our motif collection contains a number of similar motifs, the significantly enriched motifs are clustered based on motif similarity, to facilitate interpretation. Furthermore, to enable GRN reconstruction, an optimal subset of candidate target genes is selected for each motif (for details see Materials and Methods). Finally, Daphnia-cisTarget also provides a list of genes encoding TFs that putatively bind to the enriched motifs and regulate the predicted target genes, based on the motif2TF database developed by Janky et al. (2014). Daphnia-cisTarget is available via a web interface at http://daphniacistarget.aertslab.org/ (last accessed July 17, 2017).
Validation of Daphnia-cisTarget Using the Heat Shock Response in D. pulex
To validate Daphnia-cisTarget, we generated a set of coexpressed genes under heat shock (Becker et al., unpublished data). The heat shock response is controlled by the Heat Shock Factor (HSF) and evolutionary highly conserved from yeast to human (Liu et al. 1997). We acclimatized D. pulex to 20 °C, followed by an exposure to 30 °C for 2, 4, and 8 h. RNA was extracted and analyzed using a Daphnia-specific microarray platform (see Materials and Methods). Differential expression analysis (False discovery rate (FDR)≤0.05) resulted in heat shock signatures containing 314, 350, and 321 upregulated genes, respectively, for the three treatment durations (supplementary table S3, Supplementary Material online). To also test the D. magna version of Daphnia-cisTarget with these signatures, we used BLAST homology to converted the gene sets into 266, 297, and 274 D. magna genes, respectively. Given the conserved role of HSF in regulated target genes in response to heat shock, and given the presence of a HSF homologue in both Daphnia genomes (pulex: JGI_V11_213340; magna: mu8AUGepir7s01348g56; both 63% sequence identity to DNA binding domain of D. melanogaster Hsf), we expected to find the HSF motif enriched across the heat shock signatures, in agreement with observations in other species, where the HSF motif is enriched among heat shock gene signatures (e.g., nematodes GuhaThakurta et al. [2002] and mammals Mahat et al. [2016]).
As expected, Daphnia-cisTarget identified known heat shock factor motifs as the top scoring motifs (fig. 2A), both for the D. pulex gene set, and the D. magna set of homologous genes. The 300 bp version performs slightly better on this data set than the 5 kb version (supplementary fig. S1, Supplementary Material online). In D. melanogaster, the majority of genomic DNA regions bound by HSF under heat shock are located either directly within genes or within 1250 bp upstream of their TSS (Gonsalves et al. 2011). If the same holds true for Daphnia, scoring of a smaller upstream region for the 300 bp version of Daphnia-cisTarget increases the signal-to-noise ratio compared with the 5 kb version. However, for the recovery of TFs with remote binding sites, the 5 kb version might yield higher sensitivity. For the 2 h heat shock signature, the normalized enrichment scores of the best motif were 10.43 for D. pulex and 6.77 for D. magna for the 300 bp version, and 8.91 and 6.17, respectively, for the 5 kb version. The best motif clustered together with 20 other HSF motifs based on motif similarity (D. pulex, 300 bp version; fig. 2A). To construct a gene regulatory network, we combined the predicted target genes of those 21 motifs to a set of 95 candidate HSF targets (fig. 2B, supplementary table S3, Supplementary Material online). Out of the genes that are putatively regulated by HSF in D. pulex, 47% have no sequence-based homology to D. melanogaster genes thereby lacking functional annotation. However, our results are the first to provide evidence that these genes may play a role in the primary response to heat shock. First, the unknown genes are upregulated after a heat shock together with known heat shock-inducible genes such as Hsp60. Second, they are potentially directly regulated by the HSF because of the presence of HSF motifs within the search space of the gene models. With this proof of principle, we demonstrate that our method can be useful to identify relevant motifs and transcription factors, define gene regulatory networks in Daphnia gene signatures, and facilitate the work of experimentally annotating genes of ecological model species by ascribing functions based on their specific responses to ecological conditions.
Fig. 2.—

The heat shock factor as a common regulator in heat shock response. We here show Daphnia-cisTarget results for the upregulated genes in a thermal stress experiment (D. pulex exposed for 2 h to a 10 °C temperature increase, whole animal mRNA measured with microarrays). (A) Daphnia-cisTarget returns a heat shock factor (HSF) motif as the top scoring motif (the mouse motif M01244 of the TRANSFAC© Professional database [Matys et al. 2006]), followed by 14 HSF motifs derived from different species such as yeast, fruit fly and human. Those motifs are clustered as highly similar (indicated through same background color). The Daphnia-cisTarget results also show the motif logo (“enriched motif”), the Normalized Enrichment Score (NES), the recovery curve, the D. pulex HSF homologue JGI_V11_213340 derived through sequence homology as candidate transcription factor (TF) and the predicted target genes (“#targets”). The second and third best motif clusters are assigned to a basic-leucine zipper TF CrebB and a range of homeobox TFs, respectively. The remaining 122 recovered motifs that surpass the NES threshold of 2.5 are not shown. (B) The gene regulatory network controlled by the HSF in D. pulex is reconstructed by merging the 95 predicted targets of all HSF motifs recovered by Daphnia-cisTarget. Gene names are derived through homology to D. melanogaster genes; D. pulex genes mapping to the same D. melanogaster gene are numbered (e.g., slbo#1, slbo#2, etc.).
Deciphering Gene Regulatory Networks Underlying D. magna Response to Cyanobacteria
Having demonstrated the utility of Daphnia-cisTarget, we next applied our method to study the response of D. magna to cyanobacteria. In a chronic exposure experiment, Schwarzenberger et al. exposed a cyanobacteria-tolerant D. magna clone to a M. aeruginosa strain containing the toxin Microcystin-LR (termed hereafter BX, “toxic strain”), and a microcystin-free mutant strain (referred to as: BN, “less-toxic strain”) and compared both treatments to untreated control animals. The daphnids were exposed in triplicates until they reached maturity (control: 7 days, cyanobacteria treatments: 11 days), and whole-body mRNA levels were measured with RNA-Seq. We re-analyzed those sequencing data for differential expression and obtained comparable results to Schwarzenberger et al., although the absolute numbers of differentially expressed genes differed due to the different analysis methods applied (see Materials and Methods). We found 352 and 269 upregulated and 501 and 78 downregulated genes in BX and BN treatment, respectively (|log2FC| ≥ 1; FDR ≤ 0.05). In general, many genes (216) were upregulated to a similar extent in both treatments, whereas many downregulated genes (433) were stronger differentially expressed in the BX than in the BN treatment (fig. 3A). Next, we assigned functions to the differentially expressed genes using blast homology to D. melanogaster genes, and performed a Gene Ontology term enrichment analysis using these assignments. Similar to the results presented in Schwarzenberger et al. (2014), we found that the BX downregulated genes are enriched for proteolysis (adj. P value: 4.62 × 10−10), chitin catabolic process (1.56 × 10−6) and phototransduction (3.51 × 10−4). The upregulated genes are enriched for glycoprotein metabolic process (2.16 × 10−3), nonsensory hair organization (1.11 × 10−2), and proteolysis (1.27 × 10−2) (fig. 3C).
To reconstruct the gene regulatory networks that underlie the observed gene expression patterns, we performed a motif discovery analysis with Daphnia-cisTarget (using the D. magna 300 bp version). This analysis returned three main clusters of enriched motifs (fig. 3B). The upregulated gene sets in both treatments were enriched for motifs that are bound by nuclear receptors (NRs) (top motifs with NES 5.30 in BN and 4.68 in BX) and motifs corresponding to the Drosophila transcription factor Ovo/Shavenbaby (Svb) (BN: 3rd motif, NES 5.02; BX: 70th motif, NES 2.86). The downregulated genes sets were dominated by GATA motifs (top motifs in BN and BX; NES 5.37 and 8.89, respectively). Interestingly, we found upregulated genes encoding TFs that potentially bind to those motifs, namely two genes encoding nuclear receptors (DamHR3 and DamE78) and a gene that is reciprocally homologous to D. melanogaster ovo/svb (fig. 3A). The predicted protein sequences of DamHR3, DamE78, and D. magna ovo/Svb show 95%, 97%, and 90% sequence identity with the DNA binding domains of D. melanogaster Hr46, Eip78C and ovo/Svb, respectively (supplementary file S4, Supplementary Material online). Such high similarities of the DNA binding domain are strongly indicative of conserved binding sites according to Weirauch et al. (2014). Both cyanobacteria treatments elicited similar responses both in terms of differential gene expression and enriched motifs. The difference between the toxic wild-type M. aeruginosa strain and the less-toxic mutant strain is the incapability of the latter to express the phosphatase inhibitor Microcystin-LR. Nevertheless, both strains reduce the somatic growth of D. magna as compared with a control green algae diet (Schwarzenberger et al. 2014). This growth rate reduction might be attributed to the low nutritional quality of cyanobacteria (Martin-Creuzburg et al. 2008) or other cyanobacterial toxins such as the protease inhibitor microviridin J (Rohrlack et al. 2004) and digestive protease inhibitors (Schwarzenberger et al. 2010). It is therefore not surprising that the global transcriptional response to both strains is very similar. In this analysis, we did not attempt to filter for a Microcystin-LR-specific response but focused instead on the general cyanobacteria response. Therefore, we combined the Daphnia-cisTarget results of both cyanobacteria treatments to two modules, one consisting of the upregulated NR & ovo/Svb targets, and one consisting of the downregulated GATA targets (fig. 3C; the genes that are part of the cyanobacteria networks discussed in this paper are listed in table 1 and supplementary table S2, Supplementary Material online). In conclusion, Daphnia-cisTarget identifies the regulators underlying responses of D. magna to an important natural stressor and establishes links between those regulators and putative target genes, without using any functional annotation of the target genes, and relying on the homology of highly conserved transcription factors only.
Table 1.
Members of Gene Regulatory Networks That are Known to be Involved in Growth-Related Processes in Arthropods, are Conserved in Daphnia magna
| Gene Symbol D. magna | Gene ID D. magna | Gene Symbol homologue | Predicted Function | |
|---|---|---|---|---|
| 20E signaling, developmental timing | Spok | mu8AUGep24bs01592g203 | Spoka | 20E synthesis (Ono et al. 2006; Yoshiyama et al. 2006) |
| DamNvd1 | mu8PASAgasmbl_42864 | nvda | ||
| DamHR3 | mu8AUGep24bs00930g63_ mu8AUGapi5s01568g77 | Hr46a | 20E-responsive NR (King-Jones and Thummel 2005) | |
| DamE78 | mu8AUGepir7p2s02190g15 | E78a | 20E-responsive NR (King-Jones and Thummel 2005) | |
| DamE75 | mu8AUGapi5s00642g188 | E75a | 20E-responsive NR (King-Jones and Thummel 2005) | |
| Blimp-1 | mu8AUGepir2p1s00944g59 | Blimp-1a | 20E-responsive Zn-finger TF, repressor of ftz-f1 (Agawa et al. 2007) | |
| ftz-f1 | mu8AUGepir7s03025g22 | ftz-f1a | orphan NR; developmental timing, “competence factor” (King-Jones and Thummel 2005) | |
| ab | mu8AUGepir7s00662g70 | aba | attenuation of 20E-signaling (Jang et al. 2009) | |
| Trichome formation | DamSvb | mu8AUGep24b_p1s02190g182_ mu8AUGepir2s02346g268 | ovo/svba | TF; epidermis differentiation and trichome development (Delon et al. 2003; Arif et al. 2015) |
| DamPri | mu8AUGepir3s00311g138 | pria | Svb activation (Kondo et al. 2010) | |
| dyl | mu8AUGepir2s00007g44 | dyla | wing hair formation (Adler et al. 2013) | |
| m | mu8AUGapi5s01092g326 | ma | cuticle pattern formation (Chanut-Delalande et al. 2006) | |
| CG4702 | mu8AUGepir7p2s01581g65 | CG4702a | unknown | |
| DamNeo1 | mu8AUGapi5s01036g31 | neoa | cuticular structure formation (Fernandes et al. 2010) | |
| DamNeo2 | mu8AUGapi5s02861g133 | neoa | ||
| ImpE1 | mu8AUGepir2s00005g17 | ImpE1a | epithelial cell rearrangement (Natzle et al. 1988) | |
| DamPH4a1 | mu8AUGep24b_p1s00687g272 | PH4alphaEFBa | unknown | |
| DamPH4a2 | mu8AUGepir7s03057g295 | PH4alphaEFBa | unknown | |
| DamPH4a3 | mu8AUGapi5s03057g294 | PH4alphaEFBa | unknown | |
| DamPH4a4 | mu8PASAgasmbl_15376 | PH4alphaEFBa | unknown | |
| CG9095 | mu8AUGapi5p1s00024g218 | CG9095a | wing hair formation (Adler et al. 2013) | |
| CG14395 | mu8AUGapi5p1s02190g301 | CG14395a | unknown | |
| tyn | mu8AUGapi5s00190g30t1_ m8AUGepir3s00190g27 | tyna | cuticular structure formation (Fernandes et al. 2010) | |
| sha | mu8AUGepir2s03326g79 | shaa | cuticle pattern formation (Chanut-Delalande et al. 2006) | |
| f | mu8AUGepir3s00642g197 | fa | ||
| dsx-c73A | mu8AUGep24bs00781g82 | dsx-c73Aa | cuticle development (Andrew and Baker 2008) | |
| mwh | mu8AUGepir7s00311g165 | mwha | wing hair formation (Yan et al. 2008) | |
| Cuticle formation | grh | mu8AUGepir6s00018g56; Dapma7bEVm018464 | grha | adult epidermis differentiation; cuticle organization; wound healing (Lee and Adler 2004; Mace et al. 2005; Gangishetti et al. 2012) |
| pwn | mu8AUGepir7s01005g246 | pwna | wing hair formation (Adler et al. 2013) | |
| pk | mu8AUGepir7s02385g127 | pka | wing hair orientation (Hogan et al. 2011) | |
| stan | mu8AUGepir2s01005g28_ mu8AUGepir7s01005g30 | stana | wing hair formation (Lee and Adler 2004) | |
| Midgut differentiation | DamGATAe | mu8AUGep24b_ p1s02190g273t1_ m8AUGepir7p1s02190g312 | GATAea | midgut differentiation (Okumura et al. 2007; Okumura et al. 2016) |
| CG17633 | mu8AUGepir7s03311g142t1_ m8PASAgasmbl_97928 | CG17633a | Carboxypeptidase A (Okumura et al. 2007) | |
| epsilonTry | mu8AUGepir7s02861g45 | epsilonTrya | serine-protease (Ross et al. 2003) | |
| gammaTry | mu8PASAgasmbl_13209 | gammaTrya | serine-protease homologue (Ross et al. 2003) | |
| zetaTry1 | mu8AUGepir7s00868g268 | zetaTrya | gut-specific trypsin (Attrill et al. 2016) | |
| zetaTry2 | mu8AUGepir7s00868g265 | zetaTrya | gut-specific trypsin (Attrill et al. 2016) | |
| zetaTry3 | mu8AUGepir7s00868g262 | zetaTrya | gut-specific trypsin (Attrill et al. 2016) | |
| zetaTry4 | mu8AUGapi5s00868g254 | zetaTrya | gut-specific trypsin (Attrill et al. 2016) | |
| DamT152 | mu8AUGepir7s00868g263 | DamT152b | gut-specific trypsin (Von Elert et al. 2004) | |
| DamT208 | mu8PASAgasmbl_35335 | DamT208b | ||
| if | mu8AUGepir7p2s00512g264 | ifa | adhesion/signaling protein regulating cellular adhesion, migration and survival (Attrill et al. 2016) | |
| mys | mu8PASAgasmbl_83547 | mysa | ||
| Food digestion | DamT610 | mu8PASAgasmbl_87235 | Sbb | gut-specific trypsin (Von Elert et al. 2004) |
| DamCT448 | mu8AUGepir7s03102g104 | Jon65Aivb | gut-specific chymotrypsin (Von Elert et al. 2004) | |
| DamCT802 | mu8AUGepir7p1s00944g14 | CG10472b | ||
| DamCT383 | mu8PASAgasmbl_39448 | Jon65Aivb | ||
| Amy-p1 | mu8PASAgasmbl_2973 | Amy-pa | starch digestion (Attrill et al. 2016) | |
| Amy-p2 | mu8PASAgasmbl_45416 | Amy-pa | starch digestion (Attrill et al. 2016) | |
| Amy-p3 | mu8AUGapi5s00725g187 | Amy-pa | starch digestion (Attrill et al. 2016) | |
| Amy-p4 | mu8PASAgasmbl_45408 | Amy-pa | starch digestion (Attrill et al. 2016) | |
| Amy-p5 | mu8AUGapi5s00261g171 | Amy-pa | starch digestion (Attrill et al. 2016) | |
| Amy-p6 | mu8AUGapi5p1s00944g273 | Amy-pa | starch digestion (Attrill et al. 2016) | |
| Neuropeptide signaling | DamIrp2 | mu8PASAgasmbl_73983 | irp2c | insulin-related neuropeptide (Dircksen et al. 2011) |
| tobi | mu8AUGep24bs00005g33 | tobia | target of insulin-signaling; glucosidase; influences life span, growth and viability (Buch et al. 2008) | |
| ImpL2 | mu8AUGep24b_p1s02190g119 | ImpL2a | suppressor of insulin signaling (Honegger et al. 2008) | |
| DamAstB | mu8PASAgasmbl_9028 | astBc | inhibition of JH and 20E biosynthesis (Hoffmann et al. 1999; Yamanaka et al. 2010; Dircksen et al. 2011) | |
| DamAT | mu8PASAgasmbl_50343 | atc | inhibition of JH and 20E biosynthesis (Kaneko and Hiruma 2015); myotropic activity on the gut (Dircksen et al. 2011; Verlinden et al. 2015) | |
| DamRya | mu8PASAgasmbl_94734 | ryac | attenuation of feeding motivation (Dircksen et al. 2011; Ida et al. 2011; Maeda et al. 2015) | |
| DamRYa-R | mu8AUGapi5s00770g109 | RYa-Ra | RYamide receptor (Ida et al. 2011) | |
| Tequila | mu8PASAgasmbl_40324 | Tequilaa | regulates insulin-like signaling and life span; proteolytic peptide hormone activation (Huang et al. 2015) | |
| amon | mu8PASAgasmbl_17947 | amona | proteolytic peptide hormone activation (Reiher et al. 2011) | |
| 7B2 | mu8PASAgasmbl_33811 | 7B2a | required by amon for maturation (Hwang et al. 2000) | |
| JH signaling | DamJHBP | mu8PASAgasmbl_45217 | pfam06585d | JH titre promotion (Nijhout et al. 2014) |
| DamJHE | mu8AUGepir3s03135g39 | JHEb | JH degradation (Heckmann et al. 2008; Nijhout et al. 2014) |
Drosophila melanogaster.
Daphnia magna.
Daphnia pulex.
Contains haemolymph JH binding protein domain pfam06585 (BLAST CDD).
In-Depth Analysis of Chronic Cyanobacteria Response Reveals Evolutionary Conserved Pathways of Cuticle Development and Midgut Function
By in-depth literature curation and homology mapping to other species, we were able to connect the gene regulatory networks that react to chronic cyanobacteria stress to biological processes. More specifically, we found multiple lines of evidence that associate the NR/Svb module to ecdysone signaling and cuticle formation, and the GATA module to midgut and neuropeptide signaling. Our findings are summarized in figure 4 and table 1.
First, we found strong conservation between the predicted NR-driven network in D. magna and the ecdysone pathway regulating developmental timing in D. melanogaster (fig. 4A). The steroid hormone ecdysone regulates developmental programs and moulting in insects and other arthropods (Nijhout 2013) and presumably also in D. magna (Sumiya et al. 2014). By binding to the receptor dimer EcR/USP-RXR, ecdysone triggers a signaling cascade that involves the activation of other nuclear receptors. We found three nuclear receptor genes that are differentially expressed and whose D. melanogaster homologues are known targets of the ecdysone receptor: DamHR3, DamE78, and DamE75 (King-Jones and Thummel 2005). Additionally, the top enriched motif was the D. melanogaster EcR/USP motif (Down et al. 2007) with the inverted repeat of 5′-AGGTCA-3′ that is typical for nuclear receptors (Germain et al. 2006).
Second, the predicted ovo/Svb targets are strongly enriched for the Gene Ontology term “nonsensory hair organization.” This term is attributed to a group of genes whose D. melanogaster homologues are direct targets of Svb (Menoret et al. 2013) (fig. 4A). These known Svb targets are significantly overrepresented among the genes that are upregulated in the BX treatment (P = 3.2 × 10−14). In Drosophila, Svb regulates epidermis differentiation and the development of nonsensory, cuticular hairs (trichomes) (Delon et al. 2003; Arif et al. 2015). We found both the D. magna Svb orthologue DamSvb to be differentially expressed, as well as its motif to be enriched among the upregulated genes. Interestingly, in D. melanogaster, Svb is translated as a transcriptional repressor that becomes an activating TF by cleavage through short peptides encoded by polished rice (Pri) (Kondo et al. 2010). In D. magna, we find a homologous gene, DamPri (supplementary fig. S3B, Supplementary Material online), and its expression pattern clearly correlates with the expression of DamSvb and the ecdysone-inducible NRs (fig. 4B). There are thus several lines of evidence that the Pri/Svb system is conserved in D. magna and that it is activated upon cyanobacteria stress, namely: 1) DamPri and Svb sequence conservation, 2) coexpression of DamPri, DamSvb and homologues of known Svb targets, and 3) enrichment of Svb binding sites among the upregulated genes. The function of DamSvb is yet unknown, but Daphnia have numerous cuticular structures that are in the same size range as Drosophila trichomes, such as the setulae forming plumouse structures at the setae of the swimming antennae (Agar 1950) and the filter mesh at the thoracic limbs required for feeding (Fryer 1991). The size of this filter mesh has been shown to be affected by cyanobacteria in Daphnia pulicaria (Ghadouani and Pinel-Alloul 2002). Our findings suggest that DamSvb and its target genes might control the formation of those cuticular protrusions in D. magna.
Third, a homologue of the Drosophila TF Grainyhead (Grh) and several of its known target genes (Lee and Adler 2004) are upregulated in the BX treatment, although the Grh motif is not enriched among the differentially expressed genes (79% DNA binding domain conservation between D. melanogaster Grh and D. magna transcriptome-based gene model, see supplementary additional file S3, Supplementary Material online). In Drosophila, Grh is involved in adult epidermis differentiation (Lee and Adler 2004), cuticle organization (Gangishetti et al. 2012) and wound healing (Mace et al. 2005), and functions together with Svb (Menoret et al. 2013). The signal triggering the expression of this factor in insects is still unknown (Gangishetti et al. 2012). We found NR motifs in the promoter sequence of grh, indicating that in D. magna, grh is potentially regulated by one or several NRs of the ecdysone-signaling cascade.
Fourth, the predicted GATA module can be linked to the midgut epithelium and neuropeptide signaling (fig. 4C). GATA factors represent a class of Zn-finger transcription factors that bind to the DNA consensus motif 5′-HGATAR-3′ and are evolutionary conserved in eukaryotes (Lowry and Atchley 2000). GATA factors are involved in various processes in D. melanogaster, among others endoderm development (Murakami et al. 2005; Okumura et al. 2005) and immune response (Senger et al. 2006). We investigated whether any of the predicted GATA factors in D. magna qualifies as candidate regulator for the downregulated GATA module. One candidate, a homologue of D. melanogaster GATAe, has a similar expression pattern to the genes in the GATA module (supplementary fig. S4, Supplementary Material online). GATAe is a midgut-specific TF in fruit fly (Okumura et al. 2007), and 11 homologues of its targets were significantly enriched among the downregulated genes upon BX exposure (P = 9.5 × 10−9). Although the RNA for this experiment was extracted from whole-body homogenates, there is evidence that the GATA module is expressed in the midgut, as many of its genes are known to be gut-specific (table 1).
In addition to the genes related to digestive functions and the midgut, several members of the Insulin/Insulin-like growth factor signaling (IIS) and peptide hormones involved in brain-gut signaling were downregulated after chronic cyanobacteria exposure (fig. 4C and D), as well as peptidases that are necessary to process and therewith activate peptide hormones. In insects and crayfish, these genes regulate feeding motivation, nutrient storage, growth rate and starvation resistance (Ida et al. 2011; Maeda et al. 2015; Britton et al. 2002; Mirth et al. 2014; Honegger et al. 2008).
In conclusion, we were able to associate the gene regulatory networks that are affected by chronic cyanobacteria stress to ecdysone signaling, cuticle formation, midgut differentiation and neuropeptide signaling. Additionally, we provide a first indication of the putative functions of yet unannotated genes by assigning them to specific GRNs and therewith to biological processes.
The Same Gene Networks Respond to Acute and Chronic M. aeruginosa Exposure
The data set by Schwarzenberger et al. (2014), discussed in the above paragraphs, reflects a response to chronic (i.e., many days) exposure to cyanobacteria. Next, we aimed to test whether the differential expression of the moulting- and growth-related GRNs is an effect of the chronic exposure, or a direct response to cyanobacteria. To this end, we designed a follow-up experiment consisting of only 4 h (“acute”) exposure, which forms part of the study described by Orsini et al. (2016). In this experiment, we exposed two D. magna clones (termed X-clone and I-clone in the following) to two different cyanobacteria strains, a microcystin-producing (“toxic”, BX), and a microcystin-free (“less-toxic”, BN) strain. Although the cyanobacterial strains used for the chronic exposures differed from the strains used for the acute exposures, we refer to both microcystin-producing strains as “BX” or “toxic”, and to the microcystin-free strains as “BN” or “less-toxic.” This designation is justified because rather than examining condition-specific responses, we focus on the similarities in the responses to toxic and less-toxic cyanobacteria and between the chronic and acute experiment. The experimental conditions in our study differed from the experiment by Schwarzenberger et al. (2014) in terms of genotypes (of both daphnids and cyanobacteria), developmental time point (5-days-old sub-adults in our experiment vs. mature daphnids in Schwarzenberger et al. [2014]) and exposure time (4 h vs. 7–11 days, respectively). Nevertheless, a gene set enrichment analysis (GSEA) revealed that our acute experiment generated significant changes in the same gene networks that underlie the chronic response (family-wise error rate (FWER) <0.01; supplementary fig. S5B, Supplementary Material online). Surprisingly, the direction of the change (up- or downregulation) differed across the three cohorts (i.e., replicates of the acute experiment), suggesting that our networks capture a systemic cyclic response that is likely correlated with the moulting phase (supplementary fig. S5, Supplementary Material online). This finding indicates that it is important to avoid averaging across cohorts, as this would average out the upregulation in one cohort, with the downregulation of the same network in another cohort. We generated several random control networks, namely the HSF network, and a housekeeping gene network (using ribosomal genes). These control networks were stable during the treatments across the different cohorts (supplementary fig. S5B, Supplementary Material online). We therefore conclude that there is a reproducible biological response to both chronic and acute exposure to cyanobacteria in our data set.
To construct final GRNs based on the acute cyanobacteria response, we ranked each of the 12 experiments by log2FC and extracted the 500 most up- and down-regulated genes, respectively, which resulted in 24 sets comprising in total 4600 unique genes. We ran Daphnia-cisTarget on each of those sets and recovered the same motifs as in the previous analysis: GATA (in seven out of 24 runs), NR (7×), and ovo/Svb (3×). Additionally, we identified Blimp-1 (11×) and Grh (7×) motifs, two TFs that were upregulated in the chronic stress experiment, but whose motifs were not enriched. This illustrates that our increased resolution in terms of sampling time and sampling numbers, resulted in an increased motif discovery resolution. In total, 493 genes had one or more of those five motifs enriched in at least three computational runs, and were used to construct a network (fig. 5). This network consists of two subnetworks: 249 genes are predicted GATA targets, and 247 genes belong to the highly connected NR/Svb/Blimp-1/Grh-subnetwork. Although both modules derived from the chronic experiment were similarly enriched in acute data set (supplementary fig. S5B, Supplementary Material online), more genes from the midgut module than from the ecdysone/cuticle module recurred in the network (fig. 5, squared nodes).
Fig. 5.—

The moulting- and growth-related networks respond to acute as well as to chronic cyanobacteria stress. (A) The gene regulatory networks are based entirely on the acute experiment. We found two additional motifs of the transcription factors Blimp-1 and Grh. Genes encoding those factors were differentially expressed in the chronic data, but their motifs were not enriched, indicating a higher motif discover resolution in the acute data set. The midgut- and ecdysone/cuticle-related subnetworks display the same dichotomy as in the chronic data set. The subnetworks are connected by only three genes, including the egg yolk gene DamVtg2, which is known to be regulated by ecdysone and a GATA factor in mosquitoes (Martín et al. 2001; Park et al. 2006). The network displays genes that belong to the most differentially expressed genes in any of the within-cohort treatment/control comparisons and have a motif instance in at least 3 (of 12) comparisons. Edge color intensity reflects the number of comparisons in which the motif-gene connection was found, node color intensity the absolute value of log2-fold change averaged across all comparisons. Squared nodes represent genes that are significantly differentially expressed in the chronic data set. (B) The gene regulatory networks from the chronic experiment were significantly enriched in the acute experiment. The bar plot is based on gene set enrichment analysis (GSEA) enrichments in supplementary fig. S5B, Supplementary Material online. It depicts how often the NR/Svb (red) and GATA (blue) modules derived from the chronic experiment were significantly up- or downregulated (up/down) or not (nonsign.) in the 12 treatment/control comparisons of the acute experiment. The control gene sets, heat shock factor (HSF) targets from the Daphnia-cisTarget validation and ribosomal proteins (ribos), were mostly not significantly enriched (grey bars).
Interestingly, the two subnetworks are connected through three genes (DamVtg2, Est-Q, and CG13893). Vitellogenin (Vtg) is a yolk protein precursor that has been shown to be regulated by ecdysone and a GATA factor in mosquitoes (Kokoza et al. 2001; Park et al. 2006). In D. magna, Tokishita et al. (2006) found both NR and GATA motifs to be associated with DamVtg2. Our findings suggest that DamVtg2 is indeed regulated by both NRs and a GATA factor, similarly to mosquito Vtg.
Comparing the chronic and acute cyanobacteria stress experiment, we conclude that the genes in the ecdysone/cuticle-related and midgut-related networks form part of the acute response to cyanobacteria, which is probably maintained throughout days. Moreover, cyanobacteria elicit similar responses in gene expression in different life stages and clones of D. magna.
Comprehensiveness of Network Recovery
We finally wanted to investigate how much of the observed variation in gene expression can be explained by the networks described above. Therefore, we calculated which percentage of the input gene sets contains by Daphnia-cisTarget predicted targets of the moulting- and growth-related transcription factors. Under cyanobacteria exposure, those transcription factors dominate the gene expression patterns, accounting for on average 36% of differential gene expression (supplementary table S5, Supplementary Material online). About 38% of the differentially expressed genes can be grouped into putative GRNs that are not moulting- and growth-related. These networks might represent Daphnia- or tissue-specific GRNs that deserve further investigation.
Discussion
The diversity and number of high-throughput sequencing studies using ecological model species are rapidly increasing (Ekblom and Galindo 2010; Todd et al. 2016; Tagu et al. 2014). In this context, computational analysis becomes a significant bottleneck in environmental genomic studies (see e.g., Stapley et al. 2010; Alvarez et al. 2015), primarily because functional gene annotation and bioinformatic tools for such species are limited in comparison to the resources available for traditional (biomedical) genetic models (e.g., fruit fly, nematode, and mouse). For example, analytical approaches typically used to interpret Daphnia transcriptomes rely on functional gene annotation and gene ontologies (GO) that are derived from the annotation of orthologues of distantly related model species (see e.g., [Toyota et al. 2015; De Coninck et al. 2014; Rozenberg et al. 2015). Although this approach may be helpful, it makes many assumptions on the deep conservation of gene functions between species and among gene paralogues, and ignores the significant number of genes that lack homology to model species (Primmer et al. 2013). This is particularly relevant for studies in ecological model systems that contain lineage-specific genes and may also reveal putative function of genes that are shared with model species yet have no known effects on phenotypes under laboratory conditions (see e.g., Colbourne et al. 2011).
Here, we present Daphnia-cisTarget, a tool to discover gene regulatory networks by combining gene expression and genomic sequence information. Daphnia-cisTarget is an implementation of cisTargetX by Aerts et al. (2010) for D. pulex and D. magna, the two species for which there are draft genomes available. In this study, we initially demonstrate that Daphnia-cisTarget yields biologically meaningful results by validating its utility at analysing a D. pulex heat shock data set (Becker et al., unpublished data). We selected the heat shock response to validate our method because the target genes of the evolutionary conserved (Liu et al. 1997) heat shock factor (HSF) are known to contain a conserved DNA motif (Guertin et al. 2010). As predicted, this motif is highly enriched among the genes that are upregulated after a heat shock in D. pulex. Our findings confirm the efficacy of Daphnia-cisTarget at recovering the predicted heat shock response system Daphnia. Next, we used Daphnia-cisTarget to gain new insights into the response of D. magna to the cyanobacterium M. aeruginosa. Cyanobacteria threaten freshwater ecosystems trough the production of toxic algae blooms (Huisman et al. 2005) and have been shown to reduce somatic and population growth in D. magna (Schwarzenberger et al. 2014; Kuster and Von Elert 2013). Cyanobacterial toxins include among others the protein phosphatase inhibitor Microcystin-LR (DeMott and Dhawale 1995) and protease inhibitors (Agrawal et al. 2005; Rohrlack et al. 2003). The response and adaptation of Daphnia to cyanobacterial toxins is of particular interest because Daphnia, being major grazers of phytoplankton, hold the potential to suppress algae blooms (Kuster and Von Elert 2013). Recently, a series of studies has been published on the transcriptional responses of Daphnia to the cyanobacterium M. aeruginosa (Asselman et al. 2012; Schwarzenberger et al. 2014; De Coninck et al. 2014). However, the underlying gene networks and the transcriptional regulators governing those responses remain to be identified. To this end, we analyzed two transcriptome data sets covering the response after 1) chronic (Schwarzenberger et al. 2014) and 2) acute (Orsini et al. 2016) exposure. In the chronic data set, we discovered ecdysone/cuticle- and midgut/neuropeptide-related gene regulatory networks that we attribute to moulting and growth regulation. By the application of Daphnia-cisTarget, we discovered that these networks play a central role in the general response to cyanobacteria in D. magna. The networks respond both to chronic and acute cyanobacteria exposure, despite the marked differences in the designs of both experiments (i.e., different D. magna and M. aeruginosa genotypes, daphnid life stages and exposure time). First, the ecdysone/cuticle and midgut/neuropeptide-related modules from the chronic exposure experiment were strongly affected under acute stress. Second, Daphnia-cisTarget recovered similar motifs in both experiments, i.e., GATA factor, nuclear receptor and ovo/Svb motifs. The greater resolution of our acute data set revealed two additional motifs, attributed to the developmental timing factor Blimp-1 (Agawa et al. 2007) and the epidermal transcription factor Grainyhead (Lee and Adler 2004).
The ecdysteroid hormone ecdysone regulates moulting processes in arthropods, which are tightly coupled with epidermis differentiation and cuticle formation (Mitra 2013). The cooccurrence of the above-mentioned genes and motifs (i.e., nuclear receptor, ovo/Svb, Blimp-1, and Grainyhead motifs) suggests that the ecdysone/cuticle-related network regulates moulting processes in D. magna and that these are affected by exposure to cyanobacteria. The downregulated network, on the other hand, is associated with midgut-differentiation, food digestion, and neuropeptide brain-gut signaling, processes that can be related to somatic growth regulation. Given these findings, we postulate several possible links between the identified gene regulatory networks and known cyanobacteria-induced phenotypic changes.
First, Schwarzenberger et al. (2012) showed that cyanobacteria slow the growth rate and delay maturity of D. magna (supplementary fig. S5C, Supplementary Material online). Growth and moulting are tightly coupled processes in arthropods because of their rigid carapace. In insects, those processes are controlled through a complex temporal and spatial interplay of ecdysone, juvenile hormone (JH) and insulin/IGF signaling (IIS), integrating nutritional and developmental signals (Nijhout et al. 2010). When D. magna is chronically exposed to cyanobacteria, components of each of the three endocrine signaling pathways are affected and we find differentially expressed genes that are known functional links between them (fig. 6 and table 1). 1) DamNvd1 forms part of the ecdysone synthesis pathway, is crucial for moulting in D. magna, and is expressed in the gut (Sumiya et al. 2014, 2016); 2) the ecdysone-responsive (Natzle et al. 1986) gene ImpL2 counteracts IIS and is known to be overexpressed upon starvation in D. melanogaster (Honegger et al. 2008); and 3) two genes that influence juvenile hormone titres in insects (Nijhout et al. 2014), DamJHE and DamJHBP, form part of the midgut-related network. We thus established connections between ecdysone and insulin signaling that may be important for the coordination of growth and nutritional input in D. magna and underlie the observed phenotypic changes in growth rate and age at maturity.
Fig. 6.—
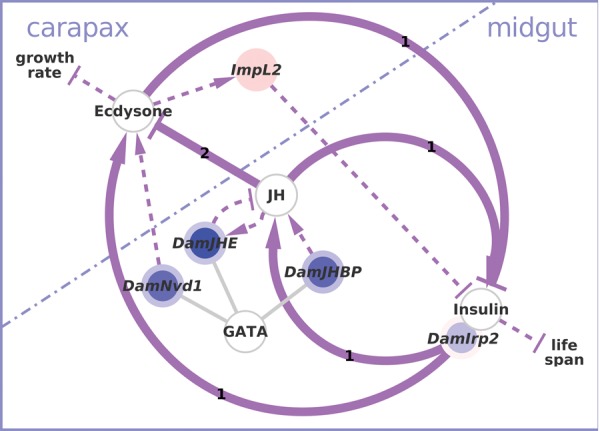
The interplay between ecdysone and insulin signaling might coordinate growth and nutritional input. Ecdysone, juvenile hormone, and insulin/IGF signaling are known to be tightly linked and to regulate growth and development in insects (purple arrows). Colored nodes represent genes from the chronic data set that can be mapped onto the insect interactions and connect the midgut GATA network containing neuropeptides and juvenile hormone-related genes, and the cuticle-related ecdysone network. References bold interactions: 1) Mirth et al. (2014) (insects); 2) Mu and Leblanc (2004) (D. magna). References for thin arrows: see figure 4A and C. Please note that this figure depicts a highly simplified model of complex stage-, tissue-, and species-specific interactions. For a comprehensive review we refer the reader to recent literature (Gruntenko and Rauschenbach 2009; Nijhout 2013; Yamanaka et al. 2013; Vafopoulou 2014; Jindra et al. 2013; Dubrovsky and Bernardo 2014).
Second, it was also shown that the Daphnia midgut epithelium is disrupted by cyanobacterial toxins (Rohrlack et al. 2005; Chen et al. 2005) and starvation (Elendt and Storch 1990). We found DamGATAe and many midgut-specific genes that contain GATA motifs differentially expressed upon cyanobacteria treatment. We therefore hypothesize that DamGATAe plays a role in midgut epithelium differentiation and maintenance, as does its Drosophila orthologue (Okumura et al. 2016), and that its differential expression in the cyanobacteria treatments reflects a response to the disruption of the midgut epithelium.
Third, the filter mesh of Daphnia’s thoracic limbs is required for feeding and changes with food conditions and cyanobacterial presence (e.g., Ghadouani and Pinel-Alloul 2002; Lampert 1994; Repka et al. 1999). This mesh is formed by cuticular structures that are of the same size range as Drosophila trichomes (cf. Fryer 1991; Chanut-Delalande et al. 2014). In Drosophila, trichome formation is controlled in an ecdysone-dependent manner by the transcription factor Svb and its activator Pri. We identified the presence of pri and svb orthologues in D. magna and their differential expression under cyanobacteria exposure, and hypothesize that DamPri and DamSvb might be important in regulation of filter mesh size in D. magna. In addition, the Daphnia exoskeleton features numerous cuticular extensions, such as spinules, denticles, and setulae required for swimming and filter feeding (Fryer 1991), that are rebuilt prior to each moult (Agar 1950). Any of those structures could be regulated by DamPri/Svb. Consequently, the DamPri/Svb system would be invoked during each moulting cycle and its expression affected by any stressor that influences the moulting cycle. Given that cyanobacteria affect both moulting (Rohrlack et al. 2004) and filter mesh size (Ghadouani and Pinel-Alloul 2002; Repka et al. 1999), a combination of both functions may be involved.
Interestingly, the direction of expression of the moulting- and growth-related networks is cohort-specific in the acute stress experiment (supplementary fig. S5, Supplementary Material online). In D. magna, ecdysone and genes involved in ecdysone signaling and moulting show cyclic expression patterns (Mu and Leblanc 2004; Martin-Creuzburg et al. 2007; Kato et al. 2007; Sumiya et al. 2014; Hannas and Leblanc 2010; Espie and Roff 1995). A possible explanation for the observed pattern is that the cohorts were sampled at different time points during the moulting cycle and that the acute exposure to cyanobacteria induces a small shift in this cycle as a general stress response. This hypothesis is in line with the statement by Chang and Mykles (2011) that external cues can inhibit moulting in Crustacea. Consequently, careful experimental design is necessary to control for such effects and to disentangle moulting-related genes from genes underlying other traits of interest (see also Vandegehuchte et al. [2010]; Alvarez et al. [2015] and supplementary note S1, Supplementary Material online).
In this study, we focused on moulting and growth regulation, processes that are conserved across arthropods (Nijhout 2013). Consequently, we found multiple homologies at the levels of transcription factors, motifs, and target genes in the Microcystis data sets presented here. It has been shown that transcription factors and their binding specificities can be highly conserved across different taxa (e.g., Liu et al. 1997), and Weirauch et al. (2014) found that transcription factors with closely related DNA-binding domains are likely to bind to similar DNA sequence motifs. The mere presence of a motif in a putatively cis-regulatory region of a gene does not necessarily imply a functional binding site. However, the fact that the gene is expressed in the experiment and that its motif is enriched among coexpressed genes provides a strong indication that the binding site is indeed functional. For each transcription factor, we examined several indications that suggest the conservation of a gene regulatory network between water fleas and other species: 1) enrichment of the motif in a set of coexpressed genes, 2) conservation of the transcription factor that has been assigned to that motif in another species, 3) coexpression of the factor with its target genes as predicted by Daphnia-cisTarget, and 4) coexpression of orthologues of known target genes of that factor in other species. By this, we were able to identify several gene expression modules in D. magna and link them to biological functions. Typical for ecological model species such as Daphnia, those modules contain many lineage-specific genes that lack functional annotation (fig. 3, supplementary table S2, Supplementary Material online). By exploiting the principle of guilt-by-association, we were able to also assign biological functions to these genes. Another network biology approach, i.e., gene coexpression clustering, has proven to be a valuable tool to obtain a mechanistic understanding of genotype–phenotype relationships in ecological model species (e.g., Weston et al. 2008; Williams et al. 2011; Filteau et al. 2013). However, clustering methods require large sample sizes (Allen et al. 2012; Altay 2012). Daphnia-cisTarget does not have this requirement and is thus better suited to small-scale gene expression studies.
Other existing methods can be used to discover motifs and gene regulatory networks in Daphnia, for example Clover (Frith et al. 2004), RSAT peak-motifs (Thomas-Chollier et al. 2012), or CisFinder (Sharov and Ko 2009). A comprehensive comparison of the different methods is beyond the scope of this article and we refer the reader to the recent review by Boeva (2016). Because of differences in the underlying algorithms and different input data requirements, it has proven difficult to directly compare the performance of motif-discovery tools, and Tran and Huang (2014) recommend the use of multiple different tools. Since all existing motif-discovery methods are not directly tailored to water fleas, some bioinformatic knowledge is required to apply those methods in the same fashion as Daphnia-cisTarget. Particularly, in other tools the user has to provide putatively cis-regulatory sequences for each gene of interest and a background set of genes (i.e., the full gene catalogue) and a library of sequence motifs. Daphnia-cisTarget offers several advantages over existing motif-discovery tools for the analysis of Daphnia gene sets: It 1) makes use of a large collection of sequence motifs, 2) comprises a precompiled full genomic background, which makes enrichment analysis quite fast, 3) provides the prediction of putative TFs and target genes, and 4) is accessible to users without bioinformatic knowledge through its web-interface.
Conclusion
We developed a tool for gene regulatory network discovery in the water flea Daphnia that combines gene expression and genomic sequence information. We demonstrated that Daphnia-cisTarget can be used to understand the structure of a gene expression data set independently of functional gene annotation. We also demonstrated how the identification of different components of conserved gene regulatory networks (transcription factors, their binding sites and target genes) can lead to new insights on the function of genes and their interactions. Therefore, Daphnia-cisTarget provides an additional layer to data interpretation and accommodates the fact that complex gene interactions rather than individual genes, determine a phenotype (Benfey and Mitchell-Olds 2008). With Daphnia-cisTarget, we presented a gene prioritization approach that yields biologically meaningful results in two genetic nonmodel species and that can be applied to any ecological model species for which a draft genome assembly and gene catalogue are available.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Author Contributions
K.I.S., L.D.M., and S.A. designed the study. K.I.S., G.H., and S.A. implemented Daphnia-cisTarget. K.I.S. analyzed and interpreted the chronic and acute cyanobacteria data and performed the network analyses. L.D.M., L.O., and M.J. designed the acute cyanobacteria experiment which was conducted by M.J., E.D., and L.O. D.B. and J.K.C. designed, conducted, and analyzed the heat shock experiment. K.I.S. and S.A. wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgments
This work was supported by the Agency for Innovation by Science and Technology in Flanders (PhD scholarship to K.I.S.); the Fund for Scientific Research Flanders (postdoc fellowship to M.J. and project STRESSFLEA-B/G061411); the Heinrich Hertz foundation (stipend to D.B.); the German Research Foundation (BE 5288/2-1 and BE 5288/3-1 to D.B.); the European Science Foundation (ESF EUROCORES Programme 09-EEFG-FP-040 STRESSFLEA); and KU Leuven Center for Excellence funding (PF/2010/07). This work benefits from and contributes to the Daphnia Genomics Consortium.
Literature Cited
- Adler PN, Sobala LF, Thom D, Nagaraj R.. 2013. dusky-like is required to maintain the integrity and planar cell polarity of hairs during the development of the Drosophila wing. Dev Biol. 379:76–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aerts S. 2012. Computational strategies for the genome-wide identification of cis-regulatory elements and transcriptional targets. Curr Top Dev Biol. 98:121–145. [DOI] [PubMed] [Google Scholar]
- Aerts S, et al. 2010. Robust target gene discovery through transcriptome perturbations and genome-wide enhancer predictions in Drosophila uncovers a regulatory basis for sensory specification. PLoS Biol. 8:e1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agar WE. 1950. The swimming setae of Daphnia carinata. Q J Microsc Sci. 91:353–368. [PubMed] [Google Scholar]
- Agawa Y, et al. 2007. Drosophila Blimp-1 is a transient transcriptional repressor that controls timing of the ecdysone-induced developmental pathway. Mol Cell Biol. 27:8739–8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal MK, et al. 2005. Characterization of proteases in guts of Daphnia magna and their inhibition by Microcystis aeruginosa PCC 7806. Environ Toxicol. 20:314–322. [DOI] [PubMed] [Google Scholar]
- Allen JD, Xie Y, Chen M, Girard L, Xiao G.. 2012. Comparing statistical methods for constructing large scale gene networks. PLoS One 7:e29348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altay G. 2012. Empirically determining the sample size for large-scale gene network inference algorithms. IET Syst Biol. 6:35–43. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. 1997. Gapped BLAST and PS I-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez M, Schrey AW, Richards CL.. 2015. Ten years of transcriptomics in wild populations: what have we learned about their ecology and evolution? Mol Ecol. 24:710–725. [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W.. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W.. 2014. HTSeq: a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew DJ, Baker BS.. 2008. Expression of the Drosophila secreted cuticle protein 73 (dsc73) requires Shavenbaby. Dev Dyn. 237:1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew RL, et al. 2013. A road map for molecular ecology. Mol Ecol. 22:2605–2626. [DOI] [PubMed] [Google Scholar]
- Arif S, Kittelmann S, McGregor AP.. 2015. From shavenbaby to the naked valley: trichome formation as a model for evolutionary developmental biology. Evol Dev. 17:120–126. [DOI] [PubMed] [Google Scholar]
- Asselman J, et al. 2012. Identification of pathways, gene networks, and paralogous gene families in Daphnia pulex responding to exposure to the toxic cyanobacterium Microcystis aeruginosa. Environ Sci Technol. 46:8448–8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attrill H, et al. 2016. FlyBase: establishing a gene group resource for Drosophila melanogaster. Nucleic Acids Res. 44:D786–D792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CR, Tuch BB, Johnson AD.. 2011. Extensive DNA-binding specificity divergence of a conserved transcription regulator. Proc Natl Acad Sci U S A. 108:7493–7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfey PN, Mitchell-Olds T.. 2008. From Genotype to Phenotype: Systems Biology Meets Natural Variation. Science 320:495–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elendt BP, Storch V.. 1990. Starvation-induced alterations of the ultrastructure of the midgut of Daphnia magna straus, 1820 (Cladocera). J Crustac Biol. 10:79–86. [Google Scholar]
- Blake JA, et al. 2015. Gene Ontology Consortium: going forward. Nucleic Acids Res. 43:D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V. 2016. Analysis of genomic sequence motifs for deciphering transcription factor binding and transcriptional regulation in eukaryotic cells. Front Genet. 7:24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton JS, Lockwood WK, Li L, Cohen SM, Edgar B. a.. 2002. Drosophila’s insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev Cell 2:239–249. [DOI] [PubMed] [Google Scholar]
- Buch S, Melcher C, Bauer M, Katzenberger J, Pankratz MJ.. 2008. Opposing effects of dietary protein and sugar regulate a transcriptional target of Drosophila insulin-like Peptide Signaling. Cell Metab. 7:321–332. [DOI] [PubMed] [Google Scholar]
- Burke M, et al. 2010. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature. 467:587–590. [DOI] [PubMed] [Google Scholar]
- Chang ES, Mykles DL.. 2011. Regulation of crustacean molting: a review and our perspectives. Gen Comp Endocrinol. 172:323–330. [DOI] [PubMed] [Google Scholar]
- Chanut-Delalande H, Fernandes I, Roch F, Payre F, Plaza S.. 2006. Shavenbaby couples patterning to epidermal cell shape control. PLoS Biol. 4:e290.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanut-Delalande H, et al. 2014. Pri peptides are mediators of ecdysone for the temporal control of development. Nat Cell Biol. 16:1035–1044. [DOI] [PubMed] [Google Scholar]
- Chen R, Xiao M, Buchberger A, Li L.. 2014. Quantitative neuropeptidomics study of the effects of temperature change in the crab Cancer borealis. J Proteome Res. 13:5767–5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Song L, Ou D, Gan N.. 2005. Chronic toxicity and responses of several important enzymes in Daphnia magna on exposure to sublethal Microcystin-LR. Environ Toxicol. 20:323–330. [DOI] [PubMed] [Google Scholar]
- Colbourne JK, et al. 2011. The ecoresponsive genome of Daphnia pulex. Science 331:555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo PF, et al. 2005. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 307:1928–1933. [DOI] [PubMed] [Google Scholar]
- Dalziel AC, Rogers SM, Schulte PM.. 2009. Linking genotypes to phenotypes and fitness: How mechanistic biology can inform molecular ecology. Mol Ecol. 18:4997–5017. [DOI] [PubMed] [Google Scholar]
- Davis NT. 2003. Localization of myoinhibitory peptide immunoreactivity in Manduca sexta and Bombyx mori, with indications that the peptide has a role in molting and ecdysis. J Exp Biol. 206:1449–1460. [DOI] [PubMed] [Google Scholar]
- De Coninck DIM, et al. 2014. Genome-wide transcription profiles reveal genotype-dependent responses of biological pathways and gene-families in Daphnia exposed to single and mixed stressors. Environ Sci Technol. 48:3513–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bene F, et al. 2007. In vivo validation of a computationally predicted conserved Ath5 target gene set. PLoS Genet. 3:e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delon I, Chanut-Delalande H, Payre F.. 2003. The Ovo/Shavenbaby transcription factor specifies actin remodelling during epidermal differentiation in Drosophila. Mech Dev. 120:747–758. [DOI] [PubMed] [Google Scholar]
- DeMott W, Dhawale S.. 1995. Inhibition of in vitro protein phosphatase activity in three zooplankton species by microcystin-LR, a toxin from cyanobacteria. Arch Für Hydrobiol. 134:417–424. [Google Scholar]
- Dircksen H, et al. 2011. Genomics, transcriptomics, and peptidomics of Daphnia pulex neuropeptides and protein hormones. J Proteome Res. 10:4478–4504. [DOI] [PubMed] [Google Scholar]
- Down TA, Bergman CM, Su J, Hubbard TJP.. 2007. Large-scale discovery of promoter motifs in Drosophila melanogaster. PLoS Comput Biol. 3:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrovsky EB, Bernardo TJ.. 2014. The juvenile hormone receptor and molecular mechanisms of juvenile hormone action. In: Ephraim Cohen, editor. Advances in insect physiology. Oxford: Vol. 46Elsevier Inc; p. 305–388. [Google Scholar]
- Ekblom R, Galindo J.. 2010. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 107:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espie PJ, Roff JC.. 1995. A biochemical index of duration of the molt cycle for planktonic Crustacea based on the chitin-degrading enzyme, chitobiase. Limnol Oceanogr. 40:1028–1034. [Google Scholar]
- Feder M, Mitchell-Olds T.. 2003. Evolutionary and ecological functional genomics. Heredity 100:649–655. [DOI] [PubMed] [Google Scholar]
- Fernandes I, et al. 2010. Zona pellucida domain proteins remodel the apical compartment for localized cell shape changes. Dev Cell 18:64–76. [DOI] [PubMed] [Google Scholar]
- Filteau M, Pavey SA, St-Cyr J, Bernatchez L.. 2013. Gene coexpression networks reveal key drivers of phenotypic divergence in lake whitefish. Mol Biol Evol. 30:1384–1396. [DOI] [PubMed] [Google Scholar]
- Frith MC, et al. 2004. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 32:1372–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Li MC, Weng Z.. 2003. Cluster-Buster: finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 31:3666–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer G. 1991. Functional morphology and the adaptive radiation of the daphniidae (Branchiopoda: Anomopoda). Philos Trans R Soc B Biol Sci. 331:1–99. [Google Scholar]
- Fu Q, Tang LS, Marder E, Li L.. 2007. Mass spectrometric characterization and physiological actions of VPNDWAHFRGSWamide, a novel B type allatostatin in the crab, Cancer borealis. J. Neurochem. 101:1099–1107. [DOI] [PubMed] [Google Scholar]
- Gangishetti U, et al. 2012. The transcription factor Grainy head and the steroid hormone ecdysone cooperate during differentiation of the skin of Drosophila melanogaster. Insect Mol Biol. 21:283–295. [DOI] [PubMed] [Google Scholar]
- Gauhar Z, et al. 2009. Genomic mapping of binding regions for the Ecdysone receptor protein complex. Genome Res. 19:1006–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, et al. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain P, Staels B, Dacquet C, Spedding M, Laudet V.. 2006. Overview of nomenclature of nuclear receptors. Am Soc Pharmacol Exp Ther. 58:685–704. [DOI] [PubMed] [Google Scholar]
- Ghadouani A, Pinel-Alloul B.. 2002. Phenotypic plasticity in Daphnia pulicaria as an adaptation to high biomass of colonial and filamentous cyanobacteria: experimental evidence. J Plankton Res. 24:1047–1056. [Google Scholar]
- Gonsalves SE, Moses AM, Razak Z, Robert F, Timothy Westwood J.. 2011. Whole-genome analysis reveals that active heat shock factor binding sites are mostly associated with non-heat shock genes in Drosophila melanogaster. PLoS One 6:e15934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green SA, Simoes-Costa M, Bronner ME.. 2015. Evolution of vertebrates as viewed from the crest. Nature 520:474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gremberghe I, et al. 2009. Influence of Daphnia infochemicals on functional traits of Microcystis strains (Cyanobacteria). Hydrobiologia 635:147–155. [Google Scholar]
- Gruntenko N, Rauschenbach I.. 2009. 20-Hydroxyecdysone, juvenile hormone and biogenic amines: mechanisms of interaction in control of Drosophila reproduction under normal and stressful conditions In: Smagghe G, editor. Ecdysone: structures and functions. The Netherlands: Springer; p. 317–332. [Google Scholar]
- Guertin MJ, Petesch SJ, Zobeck KL, Min IM, Lis JT.. 2010. Drosophila heat shock system as a general model to investigate transcriptional regulation. Cold Spring Harb Symp Quant Biol. 75:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GuhaThakurta D, et al. 2002. Identification of a novel cis-regulatory element involved in the heat shock response in Caenorhabditis elegans using microarray gene expression and computational methods. Genome Res. 12:701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannas BR, Leblanc GA.. 2010. Expression and ecdysteroid responsiveness of the nuclear receptors HR3 and E75 in the crustacean Daphnia magna. Mol Cell Endocrinol. 315:208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison RC, Taylor J.. 2012. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet. 13:469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann L-H, et al. 2008. Systems biology meets stress ecology: linking molecular and organismal stress responses in Daphnia magna. Genome Biol. 9:R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann C, Van De Sande B, Potier D, Aerts S.. 2012. i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 40:e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann KH, Meyering-Vos M, Lorenz MW.. 1999. Allatostatins and allatotropins: Is the regulation of corpora allata activity their primary function? Eur. J. Entomol. 96:255–266. [Google Scholar]
- Hogan J, Valentine M, Cox C, Doyle K, Collier S.. 2011. Two frizzled planar cell polarity signals in the Drosophila wing are differentially organized by the Fat/Dachsous pathway. PLoS Genet. 7:e1001305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honegger B, et al. 2008. Imp-L2, a putative homolog of vertebrate IGF-binding protein 7, counteracts insulin signaling in Drosophila and is essential for starvation resistance. J Biol. 7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua YJ, et al. 1999. Identification of a prothoracicostatic peptide in the larval brain of the silkworm, Bombyx mori. J Biol Chem. 274:31169–31173. [DOI] [PubMed] [Google Scholar]
- Huang C-W, et al. 2015. Tequila regulates insulin-like signaling and extends life span in Drosophila melanogaster. J Gerontol A Biol Sci Med Sci. 70:1461–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, et al. 2000. Functional discovery via a compendium of expression profiles. Cell 102:109–126. [DOI] [PubMed] [Google Scholar]
- Huisman J, Matthijs HCP, Visser PM.. 2005. Harmful cyanobacteria, 1st edn.Berlin/Heidelberg: Springer-Verlag. [Google Scholar]
- Hwang JW, Siekhaus DE, Fuller RS, Taghert PH, Lindberg I.. 2000. Interaction of Drosophila melanogaster prohormone convertase 2 and 7B2. Insect cell-specific processing and secretion. J Biol Chem. 275:17886–17893. [DOI] [PubMed] [Google Scholar]
- Ida T, et al. 2011. Identification of the novel bioactive peptides dRYamide-1 and dRYamide-2, ligands for a neuropeptide Y-like receptor in Drosophila. Biochem Biophys Res Commun. 410:872–877. [DOI] [PubMed] [Google Scholar]
- Imrichová H, Hulselmans G, Kalender Atak Z, Potier D, Aerts S.. 2015. i-cisTarget 2015 update: generalized cis-regulatory enrichment analysis in human, mouse and fly. Nucleic Acids Res. 43:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang AC-C, Chang Y-C, Bai J, Montell D.. 2009. Border-cell migration requires integration of spatial and temporal signals by the BTB protein Abrupt. Nat Cell Biol. 11:569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janky R, et al. 2014. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol. 10:e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindra M, Palli SR, Riddiford LM.. 2013. The juvenile hormone signaling pathway in insect development. Annu Rev Entomol. 58:181–204. [DOI] [PubMed] [Google Scholar]
- Kampstra P. 2008. Beanplot: a boxplot alternative for visual comparison of distributions. J Stat Softw. 28:9. [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M.. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40:109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y, Hiruma K.. 2015. Allatotropin inhibits juvenile hormone biosynthesis by the corpora allata of adult Bombyx mori. J Insect Physiol. 80:15–21. [DOI] [PubMed] [Google Scholar]
- Karasov T, Messer PW, Petrov DA.. 2010. Evidence that adaptation in Drosophila is not limited by mutation at single sites. PLoS Genet. 6:e1000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka H, et al. 1989. Identification of an allatotropin from adult Manduca sexta. Science 243:1481–1483. [DOI] [PubMed] [Google Scholar]
- Kato Y, et al. 2007. Cloning and characterization of the ecdysone receptor and ultraspiracle protein from the water flea Daphnia magna. J Endocrinol. 193:183–194. [DOI] [PubMed] [Google Scholar]
- Kethidi DR, Xi Z, Palli SR.. 2005. Developmental and hormonal regulation of juvenile hormone esterase gene in Drosophila melanogaster. J Insect Physiol. 51:393–400. [DOI] [PubMed] [Google Scholar]
- King-Jones K, Thummel CS.. 2005. Nuclear receptors: a perspective from Drosophila. Nat Rev Genet. 6:311–323. [DOI] [PubMed] [Google Scholar]
- Kinsella RJ, et al. 2011. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database 2011:bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoza VA, et al. 2001. Transcriptional regulation of the mosquito vitellogenin gene via a blood meal-triggered cascade. Gene 274:47–65. [DOI] [PubMed] [Google Scholar]
- Kondo T, et al. 2010. Small peptides switch the transcriptional activity of shavenbaby during Drosophila embryogenesis. Science 329:336–339. [DOI] [PubMed] [Google Scholar]
- Kuster CJ, Von Elert E.. 2013. Interspecific differences between D. pulex and D. magna in tolerance to cyanobacteria with protease inhibitors. PLoS One 8:e62658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhaney S, et al. 2016. A beak size locus in Darwins finches facilitated character displacement during a drought. Science 352:470–474. [DOI] [PubMed] [Google Scholar]
- Lampert W. 1994. Phenotypic plasticity of the filter screens in Daphnia: adaptation to a low-food environment. Limnol Oceanogr. 39:997–1006. [Google Scholar]
- Lampert W, Kinne O.. 2011. Daphnia: development of a model organism in ecology and evolution. Oldendorf/Luhe: International Ecology Institute. [Google Scholar]
- Lee H, Adler PN.. 2004. The grainy head transcription factor is essential for the function of the frizzled pathway in the Drosophila wing. Mech Dev. 121:37–49. [DOI] [PubMed] [Google Scholar]
- Liu XD, Liu PC, Santoro N, Thiele DJ.. 1997. Conservation of a stress response: human heat shock transcription factors functionally substitute for yeast HSF. EMBO J. 16:6466–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz MW, Kellner R, Hoffmann KH.. 1995. A family of neuropeptides that inhibit juvenile hormone biosynthesis in the cricket, Gryllus bimaculatus. J Biol Chem. 270:21103–21108. [DOI] [PubMed] [Google Scholar]
- Love M, Huber W, Anders S.. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry JA, Atchley WR.. 2000. Molecular evolution of the GATA family of transcription factors: conservation within the DNA-binding domain. J Mol Evol. 50:103–115. [DOI] [PubMed] [Google Scholar]
- Mace KA, Pearson JC, McGinnis W.. 2005. An epidermal barrier wound repair pathway in Drosophila is mediated by grainy head. Science 308:381–385. [DOI] [PubMed] [Google Scholar]
- Maeda T, et al. 2015. Suppressive effects of dRYamides on feeding behavior of the blowfly, Phormia regina. Zool Lett. 1:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M.. 2005. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21:3448–3449. [DOI] [PubMed] [Google Scholar]
- Mahat DB, Salamanca HH, Duarte FM, Danko CG, Lis JT.. 2016. Mammalian heat shock response and mechanisms underlying its genome-wide transcriptional regulation. Mol Cell 62:63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony S, Benos PV.. 2007. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 35:W253–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manceau M, Domingues VS, Mallarino R, Hoekstra HE.. 2011. The developmental role of agouti in color pattern evolution. Science 331:1062–1065. [DOI] [PubMed] [Google Scholar]
- Martín D, Wang SF, Raikhel AS.. 2001. The vitellogenin gene of the mosquito Aedes aegypti is a direct target of ecdysteroid receptor. Mol Cell Endocrinol. 173:75–86. [DOI] [PubMed] [Google Scholar]
- Martin-Creuzburg D, von Elert E, Hoffmann KH.. 2008. Nutritional constraints at the cyanobacteria–Daphnia magna interface: the role of sterols. Limnol Oceanogr. 53:456–468. [Google Scholar]
- Martin-Creuzburg D, Westerlund SA, Hoffmann KH.. 2007. Ecdysteroid levels in Daphnia magna during a molt cycle: determination by radioimmunoassay (RIA) and liquid chromatography–mass spectrometry (LC–MS). Gen Comp Endocrinol. 151:66–71. [DOI] [PubMed] [Google Scholar]
- Matys V, et al. 2006. TRANSFAC® and its module TRANSCompel®: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34:D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menoret D, et al. 2013. Genome-wide analyses of Shavenbaby target genes reveals distinct features of enhancer organization. Genome Biol. 14:R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner BE, De Meester L, Pfrender ME, Lampert W, Hairston NG.. 2012. Linking genes to communities and ecosystems: Daphnia as an ecogenomic model. Proc R Soc B Biol Sci. 279:1873–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirth CK, et al. 2014. Juvenile hormone regulates body size and perturbs insulin signaling in Drosophila. Proc Natl Acad Sci U S A. 111:7018–7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A. 2013. Cinderella’s new shoes: how and why insects remodel their bodies between life stages. Curr Sci. 104:1028–1036. [Google Scholar]
- Mu X, Leblanc GA.. 2004. Synergistic interaction of endocrine‐disrupting chemicals: model development using an ecdysone receptor antagonist and a hormone synthesis inhibitor. Environ Toxicol Chem. 23:1085–1091. [DOI] [PubMed] [Google Scholar]
- Murakami R, Okumura T, Uchiyama H.. 2005. GATA factors as key regulatory molecules in the development of Drosophila endoderm. Dev Growth Differ. 47:581–589. [DOI] [PubMed] [Google Scholar]
- Natzle JE, Fristrom DK, Fristrom JW.. 1988. Genes expressed during imaginal disc morphogenesis: IMP-E1, a gene associated with epithelial cell rearrangement. Dev Biol. 129:428–438. [DOI] [PubMed] [Google Scholar]
- Natzle JE, Hammonds AS, Fristrom JW.. 1986. Isolation of genes active during hormone-induced morphogenesis in Drosophila imaginal discs. J Biol Chem. 261:5575–5583. [PubMed] [Google Scholar]
- Naval Sanchez M. 2014. Evolutionary conservation and divergence of cis-regulatory interactions in Drosophila eye development. PhD Thesis, KU Leuven, Belgium.
- Nijhout HF. 2013. Arthropod developmental endocrinology In: Arthropod Biology and Evolution. Berlin, Heidelberg: Springer; p. 123–148. [Google Scholar]
- Nijhout HF, et al. 2014. The developmental control of size in insects. Wiley Interdiscip Rev Dev Biol. 3:113–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhout HF, Roff DA, Davidowitz G.. 2010. Conflicting processes in the evolution of body size and development time. Philos Trans R Soc B Biol Sci U S A. 365:567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta KR, et al. 2015. Conservation of transcription factor binding specificities across 600 million years of bilateria evolution. eLife 4:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura T, et al. 2016. GATAe regulates intestinal stem cell maintenance and differentiation in Drosophila adult midgut. Dev Biol. 410:24–35. [DOI] [PubMed] [Google Scholar]
- Okumura T, Matsumoto A, Tanimura T, Murakami R.. 2005. An endoderm-specific GATA factor gene, dGATAe, is required for the terminal differentiation of the Drosophila endoderm. Dev Biol. 278:576–586. [DOI] [PubMed] [Google Scholar]
- Okumura T, Tajiri R, Kojima T, Saigo K, Murakami R.. 2007. GATAe-dependent and -independent expressions of genes in the differentiated endodermal midgut of Drosophila. Gene Expr Patterns 7:178–186. [DOI] [PubMed] [Google Scholar]
- Ono H, et al. 2006. Spook and Spookier code for stage-specific components of the ecdysone biosynthetic pathway in Diptera. Dev Biol. 298:555–570. [DOI] [PubMed] [Google Scholar]
- Orsini L, et al. 2016. Daphnia magna transcriptome by RNA-Seq across 12 environmental stressors. Sci Data 3:160030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo-Diaz C, Salazar C, Jiggins CD.. 2015. Towards the identification of the loci of adaptive evolution. Methods Ecol Evol. 6:445–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Attardo GM, Hansen IA, Raikhel AS.. 2006. GATA factor translation is the final downstream step in the amino acid/target-of-rapamycin-mediated vitellogenin gene expression in the anautogenous mosquito Aedes aegypti. J Biol Chem. 281:11167–11176. [DOI] [PubMed] [Google Scholar]
- Pavey SA, Bernatchez L, Aubin-Horth N, Landry CR.. 2012. What is needed for next-generation ecological and evolutionary genomics? Trends Ecol Evol. 27:673–676. [DOI] [PubMed] [Google Scholar]
- Potier D, Kalender Atak Z, Naval Sanchez M, Herrmann C, Aerts S.. 2012. Using cisTargetX to predict transcriptional targets and networks in Drosophila. In: Gene regulatory networks. Methods in molecular biology, Vol. 786 Totowa, New Jersey: Humana Press; p. 291–314. [DOI] [PubMed] [Google Scholar]
- Primmer CR, Papakostas S, Leder EH, Davis MJ, Ragan MA.. 2013. Annotated genes and nonannotated genomes: cross-species use of Gene Ontology in ecology and evolution research. Mol Ecol. 22:3216–3241. [DOI] [PubMed] [Google Scholar]
- Protas ME, et al. 2006. Genetic analysis of cavefish reveals molecular convergence in the evolution of albinism. Nat Genet. 38:107–111. [DOI] [PubMed] [Google Scholar]
- Purdy KJ, et al. 2010. Systems biology for ecology In: Integrative ecology: from molecules to ecosystems. research, Vol. 43 Burlington: Academic Press; p. 87–149. [Google Scholar]
- Reiher W, et al. 2011. Peptidomics and peptide hormone processing in the Drosophila midgut. J Proteome Res. 10:1881–1892. [DOI] [PubMed] [Google Scholar]
- Repka S, Veen A, Vijverberg J.. 1999. Morphological adaptations in filtering screens of Daphnia galeata to food quantity and food quality. J Plankton Res. 21:971–989. [Google Scholar]
- Robinson JT, et al. 2011. Integrative genomics viewer. Nat Biotechnol. 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrlack T, et al. 2005. Ingestion of microcystins by Daphnia: intestinal uptake and toxic effects. Limnol Oceanogr. 50:440–448. [Google Scholar]
- Rohrlack T, et al. 2003. Isolation, characterization, and quantitative analysis of Microviridin J, a new Microcystis metabolite toxic to Daphnia. J Chem Ecol. 29:1757–1770. [DOI] [PubMed] [Google Scholar]
- Rohrlack T, Christoffersen K, Kaebernick M, Neilan BA.. 2004. Cyanobacterial Protease inhibitor microviridin J causes a lethal molting disruption in Daphnia pulicaria. Appl Environ Microbiol. 70:5047–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routtu J, et al. 2014. An SNP-based second-generation genetic map of Daphnia magna and its application to QTL analysis of phenotypic traits. BMC Genomics 15:1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenberg A, et al. 2015. Transcriptional profiling of predator-induced phenotypic plasticity in Daphnia pulex. Front Zool. 12:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, et al. 2003. TM4: a free, open-source system for microarray data management and analysis. BioTechniques 34:374–378. [DOI] [PubMed] [Google Scholar]
- Sayou C, et al. 2014. A promiscuous intermediate underlies the evolution of LEAFY DNA binding specificity. Science 343:645–648. [DOI] [PubMed] [Google Scholar]
- Schmidt D, et al. 2010. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328:1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzenberger A, et al. 2014. Deciphering the genetic basis of microcystin tolerance. BMC Genomics 15:776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzenberger A, Kuster CJ, Von Elert E.. 2012. Molecular mechanisms of tolerance to cyanobacterial protease inhibitors revealed by clonal differences in Daphnia magna. Mol Ecol. 21:4898–4911. [DOI] [PubMed] [Google Scholar]
- Schwarzenberger A, Zitt A, Kroth P, Mueller S, Von Elert E.. 2010. Gene expression and activity of digestive proteases in Daphnia: effects of cyanobacterial protease inhibitors. BMC Physiol. 10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger K, Harris K, Levine M.. 2006. GATA factors participate in tissue-specific immune responses in Drosophila larvae. Proc Natl Acad Sci U S A. 103:15957–15962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, et al. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov AA, Ko MSH.. 2009. Exhaustive search for over-represented DNA sequence motifs with CisFinder. DNA Res. 16:261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, et al. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapley J, et al. 2010. Adaptation genomics: the next generation. Trends Ecol Evol. 12:705–712. [DOI] [PubMed] [Google Scholar]
- Subramanian A, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumiya E, et al. 2016. Neverland regulates embryonic moltings through the regulation of ecdysteroid synthesis in the water flea Daphnia magna, and may thus act as a target for chemical disruption of molting. J Appl Toxicol. 1476–1485. [DOI] [PubMed] [Google Scholar]
- Sumiya E, et al. 2014. Roles of ecdysteroids for progression of reproductive cycle in the fresh water crustacean Daphnia magna. Front Zool. 11:60. [Google Scholar]
- Tagu D, Colbourne JK, Negre N.. 2014. Genomic data integration for ecological and evolutionary traits in non-model organisms. BMC Genomics 15:490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Chollier M, et al. 2012. RSAT peak-motifs: motif analysis in full-size ChIP-seq datasets. Nucleic Acids Res. 40:e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd EV, Black MA, Gemmell NJ.. 2016. The power and promise of RNA-seq in ecology and evolution. Mol Ecol. 25:1224–1241. [DOI] [PubMed] [Google Scholar]
- Tokishita S, et al. 2006. Organization and repression by juvenile hormone of a vitellogenin gene cluster in the crustacean, Daphnia magna. Biochem Biophys Res Commun. 345:362–370. [DOI] [PubMed] [Google Scholar]
- Toyota K, et al. 2015. NMDA receptor activation upstream of methyl farnesoate signaling for short day-induced male offspring production in the water flea, Daphnia pulex. BMC Genomics 16:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NTL, Huang C-H.. 2014. A survey of motif finding Web tools for detecting binding site motifs in ChIP-Seq data. Biol Direct. 9:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL.. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsong AE, Tuch BB, Li H, Johnson AD.. 2006. Evolution of alternative transcriptional circuits with identical logic. Nature 443:415–420. [DOI] [PubMed] [Google Scholar]
- Vafopoulou X. 2014. The coming of age of insulin-signaling in insects. Front Physiol. 5:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Loo P, et al. 2008. ModuleMiner - improved computational detection of cis-regulatory modules: are there different modes of gene regulation in embryonic development and adult tissues? Genome Biol. 9:R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandegehuchte MB, Vandenbrouck T, De Coninck DIM, De Coen WM, Janssen CR.. 2010. Can metal stress induce transferable changes in gene transcription in Daphnia magna? Aquat Toxicol. 97:188–195. [DOI] [PubMed] [Google Scholar]
- Veenstra JA. 2015. The power of next-generation sequencing as illustrated by the neuropeptidome of the crayfish Procambarus clarkii. Gen Comp Endocrinol. 224:84–95. [DOI] [PubMed] [Google Scholar]
- Veenstra JA, Agricola HJ, Sellami A.. 2008. Regulatory peptides in fruit fly midgut. Cell Tissue Res. 334:499–516. [DOI] [PubMed] [Google Scholar]
- Verlinden H, et al. 2015. The pleiotropic allatoregulatory neuropeptides and their receptors: a mini-review. J Insect Physiol. 80:2–14. [DOI] [PubMed] [Google Scholar]
- Villar D, et al. 2015. Enhancer evolution across 20 mammalian species. Cell 160:554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Elert E, et al. 2004. Protease activity in gut of Daphnia magna: Evidence for trypsin and chymotrypsin enzymes. Comp Biochem Physiol B Biochem Mol Biol. 137:287–296. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M.. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JB, et al. 2008. Systematic identification of mammalian regulatory motifs’ target genes and functions. Nat Methods 5:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, et al. 2014. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158:1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Hughes TR.. 2010. Conserved expression without conserved regulatory sequence: the more things change, the more they stay the same. Trends Genet. 26:66–74. [DOI] [PubMed] [Google Scholar]
- Weston DJ, Gunter LE, Rogers A, Wullschleger SD.. 2008. Connecting genes, coexpression modules, and molecular signatures to environmental stress phenotypes in plants. BMC Syst Biol. 2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wfleabase. wfleabase: Daphnia magna genome data Available from: http://server7.wfleabase.org/genome/Daphnia_magna/openaccess/.
- White KP, Hurban P, Watanabe T, Hogness DS.. 1997. Coordination of Drosophila metamorphosis by two ecdysone-induced nuclear receptors. Science 276:114–117. [DOI] [PubMed] [Google Scholar]
- Williams TD, et al. 2011. Towards a system level understanding of non-model organisms sampled from the environment: a network biology approach. PLoS Comput Biol. 7:e1002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka N, et al. 2010. Bombyx prothoracicostatic peptides activate the sex peptide receptor to regulate ecdysteroid biosynthesis. Proc Natl Acad Sci U S A. 107:2060–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka N, Rewitz KF, O’Connor MB.. 2013. Ecdysone control of developmental transitions: lessons from Drosophila research. Annu Rev Entomol. 58:497–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, et al. 2008. The multiple-wing-hairs gene encodes a novel GBD-FH3 Domain-containing protein that functions both prior to and after wing hair initiation. Genetics 180: 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez-Cuna JO, Kvon EZ, Stark. 2013. Deciphering the transcriptional cis-regulatory code. Trends Genet. 29:11–22. [DOI] [PubMed] [Google Scholar]
- Yoshiyama T, Namiki T, Mita K, Kataoka H, Niwa R.. 2006. Neverland is an evolutionally conserved Rieske-domain protein that is essential for ecdysone synthesis and insect growth. Development 133:2565–2574. [DOI] [PubMed] [Google Scholar]
- Zhao XL, Campos AR.. 2012. Insulin signalling in mushroom body neurons regulates feeding behaviour in Drosophila larvae. J Exp Biol. 215:2696–2702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genomic resources and databases used in this study are listed in supplementary table S4, Supplementary Material online.