

ABSTRACT
Siblings of patients with Crohn's disease (CD) have elevated risk of developing CD and display aspects of disease phenotype, including faecal dysbiosis. In our recent article we have used 16S rRNA gene targeted high-throughput sequencing to comprehensively characterize the mucosal microbiota in healthy siblings of CD patients, and determine the influence of genotypic and phenotypic factors on the gut microbiota (dysbiosis). We have demonstrated that the core microbiota of both patients with CD and healthy siblings is significantly less diverse than controls. Faecalibacterium prausnitzii contributed most to core metacommunity dissimilarity between both patients and controls and between siblings and controls. Phenotype/genotype markers of CD risk significantly influenced microbiota variation between and within groups, of which genotype had the largest effect. Individuals with elevated CD-risk display mucosal dysbiosis characterized by reduced diversity of core microbiota and lower abundance of F. prausnitzii. The presence of this dysbiosis in healthy people at-risk of CD implicates microbiological processes in CD pathogenesis.
KEYWORDS: Crohn's disease, microbiome, Faecalibacterium prausnitzii, siblings, inflammatory bowel disease, bifidobacteria
In contrast to skin, lung or eye, where barrier function is prioritised and the range of substances targeted for absorption are limited, the surface of the gut has uniquely evolved to actively interact with a wide variety of ingested constituents of the host's environment. Why the regulation of this intricate interface between humans and their environment degenerates in inflammatory bowel disease (IBD) is unclear. However, in some genetically predisposed individuals an abnormal immune reaction to gut microbes develops and results in chronic intestinal inflammation. Traditionally, 2 main clinical entities have been described: Crohn's disease (CD) and ulcerative colitis. However, genetic and microbiome studies have challenged this dogma; for example some studies support a difference between small intestinal-restricted and colonic-involving CD.1,2
A genetic predisposition to IBD is fundamental and relatives of patients are at enhanced risk of developing IBD themselves.3,4 The interaction between the gut microbiota and the gut immune system is pivotal in IBD pathogenesis5 and in patients with IBD abnormalities can be demonstrated in both the gut immune system and the gut microbiota (dysbiosis) as well as alterations in intestinal permeability and increased concentrations of neutrophil-derived calprotectin in faeces.6 However, whether dysbiosis has a role in disease pathogenesis or is merely consequent to inflammation cannot be determined by examining only individuals with established IBD. For example, increased faecal γ-proteobacteria have been widely described in CD5,7 particularly increased in Escherichia coli,8 and putative mechanisms by which E. coli may contribute to CD pathogenesis include the capacity to adhere to and invade the intestinal mucosa,9,10 as well as the persistence of these bacteria in epithelial cells and macrophages.11 However, there are also potential mechanisms by which E. coli may be increased opportunistically as a consequence of CD, such as the increased activity of nitric oxide synthases associated with inflammation,12 which could favor the survival of these nitrate-reducing bacteria, or increased intestinal luminal pH secondary to a reduction in faecal butyrate producers frequently reported in CD – potentially favoring the survival of organisms that are inhibited at acidic pH such as E. coli.13 Moreover, there is also evidence that the abundance of γ-proteobacteria may be affected by drugs such as immunosuppressant and 5-ASA drugs used to treat CD.14 There has therefore been an ongoing conundrum as to whether the dysbiosis in IBD patients is a feature of pathogenesis or whether it occurs after disease onset as a result of established inflammation. The temporal relationship between dysbiosis and disease onset is not easily examined in humans, however these factors may be more readily manipulated in animal models. There is a strong implication from animal models of IBD that the microbiota are a key part of disease pathogenesis given that in several animal models of gut inflammation, animals kept in germ-free conditions do not develop disease.15-18 Genotype-environment interactions may also be examined in animal models, such as that described between Atg16L1 and murine norovirus in a mouse model of CD.19 However, extrapolation from animal data to human disease should be approached with caution. Studying individuals before they develop CD, or individuals who share genetic and environmental exposures that predispose to IBD, but in whom the cumulative effect of these triggers is, as yet, insufficient to produce the full-blown disease phenotype, i.e. healthy relatives of IBD patients, provide a window into pathogenic pathways in the absence of the obfuscating influence of established, chronic CD.
In our recent publication “Siblings of patients with Crohn's disease exhibit biologically relevant dysbiosis in mucosal microbial metacommunities”20 we used 16S rRNA gene targeted high-throughput sequencing to test the hypothesis that dysbiosis exists in the mucosal microbiota of healthy siblings of CD patients, and is therefore not merely a consequence of established disease. In addition, we examined the influence of genotypic and phenotypic factors, on that dysbiosis. Twenty-one patients with quiescent CD and 17 of their healthy siblings were recruited via gastroenterology outpatient clinics, in addition to 19 unrelated healthy controls. Participants at the peak age when CD is diagnosed (16–35 years),21 were specifically targeted in order that data from this study is most relevant to the population in which any future pre-disease screening program is the most viable. In addition, enrolling only young relatives of CD patients increases the possibility of including individuals who will go on to develop CD. Healthy but genetically predisposed relatives may manifest biomarkers that reflect genetic risk or environmental exposures and crucially, may reveal their cumulative and combined effects. In addition, an accurate description of the ‘at risk’ state in siblings and offspring of CD patients raises the potential to predict and prevent disease. Moreover, longitudinal surveys in families who are enriched for both genetic and environmental risk factors provide a cohort with greater incidence of CD. However, the analysis of CD pathogenesis in healthy siblings is not completely straightforward. First, the degree of genetic relatedness of full-siblings is on average around 50% but detailed analysis of sibling genomes reveals that their similarity may vary between 37 to 62%.22 Secondly, expression of risk phenotypes may depend on environmental exposures that may vary between family members; for example aspirin may induce increased intestinal permeability.23 Finally, one of the advantages of family studies (the capacity to examine genotype-environment interactions) is also a limitation in that it may not be possible to determine whether a phenotype that is shared between siblings is shared because of genotype (e.g. genetic determination of gut microbiota) or shared environment (e.g., maternal microbial inoculum determining neonatal gut microbiota). Such questions may be addressed using twin cohorts. Despite these limitations, studies of healthy, at-risk relatives of patients with CD may uniquely contribute to the illumination of pathogenic pathways that are not easily discernible in studying patients with established disease.
We showed that core microbiota of both CD patients and healthy siblings were significantly less diverse compared with healthy unrelated controls. This finding confirms that dysbiosis is not merely a consequence of intestinal inflammation but is also present in at-risk, healthy individuals, clearly implicating the microbiota in CD pathogenesis. The significance of reduced microbial diversity is not fully understood but it is interesting to speculate on because this is a feature of the dysbiosis in a variety of diseased states including obesity,24,25 colorectal cancer,26 eczema,27 and in addition has been linked with smoking.28 Perhaps lower diversity is associated with incomplete occupation of ecological niches resulting in reduced resistance to colonisation by more pro-inflammatory species; alternatively a more restricted gut metagenome may contain a lower array of microbial genes that results in the loss of key functions. Currently it is not clear whether reduced diversity in itself has a specific functional consequence or if it functions as a barometer for the overall health of the gut microbiota.
Using metacommunity profiling we also showed that the sibling core microbial composition is more similar to their CD affected siblings than to matched healthy controls (Fig. 1). Moreover, reduced Faecalibacterium prausnitzii contributed most to core metacommunity dissimilarity both between siblings and controls, and between patients and controls. As a proportion of core species F. prausnitzii had a higher relative abundance in healthy controls (30.9%) than either patients with CD (22.4%) or siblings (24.2%). Interestingly, we had also previously demonstrated a similar finding in luminal samples from the same cohort (published separately): siblings had significantly lower concentrations of several Firmicute groups including Clostridia cluster IV, Roseburia spp. and F. prausnitzii which was lower in siblings (median 9.27, IQR 8.12–9.78 log10 copies/g) compared with controls (median 9.59, IQR 9.34–10.14 log10 copies/g, p = 0.048) as well as between patients (median 6.88, IQR 5.03–9.35 log10 copies/g) compared with controls (p = 0.006) (Fig. 2).29 Thus, the finding of sibling dysbiosis including reduced abundance of F. prausnitzii is robust, having been demonstrated in analyses using different techniques (454 pyrosequencing and qPCR), and in both mucosa-associated and faecal microbiota
Figure 1.
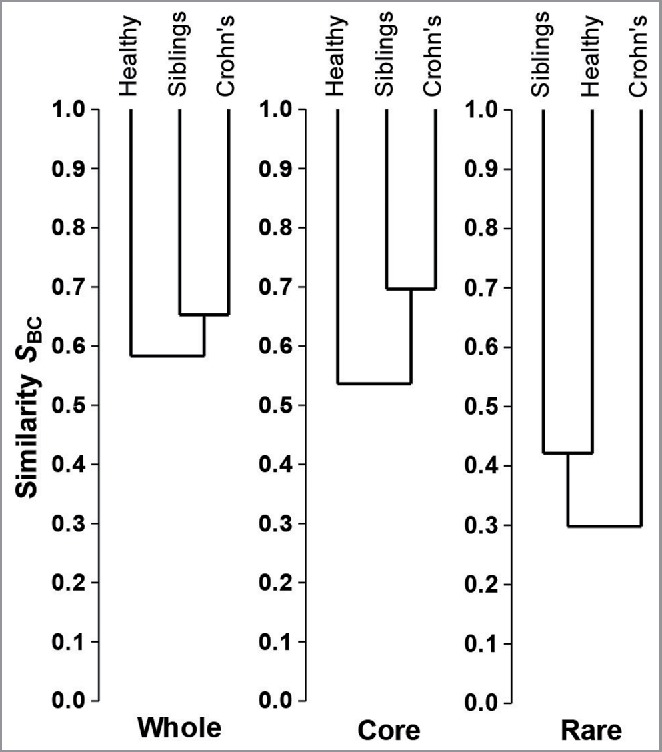
Dendrogram showing the microbial community dissimilarity between the 3 groups: The composition of the whole microbiota as determined by Bray-Curtis index, is more similar between patients and their healthy siblings than between healthy siblings and healthy controls, and this pattern is driven by similarity in the core microbiota between patients and siblings rather than the rare microbiota.
Figure 2.
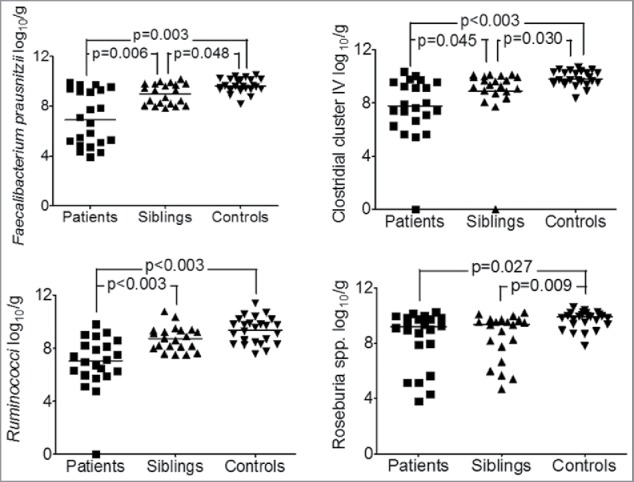
Concentrations of different Firmicute populations in faecal samples were significantly lower in patients (n = 22) and siblings, (n = 21) compared with controls, (n = 25).
There has been intense speculation regarding the role of F. prausnitzii in CD pathogenesis as it is the only microbial factor shown to be predictive of the natural history of CD30 and of the response to treatment.31 It may be speculated that loss of F. prausnitzii could result in the loss of key functions that contribute to gut health, for example the production of short-chain fatty acids, in particular butyrate,32 and NFκB-mediated effects.33 However, we would be cautious in constructing pathogenic hypotheses based on the functions of this particular species, rather interpreting these data as implicating that loss of F. prausnitzii is a sensitive indicator of a broader change in the gut microbiota. Furthermore, data are emerging demonstrating increases in F. prausnitzii in new onset pediatric Crohn's disease, indicating that the role this species plays in pathogenesis is complex.34 Interestingly, increased E. coli contributed to the dissimilarity between patients and healthy controls but not to the dissimilarity between siblings and healthy controls. Thus it may be that the CD dysbiosis comprises microbial factors that contribute to pathogenesis (as exemplified by lower F. prausnitzii), overlaid with microbial alterations that are consequent to inflammation (as exemplified by higher abundance of E. coli). Alternatively, the sibling dysbiosis may represent an incomplete version of the full CD dysbiosis, which is insufficient to lead to full-blown CD. Only longitudinal studies can answer this question.
Genotype contributes to CD pathogenesis35 and in addition the composition of the gut microbiota is partly determined by genotype.36 In our study we demonstrated that genotype relative risk (a composite score of genotypic risk across 72 loci associated with CD) was the most significant factor in explaining variance between the 3 cohorts, (patients, healthy siblings and healthy, unrelated controls) and also within each cohort. There is an evolutionary advantage to be accrued through host genetic influence over the colonisation by commensals to maximize host fitness. Furthermore, microbiota differ markedly from one host habitat to another, such as skin compared with gut,37 and this indicates that there are selection pressures, potentially under host control, that determine the differential survival of bacteria in these sites. It would therefore be surprising if the capacity to influence host microbiota had failed to evolve within the human genome. If the host can shape the microbiota, it therefore follows that due to natural variation, in some individuals a suboptimal genotype will produce a less well-adapted phenotype, and furthermore may even result in disease. Recent data in animal models supports the role for the gut immune system in shaping the microbiota and suggests that this effect may be at least in part dependent on a pathway involving both polyreactive and bacteria-specific secretory IgA,38 as well as mediated through gut epithelial cells and Hopx+ cell-derived miRNAs, which enter bacteria and regulate bacterial gene expression and growth, in turn affecting the microbiota composition and susceptibility to colitis.39 Furthermore specific SNPs have been associated with gut microbiota composition in a cohort of healthy relatives.36
We have shown that perturbations in the mucosal gut microbiota occur not only in individuals with Crohn's disease but also in otherwise healthy individuals at elevated risk of Crohn's disease, thus dysbiosis is not merely a consequence of inflammation. However, studies of pediatric IBD highlight paradoxical increases in species such as F. prausnitzii34,40 that are widely reported to be reduced in adults with IBD. While this finding might suggest that alternative pathogenic pathways exist in pediatric IBD, it might also indicate that gut microbiota composition may evolve during pathogenesis. Rather than focusing on the microbial composition at the point when the individual develops the disease, it may be that the influence of the gut microbiota occurs long before disease onset. It may be speculated that there are critical periods during immune development when dysbiosis may exert its influence. The evidence for the importance of the early childhood period comes from several sources: studies of human migration from areas of low prevalence to high prevalence and vice versa indicate that in some populations IBD risk is associated with the area of birth,41 implicating events in the perinatal or early childhood period in IBD pathogenesis. Many of the epidemiological associations with IBD link to early childhood, including breastfeeding, tonsillectomy, childhood vaccinations, childhood infections,42 birth rank43 and birth in hospital.44 In conjunction, several of these factors have been shown to influence the gut microbiota,45 and the acquisition of the gut microbiota in humans appears to occur predominantly over the first 2 y of life.46 In animal models gut immune maturation is influenced by the timing of introduction of gut microbiota,47 implying that early microbial exposure may have the capacity to condition immune responses in the long-term. Moreover, other animal studies have implicated even pre-natal effects of the maternal microbiota on the developing immune system of the fetus/neonate. Such influences may be effected through factors including microbial molecular transfer mediated by maternal immunoglobulins transmitted both trans-placentally and through lactation.48 Defining the interrelated processes of human immune development and microbial acquisition may have a significant impact on our understanding of the pathogenesis of IBD.
Given the factors discussed above, it may be hypothesized that environmental factors impact on the acquisition of the gut microbiota during the prenatal, neonatal or early childhood period, which in turn creates a persistent inflammatory immune tone, thus laying the foundations for future IBD risk. Defining the relationship between microbial acquisition, immune phenotype and IBD risk requires longitudinal studies and the results of ongoing studies including the MECONIUMstudy (Exploring MEChanisms Of disease traNsmisson In Utero through the Microbiome; a study comparing the bacterial profiles of pregnant women with and without IBD with their new-born babies and in addition assessing the influence of infant feeding practices and antibiotic use early in life on microbiota acquisition) and the GEM project (Genetics, Environment and Microbiota Project: A longitudinal study of relatives of patients with Crohn's disease)36 are eagerly awaited.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This research was funded by a research fellowship for Dr Charlotte Hedin from Core, a charity for digestive diseases.
References
- [1].Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, Andersen V, Andrews JM, Annese V, Brand S, et al., Inherited determinants of Crohn's disease and ulcerative colitis phenotypes: a genetic association study. Lancet 2016; 387(10014):156-167; PMID:26490195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Naftali T, Reshef L, Kovacs A, Porat R, Amir I, Konikoff FM, Gophna U. Distinct Microbiotas are Associated with Ileum-Restricted and Colon-Involving Crohn's Disease. Inflamm Bowel Dis 2016; 22(2):293-302; PMID:26752462; http://dx.doi.org/ 10.1097/MIB.0000000000000662 [DOI] [PubMed] [Google Scholar]
- [3].Satsangi J, Grootscholten C, Holt H, Jewell DP. Clinical patterns of familial inflammatory bowel disease. Gut 1996; 38(5): 738-41; PMID:8707121; http://dx.doi.org/ 10.1136/gut.38.5.738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Halfvarson J, Genetics in twins with Crohn's disease: less pronounced than previously believed? Inflamm Bowel Dis 2011; 17(1): 6-12; PMID:20848478; http://dx.doi.org/ 10.1002/ibd.21295 [DOI] [PubMed] [Google Scholar]
- [5].Kostic AD, Xavier RJ, and Gevers D, The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 2014; 146(6):1489-99; PMID:24560869; http://dx.doi.org/ 10.1053/j.gastro.2014.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hedin CR, Stagg AJ, Whelan K, Lindsay JO. Family studies in Crohn's disease: new horizons in understanding disease pathogenesis, risk and prevention. Gut 2012; 61(2):311-8; PMID:21561876; http://dx.doi.org/ 10.1136/gut.2011.238568 [DOI] [PubMed] [Google Scholar]
- [7].Dicksved J, Halfvarson J, Rosenquist M, Järnerot G, Tysk C, Apajalahti J, Engstrand L, Jansson JK. Molecular analysis of the gut microbiota of identical twins with Crohn's disease. ISME J 2008; 2(7):716-27; PMID:18401439; http://dx.doi.org/ 10.1038/ismej.2008.37 [DOI] [PubMed] [Google Scholar]
- [8].Li Q, Wang C, Tang C, Li N, Li J. Molecular-phylogenetic characterization of the microbiota in ulcerated and non-ulcerated regions in the patients with Crohn's disease. PLoS One 2012; 7(4): e34939; PMID:22529960; http://dx.doi.org/ 10.1371/journal.pone.0034939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Barnich N, Denizot J, and Darfeuille-Michaud A, E. coli-mediated gut inflammation in genetically predisposed Crohn's disease patients. Pathol Biol (Paris) 2013; 61(5):e65-9; PMID:20381273; http://dx.doi.org/ 10.1016/j.patbio.2010.01.004 [DOI] [PubMed] [Google Scholar]
- [10].Denizot J, Desrichard A, Agus A, Uhrhammer N, Dreux N, Vouret-Craviari V, Hofman P, Darfeuille-Michaud A, Barnich N. Diet-induced hypoxia responsive element demethylation increases CEACAM6 expression, favouring Crohn's disease-associated Escherichia coli colonisation. Gut 2015; 64(3):428-37; PMID:24898815; http://dx.doi.org/ 10.1136/gutjnl-2014-306944 [DOI] [PubMed] [Google Scholar]
- [11].Lapaquette P, Bringer MA, and Darfeuille-Michaud A, Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol 2012; 14(6):791-807; PMID:22309232; http://dx.doi.org/ 10.1111/j.1462-5822.2012.01768.x [DOI] [PubMed] [Google Scholar]
- [12].Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, et al.. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 2013; 339(6120):708-11; PMID:23393266; http://dx.doi.org/ 10.1126/science.1232467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Duncan SH, Louis P, Thomson JM, Flint HJ. The role of pH in determining the species composition of the human colonic microbiota. Environ Microbiol 2009; 11(8):2112-22; PMID:19397676; http://dx.doi.org/ 10.1111/j.1462-2920.2009.01931.x [DOI] [PubMed] [Google Scholar]
- [14].Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al., Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 2012; 13(9):R79; PMID:23013615; http://dx.doi.org/ 10.1186/gb-2012-13-9-r79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dianda L, Hanby AM, Wright NA, Sebesteny A, Hayday AC, Owen MJ. T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a microbial environment. Am J Pathol 1997; 150(1):91-7. PMID:9006326. [PMC free article] [PubMed] [Google Scholar]
- [16].Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 1998; 66(11):5224-31. PMID:9784526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Veltkamp C, Tonkonogy SL, De Jong YP, Albright C, Grenther WB, Balish E, Terhorst C, Sartor RB. Continuous stimulation by normal luminal bacteria is essential for the development and perpetuation of colitis in Tg(epsilon26) mice. Gastroenterology 2001; 120(4):900-13; PMID:11231944; http://dx.doi.org/ 10.1053/gast.2001.22547 [DOI] [PubMed] [Google Scholar]
- [18].Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, Fernández-Sueiro JL, Balish E, Hammer RE. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med 1994; 180(6):2359-64; PMID:7964509; http://dx.doi.org/ 10.1084/jem.180.6.2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell 2010; 141(7):1135-45; PMID:20602997; http://dx.doi.org/ 10.1016/j.cell.2010.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hedin C, van der Gast CJ, Rogers GB, Cuthbertson L, McCartney S, Stagg AJ, Lindsay JO, Whelan K. Siblings of patients with Crohn's disease exhibit a biologically relevant dysbiosis in mucosal microbial metacommunities. Gut 2016; 65(6):944-53; PMID:25856344; http://dx.doi.org/ 10.1136/gutjnl-2014-308896 [DOI] [PubMed] [Google Scholar]
- [21].Loftus EV Jr., Silverstein MD, Sandborn WJ, Tremaine WJ, Harmsen WS, Zinsmeister AR. Crohn's disease in Olmsted County, Minnesota, 1940-1993: incidence, prevalence, and survival. Gastroenterology 1998; 114(6):1161-8; PMID:9609752; http://dx.doi.org/ 10.1016/S0016-5085(98)70421-4 [DOI] [PubMed] [Google Scholar]
- [22].Visscher PM, Medland SE, Ferreira MA, Morley KI, Zhu G, Cornes BK, Montgomery GW, Martin NG. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet 2006; 2(3):e41; PMID:16565746; http://dx.doi.org/ 10.1371/journal.pgen.0020041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Soderholm JD, Olaison G, Lindberg E, Hannestad U, Vindels A, Tysk C, Järnerot G, Sjödahl R. Different intestinal permeability patterns in relatives and spouses of patients with Crohn's disease: an inherited defect in mucosal defence? Gut 1999; 44(1):96-100; PMID:9862833; http://dx.doi.org/ 10.1136/gut.44.1.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, et al., Richness of human gut microbiome correlates with metabolic markers. Nature 2013; 500(7464):541-6; PMID:23985870; http://dx.doi.org/ 10.1038/nature12506 [DOI] [PubMed] [Google Scholar]
- [25].Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al., A core gut microbiome in obese and lean twins. Nature 2009; 457(7228):480-4; PMID:19043404; http://dx.doi.org/ 10.1038/nature07540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, Goedert JJ, Hayes RB, Yang L. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst 2013; 105(24):1907-11; PMID:24316595; http://dx.doi.org/ 10.1093/jnci/djt300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ismail IH, Oppedisano F, Joseph SJ, Boyle RJ, Licciardi PV, Robins-Browne RM, Tang ML. Reduced gut microbial diversity in early life is associated with later development of eczema but not atopy in high-risk infants. Pediatr Allergy Immunol 2012; 23(7):674-81; PMID:22831283; http://dx.doi.org/ 10.1111/j.1399-3038.2012.01328.x [DOI] [PubMed] [Google Scholar]
- [28].Biedermann L, Brülisauer K, Zeitz J, Frei P, Scharl M, Vavricka SR, Fried M, Loessner MJ, Rogler G, Schuppler M. Smoking cessation alters intestinal microbiota: insights from quantitative investigations on human fecal samples using FISH. Inflamm Bowel Dis 2014; 20(9):1496-501; PMID:25072500; http://dx.doi.org/ 10.1097/MIB.0000000000000129 [DOI] [PubMed] [Google Scholar]
- [29].Hedin CR, McCarthy NE, Louis P, Farquharson FM, McCartney S, Taylor K, Prescott NJ, Murrells T, Stagg AJ, Whelan K, et al., Altered intestinal microbiota and blood T cell phenotype are shared by patients with Crohn's disease and their unaffected siblings. Gut 2014; 63(10):1578-86; PMID:24398881; http://dx.doi.org/ 10.1136/gutjnl-2013-306226 [DOI] [PubMed] [Google Scholar]
- [30].Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al., Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 2008; 105(43):16731-6; PMID:18936492; http://dx.doi.org/ 10.1073/pnas.0804812105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rajca S, Grondin V, Louis E, Vernier-Massouille G, Grimaud JC, Bouhnik Y, Laharie D, Dupas JL, Pillant H, Picon L, et al., Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn's disease. Inflamm Bowel Dis 2014; 20(6):978-86. PMID:24788220. [DOI] [PubMed] [Google Scholar]
- [32].Rios-Covian D, Gueimonde M, Duncan SH, Flint HJ, de los Reyes-Gavilan CG. Enhanced butyrate formation by cross-feeding between Faecalibacterium prausnitzii and Bifidobacterium adolescentis. FEMS Microbiol Lett 2015; 362(21):fnv176; http://dx.doi.org/ 10.1093/femsle/fnv176 [DOI] [PubMed] [Google Scholar]
- [33].Quevrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J, Miquel S, Carlier L, Bermúdez-Humarán LG, Pigneur B, et al., Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn's disease. Gut 2016; 65(3):415-25; PMID:26045134; http://dx.doi.org/ 10.1136/gutjnl-2014-307649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Assa A, Butcher J, Li J, Elkadri A, Sherman PM, Muise AM, Stintzi A, Mack D. Mucosa-Associated Ileal Microbiota in New-Onset Pediatric Crohn's Disease. Inflamm Bowel Dis 2016; 22(7):1533-9; PMID:27271491; http://dx.doi.org/ 10.1097/MIB.0000000000000776 [DOI] [PubMed] [Google Scholar]
- [35].McGovern DP, Kugathasan S, and Cho JH, Genetics of Inflammatory Bowel Diseases. Gastroenterology 2015; 149(5):1163–1176 e2; PMID:26255561; http://dx.doi.org/ 10.1053/j.gastro.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, Smith MI, Guttman DS, Griffiths A, Panaccione R, Otley A, et al., Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet 2016; 48(11):1413-1417; PMID:27694960; http://dx.doi.org/ 10.1038/ng.3693 [DOI] [PubMed] [Google Scholar]
- [37].Human Microbiome Project, C. , Structure, function and diversity of the healthy human microbiome. Nature 2012; 486(7402):207-14; PMID:22699609; http://dx.doi.org/ 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fransen F, Zagato E, Mazzini E, Fosso B, Manzari C, El Aidy S, Chiavelli A, D'Erchia AM, Sethi MK, Pabst O, et al., BALB/c and C57BL/6 Mice Differ in Polyreactive IgA Abundance, which Impacts the Generation of Antigen-Specific IgA and Microbiota Diversity. Immunity 2015; 43(3):527-40; PMID:26362264; http://dx.doi.org/ 10.1016/j.immuni.2015.08.011 [DOI] [PubMed] [Google Scholar]
- [39].Liu S, da Cunha AP, Rezende RM, Cialic R, Wei Z, Bry L, Comstock LE, Gandhi R, Weiner HL. The Host Shapes the Gut Microbiota via Fecal MicroRNA. Cell Host Microbe 2016; 19(1):32-43; PMID:26764595; http://dx.doi.org/ 10.1016/j.chom.2015.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hansen R, Russell RK, Reiff C, Louis P, McIntosh F, Berry SH, Mukhopadhya I, Bisset WM, Barclay AR, Bishop J, et al., Microbiota of de-novo pediatric IBD: increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn's but not in ulcerative colitis. Am J Gastroenterol 2012; 107(12):1913-22; PMID:23044767; http://dx.doi.org/ 10.1038/ajg.2012.335 [DOI] [PubMed] [Google Scholar]
- [41].Li X, Sundquist J, Hemminki K, Sundquist K. Risk of inflammatory bowel disease in first- and second-generation immigrants in Sweden: a nationwide follow-up study. Inflamm Bowel Dis 2011; 17(8):1784-91; PMID:21744434; http://dx.doi.org/ 10.1002/ibd.21535 [DOI] [PubMed] [Google Scholar]
- [42].Hansen TS, Jess T, Vind I, Elkjaer M, Nielsen MF, Gamborg M, Munkholm P. Environmental factors in inflammatory bowel disease: a case-control study based on a Danish inception cohort. J Crohns Colitis 2011; 5(6):577-84; PMID:22115378; http://dx.doi.org/ 10.1016/j.crohns.2011.05.010 [DOI] [PubMed] [Google Scholar]
- [43].Hampe J, Heymann K, Krawczak M, Schreiber S. Association of inflammatory bowel disease with indicators for childhood antigen and infection exposure. Int J Colorectal Dis 2003; 18(5):413-7; PMID:12687394; http://dx.doi.org/ 10.1007/s00384-003-0484-1 [DOI] [PubMed] [Google Scholar]
- [44].Gearry RB, Richardson AK, Frampton CM, Dodgshun AJ, Barclay ML. Population-based cases control study of inflammatory bowel disease risk factors. J Gastroenterol Hepatol 2010; 25(2):325-33; PMID:20074146; http://dx.doi.org/ 10.1111/j.1440-1746.2009.06140.x [DOI] [PubMed] [Google Scholar]
- [45].Dicksved J, Flöistrup H, Bergström A, Rosenquist M, Pershagen G, Scheynius A, Roos S, Alm JS, Engstrand L, Braun-Fahrländer C, et al., Molecular fingerprinting of the fecal microbiota of children raised according to different lifestyles. Appl Environ Microbiol 2007; 73(7):2284-9; PMID:17293501; http://dx.doi.org/ 10.1128/AEM.02223-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol 2007; 5(7):e177; PMID:17594176; http://dx.doi.org/ 10.1371/journal.pbio.0050177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hansen CH, Nielsen DS, Kverka M, Zakostelska Z, Klimesova K, Hudcovic T, Tlaskalova-Hogenova H, Hansen AK. Patterns of early gut colonization shape future immune responses of the host. PLoS One 2012; 7(3):e34043; PMID:22479515; http://dx.doi.org/ 10.1371/journal.pone.0034043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gomez de Aguero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, Steinert A, Heikenwalder M, Hapfelmeier S, Sauer U, et al., The maternal microbiota drives early postnatal innate immune development. Science 2016; 351(6279):1296-302; PMID:26989247; http://dx.doi.org/ 10.1126/science.aad2571 [DOI] [PubMed] [Google Scholar]