

Abstract
The proteasome is a vital cellular machine that maintains protein homeostasis, which is of particular importance in multiple myeloma and possibly other cancers. Targeting proteasome 20S peptidase activity with bortezomib and carfilzomib has been widely used to treat myeloma. However, not all patients respond, and those that do eventually suffer relapse. Therefore, there is an urgent and unmet need to develop novel drugs that target proteostasis through different mechanisms. We identified quinoline-8-thiol (8TQ) as a first-in-class inhibitor of the proteasome 19S subunit Rpn11. A derivative of 8TQ, capzimin, shows >5-fold selectivity for Rpn11 over the related JAMM proteases and >2 logs less activity towards metalloenzymes. Capzimin stabilized proteasome substrates, induced an unfolded protein response, and blocked proliferation of cancer cells, including those resistant to bortezomib. Proteomic analysis revealed that capzimin stabilized a subset of polyubiquitinated substrates. Identification of capzimin offers an alternative path to develop proteasome inhibitors for cancer therapy.
Introduction
The 26S proteasome is the main mediator of protein degradation in eukaryotic cells1. It is essential for multiple cellular processes including protein quality control, regulation of transcription, and cell division. A protein is targeted for proteasomal degradation when it is covalently modified on a lysine residue with a polyubiquitin (polyUb) chain, which is bound by the proteasome to initiate proteolysis1. Structurally, the proteasome is composed of the 19S regulatory particle (RP) and the 20S core particle (CP)1. The RP recognizes polyubiquitinated substrates and inserts them into the CP, which contains the proteolytic active sites. The “’omibs”, bortezomib (BTZ), carfilzomib (CFZ), and ixazomib, inhibit the β5 active sites in the CP and are approved for treatment of multiple myeloma. However, despite the clinical benefit that they provide, patients ultimately relapse2, and therefore new agents are urgently needed.
We and others previously identified an active site in the RP subunit Rpn11 that removes the polyUb chain from substrates3,4. This active site is located within Rpn11’s conserved JAMM domain, and features a catalytic Zn2+ ion5,6. The human genome encodes seven proteins that contain a JAMM domain with a complete set of conserved residues for catalysis, including: Poh1 (Rpn11), Csn5 subunit of the COP9 signalosome, AMSH, AMSH-LP, the BRCC36 subunit of BRISC, MYSM1 and MPND7. All but MPND have been shown to exhibit isopeptidase activity towards ubiquitin or ubiquitin like proteins7–10. Importantly, substrate deubiquitination by Rpn11 is essential and tightly coupled to degradation. Point mutation of the enzymatic site results in a severe decrease in substrate degradation followed by cell death3,4,11. ‘Synthetic lethality’ siRNA screens indicate that depletion of Rpn11 sensitizes lung cancers bearing EGFR mutations to killing by the EGFR inhibitor erlotinib12. Moreover, Rpn11 activity is required to sustain pluripotency13, pointing to the potential of Rpn11 inhibitors to target cancer stem cells. Together, these data identify Rpn11 as a potential alternative target to inhibit the proteasome in cancer treatment. We anticipate that Rpn11 inhibitors and the “’omib” class of β5 inhibitors might have distinct efficacy profiles because they work by different mechanisms. However, realizing this potential would require overcoming the perceived barrier to development of specific metalloprotease inhibitors.
Here, we identified quinoline-8-thiol (1, 8TQ) as an inhibitor of the essential proteasomal deubiquitinase Rpn11. Medicinal chemistry optimization of 8TQ yielded Capzimin, a potent and moderately specific Rpn11 inhibitor. Characterization of Capzimin by a combination of biochemical, cell biological, and proteomics approaches demonstrated that it inhibited proteasome function through a mechanism distinct from the classical 20S proteasome inhibitors.
Results
Two screening reveal the same moiety targeting the Rpn11
To identify Rpn11 inhibitors, we first established a fluorescence polarization assay that specifically measures the deubiquitinating activity of Rpn11. The assay features a proteasome substrate with four tandem ubiquitins (Ub4) followed by a peptide labeled with Oregon Green on a unique cysteine residue (Fig 1a). Incubation of this substrate, Ub4peptideOG, with proteasome resulted in fluorescence depolarization due to release of the peptideOG from Ub4. This activity was likely due to Rpn11, because it was not sensitive to the cysteine-based deubiquitinase (DUB) inhibitor ubiquitin aldehyde (Supplementary Results, Supplementary Fig. 1), and unlike the other DUB activities that associate with the proteasome, the cleavage we observed was dependent on ATP hydrolysis (Supplementary Fig. 2). Using this assay, we screened a first-generation in-house library containing 96 metal-binding pharmacophores (MBPs), which yielded three hits with greater than 50% inhibition at 200 μM, including quinoline-8-thiol (8TQ)(details will be published elsewhere).
Fig 1. Identification of a compound that inhibits proteasome function by binding directly to the active site zinc of Rpn11.
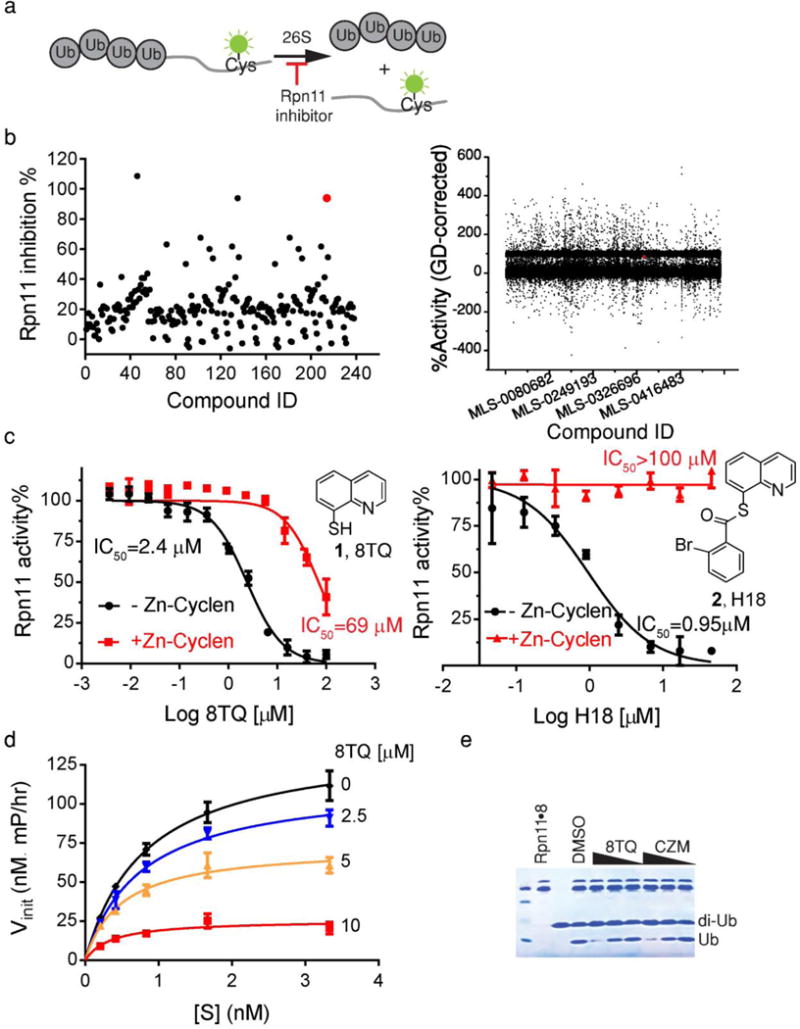
(a) Design of Rpn11 activity assay. (b) Small- and large-scale screens reveal the same Rpn11 moiety. A focused library of 255 MBPs yielded 8TQ (red dot) as the most potent Rpn11 inhibitor, whereas a >300,000 compound library yielded H18 (red dot), a thioester derivative of 8TQ. (c) 8TQ is an uncompetitive inhibitor of Rpn11. The rate of substrate cleavage is plotted as a function of substrate concentration, at 4 different concentrations of 8TQ. Error bars represent s.d., n=3 wells, from 1 representative of 3 independent experiments. (d) 8TQ and H18 inhibited Rpn11 activity by binding zinc. The IC50 for inhibition of Rpn11 activity by 8TQ (left) and H18 (right) was determined in the presence and absence of a Zn(cyclen)2+ coordination complex. Error bars represent s.d., n=4 wells, from 1 representative of 3 independent experiments. (e) 8TQ and capzimin directly inhibited Rpn11 activity. Purified Rpn11•Rpn8 was incubated with K48-linked di-ubiquitin in the presence of 40 μM, 20 μM or 10 μM of 8TQ or capzimin, and reactions were fractionated by SDS-PAGE and visualized with Coomassie Blue. Shown is a representative gel image of two independent experiments.
To survey a broader landscape of MBPs, we next screened a substantially larger second-generation library with 255 MBPs. Ten compounds showed inhibition >50% at 200 μM Further analysis indicated four of these were due to fluorescence artifacts. Another five were not reproducible (all <50%). The only remaining hit was 8TQ (Fig. 1b, c). Titration experiments revealed that the concentration of 8TQ at which 50% of the cleavage activity was inhibited (IC50) was 2.4 μM.
Encouraged by these results, we used our HTS assay to screen 330,000 compounds (each at 20 μM) in the National Institutes of Health Molecular Libraries Small Molecule Repository in the hope of finding superior inhibitors from a vast collection of different chemotypes (Supplementary Table 1). Positive hits were retested in a single-point assay and 10-point titration, followed by two secondary screens. The first screen eliminated compounds that inhibited processing of Ub4-peptideOG by thrombin and the second screen excluded compounds that blocked MMP-2 and Rpn11 activity with equivalent potency. Remaining candidates were then tested for their ability to block proteasome-dependent degradation of reporter protein UbG76V-GFP in cells14. Only one compound, H18 (2, Fig 1b, c), emerged from this winnowing process. Remarkably, this compound, S-(Quinolin-8-yl) 2-bromobenzothioate, is a thioester derivative of 8TQ. Thioesters are inherently unstable in the reducing environment of the cell, suggesting that the active moiety is 8TQ. This extraordinary confluence of results underscores the uniqueness of the 8TQ moiety for potent and specific inhibition of JAMM enzymes, and highlights the remarkable power of our approach to identify a metalloenzyme inhibitor by screening a focused library of MBPs15,16.
Based on our recovery of the 8TQ moiety in two different screens, we sought to gain a deeper understanding of its mode of inhibition. Titration experiments with different concentrations of substrate revealed a KM of 1 nM, which suggested strong binding to the proteasome. Interestingly, we observed lower KM and Vmax values in the presence of different concentrations of 8TQ (Fig. 1d and Supplementary Table 2), suggesting uncompetitive inhibition wherein 8TQ bound specifically to the enzyme•substrate complex. This mode of inhibition is consistent with the report that the coordination site of the catalytic zinc atom in Rpn11 is shielded by interaction with Asn275 of Rpn5 in the 26S proteasome holoenyzme, but becomes exposed to solvent due to a massive conformational change that occurs upon substrate binding17.
Owing to the structural similarity between 8TQ and the previously reported chelating agent 8-hydroxyquinoline, we predicted that it may inhibit Rpn11 activity through binding its catalytic Zn2+ ion. To test this hypothesis, we determined the IC50 value of H18 and 8TQ in the absence and presence of a Zn2+ coordination compound that should compete with the Rpn11 active site for binding MBPs. Zn(cyclen)2+ shifted the IC50 of both compounds to a higher value (Fig. 1c and supplementary Fig. 3), which implied that 8TQ inhibited Rpn11 activity by binding to the catalytic Zn2+ ion. Moreover, x-ray crystallography of zinc–hydrotris(5,3-methylphenylpyrazolyl) borate bound to 8TQ showed that 8TQ binds to form a five-coordinate Zn2+ center with a trigonal bipyramidal coordination geometry (details will be published elsewhere). Further confirmation that 8TQ directly blocked the activity of Rpn11 as opposed to other RP subunits was provided by the observation that it inhibited purified Rpn11•Rpn8 heterodimer (Fig. 1e)18,19. Based on these results, we concluded that 8TQ was a promising moiety for the further optimization of selective Rpn11 inhibitors.
SAR efforts yield potent and selective Rpn11 inhibitors
To improve the potency of 8TQ towards Rpn11, structure-activity relationships (SAR) were explored in a parallel study (details will be published elsewhere). This effort led to multiple compounds with improved potency and specificity such as 3021 (3) and 3027 (4, Fig 2a). The top candidate, 3027, showed seven-fold further improvement in potency toward Rpn11 compared to 8TQ (IC50 = 0.34 μM, Fig. 2b, Supplementary note for synthetic details). Most importantly, 3027 showed 80-fold selectivity for Rpn11 over Csn5, 10-fold over AMSH and 6-fold over BRCC36 (IC50 = 30 μM, 4.5 μM and 2.3 μM respectively, see Fig. 2b; note that we were not able to test the other JAMM enzymes MYSM1 and MPND due to a lack of biochemical assays). By comparison, the starting molecule 8TQ did not distinguish between Rpn11, BRCC36, and AMSH (details will be published elsewhere). Of particular note was the >2 log preference of 3027 for inhibiting Rpn11 compared to the non-JAMM domain metalloenzymes HDAC6, MMP2, MMP12, and carbonic anhydrase II (Supplementary Table 3). Of particular interest, 3027 inhibited glyoxalase I (GLO1) with an IC50 of 43 μM, which is ~20-fold less potent than 8TQ (IC50 2.4 μM). Thus, the SAR campaign enhanced specificity towards both JAMM and non-JAMM metalloenzymes. We propose naming 3027 as ‘capzimin’ (CZM), in which the affix ‘cap’ stands for 19S cap and the stem ‘zimin’ specifies the compound class as zinc metalloisopeptidase inhibitor.
Fig. 2. SAR optimization yields a potent and selective Rpn11 inhibitor.
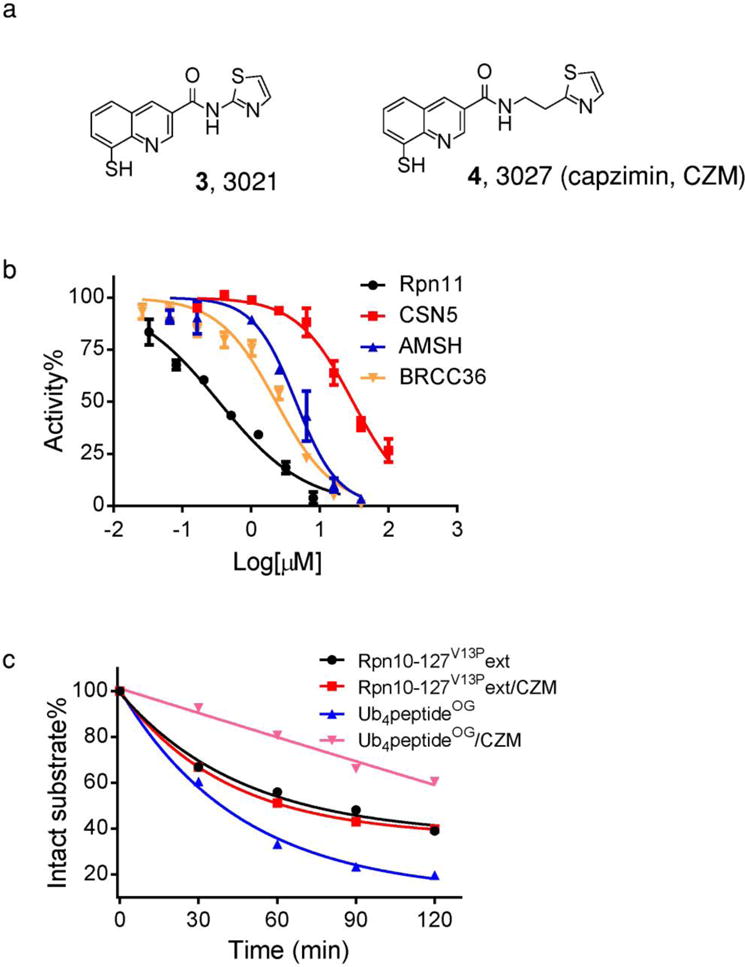
(a) Structures of 3021 and Capzimin. (b) Titration curves for inhibition of Rpn11 (black), BRCC36 (orange) AMSH (blue), and CSN 5 (red) by capzimin. Error bars represent s.d., n=4 wells, from 1 representative of 3 independent experiments. (c) Capzimin does not block ubiquitin-independent activity of proteasome. Ub4peptideOG and the ubiquitin-independent substrate Rpn10-127V13Pext were incubated with purified yeast 26S proteasome in the absence or presence of capzimin, as indicated. Note that capzimin reduced the rate of degradation of Ub4peptideOG but had no effect on Rpn10-127V13Pext. Data represents the average value for n=2.
To further address the specificity of capzimin, we performed two experiments. Because Rpn11 functions as a deubiquitinase which removes the ubiquitin chain from the substrate, capzimin should not block degradation of an ubiquitin-independent proteasome substrate. We tested this prediction using model substrate Rpn10-127V13Pext20, which was designed based on the native ubiquitin-independent proteasome substrate ornithine decarboxylase (ODC). Indeed, capzimin showed no influence on the degradation of Rpn10-127V13Pext by purified proteasomes (Fig. 2c). Second, to rule out the possibility that the 8TQ pharmacophore of capzimin acted as a nonspecific zinc chelator that titrated zinc levels in cells, we employed an optical zinc probe to measure changes in zinc levels in living cells in the presence of 10 μM capzimin21. Capzimin caused ≤5% reduction in total cellular zinc (Supplementary Fig. 4) at a concentration that strongly impedes proteasome function (see Fig. 3).
Fig. 3. Capzimin elicits cellular effects characteristic of proteasome inhibition.
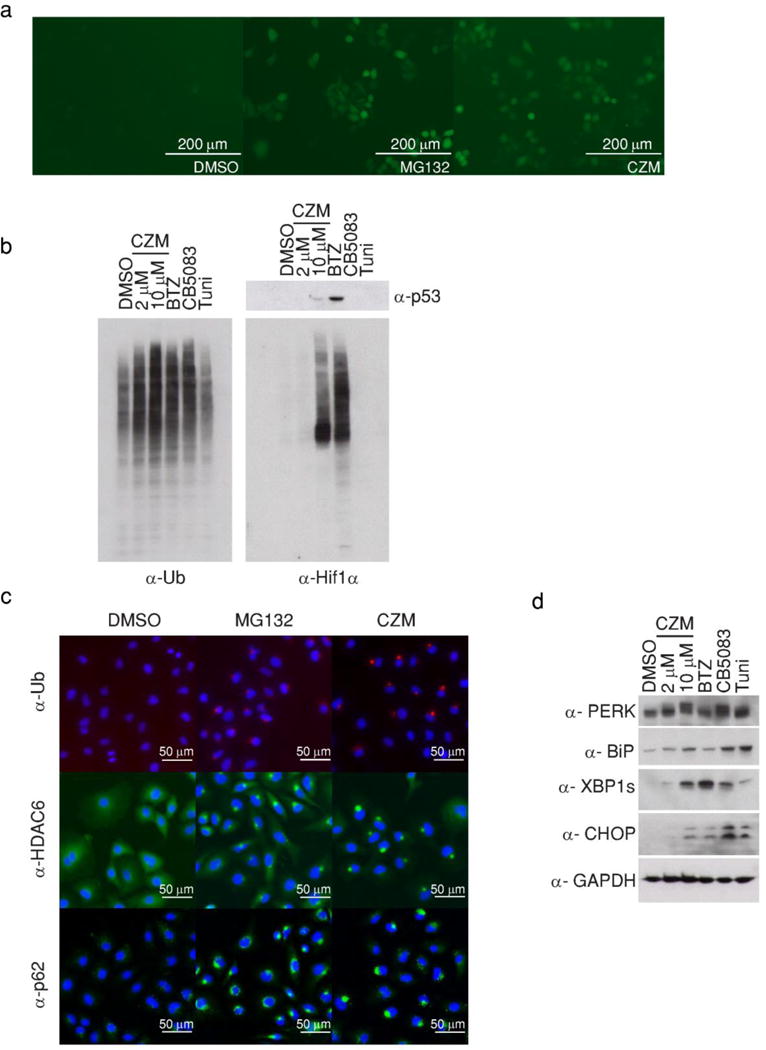
(a) Capzimin caused accumulation of UbG76V-GFP. Shown is a fluorescence image of cells stably expressing UbG76V-GFP, taken 4 hours after treatment with 5 μM of MG132 or capzimin. Here and in panel b to d, a representative image is shown from three independent experiments. (b) Capzimin treatment caused accumulation of high molecular weight ubiquitin conjugates and the UPS substrates p53 and Hif1α. HCT116 cells were treated for 6 hours with 2 or 10 μM of capzimin, BTZ (1 μM), the p97/VCP inhibitor CB5083 (10 μM), or Tunicamycin (Tuni, 2 μg/ml) and cell lysates were fractionated by SDS-PAGE and immunoblotted with antibodies against ubiquitin, p53, and Hif1α, as indicated. (c) Capzimin induced the formation of aggresomes. A549 cells were treated with DMSO, 5 μM MG132 or 10 μM capzimin for 15 hours before being fixed and stained with antibodies against ubiquitin, HDAC6, or p62/SQSTM1. (d) Same as panel b, except that antibodies against PERK, BiP, XBP1s, CHOP, and GAPDH (loading control for b and d) were used. Full gel images were shown in Supplementary Fig. 21.
We next investigated the mechanism of inhibition. Similar to 8TQ, capzimin was an uncompetitive inhibitor of the 26S proteasome (Supplementary Fig. 5 and Supplementary Table 2) and acted directly on Rpn11•Rpn8 (Fig. 1e). Interestingly, capzimin behaved as a competitive inhibitor of both AMSH and BRCC36 (Supplementary Fig. 5). This is consistent with the observation that BRCC3622 and AMSH-LP23 contain a zinc coordination site exposed to solvent that should be able to bind capzimin in the absence of substrate. Notably, these distinct modes of inhibition may enhance the specificity of capzimin in cells, where it is expected that substrates are present in high local concentrations due to the anticipated substrate-concentrating effect of ubiquitin-binding domains in proteins associated with BRCC36 and AMSH9,10.
Based on the promising in vitro properties of capzimin, we sought to test its behavior in cell-based assays. We treated Hela cells stably expressing proteasome reporter UbG76V-GFP with 5 μM of either capzimin or the canonical β5 inhibitor MG132. UbG76V-GFP is rapidly degraded by proteasome and therefore little or no GFP fluorescence is observed unless the proteasome is inhibited14,24. Both MG132 and capzimin elicited strong accumulation of UbG76V-GFP (Fig. 3a). Importantly, the inhibitory effect of capzimin was reversible after washout, which is in agreement with the result from an in vitro jump-dilution experiment (Supplementary Fig 6 and 7). A cycloheximide chase assay24 revealed that capzimin stabilized UbG76V-GFP24 with an IC50 value of 0.6 μM (Supplementary Fig. 8). Notably, earlier-generation compounds, including the closely-related 3021, blocked UbG76V-GFP degradation in the cycloheximide chase assay but had little effect on its accumulation (Supplementary Fig. 9, Supplementary note for synthetic details). Next, we tested capzimin against endogenous substrates of the ubiquitin-proteasome system. Exposure to capzimin caused accumulation of high molecular weight ubiquitin conjugates and the well-studied proteasome substrates p53, Hif1α, and Nrf2 (Fig. 3b and Supplementary Fig. 10). Importantly, capzimin had little effect on Nedd8-conjugated Cul1 (supplementary Fig. 10), indicating it did not appreciably inhibit cellular Csn5, consistent with the biochemical assay results (Fig. 2b).
Sustained inhibition of proteasome function (≥12 h) induces the formation of aggresomes, perinuclear storage depots for aggregated, misfolded proteins25. Whereas depletion of Rpn11 by ~70% was not sufficient to induce aggresome formation, the depleted cells were unable to clear aggresomes upon washout of a β5 inhibitor26. To test whether capzimin influences aggresome formation or clearance, we imaged aggresome markers ubiquitin, HDAC6 and p62/SQSTM126 in A549 cells treated with either MG132 or capzimin for 15 hours. All three markers exhibited punctate staining characteristic of aggresomes in cells treated with either MG132 or capzimin (Fig. 3c). Interestingly, although 3021 failed to cause accumulation of UbG76V-GFP, it induced aggresome formation (Supplementary Fig. 11). Aggresomes formed in the presence of MG132 were not cleared upon removal of MG132 and addition of 8TQ or 3021 (Supplementary Fig. 12 and 13). These data point to strong inhibition of Rpn11 and proteasome function by capzimin.
Previous reports revealed that perturbation of cellular proteostasis networks by inhibition of the proteasome or the p97/VCP segregase that operates upstream of the proteasome results in induction of an unfolded protein response (UPR)27,28. Consistent with its ability to target the proteasome, capzimin provoked a UPR response in both HCT116 (Fig. 3d) and 293T (Supplementary Fig. 14) cells, as judged by accumulation of phosphorylated PERK, spliced XBP1s, and the transcription factor CCAAT/enhancer-binding protein homologous protein (CHOP). Interestingly, capzimin induced a stronger UPR response than bortezomib, but less strong than the p97 inhibitor CB508327.
Capzimin causes cancer cell death
Inhibition of proteasome function results in cell death, which underlies the activity of the ‘omibs in chemotherapy of multiple myeloma. Treatment of HCT116 cell lines with capzimin or 3021 resulted in 50% inhibition of cell growth (GI50) at ~2.0 μM. The GI50 dropped to 0.6 μM in low serum (2.5% instead of 10% FBS). (Fig. 4a). Of relevance to the potential of targeting Rpn11 in ‘omib-refractory myeloma patients, both capzimin and 3021 had the same GI50 against WT and bortezomib-resistant retinal pigment epithelial cells29, indicating that 8TQ-based and ‘omib inhibitors worked through distinct mechanisms (Fig. 4b).
Fig. 4. Capzimin blocks cell growth and induces markers of apoptosis.
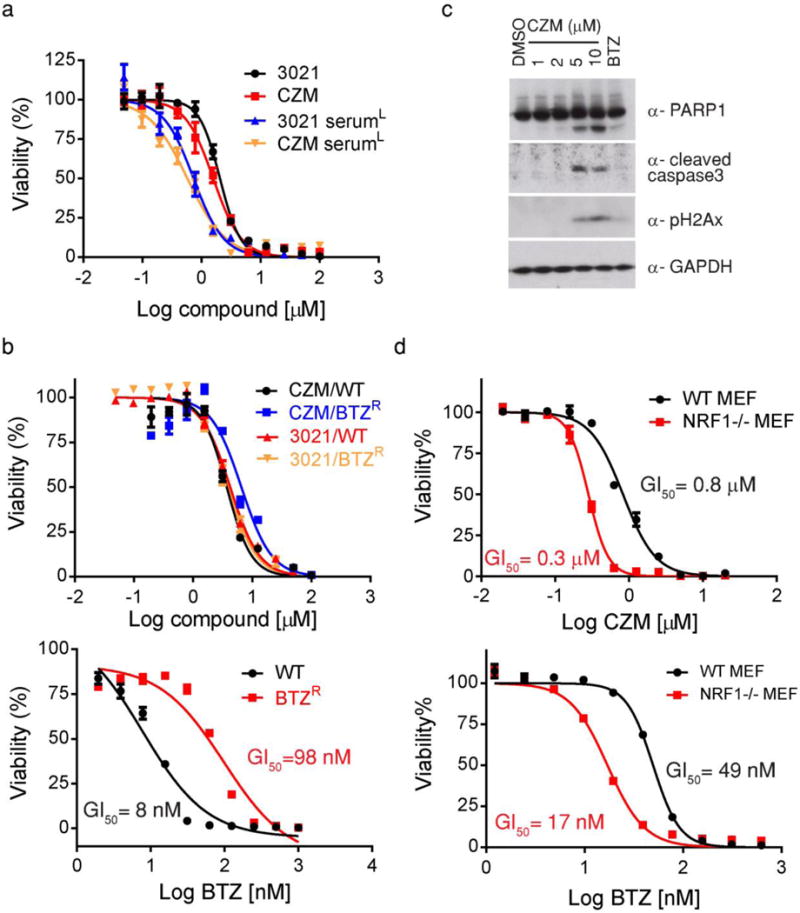
(a) Capzimin inhibited cell growth. HCT116 cells were treated with different concentrations of 3021 or capzimin for 72 hours in normal or low serum medium and then mixed with CellTiter-Glo reagent to estimate cell proliferation. Measured luminescence values were normalized to DMSO control and data were fitted to a dose-response equation to determine the GI50. Error bars here and in panels b and d represent s.d., n=3 wells, from 1 representative of 3 independent experiments. (b) Bortezomib resistant cell line was not resistant to 3021 or capzimin. WT or bortezomib-resistant RPE1 cells were treated with different concentrations of 3021, capzimin (top) or BTZ (bottom) for 72 hours and analyzed as in panel a. (c) Capzimin induced apoptosis. HCT116 cells were treated with the indicated concentrations (in μM) of capzimin or BTZ for 24 hours. Western blot analyses of cell lysates were performed with antibodies against PARP1, caspase3, pH2AX and GAPDH, as indicated. A representative image of 3 independent experiments is shown. Full gel images were shown in Supplementary Fig. 21. (d) Cells lacking Nrf1 were more sensitive to capzimin and bortezomib. WT (black) or nfe2l1−/− MEF cells (red) were treated with different concentrations of capzimin (top) or BTZ (bottom) for 72 hours and analyzed as described in a.
To gain a broader overview of its inhibitory activity, capzimin was screened against the NCI panel of 60 cancer cell lines30. The median GI50 was 3.3 μM (Supplementary Fig. 15). Capzimin exhibited promising activity in leukemia cells including the SR and K562 cell lines (GI50 values of 0.67 μM and 1 μM respectively), as well as several solid tumor cell lines including NCI-H460 (non-small cell lung cancer; GI50 = 0.7 μM) and MCF7 (breast cancer; GI50=1.0 μM) (Supplementary Fig. 15). Immunoblotting for the processed form of caspase 3 and caspase-cleaved poly ADP-ribose polymerase in HCT116 cells confirmed that capzimin not only blocked cell growth, but also induced apoptosis (Fig. 4c). Apoptosis induction was accompanied by DNA damage, as determined by accumulation of phosphorylated histone H2AX31.
To test whether cell growth inhibition caused by capzimin was likely due to proteasome inhibition, we analyzed WT and Nuclear factor erythroid derived 2-related factor 1 (NRF1) knockout MEF cell lines32. Transcription factor Nrf1 contributes to basal expression of proteasome and promotes elevated expression of proteasome subunit mRNAs in cells treated with proteasome inhibitors33,34. Notably, the Nrf1−/− cell line was more sensitive to treatment with either bortezomib34, capzimin (Fig. 4d), or 3021 (Supplementary Fig. 16). Attempts to isolate mismatch repair-deficient HCT116 cells resistant to 3021 yielded multiple lines with elevated levels of proteasome activity and cross-resistance to carfilzomib, but the phenotype was not stable, which precluded a systematic analysis (Supplementary Table 4).
Global effects of Rpn11 inhibition on protein degradation
Previous studies suggested that loss of Rpn11 function hinders the degradation of polyUb substrates leading to cell death. However, only a few proteasome substrates have been evaluated in Rpn11-deficient cells and the extent to which Rpn11 mediates the global turnover of polyUb substrates has not been studied. Given that there are at least two other proteasomal deubiquitinases (Uch37 and Usp14)35, it is possible that Rpn11 mediates deubiquitination and turnover of only a subset of polyUb substrates. We employed immunoaffinity enrichment of peptides bearing the GlyGly remnant that remains when a ubiquitin conjugate is cleaved with trypsin, coupled with quantitative SILAC mass spectrometry, to measure the impact of capzimin and bortezomib on ubiquitination site occupancy. In total, 14,325 unique sites in 4,447 proteins were identified, of which 3,930 unique sites in 1,548 proteins were quantified (Supplementary Fig. 17 and Supplementary Data 1, 2). Of these, 2556 unique sites in 1123 proteins showed >2 fold change (Supplementary Fig. 17 and Supplementary Data 1, 2). A scatter plot and histogram of the data (Fig. 5a) reveal that the effects of inhibiting CP and Rpn11 correlated, albeit only loosely (r2 = 0.3243), and that bortezomib had a more profound impact on ubiquitination site occupancy. Although we used amounts of each drug that caused strong stabilization of the UbG76VGFP reporter and roughly equivalent accumulation of polyUb as determined by immunoblotting, it is possible that the stronger effect of bortezomib was due to its greater potency. For example, there may exist ‘priviledged’ (e.g. high affinity) substrates that require more severe proteasome inhibition to accumulate, and these are differentially affected by the more potent inhibitor. To further evaluate the effect of capzimin, we determined the ubiquitination sites and proteins that showed the greatest changes after an 8-hour treatment. With the criteria of ANOVA P<0.05, there were 355 sites and 222 proteins that showed ⩾2-fold changes. Some of the most significantly affected sites and proteins are highlighted in the volcano plot in Fig. 5b and Supplementary Fig. 18–19 (for full list, see Supplementary Data 1, 2). Interestingly, multiple up-regulated proteins are associated with heat shock response such as HMOX1, DNAJB4, and DNAJB1.
Fig 5. Capzimin has a global impact on ubiquitination site occupancy.
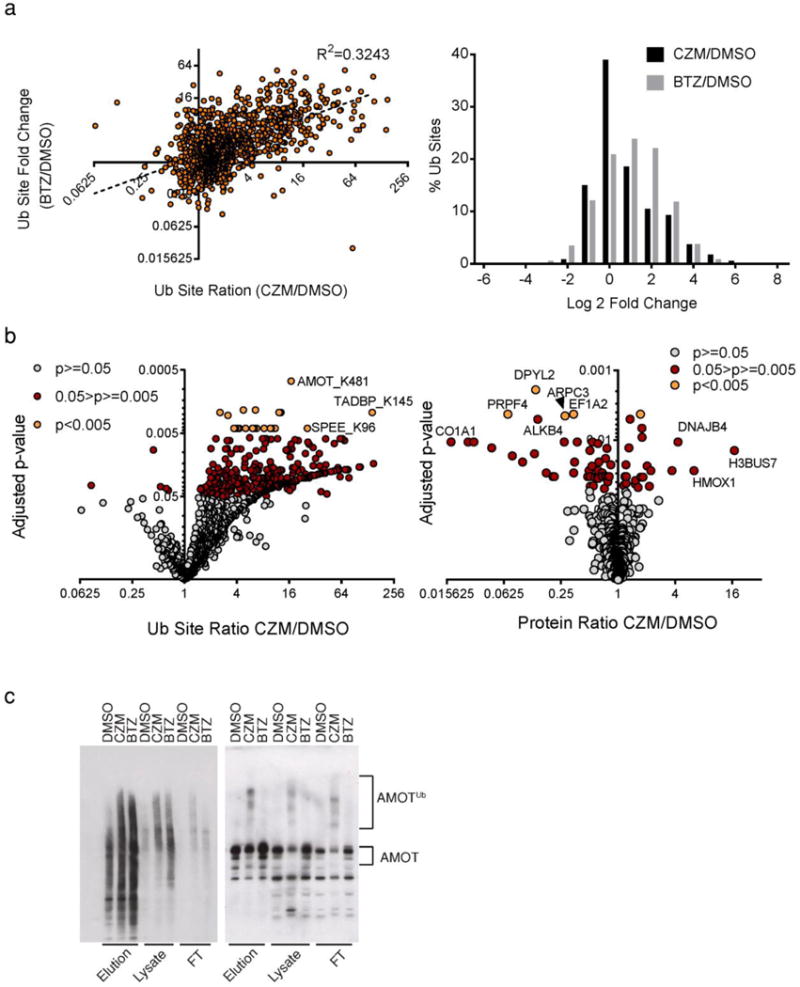
(a) Scatter plot (left) and histogram (right) analyses of data from quantitative ubiquitination site profiling performed with 293T cells treated with 10 μM capzimin (n=3) or 1 μM BTZ (n=4) for 8 hours, using an antibody that specifically captures peptides that contain the diglycine remnant that remains following digestion of an ubiquitin conjugate with trypsin. Notice that although capzimin increased ubiquitination site occupancy, it did not have as strong an effect as BTZ. (b) Volcano plot of the data from the experiment in panel a, showing changes in ubiquitination site occupancy (left) and total protein levels (right) upon treatment with capzimin. P<0.005, 0.05>p>=0.005by moderated t-test for DMSO versus CZM treated cells (n=4). (c) Capzimin promoted accumulation of ubiquitin-conjugated angiomotin. 293T cells were treated with capzimin or BTZ for 8 hours, lysed, and ubiquitinated species were enriched by adsorption to Ubiquilin 1 Tandem UBA (TUBE2) agarose beads. Western blot analyses of captured proteins were performed using antibodies to ubiquitin (left) and angiomotin (right). Result shown is a representative image from three independent experiments.
To validate the proteomic results, we focused on angiomotin and DNAJB4. These proteins were of particular interest because in contrast to most proteins, both showed a greater increase in ubiquitination site occupancy upon inhibition with capzimin compared to bortezomib. Recent studies implicate angiomotin in the pathogenesis of cancer. Angiomotin is highly expressed in breast cancer tissue and its knockdown significantly decreases cell proliferation and invasiveness36. Therefore, it is important to understand how angiomotin levels are regulated in cells. Ubiquitination at K481 was up-regulated ~16 fold by capzimin but only 2.4-fold by bortezomib. Consistent with this, immunoblotting revealed strong accumulation of high molecular weight species of angiomotin in cells treated with capzimin, but not bortezomib (Fig. 5c). For DNAJB4, Western analysis revealed that capzimin caused stronger accumulation than bortezomib (Supplementary Fig. 20). Notably, a previous study revealed that proteasome inhibition by MG132 led to heat shock response37. Our results suggest that Rpn11 inhibition by capzimin also triggered heat shock response as multiple heat shock response proteins were up-regulated (Fig. 5b and supplementary Fig. 19). Although capzimin and MG132 inhibit proteasome function through distinct mechanisms and appear to elicit distinct effects on at least some proteasome substrates, induction of the heat shock response may be a common mechanism for cells to cope with proteasome malfunction.
Discussion
It has been hypothesized that cancer cells are more dependent than normal cells on mechanisms of protein quality control, including the ubiquitin-proteasome system, to maintain protein homeostasis. This proposal is rooted in the observation that cancer cell genomes are littered with alterations that result in the production of either defective proteins encoded by mutant genes, or excess proteins arising from segmental or whole chromosome duplications. Notably, over 90% of solid tumors have an altered number of chromosomes38. Many mutated proteins cannot achieve a native conformation and therefore must be destroyed. Likewise, proteins encoded on aneuploid chromosomes are produced in excess of their binding partners and consequently may fail to assemble properly, necessitating their disposal. Together, these sources of damaged, misfolded, or unassembled proteins are proposed to place an unusually high load on the UPS. As a result, cancer cells may be more sensitive than normal cells to interference with mechanisms of protein quality control39. Use of proteasome inhibitors for the treatment of multiple myeloma and mantle cell lymphoma validates this idea. Proteasome inhibitors such as bortezomib and carfilzomib improve the clinical outcome and have been widely used for the treatment of myeloma2. The clinical success of these drugs has encouraged the development of other strategies to interfere with protein homeostasis, including targeting other components of the UPS such as Nedd8-activating enzyme, ubiquitin-activating enzyme, and p97/VCP, in addition to development of new 20S core proteasome inhibitors such as ixazomib, marizomib, and oprozomib39. However, despite the success of the ‘omibs, only a fraction of patients responds to monotherapy and essentially all of them relapse eventually. In addition, bortezomib and carfilzomib have yet to show efficacy in other types of cancer40. Given that there are already three approved CP inhibitors, greater rewards may lie in developing orthogonal approaches to inhibit the proteasome.
Several novel approaches for targeting the proteasome have been developed in the past few years including Rpn13 inhibitors, Rpt ATPase inhibitors and Usp14/UchL5 inhibitors41–44. However, there remain technical or conceptual limitations to these targets. Rpn13 serves as an ubiquitin receptor, but it is not essential in mice and its functional role in the proteasome remains uncertain45. The Rpt inhibitors identified to date are peptoids and it remains unclear how effectively these can be converted into drugs42. Finally, the mechanism by which the Usp14/UchL5 inhibitor works is ambiguous, because Usp14 is not essential for proteasome function, and it actually counteracts proteasome activity1. By contrast, Rpn11 seems like a more straightforward target. Rpn11 activity is essential for proteasome function, and its role in protein degradation is known: it removes the polyUb targeting signal so that substrate can be translocated into the CP for degradation3,4,11.
Although Rpn11 and the β5 subunit targeted by the ‘omibs are both constituents of the proteasome we anticipate that Rpn11 inhibitors and “omibs” might have distinct efficacy profiles because they work by different mechanisms. Consistent with this idea, proteome-wide analysis of ubiquitin conjugates revealed that at least two proteins, angiomotin and DNAJB4, exhibited a difference in response to capzimin vs. bortezomib. However, there are important caveats to this analysis, because it is difficult to compare the effects of inhibitors whose potencies differ by two logs. Case in point, in our prior characterization of p97 inhibitors, early-stage compounds failed to block processing of the p97 substrate Nrf1, whereas p97 knockdown or the later-stage p97 inhibitors NMS873 and CB-5083 efficiently inhibit Nrf1 processing46. Nevertheless, these data suggest that inhibiting Rpn11 versus the 20S peptidase active sites will elicit some distinct molecular effects.
Different mechanisms for inhibiting a given target, even a relatively simple target like the cyclooxygenases I and II (targeted by aspirin, acetominophen, ibuprofen, and celecoxib) can lead to significant differences in how patients respond47. One expects that for a far more complex target like the proteasome, the potential for differentiation in clinical response is even greater. Moreover, because the proteasome is an effective target for cancer therapy, the chance for success is also likely to be greater than for an unvalidated target. At the current stage, it is not resolved how inhibition of Rpn11 will differ from inhibition of β5. One possibility is suggested by reports that Rpn11 exerts regulatory functions by cleaving ubiquitin chains from proteins that are not degraded by the 20S proteasome26. Another way in which Rpn11 and β5 inhibitions may differ is that CP inhibitors should block the degradation of substrates that are targeted by an ubiquitin-independent mechanism (including those degraded by CP assembled with 13S REG complex instead of 19S RP), whereas Rpn11 inhibitors should more selectively block ubiquitin-dependent substrates. It remains to be seen whether other factors might influence Rpn11 dependence, including the position of the ubiquitin chain in the substrate and the timing of its removal relative to substrate proteolysis.
Metalloenzymes comprise a large class of potentially drugable enzymes in the human proteome. Multiple zinc metalloenzymes have been successfully targeted by marketed drugs, including carbonic anhydrase (inhibited by acetazolamide), histone deacetylases (vorinostat, romidepsin, and others), and angiotensin-converting enzyme (captopril and others)15. All of these drugs act by binding the Zn2+ ion within the active site, thereby neutralizing its catalytic activity.
A common strategy in metalloenzyme inhibitor development programs has been to identify substrate mimics that bind with high affinity to the enzyme. These compounds are then furnished with MBPs such as hydroxamic acids to bind the Zn2+ ion and thereby block enzymatic activity. Hydroxamic acid is the most common MBP in metalloprotein inhibitors and is found in the HDAC inhibitor vorinostat48. Here, we inverted the conventional wisdom for metalloenzyme inhibitor development. We first identified, in fragment-based screen of 255 MBPs, the potent and selective moiety 8TQ, which has a high ligand efficiency, and then modified it to further enhance its potency and selectivity. The validity and power of this approach was underscored by our identification of the exact same scaffold as the only high-quality hit in a screen of >300,000 compounds. Our results support the idea that ligands with different geometries, heteroatoms, and donor ability can discriminate between active site metal ions of different metalloenzymes. The extreme tropism of Rpn11 and other JAMM proteins for 8TQ and vice versa suggest there is something special about how these molecules interact. A potential concern about 8TQ is the presence of a thiol moiety, but thiols are also present in the clinically validated compounds captopril and romidepsin15.
In summary, our study identified capzimin, the first-in-class selective Rpn11 inhibitor. The parent scaffold 8TQ is a first-in-class broad spectrum JAMM enzyme inhibitor, and because it is tolerant of chemical substitutions at different positions, may be a useful starting point for the development of specific inhibitors of different JAMM enzymes, as shown here for Rpn11. Notably, 8TQ is orally bioavailable and tolerated in mice49. Given the successful development of other metalloenzyme inhibitors, including those containing thiols, capzimin is a promising scaffold for the development of a novel proteasome inhibitor for therapy of cancer.
Online methods
Protein expression and purification
The plasmid for expression of Rpn11•Rpn8 dimer was a kind gift from Dr. Andreas Martin. Expression and purification of Zn2+ bound form of Rpn11•Rpn8 dimer was as previously reported18. The plasmid for expression of ubiquitin independent substrate Rpn10-I27V13Pext was a kind gift from Dr. Philip Coffino. Expression and purification of Rpn10-I27V13Pext was as previously reported20.
Protein labeling
Ub4-peptide (MQIFVKTIKSQTSCVDKLAAALEHHHHHHH), Nedd8 and Rpn10-I27V13P-ext were covalently labeled with the fluorescent dye Oregon Green 488 or Alexa Fluor 488 (Invitrogen) by incubation with a five-fold molar excess of the maleimide-derivatized dye under the conditions recommended by the manufacturer. The reaction was quenched by the addition of 10 mM DTT and applied to a Superdex 75HR column (GE Healthcare) to separate the unincorporated dye from the labeled protein.
In vitro Rpn11 activity assay
Fluorescence polarization assays were performed in low-volume 384 well solid black plates (Molecular Devices) in quadruplicate. The assays were performed in buffer containing 50 mM Tris-HCl pH7.5, 1mM MgCl2, 50 μM ATP, 1 mM DTT and 0.01% NP-40. The components for the assay were added in the follow sequence: 1) 5 μl compound (in 3% DMSO) at different concentrations, 2) 5 μl of diluted human 26S proteasome (Enzo Life Sciences, NY. 26S proteasome was pre-incubated with epoxomicin at room temperature for 1 hour, then dilute 10-fold in 1x Assay Buffer), and 3) 5μl of substrate (3 nM Ub4-peptideOG). 100 μM Zn(cyclen)2+ was present in the titration reaction for the experiments performed with Zn(cyclen)2+. Fluorescence polarization was measured at 30°C using a PHERAstar plate reader (BMGlabtech) with excitation at 480 nm and emission at 520 nm. Collected data was normalized to DMSO control and fitted to a dose-response curve to determine the IC50 value.
In vitro Csn5 activity assay
Csn5 activity was measured using purified CSN complex and the fluorescent substrate OGNedd8–SCFSkp2, in which a unique cysteine engineered into Nedd8 was labelled with Oregon Green 488, after which the product was conjugated to SCFSkp2 as previously described50. Csn5-dependent cleavage of OGNedd8 from SCFSkp2 resulted in a decrease of fluorescence polarization. The assay was performed in buffer containing 40 mM Tris-HCl pH 7.5, 50 mM NaCl, 1mM DTT, 0.01% Triton X-100, 1% glycerol, 25 mM trehalose and 15 μg/ml ovalbumin. Polarization data was collected with a PHERAstar plate reader using black low-volume 384 well plates. Collected data were normalized to DMSO control and fitted to a dose-response curve to determine the IC50 value.
In vitro AMSH activity assay
AMSH activity was measured using 10 nM purified AMSH (E-548B, Boston Biochem) and 80 nM di-ubiquitin substrate DiUbK63TAMRA (UF-310, Boston Biochem). TAMRA fluorescence intensity was monitored using a PHERAstar plate reader with excitation at 540 nm and emission at 590 nm. Reactions were performed in black low-volume 384 well plates and analyzed as described above. Because substrate concentration was far below the KM23, the IC50 should be similar to the Ki.
In vitro BRCC36 activity assay
The BRISC complex is a kind gift from Dr Elton Zeqiraj (University of Leeds). It consists of BRCC36, KIAA0157, BRCC45 and MERIT40 was produced by co-expression in insect cells and purified to greater than 95% purity by affinity and size exclusion chromatography as described previously22. BRCC36 activity was measured using 1nM purified BRISC complex and 80 nM di-ubiquitin substrate DiUbK63TAMRA (UF-310, Boston Biochem). TAMRA fluorescence intensity was monitored using a PHERAstar plate reader with excitation at 540 nm and emission at 590 nm. Reactions were performed in black low-volume 384 well plates and analyzed as described above. Because substrate concentration was far below the KM, the IC50 should be similar to the Ki22.
HDAC 6 Activity Assay
HDAC 6 was purchased from BPS Bioscience (BPS Bioscience catalog #50006) and the assay was carried out as instructed by manufacturer. The enzyme was diluted with 25 mM Tris-Cl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.1 mg/mL BSA, pH 8.0 buffer and its activity was measured by utilizing Substrate 3 (BPS Bioscience catalog #50037). The assays were carried out in black, low binding NUNC 96-well plates. Each well contained a volume of 50 μL including buffer, HDAC (50 ng/well), inhibitor, and substrate (20 μM). Prior to adding substrate, the plate was preincubated for 5 min. Upon addition of substrate, the plate was incubated at 37 °C for 30 min. At this point, HDAC assay developer (50 μL, BPS Bioscience catalog #50030) was added to each well and the plate was incubated for 15 min at room temperature. The fluorescence was recorded with a BioTek FLx 800 or Synergy H4 microplate reader. The measured fluorescence was compared for samples versus controls containing no inhibitor (0% inhibition).
MMP Activity Assay
MMP-2, -12 and OmniMMP fluorogenic subsbtrate (P-126) were purchased from Enzo Life Sciences. The assay was carried out in white NUNC 96-well plates as previously described51. Each well contained a total volume of 90 μL including buffer (50 mM HEPES, 10 mM CaCl2, 0.05% Brij-35, pH 7.5), human recombinant MMP (1.16 U/well for MMP-2 and 0.007 U/well for MMP-12), and the fragment solution. This mixture was incubated at 37 °C for 30 min. The reaction was then initiated by the addition of 10 μL of the fluorogenic MMP substrate (4 μM final concentration, Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2-AcOH) [Mca = (7-methoxycoumarin-4-yl) acetyl; Dpa = N-3-(2,4-dinitrophenyl)-L-a,β-diaminopropionyl]. Fluorescence measurements were recorded using a BioTek Synergy H4 fluorescence plate reader every minute for 20 min with excitation and emission wavelengths at 320 and 400 nm, respectively.
Human Glyoxalase 1 Assay
Recombinant Human Glyoxalase I (GLO1) was purchased from R&D Systems (Catalog #4959-GL). Assays were carried out in 100 mM Sodium Phosphate, pH 7.0 buffer utilizing 96-well Clear UV Plate (Corning UV Transparent Microplates Catalog #3635). A fresh solution of glutathione (Pre-Substrate 1, 100 mM) as well as methylglyoxal (Pre-substrate 2, 100 mM) was prepared in deionized water. The substrate was prepared by adding 14.5 mL of buffer and 0.99 mL of each pre-substrate components. The substrate mixture was vortexed vigorously for 15 sec, then allowed to sit at room temperature for 20 min. Initial well volume was 50 μL containing GLO1 (40 ng) and inhibitor. This protein and inhibitor mixture was incubated for 15–20 min prior to addition of substrate. To this was then added substrate (150 μL) yielding a maximum amount of 5% DMSO per well. The enzyme activity was measured utilizing a Biotek Synergy H4 plate reader by measuring absorbance at 240 nm every 1 min for 8 min. The rate of absorbance increase was compared for samples versus controls containing no inhibitor (100% activity). Absorbance for background wells containing DMSO, buffer and substrate (no enzyme or inhibitor) were subtracted from the rest of the wells.
hCAII Activity Assay
hCAII was expressed and purified as previously reported52. Assays were run in 50 mM HEPES (pH 8.0). A BioTek Synergy H4 microplate sample processor was utilized. The compounds were incubated with protein (final concentrations of 100 nM for hCAII) for 10 min at 25 °C. A substrate (p-nitrophenylacetate; final concentration of 500 μM) was added, and hCAII-catalyzed cleavage was monitored by the increase in absorbance at 405 nm corresponding to formation of the p-nitrophenolate anion. The initial linear reaction rate was compared to that of wells containing no inhibitor (0% inhibition) and no protein (100% inhibition). The rate of non-hCAII-catalyzed PNPA hydrolysis in the presence of inhibitor was subtracted from each trial before determination of the percent inhibition.
Rpn11•Rpn8 di-Ub cleavage assay
Di-ubiquitin cleavage assay was performed in 40 mM HEPES, pH7.5, 100 mM NaCl, 100 mM KCl and 5% glycerol. Rpn11•Rpn8 dimer (5 μM) was incubated with DiUbK48 and different concentrations of 8TQ or capzimin at 30°C. The reactions were stopped with 2X SDS sample buffer after 2 hours and analyzed by SDS-PAGE.
Cell lines and culture conditions
A549 human lung carcinoma cell line (ATCC #CCL185) was purchased from ATCC. HEK293T cell line was a gift from David Baltimore lab (Caltech). HCT116 cell line was kindly provided by Bert Vogelstein lab (Johns Hopkins University). Cells were maintained in DMEM media supplemented with 10% FBS. All cell lines used in this study are free of mycoplasma contamination tested by MycoAlert™ mycoplasma detection kit (Lonza).
UbG76V-GFP accumulation and clearance assay
Hela cells stably expressing UbG76V–GFP14 were cultured in DMEM with 10% FBS. Eight thousand cells were seeded in black 96-well plates with clear bottom 24 hours before adding compounds. To initiate the UbG76V–GFP accumulation assay, cells were incubated with different concentrations of MG132 or capzimin. Images were taken after 4 hours.
To monitor UbG76V–GFP degradation, cells seeded into plates as described above were treated with 5 μM of MG132 for 4 hours to accumulate UbG76V–GFP, after which MG132 was washed away using PBS twice before adding different concentrations of compounds in the presence of 30 μg/ml cycloheximide. Images were taken at multiple time points using a Molecular Devices ImageXpress Micro, and the GFP fluorescence was quantified using MetaXpress software (Molecular Devices). The rate of GFP fluorescence decrease was normalized to DMSO control. Normalized data was then fitted to a dose-response curve to determine the IC50 value.
Degradation assay for Ub4peptideOG and Rpn10-I27V13Pext Alexa488
Ub4peptideOG or Rpn10-I27V13PextAlexa488 (each at 100 nM) was incubated with 10 nM purified yeast 26S proteasome in the absence or presence of 20 μM capzimin at 30°C. Samples taken at 0, 30, 60, 90 and 120 minutes were mixed with 2X SDS sample buffer and analyzed by SDS-PAGE. The gel was imaged using Typhoon™ FLA 9500 biomolecular imager (GE Healthcare) with the fluorescence filter of 488nm/520nm. The fluorescence of intact Ub4peptideOG and Rpn10-I27V13Pext Alexa488 was quantified using ImageQuant software (GE Healthcare).
Aggresome formation and clearance assay
To monitor aggresome formation, we cultured A549 cells on glass coverslips in six well plates in the absence or presence of different concentrations of MG132 or capzimin. Medium was removed after 15 hrs incubation. Cells were washed twice with PBS then fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 in PBS, blocked with 5% FBS containing 0.1% Triton X-100, and incubated with antibodies against ubiquitin, HDAC6 or p62/SQSTM1 (Supplementary Table 5). For the aggresome clearance assay, A549 cells were treated with 5 μM MG132 for 15 hrs. After washing away MG132-containing media, cells were treated in the absence or presence of different concentrations of 3021 for 24 hours. Cells were fixed and stained as described above before imaging.
Pulldown of polyubiquitylated protein
Ubiquilin 1 Tandem UBA (TUBE2) Agarose (Boston Biochem) was used to enrich polyubiquitylated proteins. Cells were treated with CZM, BTZ or DMSO for 8 hours and then lysed in buffer containing 40 mM Tris-HCl, pH7.6, 5 mM NEM, 0.1% NP-40 and bound to TUBE2 agarose at 4°C for 2 hours. Beads were washed three times with 40 mM Tris-HCl, pH7.6, 150 mM NaCl, 0.1% NP-40. Bound proteins were eluted with 2X SDS sample buffer and loaded onto 4–12% Tris-Glycine precast gel (Invitrogen). After transfer, the member was blotted with ubiquitin or AMOT antibodies (Supplementary Table 5).
Cellular imaging for Zinc sensor
Imaging experiments were performed on a Nikon Ti-E wide-field fluorescence microscope equipped with Nikon elements software, Ti-E perfect focus system, an iXon3 EMCCD camera (Andor), mercury arc lamp, and YFP FRET (434/16 excitation, 458 dichroic, 535/20 emission), CFP (434/16 excitation, 458 dichroic, 470/24 emission), and YFP (495/10 excitation, 515 dichroic, 535/20 emission) filter sets. External excitation and emission filter wheels were controlled by a Lambda 10–3 filter changer (Sutter Instruments), while dichroic mirrors were placed on cubes in the dichroic turret. Images were collected using a 40X air objective (NA 0.95), 100 ms exposure time, 5.1x conversion gain, and a neutral density filter with 12.5% light transmission. Images were collected every minute.
Sensor constructs were transfected into HEK293T cells with Lipofectamine 3000 (Thermo Fisher Scientific). Cells were imaged 24–48 hours post-transfection. Cells were maintained at 37°C and 5% CO2 in a LiveCell™ environment chamber (Pathology Devices). Cells were imaged in phosphate-free HEPES-buffered HBSS, pH = 7.4 to prevent zinc precipitation. Resting FRET data were collected for 5 min, then the cells were treated with either 0.5% DMSO (n=11 cells) or 10 μM capzimin (n=11 cells). For calibration of the sensor (n=12 cells), resting FRET data were collected for 5 min, then the cells were treated with 50 μM Tris(2-pyridylmethyl) amine (TPA) until a stable signal was achieved. The TPA solution was removed by aspiration and the cells were then treated with 0.75 μM pyrithione and 12 μM buffered zinc solution.
All data were analyzed in MATLAB (Mathworks). Images were background corrected by drawing a region of interest (ROI) in a blank area of image and subtracting the average fluorescence intensity of the background ROI from the average intensity of each cell. FRET ratios for each cell were calculated by dividing the background-corrected YFP FRET intensity by the background-corrected CFP intensity ((Icellular FRET – Ibackground FRET)/(Icellular CFP – Ibackground CFP)). Normalized FRET ratios were calculated by averaging the mean intensity of the cells before treatment with compounds (Rresting), and then dividing the experimental FRET ratios (R) by this value (R/Rresting). Error bars are the SEM of the normalized FRET ratios.
HEK293T cell growth conditions
HEK293T cells were grown in DMEM lacking arginine and lysine, to which 10% dialyzed fetal bovine serum (FBS), 1% L-glutamine, 1% Pen/Strep, 1 mM sodium pyruvate, and 10 mg/L unlabeled proline was added. Cells were then grown in either “heavy” or “light” medium for isotopic differentiation of protein components. Heavy (H) medium was supplemented with 50 mg/L of 13C6, 15N2-lysine (Lys8) and 13C6-arginine (Arg6) (Cambridge Isotope Laboratories), while light (L) was supplemented with 50 mg/L of unlabeled lysine (Lys0) and arginine (Arg0). Cells were grown in their respective media until heavy amino acid incorporation reached ~98%. Incorporation analysis was performed as described previously53. Cells were cultured at 37°C in the presence of 5% CO2 to a confluency of 80%–90% in 75 cm2 tissue culture flasks (Griener Bio-One GmbH, Frickenhausen, Germany). Once proper confluency was reached, growth medium was removed, cells 5.5–8 × 107) were washed once with 1 × PBS and then incubated for 8 h with fresh medium supplemented with 10 μM capzimin, 1 μM bortezomib (BTZ) or DMSO. After treatment cells were harvested by washing once with 1 × PBS followed by treatment with 0.25% trypsin-EDTA solution (Gibco, Life Technologies, New York, USA). Cells grown on three plates for each treatment were combined, washed twice with 1 × PBS and pelleted after each wash by centrifugation at 1,500 × g for 3 min. Immediately after harvesting, cells were frozen in liquid N2 and stored at −80°C until lysis.
Tryptic digestion, chromatography, and mass spectrometry
Cell lysis, digestion and peptide desalting procedure was done as previously described54. N-ethylmaleimide (NEM) was used as the alkylating reagent.
Peptide pre-fractionation, antibody cross-linking, and K-ε-GlyGly affinity enrichment was done using the PTMScan® Ubiquitin Remnant Motif (K-ε-GlyGly) Kit #5562 from Cell Signaling Technology with some modifications55 as described54.
K-ε-GlyGly enriched peptide samples were analysed using an EASY II nano-UPLC (Thermo Scientific) connected on-line to an Orbitrap Elite hybrid and Fusion tribrid mass spectrometers with a nanoelectrospray ion source (Thermo Scientific) using settings and instrument arrangements similar to those previously described56.
Mass spectrometry data analysis
All raw files acquired during this analysis were processed together in one MaxQuant search (v. 1.5.0.12)57. Spectra were searched against the human proteome obtained from UniProt (148,298 entries). All other settings are essentially as previously described54.
Supplementary Material
Acknowledgments
We thank Andreas Martin (University of California, Berkeley) for the plasmid that expresses Rpn11•Rpn8 dimer, Dr Elton Zeqiraj (University of Leeds) for providing purified BRISC complex, Dr. Phiip Coffino (The Rockefeller university) for providing the Rpn10-I27V13Pext plasmid, Dr. Tarun M. Kapoor (The Rockefeller university) for providing the RPE1 WT and BTZ-resistant cell lines, National Cancer Institute (Developmental therapeutics program in the Division of Cancer Treatment and Diagnosis) for screening capzimin against 60 human cancer cell lines (http://dtp.cancer.gov), Heenam Park for testing the cell lines for mycoplasma contamination and Yaru Zhang for advice on the competition studies. This work was supported by grants from the Caltech Gates Grubstake Fund and Amgen to RJD, and NIH (CA164803) to RJD and SMC. TYR and SH were supported by the Gordon and Betty Moore Gordon Foundation, through Grant GBMF775, the Beckman Institute and the NIH through Grant 1S10RR029594-01A1. RJD is an Investigator of and was supported by the Howard Hughes Medical Institute.
Footnotes
Author contributions
J.L. Design, execution, and interpretation of all experiments with capzimin. Screen of MBP library. Drafting of manuscript. T.Y.: Execution and interpretation of mass spectrometry proteomic analysis with capzimin. F.P.: Initial development of Rpn11 HTS substrate and fluorescence polarization assay. Design and execution of Rpn11 HTS. A.L.M.: Triage of Rpn11 HTS hits. C.P. Execution of HDAC6, MMP, hCAII and GLO1 assay and synthesis of 3021, 3027 (capzimin). Y.M.: Synthesis of 3027 (capzimin). S.C., G.M., B.B., Eduard S., I.P.: Developed the HTS assay. K.N., S.V., Eigo S., L.H.S.: Performed the HTS assay. I.P., A.P., Eduard S., T.C.: Evaluated and selected hits. K.C.: Design, execution, and interpretation of experiments with zinc reporter. A.E.P.: Design and interpretation of experiments with zinc reporter. S.H.: Design, interpretation and oversight of the mass spectrometric studies with capzimin. Revision of the manuscript. S.M.C.: Design and oversight of capzimin syntheses. R.J.D.: Design, interpretation, and oversight of experiments for entire study. Drafting of manuscript.
Competing Financial Interests Statement
R.J.D. is a founder, shareholder, consultant, and member of the Scientific Advisory Board of Cleave Biosciences, which is engaged in discovery and development of drugs that target enzymes involved in ubiquitin-dependent protein degradation. As required by the Conflict of Interest Office of UCSD, S.M.C. holds an equity interest in Forge Therapeutics and Cleave Biosciences and serves on the Scientific Advisory Board of these companies. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. Both companies may potentially benefit from the research results of certain projects in the laboratory of S.M.C.
Data availability
Any supplementary information, chemical compound information and source data are available in the online version of the paper.
References
- 1.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dimopoulos MA, Richardson PG, Moreau P, Anderson KC. Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat Rev Clin Oncol. 2015;12:42–54. doi: 10.1038/nrclinonc.2014.200. [DOI] [PubMed] [Google Scholar]
- 3.Verma R, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–5. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 4.Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419:403–7. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- 5.Ambroggio XI, Rees DC, Deshaies RJ. JAMM: a metalloprotease-like zinc site in the proteasome and signalosome. PLoS Biol. 2004;2:E2. doi: 10.1371/journal.pbio.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tran HJ, Allen MD, Lowe J, Bycroft M. Structure of the Jab1/MPN domain and its implications for proteasome function. Biochemistry. 2003;42:11460–5. doi: 10.1021/bi035033g. [DOI] [PubMed] [Google Scholar]
- 7.Cope GA, et al. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science. 2002;298:608–11. doi: 10.1126/science.1075901. [DOI] [PubMed] [Google Scholar]
- 8.Zhu P, et al. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol Cell. 2007;27:609–21. doi: 10.1016/j.molcel.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sobhian B, et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCullough J, Clague MJ, Urbe S. AMSH is an endosome-associated ubiquitin isopeptidase. J Cell Biol. 2004;166:487–92. doi: 10.1083/jcb.200401141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallery M, et al. The JAMM motif of human deubiquitinase Poh1 is essential for cell viability. Mol Cancer Ther. 2007;6:262–8. doi: 10.1158/1535-7163.MCT-06-0542. [DOI] [PubMed] [Google Scholar]
- 12.Bivona TG, et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471:523–6. doi: 10.1038/nature09870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buckley SM, et al. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell. 2012;11:783–98. doi: 10.1016/j.stem.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18:538–43. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- 15.Rouffet M, Cohen SM. Emerging trends in metalloprotein inhibition. Dalton Trans. 2011;40:3445–54. doi: 10.1039/c0dt01743d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobsen FE, Lewis JA, Cohen SM. The design of inhibitors for medicinally relevant metalloproteins. ChemMedChem. 2007;2:152–71. doi: 10.1002/cmdc.200600204. [DOI] [PubMed] [Google Scholar]
- 17.Dambacher CM, Worden EJ, Herzik MA, Martin A, Lander GC. Atomic structure of the 26S proteasome lid reveals the mechanism of deubiquitinase inhibition. Elife. 2016;5:e13027. doi: 10.7554/eLife.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Worden EJ, Padovani C, Martin A. Structure of the Rpn11-Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nat Struct Mol Biol. 2014;21:220–7. doi: 10.1038/nsmb.2771. [DOI] [PubMed] [Google Scholar]
- 19.Pathare GR, et al. Crystal structure of the proteasomal deubiquitylation module Rpn8-Rpn11. Proc Natl Acad Sci U S A. 2014;111:2984–9. doi: 10.1073/pnas.1400546111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henderson A, Erales J, Hoyt MA, Coffino P. Dependence of proteasome processing rate on substrate unfolding. J Biol Chem. 2011;286:17495–502. doi: 10.1074/jbc.M110.212027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carter KP, Young AM, Palmer AE. Fluorescent sensors for measuring metal ions in living systems. Chem Rev. 2014;114:4564–601. doi: 10.1021/cr400546e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeqiraj E, et al. Higher-Order Assembly of BRCC36-KIAA0157 Is Required for DUB Activity and Biological Function. Mol Cell. 2015;59:970–83. doi: 10.1016/j.molcel.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato Y, et al. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature. 2008;455:358–62. doi: 10.1038/nature07254. [DOI] [PubMed] [Google Scholar]
- 24.Chou TF, Deshaies RJ. Quantitative cell-based protein degradation assays to identify and classify drugs that target the ubiquitin-proteasome system. J Biol Chem. 2011;286:16546–54. doi: 10.1074/jbc.M110.215319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–98. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hao R, et al. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol Cell. 2013;51:819–28. doi: 10.1016/j.molcel.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson DJ, et al. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell. 2015;28:653–65. doi: 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chou TF, et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A. 2011;108:4834–9. doi: 10.1073/pnas.1015312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wacker SA, Houghtaling BR, Elemento O, Kapoor TM. Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat Chem Biol. 2012;8:235–7. doi: 10.1038/nchembio.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–23. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 31.Xu Y, Price BD. Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle. 2011;10:261–7. doi: 10.4161/cc.10.2.14543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan JY, et al. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 1998;17:1779–87. doi: 10.1093/emboj/17.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steffen J, Seeger M, Koch A, Kruger E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol Cell. 2010;40:147–58. doi: 10.1016/j.molcel.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Radhakrishnan SK, et al. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell. 2010;38:17–28. doi: 10.1016/j.molcel.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee MJ, Lee BH, Hanna J, King RW, Finley D. Trimming of ubiquitin chains by proteasome-associated deubiquitinating enzymes. Mol Cell Proteomics. 2011;10003871:R110. doi: 10.1074/mcp.R110.003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lv M, et al. Angiomotin promotes breast cancer cell proliferation and invasion. Oncol Rep. 2015;33:1938–46. doi: 10.3892/or.2015.3780. [DOI] [PubMed] [Google Scholar]
- 37.Bush KT, Goldberg AL, Nigam SK. Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem. 1997;272:9086–92. doi: 10.1074/jbc.272.14.9086. [DOI] [PubMed] [Google Scholar]
- 38.Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–87. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deshaies RJ. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014;12:94. doi: 10.1186/s12915-014-0094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson DE. The ubiquitin-proteasome system: opportunities for therapeutic intervention in solid tumors. Endocr Relat Cancer. 2015;22:T1–17. doi: 10.1530/ERC-14-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trader DJ, Simanski S, Kodadek T. A reversible and highly selective inhibitor of the proteasomal ubiquitin receptor rpn13 is toxic to multiple myeloma cells. J Am Chem Soc. 2015;137:6312–9. doi: 10.1021/jacs.5b02069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim HS, Archer CT, Kodadek T. Identification of a peptoid inhibitor of the proteasome 19S regulatory particle. J Am Chem Soc. 2007;129:7750–1. doi: 10.1021/ja072027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D’Arcy P, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 2011;17:1636–40. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- 44.Anchoori RK, et al. A bis-benzylidine piperidone targeting proteasome ubiquitin receptor RPN13/ADRM1 as a therapy for cancer. Cancer Cell. 2013;24:791–805. doi: 10.1016/j.ccr.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Al-Shami A, et al. Regulators of the proteasome pathway, Uch37 and Rpn13, play distinct roles in mouse development. PLoS One. 2010;5:e13654. doi: 10.1371/journal.pone.0013654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radhakrishnan SK, den Besten W, Deshaies RJ. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife. 2014;3:e01856. doi: 10.7554/eLife.01856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ong CK, Lirk P, Tan CH, Seymour RA. An evidence-based update on nonsteroidal anti-inflammatory drugs. Clin Med Res. 2007;5:19–34. doi: 10.3121/cmr.2007.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 49.Borg-Neczak K, Tjalve H. Effect of 8-hydroxy-, 8-mercapto- and 5-chloro-7-iodo-8-hydroxy-quinoline on the uptake and distribution of nickel in mice. Pharmacol Toxicol. 1994;74:185–92. doi: 10.1111/j.1600-0773.1994.tb01097.x. [DOI] [PubMed] [Google Scholar]
Methods-only references
- 50.Duda DM, et al. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Puerta DT, et al. Heterocyclic zinc-binding groups for use in next-generation matrix metalloproteinase inhibitors: potency, toxicity, and reactivity. J Biol Inorg Chem. 2006;11:131–8. doi: 10.1007/s00775-005-0053-x. [DOI] [PubMed] [Google Scholar]
- 52.Martin DP, Hann ZS, Cohen SM. Metalloprotein-inhibitor binding: human carbonic anhydrase II as a model for probing metal-ligand interactions in a metalloprotein active site. Inorg Chem. 2013;52:12207–15. doi: 10.1021/ic400295f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hess S, van Beek J, Pannell LK. Acid hydrolysis of silk fibroins and determination of the enrichment of isotopically labeled amino acids using precolumn derivatization and high-performance liquid chromatography-electrospray ionization-mass spectrometry. Anal Biochem. 2002;311:19–26. doi: 10.1016/s0003-2697(02)00402-5. [DOI] [PubMed] [Google Scholar]
- 54.Sung MK, et al. A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. Elife. 2016;5 doi: 10.7554/eLife.19105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Udeshi ND, et al. Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics. 2013;12:825–31. doi: 10.1074/mcp.O112.027094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Porras-Yakushi TR, Sweredoski MJ, Hess S. ETD Outperforms CID and HCD in the Analysis of the Ubiquitylated Proteome. Journal of the American Society for Mass Spectrometry. 2015;26:1580–1587. doi: 10.1007/s13361-015-1168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.