

Abstract
Due to the high cost and environmental instability of antibodies, there is precedent for developing synthetic molecular recognition agents for use in diagnostic sensors. While these materials typically have lower specificity than antibodies, their cross-reactivity makes them excellent candidates for use in differential sensing routines. In the current work, we design a set of charge-containing poly(N-isopropylacrylamide) (PNIPAM) nanogels for use as differential protein receptors in a turbidimetric sensor array. Specifically, NIPAM was copolymerized with methacrylic acid and modified via carbodiimide coupling to introduce sulfate, guanidinium, secondary amine, or primary amine groups. Modification of the ionizable groups in the network changed the physicochemical and protein binding properties of the nanogels. For high affinity protein-polymer interactions, turbidity of the nanogel solution increased, while for low affinity interactions minimal change in turbidity was observed. Thus, relative turbidity was used as input for multivariate analysis. Turbidimetric assays were performed in two buffers of different pH (i.e., 7.4 and 5.5), but comparable ionic strength, in order to improve differentiation. Using both buffers, it was possible to achieve 100% classification accuracy of eleven model protein biomarkers with as few as two of the nanogel receptors. Additionally, it was possible to detect changes in lysozyme concentration in a simulated tear fluid using the turbidimetric sensor array.
TOC Image
Introduction
Turbidity is a classic analytical tool in biochemistry used to monitor many properties and events including water quality,1 bacterial growth rate,2 precipitation of sulfates,3,4 and immunoprecipitation.5 The latter led to the development of sensors based on the increase in turbidity that occurs upon formation of large immunocomplexes (i.e., binding between multiple antibodies and antigens).6 Conjugating antibodies to particle surfaces enhances the sensitivity of these immunoturbidimetric assays by increasing the aggregate size and, consequently, scattering.7 Such particle-enhanced turbidimetric immunoassays (PETIAs) have been used since the 1920’s, and currently PETIAs for a variety of analytes are commercially available.8–10 Turbidimetric assays have the advantages of being label-free, fast (<20 min), and easy to perform on commonplace equipment (i.e., microwell plate readers).11,12 One disadvantage of common turbidimetric assays is that they require the use of antibodies, which have poor environmental stability, and the routine methods for antibody production are associated with high cost.13,14 If particle-immobilized antibodies could be replaced by synthetic receptors, the affordability and robustness of these assays could be improved, making them even more attractive as point-of-care diagnostic platforms.
As less expensive, more environmentally robust alternatives to antibodies, crosslinked hydrogels can be used as artificial protein receptors. Several researchers, particularly the Luchini, Liotta, and Shea groups, have demonstrated the utility of multifunctional nano- and microgels made from poly(N-isopropylacrylamide) (PNIPAM) for protein binding applications.15–21 In their work, it is noted that while hydrophobic functional groups and multivalent interactions alter protein binding behavior, ionizable functional groups were present in all of the best performing (i.e., high binding) particles.15–21 However, charge-containing materials are prone to cross-reactive binding of proteins with similar isoelectric points (pIs). While prohibitive for applications requiring high selectivity, such cross-reactivity is beneficial for differential sensing routines. In the current report, the utility of PNIPAM nanogels as differential receptors is explored for the first time.
Differential sensing is an approach inspired by how our chemosensory systems work. Rather than employing a single, high affinity receptor for every molecule we encounter, these systems employ multiple receptors that bind different molecules to different extents to collectively produce distinct tastes and smells. Similarly, in differential sensing, multiple cross-reactive receptors and/or multiple output signals are used to achieve analyte differentiation. Specifically, the cross-reactive receptors are combined in a sensor array and the signal output pattern generated upon analyte binding is linked to a specific analyte or mixture of analytes using multivariate analysis tools, such as linear discriminant analysis (LDA) and principal component analysis (PCA). Such sensor arrays are capable of not only differentiating multiple analytes, but also changes in analyte concentration. Overall, the advantage of differential sensing routines is that analyte differentiation can be achieved without the need for expensive, unstable receptors (e.g., antibodies).22
Original work in the field of differential sensing was done for small molecules in gases, but was later expanded to include applications in liquid systems.23 Sensor arrays have been developed to differentiate a variety of solution-based analytes, ranging from small molecules24,25 to proteins26 and whole cells.27 The first report of using differential sensing for distinguishing classes of proteins (e.g., high vs. low isoelectric point, glycosylated vs. not glycosylated, etc.) was by Wright et al. in 2005.28 Since this report, several other groups have developed sensor arrays for distinguishing proteins based on their different properties (e.g., pI, MW, glycosylation, metallo vs. non-metalloprotein, surface hydrophobicity, etc.) using a wide variety of recognition and transduction elements.29–37 Notably, until the current report, no sensor array had been developed that exclusively uses changes in relative turbidity for signal transduction/input for multivariate analysis.
In this work, we explore how the identity of the ionizable groups in PNIPAM-based nanogels affects protein binding, and then use the nanogels as cross-reactive receptors in a turbidimetric sensor array. The functional groups were introduced via post-synthesis modification of nanogels made from NIPAM and methacrylic acid (MAA) (poly(NIPAM-co-MAA)), specifically using a carbodiimide-coupling strategy to link amine-containing molecules (i.e., ligands) to the carboxyl groups of MAA.38 The five ionizable functional groups tested were carboxyl, sulfate, guanidinium, secondary amine, and primary amine groups. We discuss the ability to incorporate the ligands into the nanogels and investigate how the functional groups affect the nanogel characteristics, specifically hydrodynamic diameter, zeta potential, and protein binding properties. Then, we demonstrate the ability to utilize these nanogels in a turbidimetric sensor array. By performing the protein binding in two different buffers, it was possible to differentiate eleven proteins with a range of molecular weights and isoelectric points without needing to incorporate a colorimetric or fluorescence reporter. Lastly, we demonstrate the ability of the sensor array to differentiate changes in protein concentration in a simulated tear fluid.
Results and discussion
Nanogel synthesis and modification
The receptors for differential sensing investigated in this work are charge-containing PNIPAM nanogels. The temperature-dependent hydrophobicity of PNIPAM enables synthesis of nanogels via precipitation polymerization, an advantageous method for protein binding applications for several reasons. First, this technique results in production of nanogels with narrow size distributions, which is important for reproducibility.39 Second, the resultant nanogels are highly swollen at room temperature, a necessary feature for protein diffusion into the polymer network. Additionally, in this work, a surfactant-free precipitation polymerization was employed to eliminate potential surfactant mediated complications during protein-binding studies (e.g., surfactant-protein interactions). MAA was included in the polymerization to promote electrostatic interactions with high isoelectric point (pI) proteins and as a reactive handle for introducing different functional groups to vary affinity properties.
Unlike many other ionic co-monomers, such as 2-aminoethylmethacrylate hydrochloride40 or 2-acrylamido-2-methyl-1-propanesulfonic acid,41 incorporation of acrylic acid or MAA at high percentages (i.e., >10%) can be achieved without the use of surfactants.42,43 The upper limit of incorporation of a co-monomer is determined by the amount that can be incorporated while still allowing the polymer to be sufficiently hydrophobic to undergo collapse and nucleation. Increasing the hydrophilicity of the polymer diminishes the ability to form stable, uniform nanoparticles.44
For the reaction conditions used in this work, the polymerization solution has a pH of approximately 3.6 due to the acidity of MAA. At this pH, the MAA will be more than 90% protonated and sufficiently hydrophobic for stable nanoparticle formation. The incorporation of MAA determined by potentiometric titration (15.7 ± 0.4 weight%) was higher than the feed ratio (10.9 weight% = 17.3 mol%). Given the reactivity ratios of MAA (r1=1.13) and NIPAM (r2 = 0.89), this MAA enrichment in the polymer composition relative to the feed ratio was not surprising.45 Rather than incorporating other ionic co-monomers at low-levels during synthesis, the other functional groups were incorporated using a post-synthesis modification. More specifically, modification of the carboxylic acid groups of poly(NIPAM-co-MAA) was done using a single-step carbodiimide coupling protocol. The ionic ligands used in this work are shown in Figure 1.
Figure 1. Structures of ligands used to modify poly(NIPAM-co-MAA).
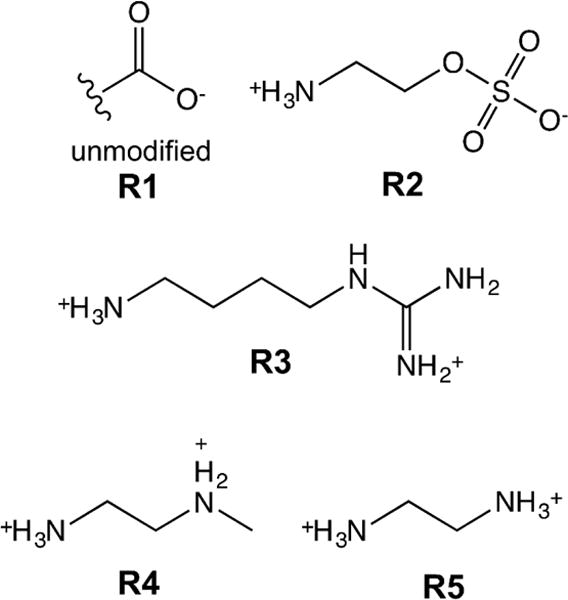
Unmodified nanogels (R1) contain carboxylic acid group. 2-aminoethylhydrogen sulfate (AEHS, R2), agmatine sulfate (AGS, R3), N-methylethylenediamine (NMEDA, R4), and ethylene diamine dihydrochloride (ED, R5) were used to introduce sulfate, guanidinium, secondary amine, and primary amine functional groups, respectively.
Throughout the remainder of the discussion, unmodified nanogels will be referred to as R1 and modified nanogels will be referred to as R2–R5 corresponding to the ligands with which they were modified. Reaction conditions (i.e., molar ratio of amine:COOH, concentration of EDC, pH) were optimized in an effort to maximize coupling efficiency. While N-hydroxysulfosuccinimide (sulfo-NHS) is often reported to improve coupling efficiency,38 under the reaction conditions tested, sulfo-NHS did not improve the coupling efficiency, and thus was excluded in order to reduce cost.
The efficiency of modification of poly(NIPAM-co-MAA) with the amines ligands was quantified by potentiometric titration (Table 1, Figure S1). With all other reaction conditions kept the same, the coupling efficiency is dependent on the properties of the ligand.46 For the ligands tested, our hypothesis was that variations in coupling efficiency were due to differences in charge character. Specifically, AEHS, the only zwitterion tested, had the lowest coupling efficiency. The combination of repulsive (from the sulfate) and attractive (from the primary amine) forces between this ligand and the anionic nanogels will reduce the electrostatic driving force for partitioning of this ligand into the R1 nanogels relative to the dicationic ligands. The coupling efficiency of AGS was significantly lower than the other dicationic ligands. Our hypothesis for this result is that the particularly high affinity interaction between the guanidinium group of AGS and sulfate ions present in solution, and on the surface of the nanogels, reduced the electrostatic partitioning of AGS into the unmodified nanogels and consequently resulted in reduced coupling efficiency.47,48 When guanidinium hydrochloride was included to interfere with sulfate-agmatine interactions, the coupling efficiency was increased from 50.4 ± 4.3% to 73.7 ± 3.3%.
Table 1.
Coupling efficiency of different ligands determined by potentiometric titration
| Modification | μmol/mg nanogel | Coupling efficiency |
|---|---|---|
| R1 (Unmodified) | 1.85 ± 0.05 | N/A |
| R2 (AEHS) | 1.33 ± 0.01 | 71.8 ± 1.5% |
| R3 (AGS) | 1.36 ± 0.09 | 73.7 ± 3.3% |
| R4 (NMEDA) | 1.66 ± 0.13 | 90.1 ± 5.4% |
| R5 (ED) | 1.68 ± 0.11 | 90.9 ± 6.5% |
Additionally, zeta potential measurements matched what would be expected based on the identity of the ionizable groups and the titration results (Table 2). Specifically, the anionic nanogels, R1 and R2, had highly negative zeta potentials, while R4 and R5 nanogels had positive zeta potentials, as the majority of the carboxyl groups were successfully converted to amines. R3 nanogels still had a net negative zeta potential because of the inefficient coupling of AGS. 1H NMR (Figure S2) and FTIR spectroscopy (Figure S3) further confirmed successful modification.
Table 2.
Dynamic light scattering results for modified poly(NIPAM-co-MAA) nanogelsa
| Nanogel | Dh (nm) | PDI | ZP (mV) |
|---|---|---|---|
| R1 (PBS) | 378.1 ± 15.4 | 0.030 ± 0.005 | −25.0 ± 0.2 |
| R2 (PBS) | 306.4 ± 14.2 | 0.058 ± 0.026 | −18.6 ± 0.7 |
| R3 (PBS) | 290.9 ± 8.7 | 0.011 ± 0.005 | −9.7 ± 0.4 |
| R4 (PBS) | 283.9 ± 10.4 | 0.022 ± 0.009 | 4.3 ± 0.3 |
| R5 (PBS) | 280.1 ± 10.2 | 0.064 ± 0.039 | 6.3 ± 0.7 |
| R1 (HBS) | 308.5 ± 9.2 | 0.111 ± 0.016 | −18.5± 1.1 |
| R2 (HBS) | 307.6 ± 3.9 | 0.147 ± 0.034 | −15.7 ± 0.7 |
| R3 (HBS) | 299.4 ± 8.1 | 0.069 ± 0.059 | −3.1 ± 0.4 |
| R4 (HBS) | 297.0 ± 3.6 | 0.095 ± 0.043 | 8.1 ± 1.0 |
| R5 (HBS) | 294.1 ± 8.4 | 0.094 ± 0.078 | 9.0 ± 0.9 |
Measurements were performed after suspension and sonication of lyophilized nanogels at a concentration of 1 mg/mL in 0.1X PBS (pH 7.4 ± 0.1) or 0.1X HBS (pH 5.5 ± 0.1). Values reported as mean ± SD (n=3).
When using nanogels as protein receptors, it is important to realize that in addition to changing the charge character, changing the type of ionizable group affects the swelling behavior of hydrogels, which can have an impact on diffusion of proteins into the networks. Thus, before protein binding, the charged nanogels were also characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM). The low polydispersity indices (PDIs) and TEM images confirmed formation of spherical nanogels with narrow size distributions (Table 2, Figure S4). The type of ionizable group significantly affected the hydrodynamic diameter of the nanogels. It is well understood that hydrogels containing weakly acidic or basic functional groups undergo pH-responsive swelling due to changes in osmotic balance as the degree of ionization changes.49 More specifically, anionic hydrogels are collapsed at low pH, but swell as the pH transitions above the pKa of the acidic group. Conversely, cationic hydrogels are collapsed at high pH, but swell as the pH drops below the pKa of the basic functional group.
Accordingly, R1 nanogels were the most highly swollen in 0.1X phosphate buffered saline (PBS, pH 7.4, ionic strength ~ 17.3 mM). However, the R2 nanogels, which also contain acidic functional groups, were significantly more collapsed than R1 nanogels. We attribute the decrease in hydrodynamic diameter to the two additional hydrogen bond acceptors present in sulfate groups relative to carboxyl groups, which only have two hydrogen bond acceptors. The polymer network has an abundance of hydrogen bond donors (i.e., protons from amide bonds of N,N’-methylenebisacrylamide (BIS) and NIPAM) that can form additional hydrogen bonds with the sulfate groups, resulting in additional tie points and, consequently, decreased mesh size.
It was expected that the cationic nanogels would be at least partially collapsed at pH 7.4. Indeed, R3–R5 all had similar hydrodynamic diameters, approximately 100 nm less than that of the unmodified nanogels in 0.1X PBS. In 0.1X histidine buffered saline (HBS, pH 5.5, ionic strength ~ 17.4 mM), R1 was significantly more collapsed than in 0.1X PBS, as would be expected for a carboxyl containing hydrogel as it goes from a near-neutral to acidic pH. For all other nanogels, a statistical difference between the hydrodynamic diameters in 0.1X HBS and 0.1X PBS was not observed.
Protein binding studies
As discussed in the introduction, several groups have demonstrated the utility of using PNIPAM based nanogels as protein receptors. In this work, protein binding studies were performed to confirm and characterize the cross-reactivity of charge-containing PNIPAM nanogels, a necessary feature for development of a differential sensor array. Specifically, we investigated how the buffer and type of ionizable groups present in the nanogels affected protein binding.
The proteins tested (Table 3) were chosen because they have a wide range of molecular weights and isoelectric points, and thus would allow us to test the sensor array’s range of performance. Furthermore, several of the proteins are high abundance proteins that serve as biomarkers for certain diseases. For example, lysozyme and lactoferrin are present at high concentrations in tears (1–3 mg/mL and 0.9–3.1 mg/mL, respectively), but are under-expressed (<1 mg/mL and < 0.9 mg/mL, respectively) in tears of patients with dry eye or Sjögren’s syndrome.50,51 As another example, the serum albumin:globulin ratio is frequently monitored in cancer patients due to its prognostic significance.52,53
Table 3.
Properties of proteins testeda
| Protein | PI | MW (KDa) |
|---|---|---|
| Lysozyme (L) | 11.35 | 14.3 |
| Cytochrome c (C) | 10.0–10.5 | 12.3 |
| Trypsin (T) | 10.1–10.5 | 24.0 |
| Lactoferrin (LF) | 8.7 | 82.4b |
| Myoglobin (M) | 7.3, 6.8 | 17.6 |
| Hemoglobin (H) | 6.8 | 64.5 |
| Gamma globulins (G) | 6.5c | 165b,c |
| Bovine serum albumin (B) | 4.8 | 66.4 |
| Ovalbumin (O) | 4.5 | 44.3b |
| Ovomucoid (TI) | 4.3d | 28.8b,d |
| Fetuin (F) | 3.3 | 48.4b |
In order to optimize the conditions for protein binding, the effects of ionic strength, temperature, and inclusion of a non-ionic surfactant on lysozyme binding to unmodified (R1) nanogels was tested. Specifically, protein binding was performed in 0.1X PBS or 1X PBS (ionic strength ~ 173 mM), with or without Tween 20, and at room temperature or 37°C (Figure S5a). When 1X PBS was used as the buffer, there was a statistically significant decrease in adsorption capacity of R1 nanogels for lysozyme compared to 0.1X PBS (p<0.005). This is not a surprising result, as the decreased mesh size and increased charge screening that result from increased ionic strength are both factors that will inhibit protein binding (Figure S5b). Addition of Tween 20, which reduces non-specific adsorption of proteins to surfaces,54 did not affect adsorption capacity.
Protein binding was also performed at 37°C, a temperature above the lower critical solution temperature (LCST) of PNIPAM where the nanogels were expected to exhibit more hydrophobic character. Interestingly, there was no observable temperature swelling behavior at pH 7.4 (Figure S5b), which we attribute to the high content of deprotonated MAA making the polymer too hydrophilic to enable hydrophobic collapse at this pH. Without a large change in mesh size or hydrophobic character, it is not surprising that there was no statistical difference between the adsorption capacity at room temperature or at 37°C.
For the remainder of this work, protein binding studies were performed in 0.1X PBS (pH 7.4) or 0.1X HBS (pH 5.5) at room temperature. The proteins tested are generally classified as low pI (pI<6.0), near-neutral pI (6.0 ≤ pI ≤ 8.0), or high pI (pI > 8.0) throughout the discussion. Adsorption capacity (i.e., mass of protein bound per mass of particle) was determined for each polymer-protein-buffer combination at a fixed protein concentration (0.5 mg/mL). The obtained adsorption capacity values were used to compare relative affinity for the eleven proteins. Specifically, a given nanogel will have a constant number of available binding sites (i.e., nanogel pores) and thus a higher adsorption capacity corresponds to a higher fractional occupancy and, in turn, lower dissociation constant, KD (i.e., higher affinity). The upper limit for adsorption capacity will depend on both the KD and the equilibrium volume swelling of the nanogels.
Overall, the protein binding trends confirmed that electrostatic interactions were the primary driving force for protein binding. In 0.1X PBS, the anionic nanogels (R1 and R2) preferentially bound high pI proteins, while the cationic nanogels (R4 and R5) preferentially bound low pI proteins, albeit with much lower overall adsorption capacity (Figure 2a). The lower adsorption capacity of proteins to R3, R4, and R5 nanogels may be attributed to several factors. First, these nanogels have smaller hydrodynamic diameters, and thus mesh size, than R1 and R2. Protein diffusion into hydrogel networks is hindered as a consequence of decreased mesh size,57 ultimately limiting protein binding to near the surface of the nanogels. Second, the presence of sulfate groups from the ammonium persulfate initiator and residual carboxyl groups could diminish the electrostatic driving force for binding of low pI proteins. Lastly, protein binding is weakened by competitive interactions with buffer ions that have the opposite charge of the nanogels, which was the case for cationic gels (R4 and R5) in 0.1X PBS.
Figure 2. Characterization of cross-reactivity of poly(NIPAM-co-MAA) nanogels.
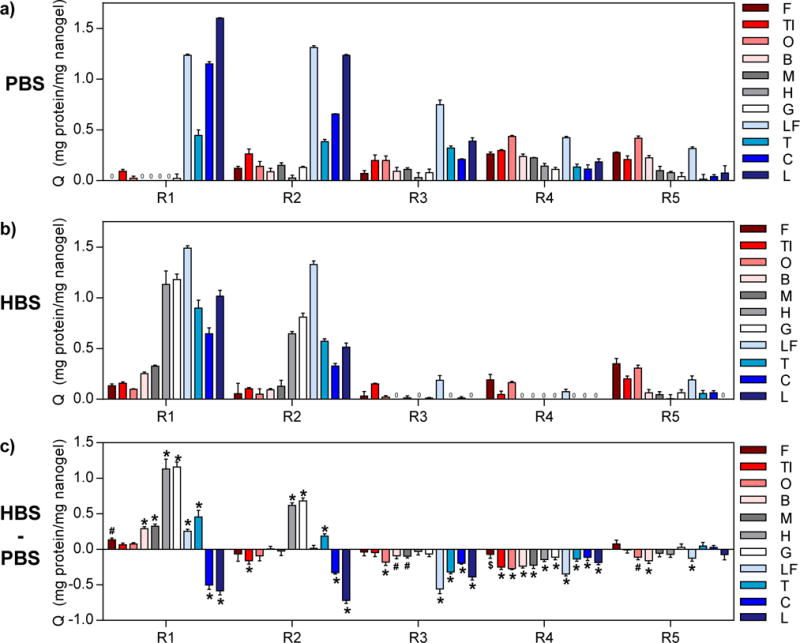
Adsorption capacity values (Q) of the eleven proteins to R1-R5 nanogels were determined from equilibrium protein binding experiments performed in (a) 0.1X PBS and (b) 0.1X HBS with initial protein concentrations of 0.5 mg/mL. (c) Difference between Q for protein binding in the two buffers (QHBS – QPBS) showing the impact of buffer on protein binding behavior. Statistical analysis was performed to determine significant differences in Q in the two buffers (n=3, Šidák’s test, $= p<0.05, # = p<0.01, * = p<0.001). Red shades = low pI proteins (pI < 6.0), gray shades = near-neutral pI proteins (6.0 ≤ pI ≤ 8.0), blue shades = high pI proteins (pI > 8.0).
When protein binding was performed in 0.1X HBS instead of 0.1X PBS, the change in degree of ionization of the nanogels and proteins was expected to significantly influence protein binding (Figure 2b–c). The most notable differences were changes in R1 adsorption capacity. At pH 5.5, the R1 nanogels are less negatively charged and have smaller mesh size than at pH 7.4, diminishing the electrostatic attraction for high pI proteins and increasing steric hindrance. Accordingly, adsorption capacities for lysozyme and cytochrome c were significantly lower in 0.1X HBS than in 0.1X PBS (p<0.005). On the other hand, near-neutral pI proteins became more positively charged as a result of this buffer change, increasing their electrostatic attraction to R1 nanogels. Consequently, the adsorption capacities for hemoglobin and gamma globulins, and, to a lesser extent, other proteins with isoelectric points between 4.8–8.7, significantly increased (p<0.005). Similarly, the adsorption capacities of R2 for hemoglobin and gamma globulins were higher, while adsorption capacities for cytochrome c and lysozyme were lower in 0.1X HBS compared to 0.1X PBS. For R2, the hydrodynamic diameter did not significantly change, so the decrease in adsorption capacities for cytochrome c and lysozyme may be attributed to protonation of residual carboxyl groups and competition with histidine ions in the buffer for nanogel binding sites. Likewise, adsorption capacity of R3 for all high pI proteins decreased in 0.1X HBS (p<0.005).
Adsorption capacity of R4 for all proteins was significantly decreased in 0.1X HBS compared to 0.1X PBS (p<0.05), which may be due to both the proteins and R4 nanogel being more positively charged at pH 5.5, increasing electrostatic repulsion. For R5, the degree of ionization of primary amine groups will not be significantly different in the two buffers and thus the changes in adsorption capacity were small or not statistically significant. All significant changes (p<0.05) were decreases in adsorption capacity, which may be explained by the proteins being more positively charged at this pH. Furthermore, the protein binding behavior of the nanogels can be altered by changing the buffer, a useful property for protein differentiation. Overall these results demonstrate that the identity and degree of ionization of the nanogel functional groups dictate protein binding behavior by affecting both electrostatic interactions and pore (i.e., binding site) size.
Turbidimetric differential sensing
Having demonstrated cross-reactive protein binding properties, we next sought to utilize these nanogels as receptors in a turbidimetric sensor array. Turbidity is a result of a combination of scattering, absorbance, and interference.58 Commercially available turbidimetric assays typically employ nanoparticles of a specific size that are functionalized with antibodies for a target analyte.59 Depending on the analyte and nanoparticle concentrations, analyte binding results in particle agglutination or aggregation. Due to the larger size of these aggregates compared to individual particles, there is increased light scattering and, consequently, increased turbidity of the solution. Turbidity changes are quick and easy to determine by measuring the absorbance of the solution.59 A turbidimetric sensor array would be advantageous for biosensing, particularly for diseases where changes in both total protein content and specific protein concentrations provide diagnostic information. For example, in dry eye and Sjögren’s syndrome, total tear protein content is significantly decreased due to down regulation of the major protein constituents, specifically lactoferrin, lysozyme, and lipocalin 1.50
Noticeable changes in turbidity were observed upon protein binding to charged PNIPAM nanogels. For high affinity protein-polymer interactions, there was a fast and visually apparent increase in turbidity, while for low affinity interactions there were insignificant changes in turbidity relative to the polymers in just buffer (Figure S6). In all cases except for R3 nanogels in HBS (for which protein binding was undetectable by MicroBCA), there was a positive correlation between adsorption capacity and turbidity (Figures S7). According to Mie theory, increasing the radius and/or refractive index of particles leads to an increase in scattering cross-section and thus turbidity.60 To understand the reason for the observed increase in turbidity (i.e., radius increase due to aggregation or refractive index increase), we performed DLS to determine if there was an increase in the hydrodynamic diameter of the nanogels after protein binding (Figure S8).
In general, increases in relative turbidity were not associated with substantial increases in hydrodynamic diameter. However, for R5, and to a much lower extent R4, the two proteins that caused turbidity increases (i.e., fetuin and ovalbumin) also increased relative hydrodynamic diameter of the nanogels. These results agree with our conclusions from the adsorption capacity experiments, that is, the high adsorption capacity of proteins to R1 and R2 (in both buffers), and R3 (in 0.1X PBS) was attributed to protein binding throughout the network while adsorption to R4 and R5 was attributed primarily to surface binding. Accordingly, for R4 and R5, DLS results suggest the main factor causing the increased turbidity is surface binding leading to particle aggregation,61 similar to standard particle-enhanced turbidimetric immunoassays.59 For R1-R3, however, the increase in turbidity was primarily due to an increase in refractive index as a result of protein molecules filling the volumes of the nanogels.62
Relative turbidity values for each protein-polymer interaction were used as input for multivariate analysis. We note that each receptor was considered as a distinct host in the different buffers, for example R1 could serve as two receptors: R1PBS and R1HBS. PCA and LDA were used to determine whether the cross-reactive nanogels were able to distinguish among the eleven proteins tested. For thorough discussions on the utility of PCA and LDA for differential sensing applications, we refer the reader to several recent reviews from the Anslyn group.22,63 Briefly, PCA is an unsupervised method (i.e., analyte classes are unknown) and reduces dimensionality by determining the directions within the data that maximize the set’s variance while ignoring class labels. In this method, the first two principal components (PC1 and PC2) contain the majority of the set’s variance. The amount of variance contained within PC1 and PC2 are often shown as percentages in parentheses on the plot axes. The loading vectors on PCA biplots show the extent that each host (i.e., receptor, nanogel) contributes to the guest (i.e., analyte) discrimination. LDA is a supervised method (i.e., analyte classes are known and are taken into consideration) that seeks to reduce the dimensionality of the data by maximizing variance between classes and minimizing variance within each class.
One of the most important properties of a sensor array is its predictive power, or the ability of the sensor array to correctly identify unknown analytes. The predictive power can be assessed using cross-validation techniques such as jack-knife (also known as “leave-one-out”) analysis, which provides information on classification accuracy for the set of receptors and analytes tested. Visually, sensor arrays with good predictive power will have large separation of proteins with different classes and tight clustering of proteins of the same identity. Confidence ellipses, which depict uncertainty in the data, are often included on LDA plots to help visualize separation. In a sensor array with good predictive power there should be tight confidence ellipses with little to no overlap.
Multivariate analysis with every combination of receptors (lowest number of receptors = 2, highest number of receptors = 10) was performed to determine which combinations provided the best classification accuracy (Table S1). Overall, receptors in 0.1X HBS provided better differentiation than receptors in 0.1X PBS. To clearly demonstrate the charge-dependent trends, the colors in the PCA and LDA plots correspond to the different pI classes, with red shades corresponding to low pI proteins, gray shades corresponding to near-neutral pI proteins, and blue shades corresponding to high pI proteins (Figures 3–4). Not surprisingly, when only 0.1X PBS was used as the binding buffer, R1, R2 and R3 (the net anionic nanogels) contributed primarily to separation of high pI proteins while R4 and R5 (the net cationic nanogels) contributed primarily to separation of low pI proteins (Figure 3a,b). LDA resulted in poor separation of near-neutral pI proteins, which led to misclassification of myoglobin (M), hemoglobin (H), and gamma globulins (G) as each other or as ovomucoid (TI) (Figure 4a). The overall classification accuracy based on jack-knife analysis was 81.8%.
Figure 3. Principal component analysis of relative turbidity changes upon protein binding to nanogels.
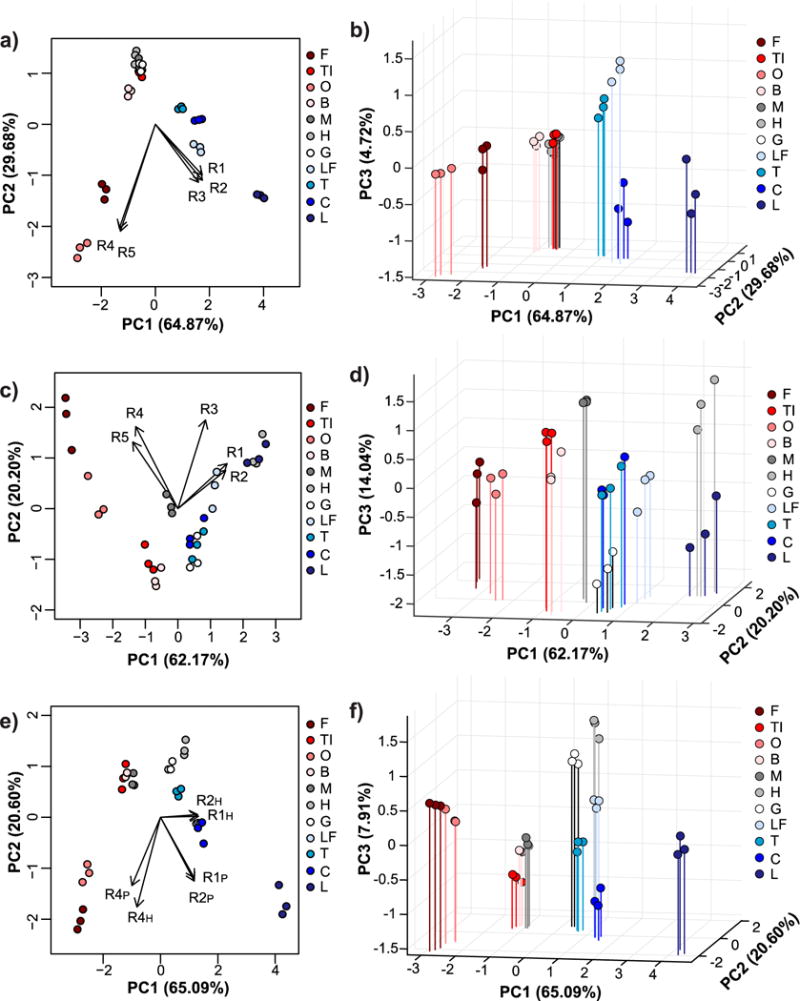
Two-dimensional (a, c, e) and three-dimensional PCA biplots (b, d, f) correspond to data for protein binding experiments performed with all five nanogels in 0.1X PBS (a and b), with all five nanogels in 0.1X HBS (c and d), or with only R1, R2, and R4 nanogels in both buffers (e and f). Red shades = low pI proteins (pI < 6.0), gray shades = near-neutral pI proteins (6.0 ≤ pI ≤ 8.0), blue shades = high pI proteins (pI > 8.0). In all cases, protein and nanogel concentrations were 0.5 mg/mL.
Figure 4. Linear discriminant analysis of relative turbidity changes upon protein binding to nanogels.
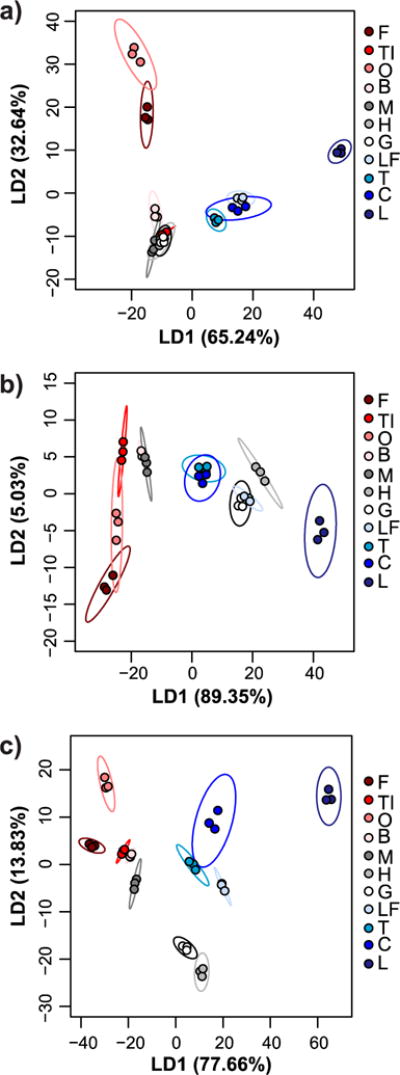
LDA plots with 95% confidence ellipses corresponding to data for protein binding experiments performed with all five nanogels in 0.1X PBS (a), with all five nanogels in 0.1X HBS (b), or with only R1, R2, and R4 nanogels in both buffers (c). While 100% classification accuracy (according to jack-knife analysis) was achieved using all five nanogels in 0.1X HBS, overlap of confidence ellipses was reduced using data from both buffers but only three of the nanogels. Red shades = low pI proteins (pI < 6.0), gray shades = near-neutral pI proteins (6.0 ≤ pI ≤ 8.0), blue shades = high pI proteins (pI > 8.0). In all cases, protein and nanogel concentrations were 0.5 mg/mL.
When only 0.1X HBS was used as the buffer, R1 and R2 contributed to separation of high and near-neutral pI proteins as both of these protein classes will be net cationic at pH 5.5. R4 and R5 still contributed mostly to separation of low pI proteins (Figure 3c,d), as would be expected because of electrostatic complementarity. The exception was myoglobin, which clustered more closely to low pI proteins than the other near-neutral pI proteins. In 0.1X HBS, low pI proteins were generally better separated than in 0.1X PBS, while the overlap between near-neutral and high pI proteins increased. Nonetheless, LDA correctly classified 100% of the proteins according to jack-knife analysis (Figure 4b). However, the significant overlap of the confidence ellipses made us wary of the true predictive power of the sensor array.
To improve the sensor array performance, turbidity data for experiments done in both 0.1X HBS and 0.1X PBS were used as input for multivariate analysis. 100% classification accuracy by LDA was achieved when as few as two receptors (either R1 + R4 or R2 + R4) were used (Table S1), but the overlap between confidence ellipses was still substantial (Figure S8). When three receptors, particularly R1 + R2 + R4 were used, better separation was achieved (Figure 3e,f). Again, the loading vectors match what would be expected based on electrostatic complementarity and the measured adsorption capacities. Specifically, R1 and R2 in 0.1X PBS contributed mostly to separation of high pI proteins, R1 and R2 in 0.1X HBS provided separation of near-neutral pI proteins, and R4 in both buffers helped differentiate low pI proteins. LDA classification accuracy was 100% by jack-knife analysis, but now the confidence ellipses had minimal overlap (Figure 4c).
Another point that is clear from the score plots is that the proteins are not lined up simply from high to low pI along one principal component. This demonstrates that, in addition to differences in net charge, the nanogels differentiate the proteins based on other distinguishing properties of the proteins. Such properties could include differences in primary, secondary, and tertiary structures of the proteins; hydrophobicity of the protein surfaces; extent of glycosylation; and molecular weight. For example, lysozyme is particularly well separated from the other high pI proteins, which we attribute to its exceptionally high arginine content compared to the other high pI proteins, which have mostly lysine residues. Arginine can form additional hydrogen bonds with the nanogel networks and has a higher pKa than lysine, likely increasing the affinity between the nanogels and lysozyme.
As another example, fetuin and ovalbumin were closely clustered together. These proteins have similar molecular weights and both are glycoproteins, but the extent of glycosylation of the two proteins is different, with fetuin being highly glycosylated and ovalbumin having very low glycosylation64. However, it is clear that it is not just a matter of glycoprotein vs. non-glycoprotein, because ovomucoid, which is also a heavily glycosylated protein64, behaved more similarly to BSA, the one low pI protein that is not a glycoprotein. Overall, while proteins of the same pI class generally clustered near one another, other structural or chemical differences between the proteins resulted in complete differentiation by the nanogels.
After demonstrating differentiation of eleven individual proteins at a single concentration (0.5 mg/mL), we sought to demonstrate the ability to detect changes in protein concentration in a simulated biological fluid, specifically simulated tear fluid. The major protein constituents of tears are lysozyme, lactoferrin, immunoglobulins (primarily IgA), lipocalin 1, and mucins. The composition of the simulated tear fluid is shown in Table S2. Tears provided a particularly interesting example because our previous studies demonstrated that lactoferrin, gamma globulins, and lysozyme all bind receptors R1 and R2 at high levels when in 0.1X HBS pH 5.5 (Figure 2b). Thus, competition between these high abundance tear proteins was anticipated.
We expected that increasing protein concentration would increase the turbidity of each nanogel receptor, but with varying sensitivity corresponding to adsorption capacity trends. Thus, little-to-no cross-reactivity related to concentration between the receptors was anticipated, which would be evident in a score plot with the majority of the variance along the first axis.63 We tested this hypothesis by increasing the concentration of lysozyme in a simulated tear fluid. Healthy levels for lysozyme in tears range between 1–3 mg/mL, with concentrations less than 1 mg/mL being suggestive of dry eye syndrome or Sjögren’s syndrome.50,51 The simulated tear fluid was diluted 1/10 in 0.1X HBS (pH 5.5) and the concentration of lysozyme was varied from 0 μg/mL to average healthy levels (200 μg/mL, for 1/10 dilution).
The relative turbidity was positively correlated with the lysozyme concentration for all 5 receptors (Figure S10), with R1 and R2 exhibiting higher sensitivity than R3–R5. As anticipated, the majority of the variance was contained within the first factor (LD1, 99.38%). Importantly, however, LDA was able to correctly classify all concentrations tested (100% by jack-knife analysis), both above and below the physiologically relevant ranges for Sjögren’s syndrome, thereby permitting detection of abnormally low levels of lysozyme that would be observed in dry eye patients (Figure 5).
Figure 5. Multivariate analysis of relative turbidity changes upon varying lysozyme concentration in a dilute (1/10) simulated tear fluid.
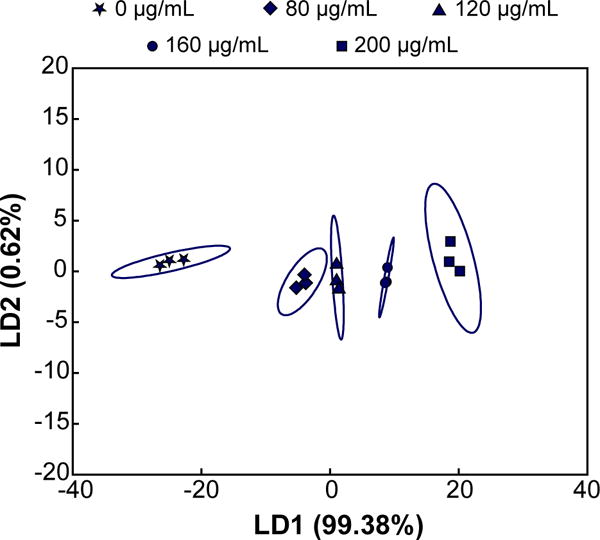
LDA plot with 95% confidence ellipses demonstrates that the majority of variance is contained within the first linear discriminant when lysozyme concentration is varied from 0 μg/mL to an average healthy concentration (200 μg/mL). Concentrations less than 100 μg/mL (i.e., 1 mg/mL in undiluted tears) are indicative of dry-eye or Sjögren’s syndrome.
Conclusions
We have designed and characterized five charge-containing PNIPAM nanogels as differential protein receptors. The identity of the ionizable groups impacted the swelling behavior, charge character, and, consequently, the protein binding behavior of the nanogels. By performing turbidimetric protein binding assays in two buffers with different pH but comparable ionic strength, it was possible to differentiate the eleven model proteins using only two or three of the charge-containing PNIPAM receptors. The hypothesized mechanisms for relative turbidity increases are aggregation for protein surface binding and refractive index increase of the nanogel when protein binding occurs throughout the bulk of the nanogel. The main driving force for protein binding was favorable electrostatic interactions, making these nanogels adaptable for differential sensing of other protein biomarker panels. Importantly, it was possible to detect changes in protein concentration even in a complex matrix.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grant R01-EB022025. During this work H.R.C. was supported by a National Science Foundation Graduate Research Fellowship and a Donald D. Harrington Dissertation Fellowship. M.E.W. was also supported by a National Science Foundation Graduate Research Fellowship. H.R.C. would like to acknowledge the generous support of the Philanthropic Educational Organization Scholar Awards. I.S. would like to acknowledge the support of an Undergraduate Research Fellowship from The University of Texas at Austin. EVA thanks the Welch Regents Chair for support (F-0045). We would also like to thank Steve Sorey and Angela Spangenberg of the UT Austin NMR Facility for technical support and David Spencer for his insightful discussions.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental details, Figures S1–S9, Tables S1–S2. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Vreeburg IJHG, Boxall DJB. Water Res. 2007;41:519–529. doi: 10.1016/j.watres.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 2.Hall BG, Acar H, Nandipati A, Barlow M. Mol Biol Evol. 2014;31:232–238. doi: 10.1093/molbev/mst187. [DOI] [PubMed] [Google Scholar]
- 3.Jackson DD. J Am Chem Soc. 1901;23:799–806. [Google Scholar]
- 4.Chesnin L, Yien CH. Soil Sci Soc Am J. 1951;15:149–151. [Google Scholar]
- 5.Ferreria P, Price CP. Clin Chim Acta. 1974;55:259–262. doi: 10.1016/0009-8981(74)90303-9. [DOI] [PubMed] [Google Scholar]
- 6.Harmoinen A, Perko M, Grönroos P. Clin Chim Acta. 1981;111:117–120. doi: 10.1016/0009-8981(81)90430-7. [DOI] [PubMed] [Google Scholar]
- 7.Price CP, Trull AK, Berry D, Gorman EG. J Immunol Methods. 1987;99:205–211. doi: 10.1016/0022-1759(87)90129-3. [DOI] [PubMed] [Google Scholar]
- 8.Newman DJ, Henneberry H, Price CP. Ann Clin Biochem. 1992;29:22–42. doi: 10.1177/000456329202900104. [DOI] [PubMed] [Google Scholar]
- 9.Conde-Sánchez M, Roldán-Fontana E, Chueca-Porcuna N, Pardo S, Porras-Gracia J. Clin Biochem. 2010;43:921–925. doi: 10.1016/j.clinbiochem.2010.04.057. [DOI] [PubMed] [Google Scholar]
- 10.Mizuno M, Shima T, Oya H, Mitsumoto Y, Mizuno C, Isoda S, Kuramoto M, Taniguchi M, Noda M, Sakai K, Koyama N, Okanoue T. Hepatol Res. 2017;47:216–225. doi: 10.1111/hepr.12710. [DOI] [PubMed] [Google Scholar]
- 11.Dasgupta A. Therapeutic drug monitoring: newer drugs and biomarkers. 1st. Elsevier/Academic Press; Waltham, Ma: 2012. [Google Scholar]
- 12.Deleo DT, Lee IR, Wetherall JD, Newman DJ, Medcalf EA, Price CP. Clin Chem. 1991;37:527–531. [PubMed] [Google Scholar]
- 13.Farid SS. J Chromatogr B. 2007;848:8–18. doi: 10.1016/j.jchromb.2006.07.037. [DOI] [PubMed] [Google Scholar]
- 14.Miller MA, Rodrigues MA, Glass MA, Singh SK, Johnston KP, Maynard JA. J Pharm Sci. 2013;102:1194–1208. doi: 10.1002/jps.23473. [DOI] [PubMed] [Google Scholar]
- 15.Luchini A, Geho DH, Bishop B, Tran D, Xia C, Dufour RL, Jones CD, Espina V, Patanarut A, Zhou W, Ross MM, Tessitore A, Petricoin EF, Liotta LA. Nano Lett. 2008;8:350–361. doi: 10.1021/nl072174l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fredolini C, Meani F, Alex Reeder K, Rucker S, Patanarut A, Botterell PJ, Bishop B, Longo C, Espina V, Petricoin EF, Liotta LA, Luchini A. Nano Res. 2008;1:502–518. doi: 10.1007/s12274-008-8054-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magni R, Espina BH, Shah K, Lepene B, Mayuga C, Douglas TA, Espina V, Rucker S, Dunlap R, Petricoin EFI, Kilavos MF, Poretz DM, Irwin GR, Shor SM, Liotta LA, Luchini A. J Transl Med. doi: 10.1186/s12967-015-0701-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamburro D, Fredolini C, Espina V, Douglas TA, Ranganathan A, Ilag L, Zhou W, Russo P, Espina BH, Muto G, Petricoin EF, Liotta LA, Luchini A. J Am Chem Soc. 2011;133:19178–19188. doi: 10.1021/ja207515j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fredolini C, Meani F, Luchini A, Zhou W, Russo P, Ross M, Patanarut A, Tamburro D, Gambara G, Ornstein D, Odicino F, Ragnoli M, Ravaggi A, Novelli F, Collura D, D’Urso L, Muto G, Belluco C, Pecorelli S, Liotta L, Petricoin EF. AAPS J. 2010;12:504–518. doi: 10.1208/s12248-010-9211-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yonamine Y, Hoshino Y, Shea KJ. Biomacromolecules. 2012;13:2952–2957. doi: 10.1021/bm300986j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoshino Y, Koide H, Furuya K, Haberaecker WW, Lee SH, Kodama T, Kanazawa H, Oku N, Shea KJ. Proc Natl Acad Sci. 2012;109:33–38. doi: 10.1073/pnas.1112828109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diehl KL, Anslyn EV. Chem Soc Rev. 2013;42:8596. doi: 10.1039/c3cs60136f. [DOI] [PubMed] [Google Scholar]
- 23.Lavigne JJ, Savoy S, Clevenger MB, Ritchie JE, McDoniel B, Yoo SJ, Anslyn EV, McDevitt JT, Shear JB, Neikirk D. J Am Chem Soc. 1998;120:6429–6430. [Google Scholar]
- 24.McCleskey SC, Griffin MJ, Schneider SE, McDevitt JT, Anslyn EV. J Am Chem Soc. 2003;125:1114–1115. doi: 10.1021/ja021230b. [DOI] [PubMed] [Google Scholar]
- 25.Umali AP, Ghanem E, Hopfer H, Hussain A, Kao Y, Zabanal LG, Wilkins BJ, Hobza C, Quach DK, Fredell M, Heymann H, Anslyn EV. Tetrahedron. 2015;71:3095–3099. [Google Scholar]
- 26.Zamora-Olivares D, Kaoud TS, Jose J, Ellington A, Dalby KN, Anslyn EV. Angew Chem. 2014;126:14288–14292. doi: 10.1002/anie.201408256. [DOI] [PubMed] [Google Scholar]
- 27.Bajaj A, Miranda OR, Kim IB, Phillips RL, Jerry DJ, Bunz UH, Rotello VM. Proc Natl Acad Sci. 2009;106:10912–10916. doi: 10.1073/pnas.0900975106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wright AT, Griffin MJ, Zhong Z, McCleskey SC, Anslyn EV, McDevitt JT. Angew Chem. 2005;117:6533–6536. doi: 10.1002/anie.200501137. [DOI] [PubMed] [Google Scholar]
- 29.Zhou H, Baldini L, Hong J, Wilson AJ, Hamilton AD. J Am Chem Soc. 2006;128:2421–2425. doi: 10.1021/ja056833c. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Wen F, Creran B, Jeong Y, Zhang X, Rotello VM. Small. 2012;8:3589–3592. doi: 10.1002/smll.201201549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pei H, Li J, Lv M, Wang J, Gao J, Lu J, Li Y, Huang Q, Hu J, Fan C. J Am Chem Soc. 2012;134:13843–13849. doi: 10.1021/ja305814u. [DOI] [PubMed] [Google Scholar]
- 32.Mao J, Lu Y, Chang N, Yang J, Zhang S, Liu Y. Biosens Bioelectron. 2016;86:56–61. doi: 10.1016/j.bios.2016.06.040. [DOI] [PubMed] [Google Scholar]
- 33.Lu Y, Kong H, Wen F, Zhang S, Zhang X. Chem Commun. 2013;49:81–83. doi: 10.1039/c2cc37293b. [DOI] [PubMed] [Google Scholar]
- 34.You CC, Miranda OR, Gider B, Ghosh PS, Kim IB, Erdogan B, Krovi SA, Bunz UHF, Rotello VM. Nat Nanotechnol. 2007;2:318–323. doi: 10.1038/nnano.2007.99. [DOI] [PubMed] [Google Scholar]
- 35.De M, Rana S, Akpinar H, Miranda OR, Arvizo RR, Bunz UHF, Rotello VM. Nat Chem. 2009;1:461–465. doi: 10.1038/nchem.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miranda OR, You CC, Phillips R, Kim IB, Ghosh PS, Bunz UHF, Rotello VM. J Am Chem Soc. 2007;129:9856–9857. doi: 10.1021/ja0737927. [DOI] [PubMed] [Google Scholar]
- 37.Takeuchi T, Goto D, Shinmori H. The Analyst. 2007;132:101–103. doi: 10.1039/b614532a. [DOI] [PubMed] [Google Scholar]
- 38.Hermanson GT. Bioconjugate techniques. 2nd. Academic Press; San Diego: 2008. [Google Scholar]
- 39.Pich A, Richtering W. In: Chemical Design of Responsive Microgels. Pich A, Richtering W, editors. Springer; Berlin Heidelberg: 2010. pp. 1–37. [Google Scholar]
- 40.Meunier F, Elaissari A, Pichot C. Polym Adv Technol. 1995;6:489–496. [Google Scholar]
- 41.Bartlett RL, Medow MR, Panitch A, Seal B. Biomacromolecules. 2012;13:1204–1211. doi: 10.1021/bm300173x. [DOI] [PubMed] [Google Scholar]
- 42.Kratz K, Hellweg T, Eimer W. Colloids Surf Physicochem Eng Asp. 2000;170:137–149. [Google Scholar]
- 43.Kondo A, Kamura H, Higashitani K. Appl Microbiol Biotechnol. 1994;41:99–105. doi: 10.1007/BF00166089. [DOI] [PubMed] [Google Scholar]
- 44.Yoshimatsu K, Koide H, Hoshino Y, Shea KJ. Nat Protoc. 2015;10:595–604. doi: 10.1038/nprot.2015.032. [DOI] [PubMed] [Google Scholar]
- 45.Brazel CS, Peppas NA. Macromolecules. 1995;28:8016–8020. [Google Scholar]
- 46.Mol NJ, Fischer MJE, editors. Surface Plasmon Resonance. Vol. 627 Humana Press; Totowa, NJ: 2010. [Google Scholar]
- 47.Wernersson E, Heyda J, Kubíčková A, Křížek T, Coufal P, Jungwirth P. J Phys Chem B. 2010;114:11934–11941. doi: 10.1021/jp1054342. [DOI] [PubMed] [Google Scholar]
- 48.Fromm JR, Hileman RE, Caldwell EEO, Weiler JM, Linhardt RJ. Arch Biochem Biophys. 1995;323:279–287. doi: 10.1006/abbi.1995.9963. [DOI] [PubMed] [Google Scholar]
- 49.Koetting MC, Peters JT, Steichen SD, Peppas NA. Mater Sci Eng R Rep. 2015;93:1–49. doi: 10.1016/j.mser.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hernández-Molina G, Sánchez-Hernández T. Semin Arthritis Rheum. 2013;42:627–639. doi: 10.1016/j.semarthrit.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 51.Ramos-Casals M, Stone JH, Moutsopoulos HM. Sjögren’s Syndrome: Diagnosis and Therapeutics. Springer Science & Business Media; 2011. [Google Scholar]
- 52.Azab B, Kedia S, Shah N, Vonfrolio S, Lu W, Naboush A, Mohammed F, Bloom SW. Int J Colorectal Dis. 2013;28:1629–1636. doi: 10.1007/s00384-013-1748-z. [DOI] [PubMed] [Google Scholar]
- 53.Chen J, Zhou Y, Xu Y, Zhu HY, Shi YQ. Tumor Biol. 2016;37:3905–3911. doi: 10.1007/s13277-015-3778-3. [DOI] [PubMed] [Google Scholar]
- 54.Duncan MR, Lee JM, Warchol MP. Int J Pharm. 1995;120:179–188. [Google Scholar]
- 55.Zhang L, Spencer HG. Desalination. 1993;90:137–146. [Google Scholar]
- 56.Lineweaver H, Murray CW. J Biol Chem. 1947;171:565–581. [PubMed] [Google Scholar]
- 57.Am Ende MT, Peppas NA. J Controlled Release. 1997;48:47–56. [Google Scholar]
- 58.Gao J, Hu Z. Langmuir. 2002;18:1360–1367. [Google Scholar]
- 59.Cölfen H, Völkel A, Eda S, Kobold U, Kaufann J, Puhlmann A, Göltner C, Wachernig H. Langmuir. 2002;18:7623–7628. [Google Scholar]
- 60.Bohren CF, Huffman DR. Absorption and Scattering of Light by Small Particles. John Wiley & Sons; 2008. [Google Scholar]
- 61.Kleinen J, Klee A, Richtering W. Langmuir. 2010;26:11258–11265. doi: 10.1021/la100579b. [DOI] [PubMed] [Google Scholar]
- 62.Vörös J. Biophys J. 2004;87:553–561. doi: 10.1529/biophysj.103.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stewart S, Ivy MA, Anslyn EV. Chem Soc Rev. 2014;43:70–84. doi: 10.1039/c3cs60183h. [DOI] [PubMed] [Google Scholar]
- 64.Yet MG, Chin CC, Wold F. J Biol Chem. 1988;263:111–117. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.