

Receptor-interacting kinase (RIPK) 1 critically controls signaling pathways triggered by the engagement of death receptors with their cognate ligands, such as tumor necrosis factor (TNF) receptor 1 and its ligand TNF. Because of its unique position at the crossroad of multiple signaling cascades, RIPK1 has an essential role in the regulation of inflammation and cell death through the activation of NF-κB-mediated gene transcription, and control of both apoptosis and necroptosis. Interestingly, RIPK1 has very distinct kinase-dependent and independent functions which can have opposite effects on the same pathway. For example, RIPK1 kinase activity promotes caspase 8-mediated apoptosis and RIPK3-dependent necroptosis, but its kinase-independent scaffolding function inhibits both forms of cell death and contributes to NF-κB activation.
The same level of complexity is found when studying the role of RIPK1 in the liver (1). RIPK1 kinase activity drives hepatocyte death and liver injury following concanavalin A injection (a model of T-cell-mediated hepatitis), but its scaffolding function protects from massive apoptosis in the same model. Similarly, depletion of RIPK1 in the liver sensitizes hepatocytes to lipopolysaccharide-induced TNF-mediated cell death. It appears that TNF-induced apoptosis in RIPK1 depleted cells is associated with degradation of cFLIP, cIAPs, and TRAF2, and activation of non-canonical NF-κB pathway. RIPK1 knockdown also exacerbates cell death in α-galactosylceramide-induced immune-mediated liver injury. It should be noted that RIPK1 has been investigated largely for its contribution to RIPK3-dependent necroptosis in various models of liver injury. However, RIPK3 is normally not expressed in hepatocytes, and even if its induction has been observed in chronic liver diseases, such as alcoholic and nonalcoholic steatohepatitis, the occurrence of RIPK3-dependent necroptosis in these diseases has not been unequivocally proven (1). Hence, the story of RIPK1 in liver biology and pathobiology is still evolving and poorly understood.
A recent publication in Cancer Cell by Schneider et al. (2) has shed new light on role of RIPK1 in liver tumorigenesis. The authors demonstrated that deletion of RIPK1 in liver parenchymal cells (LPC) in vivo sensitized hepatocytes to TNF-mediated apoptosis and acute hepatitis by promoting proteasomal degradation of TNF receptor-associated factor 2 (TRAF2) and subsequent caspase 8 activation. The anti-apoptotic effect of RIPK1 was due to its scaffolding functions as degradation of TRAF2 in response to TNF stimulation was not prevented by the use of RIPK1 kinase inhibitors or in cells expressing catalytically inactive RIPK1. Genetic deletion of RIPK1 or TRAF2 in LPC did not lead to a spontaneous phenotype, however, combined deletion of RIPK1 and TRAF2 in LPC caused excessive hepatocyte apoptosis, hepatic inflammation, compensatory proliferation and development of hepatocellular carcinoma (HCC, Figure 1). Increased apoptosis was attributed to simultaneous activation of caspase 8 and impaired activation of NF-κB canonical pathway, which resulted in decreased levels of the anti-apoptotic proteins cFLIP and cIAP1. These observations suggested that RIPK1 and TRAF2 have partially redundant functions in controlling apoptotic pathway and NF-κB activation. Unfortunately, the authors do not report whether the non-canonical NF-κB pathway was affected in these mice as this alternative pathway is activated in TRAF2−/− mice (3). Finally, analysis of RIPK1 and TRAF2 expression in 99 human HCC samples demonstrated that lower expression of these two proteins correlated with worse prognosis and survival.
Figure 1.
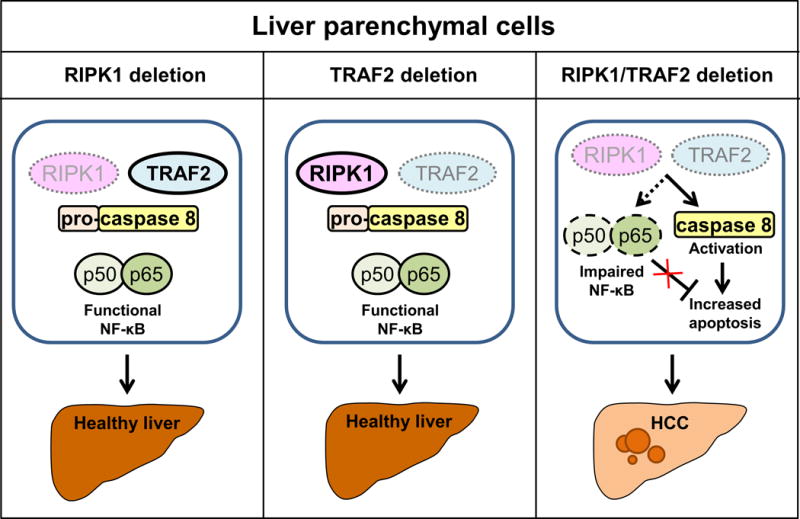
Schematic overview of receptor-interacting kinase 1 (RIPK1) and TNF receptor-associated factor 2 (TRAF2) role in spontaneous liver carcinogenesis. Simplified illustration demonstrates that single RIPK1 or TRAF2 deletion in liver parenchymal cells does not lead to liver carcinogenesis. In contrast, simultaneous loss of both RIPK1 and TRAF2 in liver parenchymal cells results in impaired canonical NF-κB signaling, caspase 8 activation, increased apoptosis and spontaneous development of hepatocellular carcinoma (HCC).
The study by Schneider et al. (2) is an important confirmation that RIPK1 controls LPC cell fate in terms of life and death and highlights the diverse roles of RIPK1 mediated by either its kinase activity or scaffolding functions, a dichotomy clearly demonstrated in genetically modified mice. Mice expressing kinase-dead RIPK1 (K45A or D138N point mutation) are viable and develop normally. In contrast, RIPK1−/− mice die shortly after birth due to massive cell death and inflammation in several tissues (3). This perinatal lethality can be rescued by combined deletion of caspase 8 and RIPK3, suggesting this phenotype is a consequence of aberrant activation of extrinsic apoptosis and necroptosis. In conditional knockout mice, RIPK1 deletion in the intestine or skin results in a phenotype characterized by increased tissue apoptosis and inflammation (3). In contrast, mice lacking RIPK1 specifically in the LPC do not display any abnormalities, suggesting RIPK1 is dispensable for liver homeostasis under steady-state conditions (2). The distinct phenotypes found in conditional knockout mice underscore that RIPK1 functions in tissue homeostasis are cell type/tissue-specific. Future scientific inquiries will be needed to provide mechanistic explanations for these differences.
According to the current study, RIPK1 expressed in LPC serves a prosurvival function and, via regulation of cell death, controls liver cancer development in mice. In human HCC samples, low or absent expression of both RIPK1 and TRAF2 was associated with worse prognosis, suggesting that RIPK1 have anti-carcinogenic role in HCC. The mechanisms responsible for dysregulated RIPK1 and TRAF2 expression in human HCC are currently unknown. Of note, low expression of RIPK1 was not necessarily associated with low TRAF2 expression and vice versa, but approximately 40% of HCC cases studied displayed simultaneous downregulation of these two proteins. In contrast, several other studies in other malignancies, such as melanoma, breast and pancreatic cancer, have described rather a tumorigenic role of RIPK1, and thus RIPK1 has been suggested as a potential therapeutic target (2). Therefore, further studies are warranted to better understand the role of RIPK1 in specific cancers and its potential for therapeutic targeting.
The present study also provides experimental evidence that clearly demonstrates that excessive hepatocyte cell death can drive liver tumorigenesis. Cell death and tumorigenesis are intricately linked; however, the relationship is often attributed to malignant cell resistance to apoptotic stimuli generated by oncogenic signaling networks. In contrast, cell death may also drive malignant transformation by several possible mechanisms including, but not limited to, promoting compensatory proliferation or activating the immune system. Among various cancers, HCC has accumulated the strongest evidence for cell death-driven proliferation and tumorigenesis. This has been well established in preclinical models in the context of dysregulated expression of BCL-2 family proteins, which control the mitochondrial pathway of apoptosis (4). For example, liver-specific deletion of anti-apoptotic protein Mcl-1 leads to spontaneous hepatocyte apoptosis, compensatory proliferation and eventually HCC, even in the absence of overt inflammation. On the other hand, inhibition of apoptosis by deletion of proapoptotic mediator PUMA can actually prevent development of carcinogen-induced HCC. The discussed study by Schneider et al. is thus an important confirmation of the relevance of cell death in liver carcinogenesis. In addition, this study highlights that perturbation of extrinsic apoptotic pathway may lead to increased apoptosis, proliferation and HCC similarly to previously reported dysregulation of the mitochondrial pathway.
Recent studies describing involvement of apoptosis in liver cancer have raised some further questions. For example, is it possible that the detection of apoptotic cells in liver tissue is just the tip of the iceberg of a large population of “stressed” cells in which proapoptotic signaling is activated? Apoptotic cells would thus represent just a portion of cells in which proapoptotic signaling was able to execute the cellular demise. It appears that not all cells that activate proapoptotic signaling actually undergo cell death per se (5, 6). Rather, in these cells proapoptotic sub-lethal signaling can promote inflammation and genomic instability that can, in turn, lead to malignant transformation. In particular, sub-lethal caspase signaling can activate caspase-activated DNase (CAD). CAD activation results in DNA breaks and DNA damage repair response, introducing potentially oncogenic genomic changes and cellular transformation (6). The role of sub-lethal apoptotic signaling has been recently described in therapy-related leukemia, however, very little is known about liver cancer (6). It might be possible that, besides apoptotic cells, live cells with sub-lethal caspase activation may contribute to liver carcinogenesis. In this scenario, apoptotic cells may represent rather a biomarker of perturbation in tissue homeostasis, a concept we have recently proposed for nonalcoholic steatohepatitis (7).
In conclusion, the novel findings by Schneider and colleagues strengthen the link between chronic apoptosis and carcinogenesis in the liver and advance our knowledge about the role of RIPK1 in the liver as an important suppressor of hepatocyte cell death, thereby bringing us one step closer to understanding the complex nature of RIPK1.
Acknowledgments
Grant support: This work was supported by NIH grant DK41876, DK59427, DK63947 (to GJG), the Chris M. Carlos and Catharine N. Jockisch Carlos Foundation for PSC, the PSC Partners Seeking a Cure Foundation (to MEG), Edward C. Kendall Research Fellowship Award (to PH) and the Mayo Foundation.
Abbreviations
- CAD
caspase-activated DNase
- HCC
hepatocellular carcinoma
- LPC
liver parenchymal cells
- NF-κB
nuclear factor-κB
- RIPK
receptor-interacting kinase
- TNF
tumor necrosis factor
- TRAF2
TNF receptor-associated factor 2
Footnotes
Disclosures: The authors have no financial or personal disclosures relevant to this manuscript.
References
- 1.Dara L, Liu ZX, Kaplowitz N. Questions and controversies: the role of necroptosis in liver disease. Cell Death Discov. 2016;2:16089. doi: 10.1038/cddiscovery.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schneider AT, Gautheron J, Feoktistova M, Roderburg C, Loosen SH, Roy S, Benz F, et al. RIPK1 Suppresses a TRAF2-Dependent Pathway to Liver Cancer. Cancer cell. 2017;31:94–109. doi: 10.1016/j.ccell.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Weinlich R, Green DR. The two faces of receptor interacting protein kinase-1. Mol Cell. 2014;56:469–480. doi: 10.1016/j.molcel.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Labi V, Erlacher M. How cell death shapes cancer. Cell Death Dis. 2015;6:e1675. doi: 10.1038/cddis.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirsova P, Ibrabim SH, Gores GJ, Malhi H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res. 2016;57:1758–1770. doi: 10.1194/jlr.R066357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larsen BD, Sorensen CS. The caspase-activated DNase: apoptosis and beyond. FEBS J. 2017;284:1160–1170. doi: 10.1111/febs.13970. [DOI] [PubMed] [Google Scholar]
- 7.Hirsova P, Ibrahim SH, Malhi H, Gores GJ. Hepatocyte Lethal and Nonlethal Lipotoxic Injury. In: Ding WX, Yin XM, editors. Cellular Injury in Liver Diseases. 1st. Springer International Publishing; 2017. [Google Scholar]