

ABSTRACT
Francisella tularensis is a highly infectious Gram-negative intracellular pathogen that causes tularemia. Because of its potential as a bioterrorism agent, there is a need for new therapeutic agents. We therefore developed a whole-animal Caenorhabditis elegans-F. tularensis pathosystem for high-throughput screening to identify and characterize potential therapeutic compounds. We found that the C. elegans p38 mitogen-activate protein (MAP) kinase cascade is involved in the immune response to F. tularensis, and we developed a robust F. tularensis-mediated C. elegans killing assay with a Z′ factor consistently of >0.5, which was then utilized to screen a library of FDA-approved compounds that included 1,760 small molecules. In addition to clinically used antibiotics, five FDA-approved drugs were also identified as potential hits, including the anti-inflammatory drug diflunisal that showed anti-F. tularensis activity in vitro. Moreover, the nonsteroidal anti-inflammatory drug (NSAID) diflunisal, at 4× MIC, blocked the replication of an F. tularensis live vaccine strain (LVS) in primary human macrophages and nonphagocytic cells. Diflunisal was nontoxic to human erythrocytes and HepG2 human liver cells at concentrations of ≥32 μg/ml. Finally, diflunisal exhibited synergetic activity with the antibiotic ciprofloxacin in both a checkerboard assay and a macrophage infection assay. In conclusion, the liquid C. elegans-F. tularensis LVS assay described here allows screening for anti-F. tularensis compounds and suggests that diflunisal could potentially be repurposed for the management of tularemia.
KEYWORDS: Caenorhabditis elegans, high-throughput screen, tularemia, Francisella, antibiotic, diflunisal, drug repurposing
INTRODUCTION
Francisella tularensis, the causative agent of tularemia, is a Gram-negative facultative intracellular pathogen (1). Multiple Francisella species and subspecies have been identified, among which the Francisella tularensis subsp. tularensis and F. tularensis subsp. holarctica biovars are most commonly associated with human disease (1–3) and are associated with significant mortality (4, 5). F. tularensis infects a variety of mammalian hosts, including squirrels, rabbits, and voles (1). In areas where F. tularensis is endemic, outbreaks in animal populations are often followed by a similar outbreak among humans (1, 6). The mechanism of disease transmission is unknown, but a number of arthropod vectors have been implicated, including ticks, biting flies, and mosquitoes (5, 7). Due to ease of dissemination and a high level of virulence, F. tularensis is classified as a category A bioterrorism agent by the Centers for Disease Control and Prevention (1–3, 5). An attenuated live vaccine strain (LVS) has been developed from F. tularensis subsp. holarctica (type B) (4, 5, 12) and has been used to study the organism and vaccinate individuals at risk (1, 13–16).
F. tularensis can be treated with a number of known antibiotics, including the aminoglycosides streptomycin and gentamicin (1, 3, 6, 13). A high frequency of relapse is associated after use of tetracyclines (5, 7, 19, 20) and β-lactams; macrolides, lincosamides, and co-trimoxazole are not reliable options (21). Oral ciprofloxacin is currently used for uncomplicated tularemia because it is associated with a low therapeutic failure rate (6, 23) and is suitable for use in children and pregnant women (24).
The potential use of F. tularensis as a biological weapon has led to a renewed interest in elucidating the virulence traits of the bacterium, as well as in the discovery of new antibiotics and anti-infective agents for the treatment of tularemia. The model nematode Caenorhabditis elegans has been used to study many human bacterial pathogens, including Salmonella enterica (25, 26), S. enterica serovar Typhimurium (27, 28), Pseudomonas aeruginosa (29–31), Staphylococcus aureus (32), Enterococcus faecium (33), and Enterococcus faecalis (34). More recently, C. elegans has been used for the evaluation of libraries of low-molecular-weight compounds in order to identify those that have antimicrobial or immunomodulatory efficacy while performing a toxicity assessment in the same assay (9, 10).
In this paper, we describe the development of a suitable C. elegans pathosystem for the study of the F. tularensis live vaccine strain (LVS) in liquid medium. Furthermore, we demonstrate that the C. elegans-F. tularensis LVS host-pathogen system can be utilized in a high-throughput, whole-animal assay to screen for compounds that can extend survival of the host. Using the C. elegans-F. tularensis LVS liquid assay, we screened a library of FDA-approved therapeutics consisting of 1,760 small molecules. Interestingly, we found that the nonsteroidal anti-inflammatory drug (NSAID) diflunisal, a salicylic derivative, is effective against F. tularensis LVS and that the action of diflunisal is synergistic with that of ciprofloxacin. Diflunisal displayed no significant toxicity to mammalian cells and human erythrocytes. The findings from this study outline a new screening model for the identification and study of anti-F. tularensis compounds that can be performed in standard research laboratories without high-level biological containment facilities. This work suggests that the salicylic derivative drug diflunisal should be studied further as a potential repurposed drug for treating F. tularensis infections.
RESULTS AND DISCUSSION
The p38 MAP kinase cascade is involved in the C. elegans immune response to F. tularensis.
We extended the use of the C. elegans model of microbial pathogenesis to study the pathogenesis of F. tularensis. First, we found that wild-type C. elegans animals (strain N2) feeding on lawns of F. tularensis LVS had longevity similar to that of animals feeding on lawns of Escherichia coli strain OP50 (the standard C. elegans laboratory food source) (Fig. 1). In contrast, a C. elegans sek-1 immunocompromised mutant, in which the p38 mitogen-activated protein kinase (MAPK) pathway is abrogated (36), was susceptible to F. tularensis LVS-mediated killing (Fig. 1) (P < 0.001). This finding suggests that F. tularensis LVS is a weak C. elegans pathogen and that survival of the nematodes in the presence of F. tularensis LVS depends on the p38 MAPK immune response pathway.
FIG 1.
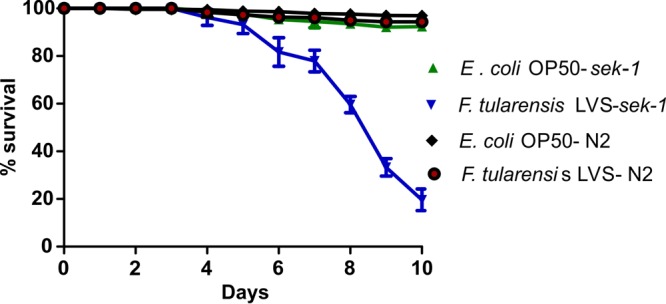
The MAP kinase pathway is essential for survival of C. elegans challenged with F. tularensis. Percent survival of wild-type N2 or mutant sek-1 (kinase involved in the p38 MAP kinase cascade) C. elegans worms grown on lawns of E. coli or F. tularensis LVS was determined. Data represent the means ± standard errors of the means (n = 3).
A high-throughput screening (HTS) assay.
Based on the agar-based killing assay shown in Fig. 1, we developed a high-throughput killing assay that can be performed in liquid medium (Fig. 2a). As previously described, C. elegans pathogen-mediated liquid killing assays can be used for automated large-scale screening for low-molecular-weight compounds that block the ability of a pathogen to kill the nematodes (8–10). We determined the optimal F. tularensis LVS inoculum, time of incubation, temperature, and medium conditions empirically. For example, for worms exposed to an initial dose of F. tularensis LVS at an optical density at 600 nm (OD600) of 0.06, the survival rate after 96 h in 10% Trypticase soy broth (TSB) with 0.1% l-cysteine HCl was 5% in contrast to 75% survival after exposure to E. coli OP50 (Fig. 2b). At the end of the assay, the wells were washed to remove bacteria, and the worms were stained with Sytox orange, which preferentially stains the dead worms (Fig. 2c).
FIG 2.

Development of a high-throughput assay for anti-Francisella compounds. (a) Flowchart representing the work flow of the C. elegans-F. tularensis high-throughput screening assay. (b) Optimization of F. tularensis LVS inoculum for the liquid killing assay. (c) C. elegans-F. tularensis liquid killing assay: Sytox-stained images of C. elegans with or without ciprofloxacin.
Robustness and quality assessment of the coinoculation assay.
The Z′ factor, which is widely used in high-throughput screens to determine if an assay is sufficiently robust, was used to evaluate the reproducibility and robustness of the F. tularensis-C. elegans killing assay (11). Dimethyl sulfoxide (DMSO) was used as a negative control (background), and ciprofloxacin and gentamicin were used as the positive controls. The Z′ factor values from three independent experiments were calculated with a range of ciprofloxacin (0.5, 0.75, 1, 1.25, 1.5, and 1.75 μg/ml) and gentamicin (2, 4, 8, 16, 32, and 64 μg/ml) concentrations. Ciprofloxacin or gentamicin at 0.1 μg/ml or 2.0 μg/ml, respectively, enabled the survival of 93 to 100% of the worms, and in three independent experiments, the Z′ factor values using these concentrations ranged from 0.59 to 0.64, with an average value of 0.63.
Identification of antibacterial compounds.
The C. elegans-F. tularensis LVS high-throughput liquid killing assay pathosystem was implemented in a screen of known bioactive compounds to identify compounds that prolonged nematode survival. A total of 1,760 known bioactive and FDA-approved compounds, including many antibiotics, were screened. A total of 31 hits were identified in the primary screen (see Table S1 in the supplemental material). Among these hits, a total of 13 compounds that prolonged survival of ≥60% of the nematodes in the assay were chosen for further study (Table 1). Four of these compounds were quinolones, and two were tetracyclines, two families of compounds that have been used in the clinical treatment of tularemia (Table 1), thereby providing proof of principle that the assay could identify bona fide anti-F. tularensis therapeutics.
TABLE 1.
Hit compounds and their corresponding percentage of survival in the C. elegans-F. tularensis infection assaya
| Compound group and name (2 μg/ml) | Reference(s) | % survival |
|---|---|---|
| Quinolone antibiotics | ||
| Oxolinic acid | 48 | 87 |
| Ofloxacin | 35 | 87 |
| Flumequine | 35 | 80 |
| Lomefloxacin hydrochloride | 35 | 80 |
| Tetracycline antibiotics | ||
| Tetracycline hydrochloride | 49 | 67 |
| Meclocycline sulfosalicylate | 49 | 67 |
| Other antibiotics | ||
| Benzalkonium chloride | 50 | 60 |
| Other compounds | ||
| Oxibendazole | 51 | 67 |
| Paroxetine hydrochloride | 37 | 73 |
| Mefloquine | 52 | 67 |
| Ethidium bromide | 53, 54 | 60 |
| Diflunisal | 55 | 60 |
| Acetaminophen | 56 | 60 |
Survival with the compound was compared to that with the positive control, ciprofloxacin.
In addition to the quinolone and tetracycline antibiotics, the screen identified seven additional bioactive drugs that prolonged the survival of worms by >60%. Three of these hit compounds, benzalkonium chloride, paroxetine hydrochloride, and oxibendazole, were previously shown to have antibacterial activity (37–39). These three known antimicrobial compounds, as well as two additional hits, ethidium bromide (because it is associated with broad toxicity) and acetaminophen (because it was a false hit that was not confirmed in secondary screens), were not investigated further. Mefloquine and diflunisal showed MICs of 16 μg/ml and 8 μg/ml, respectively, suggesting that they rescued C. elegans from F. tularensis killing by blocking bacterial replication. Interestingly, standard broth MIC assays of diflunisal and mefloquine did not indicate any antimicrobial activity against the Gram-positive species E. faecium or the Gram-negative species Klebsiella pneumoniae, Acinetobacter baumannii, P. aeruginosa, or Enterobacter aerogenes (MICs of >64 μg/ml). Because diflunisal exhibited a lower MIC (8 μg/ml) than mefloquine (16 μg/ml) against F. tularensis LVS, we focused our further studies on the evaluation of diflunisal for its antibacterial activity and synergy with antibiotics. Moreover, the MIC (8 μg/ml) of diflunisal is a lower dose than the plasma concentration of diflunisal (41 μg/ml) required when it is used at the recommended dosage as an NSAID (40).
Enhanced efficacy of diflunisal in combination with ciprofloxacin.
Although quinolones and especially ciprofloxacin have been successfully used in clinical treatment of tularemia, efficacy against intracellular bacteria has been a concern (19, 20). We evaluated the hypothesis that diflunisal might be able to function synergistically with ciprofloxacin. A broth microplate assay was used to test for growth inhibition of the F. tularensis LVS in the presence of diflunisal, adjusted to a final concentration between 0.25 and 16 μg/ml, in combination with ciprofloxacin, adjusted to final concentrations between 0.03 and 2 μg/ml. The MICs at which the test compounds inhibited bacterial growth both alone and in combinations were determined to calculate the fractional inhibitory concentration (FIC) index according to the equation shown in Materials and Methods. The FIC index was 0.5 for the diflunisal-ciprofloxacin combination (Fig. 3a). Synergy was also observed in a time-kill assay where the combination of diflunisal and ciprofloxacin at low concentrations had the same effect as a higher dose of ciprofloxacin alone (0.25 μg/ml) (Fig. 3b). The combination of diflunisal and ciprofloxacin allows the treatment of F. tularensis infections at lower concentrations of ciprofloxacin, reducing the probability of the development of antibiotic resistance (41).
FIG 3.

Checkerboard and time-to-kill assay for diflunisal with ciprofloxacin. (a) Diflunisal was adjusted to a final concentration between 0.25 and 16 μg/ml, in combination with ciprofloxacin, which was adjusted to a final concentration between 0.03 and 2 μg/ml. The FIC index was 0.5 for the diflunisal-ciprofloxacin combination. The scale represents the optical density of bacterial growth of F. tularensis LVS. (b) Time-to-kill curve shows that the efficacy of diflunisal (2 μg/ml) is enhanced in combination with ciprofloxacin (0.06 μg/ml) compared to that of diflunisal alone (8 μg/ml) or ciprofloxacin alone (0.25 μg/ml). Data are means ± standard errors of the means (n = 3).
Evaluation of diflunisal toxicity.
Although diflunisal is an FDA-approved drug and has been in clinical use for several decades, it is important to test the toxicity of diflunisal at concentrations that are effective against F. tularensis LVS. Diflunisal did not cause any detectable hemolysis of human erythrocytes at concentrations of up to 64 μg/ml, suggesting that it would be nontoxic at twice the concentration that inhibits the growth of F. tularensis LVS in phagocytic and nonphagocytic assays (Fig. 4a). Triton X-100 was used as a positive control. Consistent with the hemolysis assay, diflunisal did not exhibit cytotoxicity toward HepG2 cells, a cell line derived from human liver carcinoma cells, at concentrations of up to 32 μg/ml (Fig. 4b).
FIG 4.

Hemolysis and cytotoxicity testing for diflunisal. (a) Human erythrocytes were treated with serial dilutions of Triton X-100 (0.00 to 1%) or diflunisal (0.06 to 64 μg/ml). (b) The viability of HepG2 cells was measured after treatment with serially diluted concentrations (1 to 64 μg/ml) of diflunisal. Data represent the means ± standard errors of the means (n = 3).
Diflunisal limits intracellular replication of F. tularensis in phagocytic cells and nonphagocytic cells and shows synergy with ciprofloxacin in macrophages.
Because F. tularensis LVS replicates in macrophages (13, 15), we evaluated the activity of diflunisal to limit intracellular growth of the bacterium. In this series of experiments, we used RAW 264.7 macrophages infected with F. tularensis LVS and quantified the viable intracellular bacteria using a gentamicin protection assay (13, 17, 18). The diflunisal treatment resulted in a 100-fold reduction of the intracellular bacterial load within the macrophage cells (Fig. 5a) (P < 0.001). Consistent with the macrophage assay, diflunisal inhibited the growth of F. tularensis LVS in nonphagocytic 293T/17 human kidney epithelial cells by 10,000-fold (Fig. 5b) (P < 0.001). These data show that diflunisal displays antimicrobial activity against intracellular F. tularensis LVS. In addition, we observed that a low concentration of ciprofloxacin (0.06 μg/ml) in combination with diflunisal (2 μg/ml) was able to reduce the intracellular bacterial load in macrophages significantly more than 0.25 μg/ml ciprofloxacin alone (Fig. 6) (P < 0.001). We also tested the diflunisal activity against the intracellular pathogen Salmonella Typhimurium. Interestingly, diflunisal did not display significant antimicrobial activity against S. Typhimurium (>32 μg/ml) and did not inhibit the intracellular replication of S. Typhimurium inside macrophage cells (Fig. S1). These data suggest that the antimicrobial activity of diflunisal may be specific for F. tularensis.
FIG 5.

Diflunisal inhibits intracellular growth of F. tularensis LVS in macrophages and 293T/17 cells. Macrophages (a) and 293T/17 (b) cells were infected with F. tularensis LVS bacteria for 2 h, treated with gentamicin to kill extracellular bacteria, and then cocultured with 4× MIC of diflunisal for 22 h. ***, P < 0.001, comparing results with diflunisal to those with the DMSO control at the 24-h time point using two-way ANOVA with a Bonferroni posttest correction.
FIG 6.
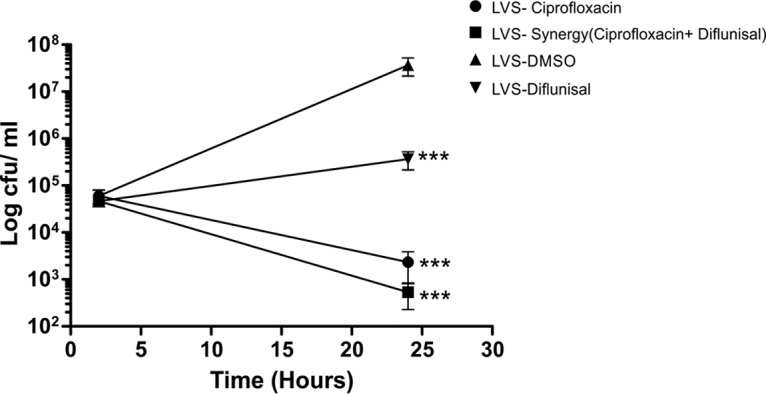
Diflunisal and ciprofloxacin show synergy in macrophages. Macrophages were infected with F. tularensis LVS bacteria for 2 h, followed by gentamicin treatment to kill extracellular bacteria, and cocultured with ciprofloxacin (0.25 μg/ml), diflunisal (8 μg/ml), or a combination of diflunisal (2 μg/ml) and ciprofloxacin (0.06 μg/ml) for 22 h. Data shown are CFU counts of intracellular F. tularensis LVS in macrophages. ***, P < 0.001, comparing results with diflunisal to those with the DMSO control at the 24-h time point using two-way ANOVA with a Bonferroni posttest correction.
Conclusions.
In conclusion, we developed a whole-animal screening platform using the model host C. elegans and screened 1,760 FDA-approved compounds that blocked the ability of F. tularensis to kill the nematodes. In addition to known antibiotics, our screen identified FDA-approved drugs that were confirmed in secondary screens. We further investigated diflunisal, one of the hits, and found that it blocks intracellular replication of F. tularensis, exhibits synergy with ciprofloxacin, and has limited toxicity against mammalian cells. Diflunisal is a good example of the possibility of repurposing a drug already used in the clinic for other therapies. Importantly, diflunisal and ciprofloxacin appear to work synergistically against F. tularensis LVS in vitro as well as in clearing an intracellular infection in macrophages. Thus, the combination therapy of ciprofloxacin and diflunisal to treat tularemia may be an attractive option and may help to reduce the chance of ciprofloxacin resistance in tularemia patients.
MATERIALS AND METHODS
Bacterial, worm strains, media, and preparation of inoculum.
The live vaccine strain (LVS) of F. tularensis was obtained from the Centers for Disease Control and Prevention (Atlanta, GA) and was used throughout this work. Bacterial cultures of F. tularensis LVS were maintained on cysteine heart agar plates with 2% hemoglobin (CHA-H) (Difco, NJ, USA). Prior to each assay, F. tularensis LVS was grown on CHA-H plates at 37°C for 72 h, and then a single colony was transferred to 50 ml of brain heart infusion (BHI) medium (Difco, NJ, USA) containing 200 μg/ml ampicillin and 0.5 μg/ml amphotericin B. The liquid culture was incubated for 48 h at 30°C with shaking and diluted with fresh BHI medium (containing the same antibiotics as above) to an optical density of 0.6 at 600 nm, yielding approximately 2 × 106 CFU/ml. This was the inoculum used for the assay and confirmed by plating on CHA-H plates at 30°C and by counting CFU 72 to 96 h later. Screening was performed using BHI medium containing 200 μg/ml ampicillin and 0.5 μg/ml amphotericin B.
A C. elegans glp-4; sek-1 double mutant strain was used throughout this work. The glp-4(bn2) mutation renders the strain unable to produce progeny at 25°C (42), and the sek-1(km4) mutation leads to enhanced sensitivity to various pathogens (36), thereby reducing the length of the assay period. Nematodes were propagated on nematode growth medium (NGM) (8). Escherichia coli strains HB101 and OP50 were used as the food source for C. elegans and are the strains normally used for large-scale propagation of worms (9). During the assay, M9 buffer was used to wash the worms, as previously described (9).
Statistical analysis of high-throughput assay.
The Z′ factor was used to evaluate the overall quality of our assay using positive- and negative-control data based on the following equation: Z′ = 1 − [(3σc+ + 3σc−)/(∣μc+ − μc−∣)], where μc+ and μc− specify the averages of test sample signals for both the positive and negative controls, respectively, and σc+ and σc− refer to the standard deviations for the positive and negative controls, respectively (11). The negative control (background) was 1% dimethyl sulfoxide (DMSO), and the positive control was ciprofloxacin (0.1 μg/ml) or gentamicin (1.0 μg/ml). The MICs of ciprofloxacin and gentamicin for LVS have been previously determined to be 0.004 to 0.06 μg/ml and 0.03 to 0.5 μg/ml, respectively (43, 44). One column (eight wells) in each 96-well plate was used for either ciprofloxacin or gentamicin (positive control), and one column (eight wells) was used for DMSO (negative control). The ratio of the Sytox-stained worm area to the bright-field worm area and the resultant percent survival data were calculated using CellProfiler software for each well of the assay plates (8). To identify hits, the Z score was calculated from the ratio data. The Z score is defined as the number of standard deviations an observation is separated from the mean: Z = (x − μ)/σ, where x is the raw sample score, μ is the mean of the population, and σ is the standard deviation of the population. Wells with Z scores of >2σ were considered hits. Screening wells with Z scores above 0.6 were considered valid hits.
C. elegans-LVS antimicrobial screen assays.
Prior to the assay, C. elegans glp-4(bn2); sek-1(km4) worms were propagated at 25°C for 52 h in order for them to be at larval stage 4 (L4) on the day of the assay. The HTS assay was performed in 96-well half-area plates (Corning, NY, USA) by dispensing 70 μl of the screen medium, using a MultiDrop Combi plate filler (Thermo Scientific, MA, USA) equipped with a standard dispensing cassette (Thermo Scientific, MA, USA). In the 96-well assay plates, 0.1 μl of each of the compounds to be tested at 2 mg/ml in DMSO was pin transferred to each well from 384-well compound stock plates (containing the test compounds) into individual wells (containing 70 μl of screening medium) to yield a final concentration of 20 μg/ml in the assay. As noted above, positive-control assay wells contained either 0.1 μg/ml ciprofloxacin or 2.0 μg/ml gentamicin, and negative-control wells contained 1% DMSO. Worms were washed three times with M9 buffer, and a commercial robot (COPAS Biosort; Union Biometrica, MA, USA) was used to automatically dispense 15 ± 1 worms into each well. Ten microliters of an F. tularensis inoculum (in BHI medium at ∼2 × 106 CFU/ml) was then manually pipetted into each assay well. Plates were sealed with Breathe-Easy membranes (Diversified Biotech, MA, USA) and incubated at 25°C for 96 h. The assay wells were washed to remove bacteria. Finally, 60 μl of 0.9 μM Sytox orange (Fisher Scientific, MA, USA) in M9 buffer was added by using a MultiDrop Combi dispenser into each well to a final Sytox concentration of 0.7 μM, and the plates were incubated overnight. Worm plates were imaged using a IX-Micro automated microscope (Molecular Devices, CA, USA) with bright-field and Cy3 channels.
Antimicrobial susceptibility testing.
Compounds (5 mg/ml stock solutions in DMSO) were tested by broth microdilution assays in triplicate, according to a procedure adapted from established protocols (45). Broth microdilution was performed in triplicate in 96-well plates using Trypticase soy broth (TSB) (BD Biosciences, NJ, USA) containing 0.1% l-cysteine hydrochloride monohydrate (Fisher Scientific, MA, USA). The assay volume was 100 μl, and 2-fold serial dilutions were carried out to dilute compounds in the concentration range of 0.0625 to 64 μg/ml, with a bacterial concentration (measured by the OD600) of 0.03. The bacterial OD after overnight incubation at 37°C was measured to determine antimicrobial activity. The minimum bactericidal concentration (MBC) was determined by plating 10 μl of culture volume from the MIC assay onto Mueller-Hinton agar, and colony formation was examined after 48 h at 37°C. The lowest concentration at which colonies were not observed was regarded as the MBC.
Checkerboard assay.
The antimicrobial activity of a combination of diflunisal with ciprofloxacin was determined using a checkerboard assay (4). Briefly, compounds were arrayed in serial concentrations, vertically for one compound and horizontally for the other compound, in the same 96-well microplate. The addition of bacteria and measurement of growth were carried out as described in the previous section for measurement of MIC. The fractional inhibitory concentration (FIC) index for two compounds A and B is defined using the following equation: FIC = (A/MICA) + (B/MICB), where MICA and MICB are the MICs of compounds A and B, respectively, and (A) is the lowest concentration of compound A in combination with compound B that inhibits bacterial growth and vice versa for (B). An FIC of <0.5 indicates synergism between the compounds being tested.
Time-to-kill study.
Time-to-kill assays were carried out in 10-ml tubes to determine the bactericidal or static nature of the compound. An overnight culture of F. tularensis LVS was diluted in fresh TSB containing 0.1% l-cysteine hydrochloride monohydrate to a density of 108 cells/ml. The test compound, at 4× MIC (32 μg/ml), was added to the tube and incubated at 37°C with shaking. At periodic intervals, aliquots sampled from each tube were diluted serially and plated on cysteine heart agar plates with 2% hemoglobin. The plates were incubated at 37°C in 5% CO2 for 48 h to observe the viable counts.
Human blood hemolysis assay.
A hemolysis assay of human erythrocytes (Rockland Immunochemicals, PA, USA) was adapted from Rosch et al. (46) and Rajamuthiah et al. (47). The assay was carried out in 96-well plates with 50 μl of 2% human erythrocytes suspended in phosphate-buffered saline (PBS) added to 50 μl of compounds serially diluted in PBS and incubated at 37°C for 1 h. Plates were then centrifuged at 500 × g for 5 min, and 50 μl of the supernatant from each well of the assay plate was transferred to a fresh 96-well plate. Erythrocyte lysis was monitored by both visual observation and measuring the absorbance at 540 nm. The hemolysis experiments were performed in triplicate.
Cytotoxicity assay.
A cytotoxicity assay protocol was adopted from Kwon et al. (2). HepG2 (ATCC HB 8065) cells were cultured in Dulbecco's modified Eagle medium (DMEM; Life Technologies, CA, USA) containing 10% fetal bovine serum (FBS), 25 mM d-glucose, 2 mM l-glutamine, 1 mM sodium pyruvate, and 1% penicillin-streptomycin and maintained at 37°C in 5% CO2. For the toxicity test, HepG2 cells were cultured at 70 to 80% confluence in 96-well plates in a volume of 100 μl/well culture medium. Serially diluted diflunisal was incubated with the cells at 37°C in 5% CO2 for 24 h. Ten microliters of 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium (WST-1) solution (Roche, Mannheim, Germany) was added per well for the last 4 h of the 24-h incubation period. WST-1 reduction was detected at 490 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA). Percent fluorescence was calculated by comparing signals of treated cells to those of control untreated cells. The assay was done in triplicate.
F. tularensis LVS growth inhibition assay in macrophages and 293T/17 cells.
RAW 264.7 macrophages were grown in Dulbecco's modified Eagle medium (DMEM; Gibco, NY, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, NY, USA) and 1% penicillin-streptomycin (Gibco) and maintained at 37°C in 5% CO2. Cells of the nonphagocytic kidney epithelial cell line 293T/17 (ATCC CRL-11268) were cultured in DMEM supplemented with 10% fetal bovine serum (Gibco, NY, USA). To assess intracellular growth, gentamicin protection assays were performed as described previously (13). Macrophages and 293T/17 cells were seeded in 96-well culture dishes (BD Biosciences, CA, USA) at a density of 5 × 104 cells/well. Overnight-grown F. tularensis LVS bacteria were adjusted to an OD600 of 0.3 and diluted to achieve a multiplicity of infection (MOI) of ∼500. After the infection period, cells were incubated with gentamicin (100 μg/ml) for 30 min to kill extracellular bacteria and then washed twice with warm Hanks balanced salt solution (Cellgro, VA, USA). Fresh culture medium with or without diflunisal was then added, and cells were incubated for another 22 h at 37°C with 5% CO2. At the indicated time points, cells were lysed with 0.02% sodium dodecyl sulfate, and the numbers of viable CFU were determined.
Supplementary Material
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00310-17.
REFERENCES
- 1.Ellis J, Oyston PCF, Green M, Titball RW. 2002. Tularemia. Clin Microbiol Rev 15:631–646. doi: 10.1128/CMR.15.4.631-646.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwon B, Kumar P, Lee H-K, Zeng L, Walsh K, Fu Q, Barakat A, Querfurth HW. 2014. Aberrant cell cycle reentry in human and experimental inclusion body myositis and polymyositis. Hum Mol Genet 23:3681–3694. doi: 10.1093/hmg/ddu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oyston PCF. 2008. Francisella tularensis: unravelling the secrets of an intracellular pathogen. J Med Microbiol 57:921–930. doi: 10.1099/jmm.0.2008/000653-0. [DOI] [PubMed] [Google Scholar]
- 4.Draper LA, Cotter PD, Hill C, Ross RP. 2013. The two peptide lantibiotic lacticin 3147 acts synergistically with polymyxin to inhibit Gram-negative bacteria. BMC Microbiol 13:212. doi: 10.1186/1471-2180-13-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Layton M, Lillibridge SR, McDade JE, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Tonat K, Working Group on Civilian Biodefense. 2001. Tularemia as a biological weapon: medical and public health management. JAMA 285:2763–2773. doi: 10.1001/jama.285.21.2763. [DOI] [PubMed] [Google Scholar]
- 6.Pérez-Castrillón JL, Bachiller-Luque P, Martín-Luquero M, Mena-Martín FJ, Herreros V. 2001. Tularemia epidemic in northwestern Spain: clinical description and therapeutic response. Clin Infect Dis 33:573–576. doi: 10.1086/322601. [DOI] [PubMed] [Google Scholar]
- 7.Hepburn MJ, Simpson AJ. 2008. Tularemia: current diagnosis and treatment options. Expert Rev Anti Infect Ther 6:231–240. doi: 10.1586/14787210.6.2.231. [DOI] [PubMed] [Google Scholar]
- 8.Moy TI, Conery AL, Larkins-Ford J, Wu G, Mazitschek R, Casadei G, Lewis K, Carpenter AE, Ausubel FM. 2009. High-throughput screen for novel antimicrobials using a whole animal infection model. ACS Chem Biol 4:527–533. doi: 10.1021/cb900084v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajamuthiah R, Fuchs BB, Jayamani E, Kim Y, Larkins-Ford J, Conery A, Ausubel FM, Mylonakis E. 2014. Whole-animal automated platform for drug discovery against multi-drug resistant Staphylococcus aureus. PLoS One 9:e89189. doi: 10.1371/journal.pone.0089189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayamani E, Rajamuthiah R, Larkins-Ford J, Fuchs BB, Conery AL, Vilcinskas A, Ausubel FM, Mylonakis E. 2015. Insect-derived cecropins display activity against Acinetobacter baumannii in a whole-animal high-throughput Caenorhabditis elegans model. Antimicrob Agents Chemother 59:1728–1737. doi: 10.1128/AAC.04198-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Chung T, Oldenburg K. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 12.Eigelsbach HT, Downs CM. 1961. Prophylactic effectiveness of live and killed tularemia vaccines. I. Production of vaccine and evaluation in the white mouse and guinea pig. J Immunol 87:415–425. [PubMed] [Google Scholar]
- 13.Schmitt DM, O'Dee DM, Cowan BN, Birch JW-M, Mazzella LK, Nau GJ, Horzempa J. 2013. The use of resazurin as a novel antimicrobial agent against Francisella tularensis. Front Cell Infect Microbiol 3:93. doi: 10.3389/fcimb.2013.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saslaw S, Eigelsbach HT, Wilson HE, Prior JA, Carhart S. 1961. Tularemia vaccine study. I. Intracutaneous challenge. Arch Intern Med 107:689–701. doi: 10.1001/archinte.1961.03620050055006. [DOI] [PubMed] [Google Scholar]
- 15.Elkins KL, Cowley SC, Bosio CM. 2007. Innate and adaptive immunity to Francisella. Ann N Y Acad Sci 1105:284–324. doi: 10.1196/annals.1409.014. [DOI] [PubMed] [Google Scholar]
- 16.Saslaw S, Eigelsbach HT, Prior JA, Wilson HE, Carhart S. 1961. Tularemia vaccine study. II. Respiratory challenge. Arch Intern Med 107:702–714. [DOI] [PubMed] [Google Scholar]
- 17.Horzempa J, Tarwacki DM, Carlson PE. 2008. Characterization and application of a glucose-repressible promoter in Francisella tularensis. Appl Environ Microbiol 74:2161–2170. doi: 10.1128/AEM.02360-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horzempa J, Carlson PE, O'Dee DM. 2008. Global transcriptional response to mammalian temperature provides new insight into Francisella tularensis pathogenesis. BMC Microbiol 8:172. doi: 10.1186/1471-2180-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheel O, Reiersen R, Hoel T. 1992. Treatment of tularemia with ciprofloxacin. Eur J Clin Microbiol 11:447–448. doi: 10.1007/BF01961860. [DOI] [PubMed] [Google Scholar]
- 20.Russell P, Eley SM, Fulop MJ, Bell DL, Titball RW. 1998. The efficacy of ciprofloxacin and doxycycline against experimental tularaemia. J Antimicrob Chemother 41:461–465. doi: 10.1093/jac/41.4.461. [DOI] [PubMed] [Google Scholar]
- 21.La Scola B, Elkarkouri K, Li W, Wahab T, Fournous G, Rolain JM, Biswas S, Drancourt M, Robert C, Audic S, Lofdahl S, Raoult D. 2008. Rapid comparative genomic analysis for clinical microbiology: the Francisella tularensis paradigm. Genome Res 18:742–750. doi: 10.1101/gr.071266.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reference deleted.
- 23.Johansson A, Berglund L, Sjöstedt A, Tärnvik A. 2001. Ciprofloxacin for treatment of tularemia. Clin Infect Dis 33:267–268. doi: 10.1086/321825. [DOI] [PubMed] [Google Scholar]
- 24.Johansson A, Berglund L, Gothefors L, Sjöstedt A, Tärnvik A. 2000. Ciprofloxacin for treatment of tularemia in children. Pediatr Infect Dis J 19:449–453. doi: 10.1097/00006454-200005000-00011. [DOI] [PubMed] [Google Scholar]
- 25.Ikeda T, Yasui C, Hoshino K, Arikawa K, Nishikawa Y. 2007. Influence of lactic acid bacteria on longevity of Caenorhabditis elegans and host defense against Salmonella enterica serovar Enteritidis. Appl Environ Microbiol 73:6404–6409. doi: 10.1128/AEM.00704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Gerven N, Derous V, Hernalsteens J-P. 2008. Expression of in vivo-inducible Salmonella enterica promoters during infection of Caenorhabditis elegans. FEMS Microbiol Lett 278:236–241. doi: 10.1111/j.1574-6968.2007.01001.x. [DOI] [PubMed] [Google Scholar]
- 27.Aballay A, Yorgey P, Ausubel FM. 2000. Salmonella typhimurium proliferates and establishes a persistent infection in the intestine of Caenorhabditis elegans. Curr Biol 10:1539–1542. doi: 10.1016/S0960-9822(00)00830-7. [DOI] [PubMed] [Google Scholar]
- 28.Tenor JL, McCormick BA, Ausubel FM, Aballay A. 2004. Caenorhabditis elegans-based screen identifies Salmonella virulence factors required for conserved host-pathogen interactions. Curr Biol 14:1018–1024. doi: 10.1016/j.cub.2004.05.050. [DOI] [PubMed] [Google Scholar]
- 29.Mahajan-Miklos S, Tan MW, Rahme LG, Ausubel FM. 1999. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 96:47–56. doi: 10.1016/S0092-8674(00)80958-7. [DOI] [PubMed] [Google Scholar]
- 30.Tan MW, Mahajan-Miklos S, Ausubel FM. 1999. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci U S A 96:715–720. doi: 10.1073/pnas.96.2.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan MW, Rahme LG, Sternberg JA, Tompkins RG, Ausubel FM. 1999. Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc Natl Acad Sci U S A 96:2408–2413. doi: 10.1073/pnas.96.5.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sifri CD, Begun J, Ausubel FM, Calderwood SB. 2003. Caenorhabditis elegans as a model host for Staphylococcus aureus pathogenesis. Infect Immun 71:2208–2217. doi: 10.1128/IAI.71.4.2208-2217.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moy TI, Mylonakis E, Calderwood SB, Ausubel FM. 2004. Cytotoxicity of hydrogen peroxide produced by Enterococcus faecium. Infect Immun 72:4512–4520. doi: 10.1128/IAI.72.8.4512-4520.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maadani A, Fox KA, Mylonakis E, Garsin DA. 2007. Enterococcus faecalis mutations affecting virulence in the Caenorhabditis elegans model host. Infect Immun 75:2634–2637. doi: 10.1128/IAI.01372-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drlica K, Zhao X. 1997. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev 61:377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan M-W, Ausubel FM. 2002. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297:623–626. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- 37.Lass-Flörl C, Dierich MP, Fuchs D, Semenitz E, Jenewein I, Ledochowski M. 2001. Antifungal properties of selective serotonin reuptake inhibitors against Aspergillus species in vitro. J Antimicrob Chemother 48:775–779. doi: 10.1093/jac/48.6.775. [DOI] [PubMed] [Google Scholar]
- 38.Argüello-García R, Cruz-Soto M, Romero-Montoya L, Ortega-Pierres G. 2004. Variability and variation in drug susceptibility among Giardia duodenalis isolates and clones exposed to 5-nitroimidazoles and benzimidazoles in vitro. J Antimicrob Chemother 54:711–721. doi: 10.1093/jac/dkh388. [DOI] [PubMed] [Google Scholar]
- 39.Mosca A, Russo F, Miragliotta G. 2006. In vitro antimicrobial activity of benzalkonium chloride against clinical isolates of Streptococcus agalactiae. J Antimicrob Chemother 57:566–568. doi: 10.1093/jac/dki474. [DOI] [PubMed] [Google Scholar]
- 40.Stone CA, Arman CG, Lotti VJ, Minsker DH, Risley EA, Bagdon WJ, Bokelman DL, Jensen RD, Mendlowski B, Tate CL, Peck HM, Zwickley RE, McKinney SE. 1977. Pharmacology and toxicology of diflunisal. Br J Clin Pharmacol 4(Suppl 1):19S–29S. doi: 10.1111/j.1365-2125.1977.tb04510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zabawa TP, Pucci MJ, Parr TR, Lister T. 2016. Treatment of Gram-negative bacterial infections by potentiation of antibiotics. Curr Opin Microbiol 33:7–12. doi: 10.1016/j.mib.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 42.Beanan MJ, Strome S. 1992. Characterization of a germ-line proliferation mutation in C. elegans. Development 116:755–766. [DOI] [PubMed] [Google Scholar]
- 43.Ikäheimo I, Syrjälä H, Karhukorpi J, Schildt R, Koskela M. 2000. In vitro antibiotic susceptibility of Francisella tularensis isolated from humans and animals. J Antimicrob Chemother 46:287–290. doi: 10.1093/jac/46.2.287. [DOI] [PubMed] [Google Scholar]
- 44.Urich SK, Petersen JM. 2008. In vitro susceptibility of isolates of Francisella tularensis types A and B from North America. Antimicrob Agents Chemother 52:2276–2278. doi: 10.1128/AAC.01584-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jorgensen JH, Ferraro MJ. 2009. Antimicrobial susceptibility testing: a review of general principles and contemporary practices. Clin Infect Dis 49:1749–1755. doi: 10.1086/647952. [DOI] [PubMed] [Google Scholar]
- 46.Rosch JW, Boyd AR, Hinojosa E, Pestina T, Hu Y, Persons DA, Orihuela CJ, Tuomanen EI. 2010. Statins protect against fulminant pneumococcal infection and cytolysin toxicity in a mouse model of sickle cell disease. J Clin Invest 120:627–635. doi: 10.1172/JCI39843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajamuthiah R, Jayamani E, Conery AL, Fuchs BB, Kim W, Johnston T, Vilcinskas A, Ausubel FM, Mylonakis E. 2015. A defensin from the model beetle Tribolium castaneum acts synergistically with telavancin and daptomycin against multidrug resistant Staphylococcus aureus. PLoS One 10:e0128576. doi: 10.1371/journal.pone.0128576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gellert M, Mizuuchi K, O'Dea MH, Itoh T, Tomizawa JI. 1977. Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc Natl Acad Sci U S A 74:4772–4776. doi: 10.1073/pnas.74.11.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chopra I, Roberts M. 2001. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev 65:232–260. doi: 10.1128/MMBR.65.2.232-260.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mc Cay PH, Ocampo-Sosa AA, Fleming GTA. 2010. Effect of subinhibitory concentrations of benzalkonium chloride on the competitiveness of Pseudomonas aeruginosa grown in continuous culture. Microbiology 156:30–38. doi: 10.1099/mic.0.029751-0. [DOI] [PubMed] [Google Scholar]
- 51.Solaymani-Mohammadi S, Genkinger JM, Loffredo CA, Singer SM. 2010. A meta-analysis of the effectiveness of albendazole compared with metronidazole as treatments for infections with Giardia duodenalis. PLoS Negl Trop Dis 4:e682. doi: 10.1371/journal.pntd.0000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunin CM, Ellis WY. 2000. Antimicrobial activities of mefloquine and a series of related compounds. Antimicrob Agents Chemother 44:848–852. doi: 10.1128/AAC.44.4.848-852.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. 2013. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee Y-S, Han S-H, Lee S-H, Kim Y-G, Park C-B, Kang O-H, Keum J-H, Kim S-B, Mun S-H, Shin D-W, Kwon D-Y. 2011. Synergistic effect of tetrandrine and ethidium bromide against methicillin-resistant Staphylococcus aureus (MRSA). J Toxicol Sci 36:645–651. doi: 10.2131/jts.36.645. [DOI] [PubMed] [Google Scholar]
- 55.Wallace JL. 2008. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn't the stomach digest itself? Physiol Rev 88:1547–1565. doi: 10.1152/physrev.00004.2008. [DOI] [PubMed] [Google Scholar]
- 56.Al-Janabi AA. 2010. In vitro antibacterial activity of ibuprofen and acetaminophen. J Glob Infect Dis 2:105–108. doi: 10.4103/0974-777X.62880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.