

Abstract

As the central figure of the cellular protein degradation machinery, the proteasome is critical for cell survival. Having been extensively targeted for inhibition, the constitutive proteasome has proven its role as a highly valuable drug target. However, recent advances in the protein homeostasis field suggest that additional chapters can be added to this successful story. For example, selective immunoproteasome inhibition promises high clinical efficacy for autoimmune disorders and inflammation, and proteasome inhibitors might serve as novel therapeutics for malaria or other microorganisms. Furthermore, utilizing the destructive force of the proteasome for selective degradation of essential drivers of human disorders has opened up a new and exciting area of drug discovery. Thus, the field of proteasome drug discovery still holds exciting questions to be answered and does not simply end with inhibiting the constitutive proteasome.
Short abstract
Proteasome inhibition is a successful branch of modern drug discovery. This outlook summarizes recent advances and achievement in the field focusing on developments beyond the constitutive proteasome.
Introduction
Homeostasis between protein synthesis and degradation is a pivotal cellular process involving a multitude of precise and highly complex regulatory processes. The predominant system responsible for the degradation of ∼80% of all cellular proteins is the ubiquitin proteasome system (UPS).1 At the heart of this eukaryotic protein degradation machinery is the proteasome, a large, tightly regulated protein complex with a total molecular weight of about 2.5 MDa.2,3 Proteins are targeted for proteasomal degradation via the covalent attachment of the 8.5 kDa protein ubiquitin.4 Ubiquitination occurs via three different enzymes.5 Ubiquitin is activated by a ubiquitin-activating enzyme (E1) and subsequently transferred to a ubiquitin-conjugating enzyme (E2) before it is finally coupled to the substrate protein by means of a ubiquitin-protein ligase (E3). The typical ubiquitination pattern for recognition by the proteasome comprises a chain of at least four ubiquitins, with the first one being attached to a surface Lys of the target protein via an isopeptide bond.4,6
As mentioned above, the proteasome is at the center of the protein degradation regulatory network and can be found in the cytoplasm as well as the nucleus of eukaryotic cells. It is a highly complex molecular machine, consisting of various complexes, all possessing the 20S core particle (CP).7,8 The 20S CP has a mass of ∼700 kDa and comprises 28 protein subunits that are stacked in four homologous rings of seven, forming a hollow cylindrical structure. The two inner rings each formed by seven β subunits (β1–7) are enclosed by the two outer rings assembled from seven α subunits (α1–7) (Figure 1A).9,10 The proteolytic chamber is formed by the β-rings, which harbor the three catalytically active subunits β1, β2, and β5 that exhibit caspase-like (CL), trypsin-like (TL), and chymotrypsin-like (ChTL) activities, respectively (Figure 1B). The two α-rings regulate access to the proteolytic chamber by limiting entry to unfolded polypeptide chains. In vertebrates, three different CPs have been identified. The highly abundant constitutive proteasome (cCP) is present in all tissues, whereas the immunoproteasome (iCP) appears predominantly in monocytes and lymphocytes and the thymoproteasome (tCP) is exclusively found in cortical thymic epithelial cells (Figure 1B).11−13 Each of the three CPs harbors a unique set of catalytic β-subunits resulting in slightly modified cleavage preferences. While the cCP contains the proteolytic β-subunits β1c, β2c, and β5c, the iCP incorporates β1i, β2i, and β5i, while the tCP holds subunits β1i, β2i, and β5t. Due to modified substrate binding pockets, the proteolytic subunits of the iCP and tCP generate substrate epitopes for the antigen presenting major histocompatibility complex-I (MHC-I) receptors of the immune system at a considerably higher rate.14−16
Figure 1.

The proteasome. (A) α- and β-subunits are arranged in rings of seven. The catalytically active subunits are β1 (CL), β2 (TL), and β5 (ChTL). (B) The 20S CP comprises 28 subunits grouped into four rings stacked in an αββα pattern and forming the catalytic chamber. The three different 20S CPs are the cCP, iCP, and tCP and vary by their catalytic subunits. (C) Schematic assembly of the two proteasome lids, the 11S cap and the 19S RP. The 11S cap is formed out of seven subunits and acts in a ubiquitin- and ATP-independent manner. The 19S RP can be divided into the base (10 subunits) and the lid (9 subunits) which inherits the deubiquinating enzyme Rpn11. (D) Different proteasome assemblies have been identified, thus far. The 26S proteasome comprises the 20S CP capped with two 19S RP. The 11S cap can either associate with the free end of a 19S–20S complex to form a hybrid proteasome or bind to both sides of the 20S CP.
To prevent uncontrolled degradation of cellular proteins, access to the 20S CP is tightly regulated. Three different caps, the bleomycin-sensitive 10 cap (Blm10), the 11S cap, and the 19S regulatory particle (RP), have been identified to dock onto the 20S CP and gate admission to the proteolytic chamber (Figure 1C).1,7,8 Gating requires controlled opening of the α-ring to allow for proteolytic breakdown of the administered substrate. The two proteasome activators the 11S cap (proteasome activator 28, PA28) and Blm10 (also PA200 in humans) open the proteasome for substrate degradation in an ATP- and ubiquitin-independent manner. While their structures have been solved, their exact mode of action and regulation is still not fully understood.7,17 Blm10 is composed of a single-chain ∼250 kDa cap that wraps around the 20S CP and forms multiple HEAT repeats.18 The 11S cap is assembled from a ring of seven subunits that interact with the α-ring via their C-termini in a similar fashion as the 19S RP. It is primarily found in the immunoproteasome in an 11S–20S–11S assembly or as a hybrid proteasome in combination with the 19S RP (19S–20S–11S) (Figure 1D).8 The 19S RP is the best characterized proteasome activator and complexes with the 20S CP as the prominent constitutive 26S proteasome harboring a 19S–20S–19S setup (Figure 1D). The 19S RP is an ∼900 kDa complex of 19 individual subunits that activates the proteasome in an ATP-dependent manner and recognizes and cleaves ubiquitin chains from the substrate. Structure elucidation has divided the 19S RP into two subcomplexes: the base and the lid.19 The base is assembled from ten subunits including six ATPases (Rpt 1–6), two organizing subunits (regulatory particle non-ATPase 1 (Rpn1) and Rpn2), and two ubiquitin receptors (Rpn10 and Rpn13). The lid is composed of nine subunits (Rpn3, 5–9, 11, 12, and 15) with Rpn11 as the only deubiquitylating enzyme of the 19S RP and the whole proteasome (Figure 1C).20,21
The proteasome is pivotal for intracellular protein homeostasis as it eliminates misfolded proteins. Proteasome inhibition results in a multitude of cellular responses such as endoplasmatic reticulum (ER) stress, unfolded protein response, NFκB inhibition, cell cycle arrest, inhibition of angiogenesis, or an increase in proapoptotic factors and tumor suppressors.22−24 Consequently, the proteasome is a highly interesting and long-established drug target with three FDA approved drugs on the market (bortezomib, carfilzomib, and ixazomib) that inhibit its proteolytic activity. Proteasome inhibition has been extensively reviewed in previous articles.22,23,25−30 This review will focus only on the recent advances in the field, especially in targeting the immunoproteasome, proteasome inhibitors as potential antimalaria agents, and the novel Rpn11 inhibitor capmizin.
Proteasome Inhibition
The Constitutive Proteasome
In early studies proteasome inhibitors were primarily used to uncover and study the proteasome’s catalytic activity.31−33 Although these compounds were only limited to proof-of-principle studies due to a lack of efficacy, stability, or specificity, they revealed the essential role of the proteasome for cell function and survival. It was observed that proteasome inhibitors induced apoptosis in leukemic cell lines and were even effective against hematological and solid tumors.34−36 The substrate binding channel with its specificity pockets (S) as well as the N-terminal Thr (Thr1) in the active site represents the central leverage point for proteasome inhibition.25 Most proteasome inhibitors are peptide-inspired compounds whose side chains (P sites) are tailored to engage the S pockets in order to gain subunit selectivity (Figure 2A). To do so, these peptide-like inhibitors imitate the binding mode of natural proteasome substrates. Most proteasome inhibitors target the ChTL β5-subunit because inhibition of β5 results in the greatest reduction of protein breakdown rates, whereas inactivation of β1 and β2 has a smaller impact on general proteolysis.8 The additional affinity of most β5 inhibitors for β1 and β2 is primarily coincidental.28 In order to inhibit the catalytic activity of the β subunit active site, most inhibitors are equipped with an electrophilic headgroup that either reversibly or irreversibly engages the N-terminal active site Thr1. The majority of current proteasome inhibitors comprise a boronic acid, an epoxyketone, or a β-lactone as electrophilic warhead.
Figure 2.

Proteasome inhibition. (A) Schematic representation of the binding channel of the constitutive proteasome (left) and the immunoproteasome (right) containing a representative peptide sequence. The catalytically active Thr1 and the scissile peptide bond are highlighted in red. The selectivity pockets are depicted in blue. Met45 adopts a different conformation in the immunoproteasome widening the S1 pocket. The unique Cys48 in the immunoproteasome S4 pocket is shown explicitly. (B) Chemical structures of known proteasome inhibitors (cCP). P sites have been matched with the corresponding S pockets.
The first proteasome inhibitor, bortezomib (Velcade, Millennium Pharmaceuticals), was approved by the FDA in 2003 for the treatment of multiple myeloma (Figure 2B).37 Bortezomib is a reversible dipeptide boronate inhibitor targeting the ChTL β-subunits β5c and β5i with low nanomolar half maximal inhibitory concentration (IC50) values of 7 nM and 4 nM, respectively, while showing reduced affinity for the β1c subunit (74 nM) and negligible affinity for the remaining β-subunits.38 Since its initial approval for multiple myeloma in 2003 bortezomib has been additionally approved for the treatment of mantle cell lymphoma and is currently under investigation in a multitude of clinical trials in combination with various other chemotherapeutic agents.39 Although bortezomib is approved for the treatment of blood cancer, its initially promising results against solid tumors did not translate into clinical trials, and the amount administered is restricted by a narrow therapeutic window.22 Furthermore, bortezomib needs to be administered intravenously and exhibits considerable side effects such as peripheral neuropathy, thrombocytopenia, and gastrointestinal disorders.40
The success and shortcomings of bortezomib prompted the hunt for novel proteasome inhibitors with reduced off-target effects. Based on the natural product epoxomicin the tetrapeptide carfilzomib (Kyprolis, Proteolix Inc.) was evolved as an irreversible proteasome inhibitor (Figure 2B). Carfilzomib belongs to the epoxyketone family of proteasome inhibitors and covalently attacks active site Thr1 under the formation of a morpholine ring.41,42 It targets the β5c and β5i subunits of the 20S CP with IC50 values of 6 nM and 33 nM, respectively, and shows an improved selectivity profile with fewer off-target effects compared to bortezomib.38 Carfilzomib, showing a broader therapeutic window, was approved for treatment of multiple myeloma by the FDA in 2012.43In vitro, carfilzomib proved active even against bortezomib-resistant multiple myeloma cell lines.44 However, like bortezomib, carfilzomib has to be administered intravenously and has a short half-life of roughly 30 min.45 An orally available carfilzomib analogue, oprozomib (ONX0912, Onyx Pharmaceuticals), is currently evaluated in clinical trials (Figure 2B).46 Oprozomib appears to be almost as potent as carfilzomib and inhibits β5c and β5i with IC50 values of 36 and 82 nm, respectively.47,48
The third proteasome inhibitor approved by the FDA in 2015 is the second generation peptide boronic acid ixazomib (MLN2238, Millenium Pharmaceuticals, Figure 2B). Ixazomib is the first orally available proteasome inhibitor and is administered as a prodrug (MLN9708) which rapidly hydrolyzes into the bioactive boronate.49 Ixazomib shows an IC50 value of 3.4 nM toward β5c and 31 nM for β1c, respectively, with no reported data on β5i.49 However, the half-life of ixazomib is substantially shorter than that of bortezomib.22,30
The only nonpeptidic proteasome inhibitor in advanced clinical trials for multiple myeloma is the natural product salinosporamide A, also known as marizomib (Nereus Pharmaceuticals, Figure 2B). Marizomib is orally available and inhibits the proteasome irreversibly via an ester and tetrahydrofuran formation.50 It is the smallest proteasome inhibitor identified thus far and has the lowest IC50 value among all previous reported β5c inhibitors with 2.5 nM, while additionally engaging β2c (IC50 = 26 nM) and β1c (IC50 = 330 nM).50 However, its very short half-life of less than 15 min and its ability to penetrate the blood–brain barrier might hamper its therapeutic success.51
The Immunoproteasome
Selective inhibition of the immunoproteasome has recently gained substantial interest as the immunoproteasome has been associated with the development and progression of neurodegenerative diseases, autoimmune disorders, inflammation, and certain types of cancer.15,52,53 In particular, inhibition of the β5i subunit of the immunoproteasome has been associated with beneficial effects for the treatment of arthritis and colorectal carcinoma.54,55 Crystal structures of the murine cCP and iCP revealed structural differences between β5c and β5i in the S1 pocket, which indicates that inhibitors with larger P1 residues favor β5i over β5c (Figure 2A).56 Additionally, differences in the amino acid sequence between the cCP and iCP can be used for selective targeting of the immunoproteasome.57
The most advanced immunoproteasome inhibitors thus far are the β1i selective IPSI-001 and the β5i selective ONX0914 (Figure 3). The peptide aldehyde inhibitor IPSI-001 shows an over 100-fold increased selectivity for β1i compared to β1c. Treatment with IPSI-001 results in an accumulation of ubiquitin–protein conjugates and proapoptotic proteins, as well as causing caspase-mediated apoptosis in in vitro models of hematological malignancies.58 However, due to its high Ki of 1.03 μM (no IC50 reported) and the well-known cross-reactivity of aldehydes with Cys residues, off-target effects are highly likely. ONX0914 is a β5i selective epoxyketone with low nanomolar activity (IC50 = 28 nM) and more than 10-fold selectivity over β5c.55 It reduces the production of proinflammatory cytokines and the expression of MHC-I receptors on the cell surface without significant toxicity. ONX0914 capitalizes on the slightly more spacious S1 pocket in β5i to gain immunoproteasome selectivity.56 Recentyl, the ONX0914 derivative KZR-616 (Kezar Life Sciences) has entered phase 1a clinical trials (August 2016) as the first immunoproteasome inhibitor and shall be tested against a number of autoimmune and inflammatory diseases. Despite their initially promising data, only one clinical trial for immunoproteasome inhibitors has been launched thus far (KZR-616), and various recent studies are still trying to elucidate the structural requirements for selective immunoproteasome targeting and to identify novel inhibitors. This effort is paired with the ability to detect cCP and iCP subunit binding in a feasible assay. A recently reported method utilizing fluorescently labeled activity-based probes followed by SDS–PAGE separation allows for simultaneous detection of all six cCP and iCP catalytic subunits and might prove useful to fully evaluate inhibitor binding in the future.59
Figure 3.

Inhibitors of the immunoproteasome. If applicable, P sites have been matched with corresponding selectivity pockets.
The structural differences in the S1 binding pocket between β5i and β5c arise from a different orientation of Met45 and have been elucidated in different studies to understand and develop selective immunoproteasome inhibitors (Figure 2A).60,61 Based on the natural product belactosin C it was observed that the difference between an isoleucine versus a valine residue is already sufficient to achieve subtype selectivity between β5i and β5c due to deeper penetration into the S1 pocket of the isoleucine side chain (lactone 3, Figure 3).60 The same principle was used to increase subtype selectivity of ONX0914 by replacing the P1 Phe residue with a cyclohexyl moiety.61 Besides harnessing the structural differences of the S1 pocket to achieve selectivity for β5i, exploitation of the S4 pocket provides an additional possibility. Superimposition of the murine mβ5i and mβ5c subunits in combination with sequence alignment identified a noncatalytic Cys residue (Cys48) exclusively present in the S4 binding pocket of the β5i subunit (Figure 2A). The nucleophilic nature of Cys48 was exploited to covalently attack an α-chloroacetamide-modified side chain of the decarboxylated tetrapeptide 4-CA (Figure 3).57 The optimized peptide 4-CA shows more than 150-fold selectivity for β5i over β5c and decreases the production of inflammatory cytokines. Other nonpeptidic, selective inhibitors of β5i have been identified using a structure-guided virtual screen.62 The initially identified reversible binders could be evolved into irreversible inhibitors bearing a oxathiazolone warhead (compound 42), which was recently identified as selective for Thr modification (Figure 3).63 Due to structural similarities with another nonpeptidic β5i selective inhibitor, it is likely that 42 engages the β5i subunit in a unique binding mode utilizing subpockets outside of the natural substrate binding channels.64 These subpockets might be exploited for the development of novel selective immunoproteasome inhibitors that are not dependent on the traditional peptide binding sites. Besides alternating the side chains (P sites) of the different inhibitors, the introduction of a peptide sulfonyl fluoride (PSF) warhead showed selective modification of the β5i subunit while having no effect on β5c.65 Treatment of β5i with PSF peptide 3 induced irreversible deactivation of the proteolytic active site via polarity inversion and intramolecular cross-linking between Thr1 and K33 (Figure 3). This resulted in a catalytically dead β5i subunit and identified a novel mechanism of proteasome deactivation.
Proteasome Inhibitors in Malaria
Not only have proteasome inhibitors been evaluated for inhibition of the human proteasome, but likewise, they have proven to be effective against the malaria parasite Plasmodium falciparum. As the parasite depends on a rapid protein turnover while dividing inside host cells, the proteasome offers a valid target for antimalarial drugs.66,67 Early studies identified inhibition of the P. falciparum proteasome as a valuable strategy; however, the tested compounds also inhibited the mammalian proteasome hampering their use as pharmaceutical agents. Moreover, the lack of structural data restricted the identification of suitable inhibitors solely to screening trials.68−70 A carfilzomib analogue was identified as effective in killing parasites while having only minor effects on host cells.71 Interestingly, this compound owes its therapeutic window not to selective inhibition of the parasite proteasome but to insufficient inhibition of the human β2 subunit. To assess subunit dependency within the P. falciparum life cycle, an active site probe labeling the catalytic subunits β1, β2, and β5 was designed that identified β5 inhibition as effective during the replication stage (schizogony), while simultaneous β2 and β5 inhibition resulted in enhanced parasite killing at all stages.72 Further investigation led to the assumption that, as previously identified for Mycobacterium tuberculosis,73 the P1 and P3 amino acid residues of the inhibitor are especially important for selective targeting of the P. falciparum proteasome.74 This hypothesis was verified in 2016 by the first structural insight into the P. falciparum 20S CP using cryo-electron microscopy combined with single particle analysis.75 This groundbreaking study identified several tripeptide vinyl sulfones containing sterically demanding Trp residues as selective inhibitors which favor the parasite β2 subunit over human β2 (WLW-vs, Figure 4A). Changing the P1 side chain to Leu (WLL-vs) results in simultaneous inhibition of parasite subunits β2 and β5 as well as human β5.75 Structural analysis indicated a narrower binding pocket of human β2 with reduced accessibility for Trp in positions P1 and P3 as observed for WLW-vs (Figure 4B). Effective killing of artemisinin-resistant parasites was achieved via cotreatment with the β2-selective inhibitor WLW-vs and dihydroartemisinin at concentrations where WLW-vs selectively inhibits the parasite β2 subunit. Furthermore, the β2/β5 selective inhibitor WLL-vs showed a broad therapeutic window and was highly efficient in a Plasmodium chabaudi mouse model where a single dosage of WLL-vs resulted in almost complete parasite clearance without any significant side effects.
Figure 4.

Plasmodium falciparum proteasome. (A) Recently identified irreversible inhibitors of the P. falciparum proteasome. (B) Crystal structure of WLW-vs bound to the active site of the β2 subunit (PDB: 5FMG). P sites have been matched with corresponding selectivity pockets.
Inhibition of Rpn11
In contrast to targeting the proteolytic β subunits of the 20S CP—the mode of action for all the previously described compounds—a recently published study pursued the idea of clogging the proteasome by inhibiting its deubiquitinase activity of Rpn11.76 Rpn11 is a metalloisopeptidase located in the lid of the 19S RP that cleaves polyubiquitin chains from the substrates (Figure 5A), thus allowing ubiquitin recycling as well as substrate access to the 20S CP. Rpn11 is the only deubiquitylating enzyme present in the 26S proteasome, and its catalytically active JAMM domain with its bound Zn2+ cation was validated as a potential target for proteasome inhibition. Point mutations of its active site resulted in a severe decrease in proteolysis and ultimately cell death.20,21 A fragment-based drug discovery (FBDD) approach that screened more than 330,000 compounds including metal-binding pharmacophores yielded a moderate Rpn11 inhibitor with an IC50 value of ∼2.5 μM. Further lead optimization and SAR studies resulted in the identification of capzimin, a Rpn11 inhibitor with an IC50 of 300 nM and a more than 10-fold preference for Rpn11 over other JAMM isopeptidases (Figure 5B).76,77 Capzimin proved active against several cancer cell lines, including bortezomib-resistant cell lines, induces the unfolded protein response, and blocks cell proliferation. Although capzimin needs to be further optimized to gain more drug-like properties, its orthogonal mode of action identified a novel approach for proteasome inhibition, which is especially interesting considering the emergence of resistances toward the classic “omib” therapeutics.
Figure 5.
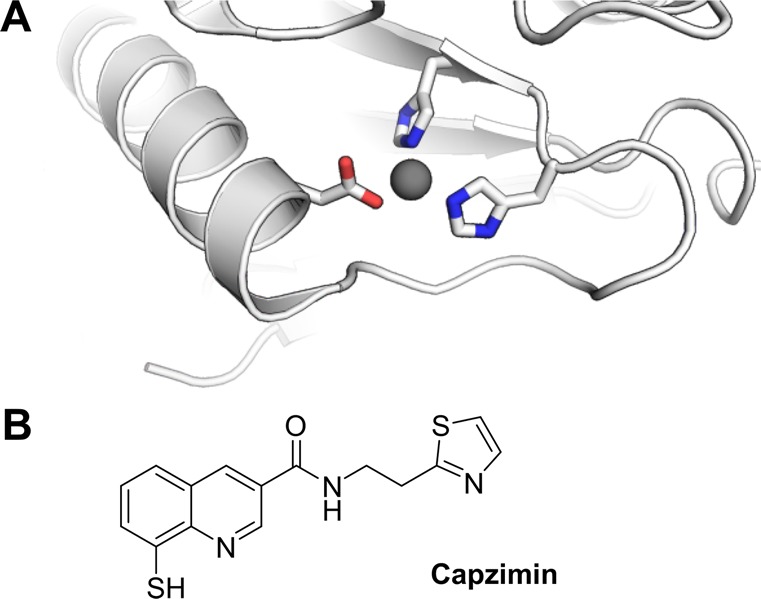
Inhibition of Rpn11. (A) Structural representation of the Rpn11 active site. The active site Zn2+ is highlighted in gray, and the complexing amino acids are shown explicitly (PDB: 4OWP). (B) Chemical structure of capzimin, the first inhibitor of the deubiquinating enzyme Rpn11 that is part of the 19S RP.
Conclusion
The proteasome is the key player of the cellular protein degradation machinery and is pivotal for protein homeostasis to ensure cell proliferation and survival. The 20S constitutive catalytic core of the proteasome represents a valid drug target with three FDA approved drugs and many compounds in clinical trials. Despite their huge success, proteasome inhibitors may be limited to nonsolid tumors, especially blood cancer.78 As observed for numerous anticancer agents or antibiotics, drug resistance emerges after long-term treatment, hampering clinical efficacy. Extensive structural analysis has pinpointed bortezomib resistance to different mutations in the β5 subunit that restrict inhibitor access to the active site.79 However, carfilzomib binding is less affected than bortezomib or ONX0914, owing its reduced susceptibility to (1) its irreversible mode of action and (2) its tetrapeptide structure allowing for better anchoring in the β5 binding channel compared to the dipeptide bortezomib or the tripeptide ONX0914. The emerging resistances and still severe off-target and side effects of proteasome inhibitors fuel the need for novel and more selective therapeutics. Additionally, a deeper understanding of the emerging resistance mechanisms might guide the design of next generation proteasome inhibitors.
Accumulated evidence suggests that selective targeting of the immunoproteasome will bear distinct clinical benefits in the treatment of inflammatory and autoimmune disorders.25 As KZR-616 is the only immunoproteasome inhibitor that has advanced to clinical trials, various different strategies to develop selective immunoproteasome inhibitors are still being pursued. Exploiting structural differences of the iCP catalytic subunits or unique reactivities due to sequence differences led to the identification of selective iCP inhibitors. However, it appears that selectivity for murine β5i does not easily translate across species for selectivity against human proteasomes. Therefore, further studies are needed to evaluate if the mouse immunoproteasome can function as a suitable mimic for the human immunoproteasome. Accumulating evidence suggests that the rat model might be more suitable than the mouse model in this regard. As there is still a considerable lack of structural and biological information on the immunoproteasome, further studies are necessary to fill the gaps. Even less is known about the thymoproteasome and its role in human disorders. Nonetheless, the expected therapeutic benefits of immunoproteasome inhibition make this field a current focus of proteasome drug discovery.
The recent insight on proteasome inhibitors as selective antimalaria agents represents another highly interesting branch of proteasome research. Today, technical advances in electron microscopy allow detailed studies of huge molecular machines and facilitated the first structural insight into the proteasome of P. falciparum.75 This study constitutes a breakthrough for the development of more selective proteasome inhibitors as antimalaria therapeutics. However, this field is still in its infancy, and a better understanding of the underlying processes will allow for therapeutic advancement. It highlights the essential role of the proteasome in all forms of life, and how proteasome inhibition might allow selective targeting of other organisms as well.
Instead of blocking the proteasome to achieve therapeutic benefit, small molecules that specifically induce proteasomal degradation are able to exploit its unique ability to degrade almost every cellular protein. These bifunctional molecular entities, known as proteolysis-targeting chimeras (PROTACs), have emerged as a highly interesting approach in drug discovery.80−82 PROTACs have successfully reduced cellular levels of highly interesting protein targets and are capable of reaching beyond the limits imposed by traditional drug discovery as target engagement is already sufficient for proteasomal degradation. Furthermore, PROTAC activity might even be enhanced by cotreatment with certain recently identified proteasome activators.83
In summary, the proteasome constitutes a well-established drug target that has advanced the treatment of various forms of blood cancer. However, the therapeutic potential of proteasome inhibition does not seem to be exhausted, yet. Especially the immunoproteasome and the proteasome of various parasites and microorganisms depict promising targets to continue the success story of proteasome inhibition.
Acknowledgments
P.M.C. is thankful to the Alexander von Humboldt Foundation for a Feodor Lynen research fellowship. C.M.C. gratefully acknowledges the US National Institutes of Health for their support (R35CA197589).
The authors declare no competing financial interest.
References
- Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coux O.; Tanaka K.; Goldberg A. L. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996, 65, 801–847. 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S.; Yu H.; Mim C.; Matouschek A. Regulated protein turnover: snapshots of the proteasome in action. Nat. Rev. Mol. Cell Biol. 2014, 15, 122–133. 10.1038/nrm3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D.; Rape M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Pickart C. M.; Eddins M. J. Ubiquitin: structures, functions, mechanisms. Biochim. Biophys. Acta, Mol. Cell Res. 2004, 1695, 55–72. 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Yau R.; Rape M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. 10.1038/ncb3358. [DOI] [PubMed] [Google Scholar]
- Kish-Trier E.; Hill C. P. Structural biology of the proteasome. Annu. Rev. Biophys. 2013, 42, 29–49. 10.1146/annurev-biophys-083012-130417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung T.; Catalgol B.; Grune T. The proteasomal system. Mol. Aspects Med. 2009, 30, 191–296. 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Groll M.; Ditzel L.; Lowe J.; Stock D.; Bochtler M.; Bartunik H. D.; Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997, 386, 463–471. 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Lowe J.; Stock D.; Jap B.; Zwickl P.; Baumeister W.; Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995, 268, 533–539. 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- Murata S.; Sasaki K.; Kishimoto T.; Niwa S.-I.; Hayashi H.; Takahama Y.; Tanaka K. Regulation of CD8+ T cell development by thymus-specific proteasomes. Science 2007, 316, 1349–1353. 10.1126/science.1141915. [DOI] [PubMed] [Google Scholar]
- Kunjappu M. J.; Hochstrasser M. Assembly of the 20S proteasome. Biochim. Biophys. Acta, Mol. Cell Res. 2014, 1843, 2–12. 10.1016/j.bbamcr.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groettrup M.; Kirk C. J.; Basler M. Proteasomes in immune cells: more than peptide producers?. Nat. Rev. Immunol. 2010, 10, 73–78. 10.1038/nri2687. [DOI] [PubMed] [Google Scholar]
- Murata S.; Takahama Y.; Tanaka K. Thymoproteasome: probable role in generating positively selecting peptides. Curr. Opin. Immunol. 2008, 20, 192–196. 10.1016/j.coi.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Basler M.; Kirk C. J.; Groettrup M. The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol. 2013, 25, 74–80. 10.1016/j.coi.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Kruger E.; Kloetzel P.-M. Immunoproteasomes at the interface of innate and adaptive immune responses: two faces of one enzyme. Curr. Opin. Immunol. 2012, 24, 77–83. 10.1016/j.coi.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Rechsteiner M.; Hill C. P. Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 2005, 15, 27–33. 10.1016/j.tcb.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Iwanczyk J.; Sadre-Bazzaz K.; Ferrell K.; Kondrashkina E.; Formosa T.; Hill C. P.; Ortega J. Structure of the Blm10–20 S proteasome complex by cryo-electron microscopy. Insights into the mechanism of activation of mature yeast proteasomes. J. Mol. Biol. 2006, 363, 648–659. 10.1016/j.jmb.2006.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman M. H.; Rubin D. M.; Coux O.; Wefes I.; Pfeifer G.; Cjeka Z.; Baumeister W.; Fried V. A.; Finley D. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell 1998, 94, 615–623. 10.1016/S0092-8674(00)81603-7. [DOI] [PubMed] [Google Scholar]
- Yao T.; Cohen R. E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407. 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- Verma R.; Aravind L.; Oania R.; McDonald W. H.; Yates J. R. 3.; Koonin E. V.; Deshaies R. J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002, 298, 611–615. 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- Crawford L. J.; Walker B.; Irvine A. E. Proteasome inhibitors in cancer therapy. J. Cell Commun. Signal 2011, 5, 101–110. 10.1007/s12079-011-0121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau P.; Richardson P. G.; Cavo M.; Orlowski R. Z.; San Miguel J. F.; Palumbo A.; Harousseau J.-L. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012, 120, 947–959. 10.1182/blood-2012-04-403733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinova I. M.; Tsimokha A. S.; Mittenberg A. G. Role of Proteasomes in Cellular Regulation. Int. Rev. Cell Mol. Biol. 2008, 267, 59–124. 10.1016/S1937-6448(08)00602-3. [DOI] [PubMed] [Google Scholar]
- Huber E. M.; Groll M. Inhibitors for the immuno- and constitutive proteasome: current and future trends in drug development. Angew. Chem., Int. Ed. 2012, 51, 8708–8720. 10.1002/anie.201201616. [DOI] [PubMed] [Google Scholar]
- Dick L. R.; Fleming P. E. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discovery Today 2010, 15, 243–249. 10.1016/j.drudis.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Johnson D. E. The ubiquitin-proteasome system: opportunities for therapeutic intervention in solid tumors. Endocr.-Relat. Cancer 2015, 22, T1–17. 10.1530/ERC-14-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev A. F.; van der Linden W. A.; Overkleeft H. S. Proteasome Inhibitors: An Expanding Army Attacking a Unique Target. Chem. Biol. 2012, 19, 99–115. 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentsch A.; Landsberg D.; Brodmann T.; Bulow L.; Girbig A.-K.; Kalesse M. Synthesis and pharmacology of proteasome inhibitors. Angew. Chem., Int. Ed. 2013, 52, 5450–5488. 10.1002/anie.201207900. [DOI] [PubMed] [Google Scholar]
- Teicher B. A.; Tomaszewski J. E. Proteasome inhibitors. Biochem. Pharmacol. 2015, 96, 1–9. 10.1016/j.bcp.2015.04.008. [DOI] [PubMed] [Google Scholar]
- Adams J. The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349–360. 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- Vinitsky A.; Michaud C.; Powers J. C.; Orlowski M. Inhibition of the chymotrypsin-like activity of the pituitary multicatalytic proteinase complex. Biochemistry 1992, 31, 9421–9428. 10.1021/bi00154a014. [DOI] [PubMed] [Google Scholar]
- Vinitsky A.; Cardozo C.; Sepp-Lorenzino L.; Michaud C.; Orlowski M. Inhibition of the proteolytic activity of the multicatalytic proteinase complex (proteasome) by substrate-related peptidyl aldehydes. J. Biol. Chem. 1994, 269, 29860–29866. [PubMed] [Google Scholar]
- Orlowski R. Z.; Kuhn D. J. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin. Cancer Res. 2008, 14, 1649–1657. 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- Shinohara K.; Tomioka M.; Nakano H.; Toné S.; Ito H.; Kawashima S. Apoptosis induction resulting from proteasome inhibition. Biochem. J. 1996, 317, 385–388. 10.1042/bj3170385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imajohohmi S.; Kawaguchi T.; Sugiyama S.; Tanaka K.; Omura S.; Kikuchi H. Lactacystin, a Specific Inhibitor of the Proteasome, Induces Apoptosis in Human Monoblast U937 Cells. Biochem. Biophys. Res. Commun. 1995, 217, 1070–1077. 10.1006/bbrc.1995.2878. [DOI] [PubMed] [Google Scholar]
- Kane R. C.; Farrell A. T.; Sridhara R.; Pazdur R. United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin. Cancer Res. 2006, 12, 2955–2960. 10.1158/1078-0432.CCR-06-0170. [DOI] [PubMed] [Google Scholar]
- Demo S. D.; Kirk C. J.; Aujay M. A.; Buchholz T. J.; Dajee M.; Ho M. N.; Jiang J.; Laidig G. J.; Lewis E. R.; Parlati F.; et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67, 6383–6391. 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]
- Kane R. C.; Dagher R.; Farrell A.; Ko C.-W.; Sridhara R.; Justice R.; Pazdur R. Bortezomib for the treatment of mantle cell lymphoma. Clin. Cancer Res. 2007, 13, 5291–5294. 10.1158/1078-0432.CCR-07-0871. [DOI] [PubMed] [Google Scholar]
- Orlowski R. Z.; Stinchcombe T. E.; Mitchell B. S.; Shea T. C.; Baldwin A. S.; Stahl S.; Adams J.; Esseltine D.-L.; Elliott P. J.; Pien C. S.; et al. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J. Clin. Oncol. 2002, 20, 4420–4427. 10.1200/JCO.2002.01.133. [DOI] [PubMed] [Google Scholar]
- Groll M.; Kim K. B.; Kairies N.; Huber R.; Crews C. M. Crystal Structure of Epoxomicin: 20S Proteasome Reveals a Molecular Basis for Selectivity of α‘,β‘-Epoxyketone Proteasome Inhibitors. J. Am. Chem. Soc. 2000, 122, 1237–1238. 10.1021/ja993588m. [DOI] [Google Scholar]
- Harshbarger W.; Miller C.; Diedrich C.; Sacchettini J. Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure 2015, 23, 418–424. 10.1016/j.str.2014.11.017. [DOI] [PubMed] [Google Scholar]
- Moreau P. The emerging role of carfilzomib combination therapy in the management of multiple myeloma. Expert Rev. Hematol. 2014, 7, 265–290. 10.1586/17474086.2014.873699. [DOI] [PubMed] [Google Scholar]
- Kuhn D. J.; Chen Q.; Voorhees P. M.; Strader J. S.; Shenk K. D.; Sun C. M.; Demo S. D.; Bennett M. K.; van Leeuwen F. W. B.; Chanan-Khan A. A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Wang Z.; Fang Y.; Jiang J.; Zhao F.; Wong H.; Bennett M. K.; Molineaux C. J.; Kirk C. J. Pharmacokinetics, pharmacodynamics, metabolism, distribution, and excretion of carfilzomib in rats. Drug Metab. Dispos. 2011, 39, 1873–1882. 10.1124/dmd.111.039164. [DOI] [PubMed] [Google Scholar]
- Chauhan D.; Singh A. V.; Aujay M.; Kirk C. J.; Bandi M.; Ciccarelli B.; Raje N.; Richardson P.; Anderson K. C. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 2010, 116, 4906–4915. 10.1182/blood-2010-04-276626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurchla M. A.; Garcia-Gomez A.; Hornick M. C.; Ocio E. M.; Li A.; Blanco J. F.; Collins L.; Kirk C. J.; Piwnica-Worms D.; Vij R.; et al. The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects. Leukemia 2013, 27, 430–440. 10.1038/leu.2012.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.-J.; Aujay M. A.; Bennett M. K.; Dajee M.; Demo S. D.; Fang Y.; Ho M. N.; Jiang J.; Kirk C. J.; Laidig G. J.; et al. Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). J. Med. Chem. 2009, 52, 3028–3038. 10.1021/jm801329v. [DOI] [PubMed] [Google Scholar]
- Kupperman E.; Lee E. C.; Cao Y.; Bannerman B.; Fitzgerald M.; Berger A.; Yu J.; Yang Y.; Hales P.; Bruzzese F.; et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010, 70, 1970–1980. 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]
- Groll M.; Huber R.; Potts B. C. M. Crystal structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of beta-lactone ring opening and a mechanism for irreversible binding. J. Am. Chem. Soc. 2006, 128, 5136–5141. 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- Williamson M. J.; Blank J. L.; Bruzzese F. J.; Cao Y.; Daniels J. S.; Dick L. R.; Labutti J.; Mazzola A. M.; Patil A. D.; Reimer C. L.; et al. Comparison of biochemical and biological effects of ML858 (salinosporamide A) and bortezomib. Mol. Cancer Ther. 2006, 5, 3052–3061. 10.1158/1535-7163.MCT-06-0185. [DOI] [PubMed] [Google Scholar]
- Kaur G.; Batra S. Emerging role of immunoproteasomes in pathophysiology. Immunol. Cell Biol. 2016, 94, 812–820. 10.1038/icb.2016.50. [DOI] [PubMed] [Google Scholar]
- McCarthy M. K.; Weinberg J. B. The immunoproteasome and viral infection: a complex regulator of inflammation. Front. Microbiol. 2015, 6, 21. 10.3389/fmicb.2015.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerner J.; Brunner T.; Groettrup M. Inhibition and deficiency of the immunoproteasome subunit LMP7 suppress the development and progression of colorectal carcinoma in mice. Oncotarget 2017, 10.18632/oncotarget.15141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchamuel T.; Basler M.; Aujay M. A.; Suzuki E.; Kalim K. W.; Lauer C.; Sylvain C.; Ring E. R.; Shields J.; Jiang J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. 10.1038/nm.1978. [DOI] [PubMed] [Google Scholar]
- Huber E. M.; Basler M.; Schwab R.; Heinemeyer W.; Kirk C. J.; Groettrup M.; Groll M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148, 727–738. 10.1016/j.cell.2011.12.030. [DOI] [PubMed] [Google Scholar]
- Dubiella C.; Baur R.; Cui H.; Huber E. M.; Groll M. Selective Inhibition of the Immunoproteasome by Structure-Based Targeting of a Non-catalytic Cysteine. Angew. Chem., Int. Ed. 2015, 54, 15888–15891. 10.1002/anie.201506631. [DOI] [PubMed] [Google Scholar]
- Kuhn D. J.; Hunsucker S. A.; Chen Q.; Voorhees P. M.; Orlowski M.; Orlowski R. Z. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 2009, 113, 4667–4676. 10.1182/blood-2008-07-171637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin G.; Xin B. T.; Kraus M.; van der Stelt M.; van der Marel G. A.; Kisselev A. F.; Driessen C.; Florea B. I.; Overkleeft H. S. A Set of Activity-Based Probes to Visualize Human (Immuno)proteasome Activities. Angew. Chem., Int. Ed. 2016, 55, 4199–4203. 10.1002/anie.201509092. [DOI] [PubMed] [Google Scholar]
- Groll M.; Korotkov V. S.; Huber E. M.; de Meijere A.; Ludwig A. A Minimal beta-Lactone Fragment for Selective beta5c or beta5i Proteasome Inhibitors. Angew. Chem., Int. Ed. 2015, 54, 7810–7814. 10.1002/anie.201502931. [DOI] [PubMed] [Google Scholar]
- de Bruin G.; Huber E. M.; Xin B.-T.; van Rooden E. J.; Al-Ayed K.; Kim K.-B.; Kisselev A. F.; Driessen C.; van der Stelt M.; van der Marel G. A.; et al. Structure-based design of beta1i or beta5i specific inhibitors of human immunoproteasomes. J. Med. Chem. 2014, 57, 6197–6209. 10.1021/jm500716s. [DOI] [PubMed] [Google Scholar]
- Sosic I.; Gobec M.; Brus B.; Knez D.; Zivec M.; Konc J.; Lesnik S.; Ogrizek M.; Obreza A.; Zigon D.; et al. Nonpeptidic Selective Inhibitors of the Chymotrypsin-Like (beta5 i) Subunit of the Immunoproteasome. Angew. Chem., Int. Ed. 2016, 55, 5745–5748. 10.1002/anie.201600190. [DOI] [PubMed] [Google Scholar]
- Fan H.; Angelo N. G.; Warren J. D.; Nathan C. F.; Lin G. Oxathiazolones Selectively Inhibit the Human Immunoproteasome over the Constitutive Proteasome. ACS Med. Chem. Lett. 2014, 5, 405–410. 10.1021/ml400531d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H.; Baur R.; Le Chapelain C.; Dubiella C.; Heinemeyer W.; Huber E. M.; Groll M. Structural elucidation of a nonpeptidic inhibitor specific for the human immunoproteasome. ChemBioChem 2017, 18, 523–526. 10.1002/cbic.201700021. [DOI] [PubMed] [Google Scholar]
- Dubiella C.; Cui H.; Gersch M.; Brouwer A. J.; Sieber S. A.; Kruger A.; Liskamp R. M. J.; Groll M. Selective inhibition of the immunoproteasome by ligand-induced crosslinking of the active site. Angew. Chem., Int. Ed. 2014, 53, 11969–11973. 10.1002/anie.201406964. [DOI] [PubMed] [Google Scholar]
- Aminake M. N.; Arndt H.-D.; Pradel G. The proteasome of malaria parasites: A multi-stage drug target for chemotherapeutic intervention?. Int. J. Parasitol.: Drugs Drug Resist. 2012, 2, 1–10. 10.1016/j.ijpddr.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Chapelain C.; Groll M. Rational Design of Proteasome Inhibitors as Antimalarial Drugs. Angew. Chem., Int. Ed. 2016, 55, 6370–6372. 10.1002/anie.201602519. [DOI] [PubMed] [Google Scholar]
- Czesny B.; Goshu S.; Cook J. L.; Williamson K. C. The proteasome inhibitor epoxomicin has potent Plasmodium falciparum gametocytocidal activity. Antimicrob. Agents Chemother. 2009, 53, 4080–4085. 10.1128/AAC.00088-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreidenweiss A.; Kremsner P. G.; Mordmuller B. Comprehensive study of proteasome inhibitors against Plasmodium falciparum laboratory strains and field isolates from Gabon. Malar. J. 2008, 7, 187. 10.1186/1475-2875-7-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantt S. M.; Myung J. M.; Briones M. R.; Li W. D.; Corey E. J.; Omura S.; Nussenzweig V.; Sinnis P. Proteasome inhibitors block development of Plasmodium spp. Antimicrob. Agents Chemother. 1998, 42, 2731–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Ponder E. L.; Verdoes M.; Asbjornsdottir K. H.; Deu E.; Edgington L. E.; Lee J. T.; Kirk C. J.; Demo S. D.; Williamson K. C.; et al. Validation of the proteasome as a therapeutic target in Plasmodium using an epoxyketone inhibitor with parasite-specific toxicity. Chem. Biol. 2012, 19, 1535–1545. 10.1016/j.chembiol.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; van der Linden W. A.; Verdoes M.; Florea B. I.; McAllister F. E.; Govindaswamy K.; Elias J. E.; Bhanot P.; Overkleeft H. S.; Bogyo M. Assessing subunit dependency of the Plasmodium proteasome using small molecule inhibitors and active site probes. ACS Chem. Biol. 2014, 9, 1869–1876. 10.1021/cb5001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G.; Chidawanyika T.; Tsu C.; Warrier T.; Vaubourgeix J.; Blackburn C.; Gigstad K.; Sintchak M.; Dick L.; Nathan C. N,C-Capped dipeptides with selectivity for mycobacterial proteasome over human proteasomes: role of S3 and S1 binding pockets. J. Am. Chem. Soc. 2013, 135, 9968–9971. 10.1021/ja400021x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Tsu C.; Blackburn C.; Li G.; Hales P.; Dick L.; Bogyo M. Identification of potent and selective non-covalent inhibitors of the Plasmodium falciparum proteasome. J. Am. Chem. Soc. 2014, 136, 13562–13565. 10.1021/ja507692y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; O’Donoghue A. J.; van der Linden W. A.; Xie S. C.; Yoo E.; Foe I. T.; Tilley L.; Craik C. S.; da Fonseca P. C. A.; Bogyo M. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530, 233–236. 10.1038/nature16936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Yakushi T.; Parlati F.; Mackinnon A. L.; Perez C.; Ma Y.; Carter K. P.; Colayco S.; Magnuson G.; Brown B.; et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat. Chem. Biol. 2017, 13, 486–493. 10.1038/nchembio.2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C.; Li J.; Parlati F.; Rouffet M.; Ma Y.; Mackinnon A. L.; Chou T.-F.; Deshaies R. J.; Cohen S. M. Discovery of an Inhibitor of the Proteasome Subunit Rpn11. J. Med. Chem. 2017, 60, 1343–1361. 10.1021/acs.jmedchem.6b01379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manasanch E. E.; Orlowski R. Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber E. M.; Heinemeyer W.; Groll M. Bortezomib-resistant mutant proteasomes: structural and biochemical evaluation with carfilzomib and ONX 0914. Structure 2015, 23, 407–417. 10.1016/j.str.2014.11.019. [DOI] [PubMed] [Google Scholar]
- Lai A. C.; Crews C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16, 101–114. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salami J.; Crews C. M. Waste disposal—An attractive strategy for cancer therapy. Science 2017, 355, 1163–1167. 10.1126/science.aam7340. [DOI] [PubMed] [Google Scholar]
- Cromm P. M.; Crews C. M. Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell. Chem. Biol. 2017, 10.1016/j.chembiol.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leestemaker Y.; de Jong A.; Witting K. F.; Penning R.; Schuurman K.; Rodenko B.; Zaal E. A.; van de Kooij B.; Laufer S.; Heck A. J. R.; et al. Proteasome Activation by Small Molecules. Cell Chem. Biol. 2017, 24, 725–736.e7. 10.1016/j.chembiol.2017.05.010. [DOI] [PubMed] [Google Scholar]