

Abstract
Motivation
Despite considerable advancements in sequencing and computing technologies, de novo assembly of whole eukaryotic genomes is still a time-consuming task that requires a significant amount of computational resources and expertise. A targeted assembly approach to perform local assembly of sequences of interest remains a valuable option for some applications. This is especially true for gene-centric assemblies, whose resulting sequence can be readily utilized for more focused biological research. Here we describe Kollector, an alignment-free targeted assembly pipeline that uses thousands of transcript sequences concurrently to inform the localized assembly of corresponding gene loci. Kollector robustly reconstructs introns and novel sequences within these loci, and scales well to large genomes—properties that makes it especially useful for researchers working on non-model eukaryotic organisms.
Results
We demonstrate the performance of Kollector for assembling complete or near-complete Caenorhabditis elegans and Homo sapiens gene loci from their respective, input transcripts. In a time- and memory-efficient manner, the Kollector pipeline successfully reconstructs respectively 99% and 80% (compared to 86% and 73% with standard de novo assembly techniques) of C.elegans and H.sapiens transcript targets in their corresponding genomic space using whole genome shotgun sequencing reads. We also show that Kollector outperforms both established and recently released targeted assembly tools. Finally, we demonstrate three use cases for Kollector, including comparative and cancer genomics applications.
Availability and Implementation
Kollector is implemented as a bash script, and is available at https://github.com/bcgsc/kollector
Supplementary information
Supplementary data are available at Bioinformatics online.
Introduction
Production of high quality reference genome sequences for non-model organisms remains a challenging undertaking, especially for large (>1 Gbp) genomes. For such projects, de novo whole-genome assembly typically requires billions of sequencing reads from several different types of DNA libraries. Processing these large volumes of data, and using them to assemble a genome, usually necessitates access to a high-performance computing environment, significant expertise and specialized software (Nagarajan and Pop, 2013). An attractive alternative for generating reference sequences can be achieved through targeted assembly of gene/transcript sequences of interest. Even for species with scant transcriptomic sequence information, there are likely existing sequences that could be used to aid de novo assembly, such as homologous gene sequences from a related organism. Utilization of these data helps to localize the assembly problem, and ensures that the desired sequences (e.g. genic regions) are fully reconstructed. A favorable consequence of this localization is a reduction of the complexity and computational cost relative to that of a whole genome assembly. In practice, however, the computational cost of identifying reads related to target sequences has remained challenging due variation and novel sequences not found within a target.
The first solution for the reconstruction of specific targets was accomplished with TASR, an alignment-free targeted de novo assembly software (Warren and Holt, 2011). This method was followed by Mapsembler (Peterlongo and Chikhi, 2012), which uses read alignments to guide the process, and presented a more memory-efficient and faster alternative. These pioneering targeted assembly technologies were originally designed to reconstruct specific transcript variants, fusion transcripts or genes from large consortium shotgun data, and have now found applications in human health research (Brown et al., 2014; Warren et al., 2012). These method are notably very efficient as they pair sequence recruitment with built in de novo assembly algorithms that utilizes internal data structures directly. Unfortunately, these targeted assembly tools have limited utility when an incomplete sequence bait is used to localize reads for assembly, such as using a transcript to reconstruct a whole genic region or the use of homologous, yet divergent, sequences as bait.
To reconstruct incomplete regions, most modern methods utilize an iterative read recruitment process to fill in gaps. MITObim (Hahn et al., 2013), GRAbB (Brankovics et al., 2016) and aTRAM (Allen et al., 2015) recruit reads based on sequences derived from read recruited in earlier iterations to extend novel regions of previously incomplete sequences. MITObim was designed to assemble mitochondrial sequences, and works by recruiting reads that share a 31-mer (subsequence of length 31) in common with the target, cycling through the read set multiple times until the target is reconstructed. GRAbB, works in a similar fashion, however it is designed to recruit reads for multiple targets at a time, and thus had been shown to computationally out-perform MITObim (Brankovics et al., 2016). Finally, aTRAM is designed for assembling orthologs from a related genome, and utilizes BLAST (Altschul et al., 1990) to index portions of the sequence reads so multiple iterations do not require multiple passes through the raw reads during the recruitment process at the cost of a higher memory usage. For each of these tools, after each cycle of recruitment, an assembly using an established assembly tools (e.g. Velvet; Zerbino and Birney, 2008) is performed and fed into later iterations to extend the bait sequence.
Advances in RNA-Seq technology and de novo assembly tools (Grabherr et al., 2011; Peng et al., 2013; Robertson et al., 2010) have made high-quality transcriptomes from non-model organisms increasingly available, which can provide a valuable resource to inform targeted assembly of genic regions. In this manuscript we introduce Kollector, an alignment-free targeted assembly pipeline that can use whole transcriptome assemblies to filter and classify whole genome shotgun sequencing reads, thereby localizing the de novo assembly of corresponding genic loci. The pipeline collects genomic reads related to target loci using a novel data structure called progressive Bloom filters implemented within BioBloom Tools (BBT) (Chu et al., 2014), and assembles them with ABySS (Simpson et al., 2009), a de Bruijn graph (Pevzner et al., 2001) short read de novo assembler. Kollector is able to expand intronic regions iteratively, but in practice requires fewer number of iterations than previous methods by greedily populating progressive Bloom filters. We demonstrate efficient targeted assembly of Caenorhabditis elegans and Homo sapiens genes by Kollector, and its relative effectiveness compared to four published targeted assembly tools. We also show applications of Kollector in comparative and cancer genomics use cases.
2 Materials and methods
2.1 Progressive Bloom filters
Bloom filters are memory-efficient probabilistic data structures with tunable false positives rates (Bloom, 1970). They are effective in storing sequences for use in fast, specific and sensitive sequence classification (Chu et al., 2014; Stranneheim et al., 2010). This works by shredding (k-merizing) a reference sequence, such as transcript sequences, into its constituent k-mers (subsequences of a uniform length k), and seeding a Bloom filter with these k-mers. This seeding sequence is also referred to as a bait sequence. One can then query the Bloom filter using k-mers derived from whole genome sequence reads, and test for sequence similarity at a given threshold. Within this paper, our match criteria is based on a similarity score threshold (a value between 0 and 1) described in the BioBloom Tools (BBT) manuscript (Chu et al., 2014). Reads/queries are then categorized as match or no match.
Implemented within BBT, we introduce efficient read recruitment using a novel data structure called a progressive Bloom filter. In a progressive Bloom filter, we scan a read set and greedily add additional k-mers from matching reads (and their pairs, when available) after each filter query. Because the sequence similarity/k-mer overlap threshold (r parameter) can result in partial overlaps, it allows for addition of new k-mers in the Bloom filter. Consequently, the contents of our Bloom filter will expand into genomic regions not found in our original seed sequences, such as intronic regions. This method works particularly well when read pairs are used, by incorporating the entire k-mer content of a read when its pair registers a positive match (Supplementary Algorithm S1). After the k-mer space is expanded sufficiently, the resulting Bloom filter can be used to again scan through the reads to recruit all relevant reads within the expanded regions of interest.
2.2 Kollector pipeline
In Kollector we first use BBT in progressive Bloom filter mode seeded with input transcript (target) sequences. We scan a set of genomic reads for reads pairs that share a user-defined amount of k-mer overlap based on the (r parameter) referred to as the tagging stage of Kollector. The threshold parameter r should remain high (0.5–0.9) to minimize off-target k-mer recruitment, but in the case of high read error rates, divergent or low coverage regions, a low value of r (0.2–0.5) might be used instead. Read tagging continues until the progressive Bloom filter reaches a user-defined maximum number of k-mers (n parameter), or until all the genomic reads are processed. The n parameter determines the maximum number of k-mers the progressive Bloom filter can contain to maintain a maximum FPR before the second stage mentioned below. The maximum FPR is determined by the f parameters in BBT, set to 0.001 by Kollector. In practice, n should be set to the maximum expectedsize of the total genic space being reconstructed. At this FPR the memory usage of the progressive Bloom filter will not exceed 15n bits.
A real-data illustration of this process for a single C.elegans transcript C17E4.10 is shown in Figure 1. As one would expect, initially tagged reads are derived from exonic regions, but as those reads are added to the filter it allows for reads from intronic regions to be tagged as well (from recruiting their pair), until reads spanning the entire gene have been added to the filter. This progressive Bloom filter is used to recruit reads that share a k-mer overlap with the filter based on the length of the read (s parameter, defined in BBT (Chu et al., 2014)) within the entire read set in the second stage of the pipeline (All Recruited reads in Fig. 1). The threshold parameter s uses the same similarity metric as r when evaluating reads, however s is not as critical since off-target read classifications will not propagate, and can remain safely set to values between 0.2 and 0.5.
Fig. 1.
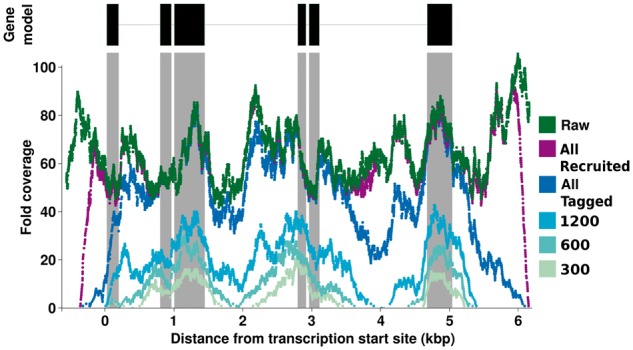
Genomic read tagging by progressive Bloom filter. Fold coverage of tagged reads is shown at points in time (first 300, 600 and 1200 and all tagged reads) during a single run with the C.elegans C17E4.10 gene as target. The raw read coverage is indicated solely as a baseline, to show the tagging process of the progressive Bloom filter. The final recruited coverage is shown in purple after the second stage of Kollector has collected all the reads. The fold coverage was calculated after read alignment to the reference transcript. In the Gene Model track, the black rectangles depict the exons and the connecting grey line depicts the introns
In the third stage, the recruited reads are assembled with ABySS (v1.5.2; np = 12 CPUs, k = 96), and finally the input transcripts are aligned to the assembly with GMAP (version 2016-05-01; t = 10) (Wu and Watanabe, 2005) to report assembled scaffolds that contain the targeted loci. The peak memory of Kollector is dominated by of the downstream assembly algorithm used. By default, our pipeline uses ABySS 1.5.2 but in principle other assembly algorithms may be used as well.
This pipeline can also be run in an iterative mode. Un- or partially-assembled targets from an earlier iteration may be selected as input, along with other targets, and fed back to the pipeline, as the read localization process and the resulting de Bruijn graph complexity are dependent on the context represented in the Bloom filters. Therefore, iterations may result in successful reconstructions for targets that had failed previous attempts.
To improve the performance of Kollector in complex genomes, BBT can also take a Bloom filter of repeat sequences as an additional input, and use it to tag repeats while extending the progressive Bloom filter. Sequences that are tagged as repeats are not used for the expansion of the k-mer space within the filter, thus preventing the recruitment of off-target regions (Fig. 2).
Fig. 2.
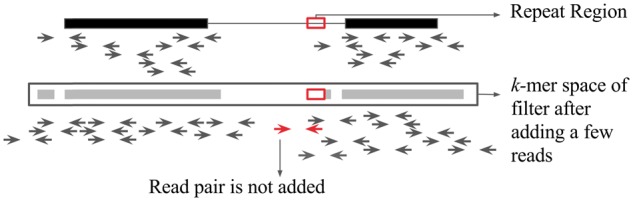
Graphic of progressive Bloom filter with repeat filtering. The black bars depict k-mers derived from the input transcripts. The grey bars depict the k-mer space after a few reads have been tagged and their constituent k-mers added to the Bloom filter
3 Results
We tested our tool on a variety of datasets (Table 1), and in different targeted assembly contexts to show the utility and efficacy of Kollector. All benchmarks described below were obtained using a single high performance computer with 48 GB of RAM and 12 dual Intel Xeon X5650 2.66 GHz CPUs.
Table 1.
Datasets used in targeted assembly experiments
| # | Species | Datatype | Read lengths | Total bases | Raw Cov. | Source |
|---|---|---|---|---|---|---|
| 1 | C.elegans | WGS | 110 bp | 7.5 Gbp | 75x | SRA Accession: DRR008444 |
| C.elegans | Transcripts | – | 27 Mbp | – | RefSeq mRNA (>1kb) | |
| 2 | H.sapiens | WGS | 250 bp | 229 Gbp | 70x | SRA Accession: ERR309932 |
| H.sapiens | Transcripts | – | 138 Mbp | – | TCGA barcode: 22-4593-01 assembled using Trans-ABySS (v1.5.1; k = 42) | |
| 3 | P.glauca | WGS | 150–300 bp | 1.2 Tbp | 48x | SRA Accession: SRR1982100 |
| P.glauca | Transcripts | – | 23 Mbp | – | Genome Annotation: GCA_000966675.1 (high confidence genes) | |
| 4 | P.schaeffi | WGS | 100 bp | 12.8 Gbp | 128x | SRA Accession: SRX390495 |
| P.Humanus | Transcripts | – | 2.44 Mbp | – | Dryad DOI: http://dx.doi.org/10.5061/dryad.9fk1s | |
| 5 | M.musculus | WGS | 150 bp | 116 Gbp | 41x | SRA Accession: SRX1595526 |
| H.sapiens | Transcripts | – | 57 Mbp | – | Ensembl bioMART | |
| 6 | H.sapiens | WGS | 100 bp | 13.8 Gbp | 4x | TCGA barcode: TCGA-BA-4077 (subset) |
| HPV 16 | Ref. Genome | – | 8 Kbp | – | Papillomavirus Episteme |
The white spruce whole genome shotgun sequence read data reported in Table 1 dataset #3 is available at the SRA under accession SRP041401 and Genome Annotation from the high-confidence genes are available at ftp://ftp.bcgsc.ca/supplementary/PG29_20140822/high_confidence_genes.fasta.
3.1 Targeted gene assembly using transcript sequences
3.1.1 Kollector assemblies
Kollector performs incrementally better when used iteratively, where genes not assembled in initial stages are provided as input for the next iteration. After each iteration, the target transcript sequences are aligned to the Kollector output with GMAP (Wu and Watanabe, 2005), and those that have a unique match to a single genomic contig along a certain sequence identity (default = 90%) are deemed to have been successfully reconstructed. Transcripts that do not satisfy this criterion are re-tried in the subsequent iteration by setting a lower specificity for sequence selection during the read collection phase. This is achieved by lowering the r parameter in each iteration (e.g. 0.90, 0.70, 0.50, 0.30, 0.20), while keeping the other parameters constant (s = 0.50, n = 100 000 000 in our experiments).
Before running Kollector on H.sapiens, we randomly divided the transcriptome (Table 1, #2) into five bins of ∼10k transcripts each to prevent the memory usage of ABySS v1.5.2 from exceeding 48 GB of RAM. Each bin is used to initiate a single progressive Bloom filter in a separate Kollector run with a k-mer cap (-n) of 100 000 000. Each bin took 29 GB of RAM and completed within 12 h. Isoforms split across different bins may assemble the same genic region multiple times. Similarly, isoforms within the same bin will help reinforce the assembly of their common target locus from the improved read tagging. Highly similar transcripts originating from multi-copy genes may be collapsed into a single sequence, however, the extent of this depends on the assembly algorithm used by Kollector.
Because progressive Bloom filters are also sensitive to the order of reads in the input file, Kollector has the option to shuffle the genomic reads before each iteration to reduce any potential bias created by read order (Fig. 3). In our tests, read shuffling led to significant gains in the overall assembly success for both species, reaching 98.7% in C.elegans (13 378 out of 13 556 assembled) and 80.1% (41 631 out of 52 006 assembled) in H.sapiens.
Fig. 3.

Performance of Kollector for assembling target sequences in (A) C. elegans and (B) H. sapiens. Genomic reads were randomly shuffled prior to being provided as input to the pipeline (solid lines, mean of 10 independent experiments). For comparison, runs with no shuffling are also depicted (dashed lines). The success rate was calculated as the proportion of target genes successfully assembled over five iterations
To determine the efficacy of our targeted approach compared to a regular de novo assembly in reconstructing the genic space, we performed a whole genome assembly of the C.elegans and H.sapiens datasets discussed above. We used ABySS v1.5.2 to assemble both genomes, utilizing the same assembly k-mer size used within our targeted assemblies. This value of k was used because it yielded the highest N50 in a set of k-mer sweeps on the whole genome assembly. We found that only 85.7% and 72.9% of all genic regions were completely captured (within a single contig) in the C.elegans and H.sapiens whole genome de novo assemblies, respectively. Compared to 98.7% and the 80.1% with Kollector, these results show that performing targeted assembly increases reconstruction contiguity in genic regions. In addition, each complete whole genome assembly required more computational resources than Kollector, requiring 3 hours with 8 GB (single node) and 41 hours with 857 GB (18 nodes) for the C.elegans and H.sapiens assemblies, respectively.
We investigated the failed reconstruction of 199 C.elegans transcripts (unshuffled experiment, Fig 3A dashed line), and found that the failed targets had on average a longest intron length significantly larger than the successfully assembled ones (P = 1.5 × 10 − 8; Student’s t-test; Fig. 4A), indicating that the failure was due in part to the lengths of the longest introns. For targets with maximum intron lengths of approximately 20 kbp, we expect 50% of the reconstructions to fail reconstruction. However, we note that these large intron genes make-up a very small proportion of the total dataset (Fig. 4B). Although the distributions suggest substantial overlap, and there were many long targets with successful assembly, with lengths comparable to the failed targets, our statistical test suggests that Kollector has a bias towards assembling smaller genes, likely due to the challenge of identifying enough genomic reads to connect exons separated by long introns. We expect that the use of longer reads and insert sizes (possibly mate-pair reads) could help alleviate some of these issues.
Fig. 4.

(A) Longest intron length comparison between the C.elegans target genes that are successfully assembled (top) versus those that failed (bottom). Notches in the boxes represent a 95% confidence interval around the median. The length difference between two groups, represented on a logarithmic scale on the x-axis, is found to be statistically significant by t-test (P = 1.5 × 10 − 8). This analysis yields the same result for gene length comparison (Supplementary Fig. S2) B) Proportion of successful gene assemblies versus longest introns (bars) with number of gene in each bin (lines). The first and second bins make up 10 237 and 1496 genes respectively, and make up 86% of all genes in the dataset (Supplementary Fig. S1)
To evaluate the accuracy of the genomic contigs produced by Kollector, we aligned the output of a Kollector run from both species to their respective reference genome with BLASTn (Altschul, 1990), calling a correct alignment at a threshold of 95% query coverage and 95% sequence identity. Doing so, we find that 99.7% or more of the assembled genes satisfy these criteria in both species (Table 2).
Table 2.
Accuracy of Kollector-assembled genes
| Species | Number of genes assembled | Number of genes aligned | Success rate |
|---|---|---|---|
| C.elegans | 13 378 | 13 356 | 99.8 |
| H.sapiens | 41 631 | 41 525 | 99.7 |
Due to the greedy nature of our algorithm it is possible to recruit off-target regions. The primary source of error is due to repeats, with minor contributions from false positives and read errors. We investigated these off-target events by aligning H.sapiens transcripts to the genome. We extracted the regions based on the start and end coordinates of all contigs produced by Kollector that aligned. We found that for contigs >500bp, only 40% were on target. Though this may seem low, this assembly still represents a significantly smaller subset of the whole genome, and this analysis does not take into account contigs generated directly upstream of each gene.
3.1.2 Comparisons against mapsembler, TASR, aTRAM and GRAbB
Although not originally and specifically designed for reconstructing genomic loci from transcript sequences, we tested Mapsembler2 (v2.2.4) and TASR (v1.5.1) using the C.elegans genomic dataset and input transcripts. For Mapsembler2, we used the default k-mer size (k = 31) and the consensus sequence mode (-i 2), which means extensions of the target sequence are collapsed. We ran TASR using the independent, de novo assembly mode (-i 1) with default parameters, meaning targets are only used for read recruitment and not for seeding the assembly. We evaluated results of these tools with the same GMAP alignment criteria that we reported for Kollector. In our tests, Mapsembler2 and TASR assembled 28% and 18% of the targets, respectively (Table 3). In contrast, Kollector was able to assemble 97% of the target gene set in the first iteration alone. This is because Mapsembler2 and TASR are designed for re-assembling input targets within their respective sequence boundaries, with limited extension capability in the flanking regions, limiting their utility beyond single exon genes. Kollector’s progressive filtering algorithm, unlike the former approaches, can incorporate reads derived from intronic regions for assembly, as discussed.
Table 3.
Comparison with Mapsembler2 and TASR
| Method | Number of targets attempted | Number of targets assembled | Percentage of targets assembled | Wall clock time (h) | Peak memory (GB) |
|---|---|---|---|---|---|
| Kollector | 13 556 | 13 144 | 96.96 | 2 | 4.8 |
| Mapsembler2 | 13 556 | 3 742 | 27.60 | 2 | 9.3 |
| TASR | 13 556 | 2418 | 17.83 | 3 | 15.6 |
| aTram | 1000 | 731 | 73.1 | 38 | 2.4 |
| GRAbB | 1000 | 425 | 42.5 | 48a | 3.1 |
Time of termination.
We also tested iterative read recruitment methods aTRAM and GRAbB on a random subset of 1000 transcripts from our C.elegans dataset. A smaller subset was chosen as the methods proved intractable on larger datasets. To complete the computation of GRAbB, it requires stopping conditions such as a minimum length of assembly for each target. We provided GRAbB with the exact genomic lengths specified by the reference annotations. Despite this, we were unable to finish computation of GRAbB in a tractable amount of time, and instead took the intermediate results of the method after running GRAbB for 48 hours. We found that on these read subsets, aTRAM and GRAbB assembled 73.1% and 42.5% of the targets respectively. Despite the smaller size of the subset, Kollector outperformed both tools in speed, while utilizing a comparable amount of memory.
3.1.3 Scaling to large genomes: gene assembly in white spruce
Assembling complex and very large eukaryotic genomes is a computationally demanding process. Therefore, a targeted assembly approach for retrieving biologically important gene sequences remains an attractive option for most researchers. We have tested such a use case for Kollector using Picea glauca (white spruce), which has a genome size in the 20 Gbp range (Birol et al., 2013; Warren et al., 2015). For the species, Warren and colleagues derived a high-confidence set of 16 386 gene predictions and associated transcripts. Using these transcript sequences, Kollector was able to reconstruct 13 277 (80.4%) of the original target genes in a single iteration, requiring five processes, each using 43.3 GB of RAM and running for 24 hours.
Researchers are often also interested in the regions immediately upstream and downstream of genes, which contain promoters and other regulatory elements. Due to the nature of the progressive filtering algorithm, Kollector assemblies may extend into these regions. In order to demonstrate this, we aligned the aforementioned high-quality transcript models to the resulting Kollector assemblies, and quantified the amount of sequence upstream and downstream with respect to the transcript. We show that, in addition to a gene’s exonic and intronic sequences, Kollector typically reconstructs approximately 1 kb of sequence beyond the 5’ and 3’ ends of the target transcript (Fig. 5). Such extensions would enable characterization of the regulatory factors and complexes in the proximal promoter of genes of interest by chromatin immunoprecipitation, and would be especially empowering to studies of non-model organisms without available reference genome sequences.
Fig. 5.

Length distribution of flanking regions, after Kollector assembly of P. glauca genes. In order to define the flanking regions, we aligned the high-confidence transcript models of white spruce to our Kollector assemblies and quantified the length of the sequence upstream (dashed line, upper x axis and right-side y axis) and the downstream (solid line, lower x axis and left y axis) of the input transcript alignment
3.2 Targeted cross-species gene assembly
Non-model organisms might not have extensive and well-annotated transcriptomes available to researchers. In such cases, Kollector can use transcript sequences from one species to reconstruct the genic regions of a related species.
3.2.1 Cross-species assembly using kollector
We tested Kollector using H.sapiens transcript sequences as bait to assemble orthologous (>70% sequence identity) Mus musculus genes (Table 1, #4). Despite being separated by approximately 90 million years of evolution (http://timetree.org), Kollector was able to assemble 3295 of 4025 target genes in a single iteration (r = 0.90, assembly k = 96), corresponding to an 81.9% success rate, as assessed using the orthologous M. musculus transcripts. This single iteration took 4 hours using 18.1 GB of RAM.
3.2.2 Comparisons to aTRAM
We assessed Kollector’s performance against aTRAM (Allen et al., 2015), which to our knowledge, is the only tool designed to assemble entire genes (including introns) guided by an input protein or DNA gene sequence with or without introns. In their study, Allen and co-workers (2015) used a dataset of 1534 conserved proteins from human head lice (Pediculus humanus) (Johnson et al., 2014) to assemble orthologous genes in the chimpanzee lice (Pediculus schaeffi) genome. We ran Kollector using the same whole genome shotgun reads and corresponding cDNA sequences for each orthologous gene, and compared the results using two metrics: proportion of the target gene that aligned to the assembled scaffold, and number of assembled genes that passed a reciprocal best BLASTn hit test with each target gene.
In our tests Kollector slightly outperformed aTRAM on both metrics (on average 99.3% to aTRAM’s 98.6%, and 1552 genes passing the reciprocal BLASTn test compared with 1530 in aTRAM). Kollector achieved this task in less than one tenth of aTRAM’s run time (Table 4). The markedly greater speed of Kollector is mainly due to its use of alignment free classification with k-mers and Bloom filters, allowing it to process thousands of transcripts in a single run. Whereas aTRAM relies on iterative alignments to individual targets to recruit enough reads for their assembly. For applications on this relatively small scale, the progressive read filtering algorithm used by Kollector eliminates the need for iterations.
Table 4.
Comparison with aTRAM
| Method | Proportion | Reciprocal Best-BLASTn | Time (h) |
|---|---|---|---|
| Kollector | 99.3 | 1532 | 2 |
| aTRAM | 98.6 | 1530 | 25 |
For these experiments parameterized Kollector with r = 0.9, s = 0.5, k = 48, and n = 10 000 000, values similar to our other experiments. Our success rate suggests that this method is robust in capturing relatively divergent sequences. We think this is due to our progressive filtering algorithm, which recruits reads from more conserved regions first and then uses them to extend the sequence in more divergent regions. Furthermore, the sensitivity of reconstruction of more divergent sequences may be increased by using a low r parameter (the specificity required for read tagging), which may be preferable if evolutionary distance between two species is considerable.
3.3 Whole genome targeted assembly
A prominent application of de novo assembly is the detection of specific, yet novel, sequences, where a mapping approach can introduce bias (Alkan et al., 2011). However, the large sample size of many studies, such as those of cancer genomics consortia (e.g. ICGC, TCGA), can put a strain on computational resources when de novo whole genome assembly is required. Therefore, a fast and reliable targeted assembler, like Kollector, might be an attractive option for most researchers, especially when considering its ability to extend incomplete input sequences.
In order to demonstrate the extent of its utility, we have used Kollector for the targeted assembly of Human Papilloma Virus (HPV) in a cancer sample. The Cancer Genome Atlas consortium has profiled 279 head and neck squamous cell carcinomas (HSNCs), and detected many HPV positive samples, which were experimentally confirmed with immunohistochemistry and in situ hybridization (Cancer Genome Atlas, 2015). We ran Kollector on the genomic data from one of the confirmed samples [TCGA-BA-4077] with HPV type -33. Kollector does not need the bait to match the exact target sequence, as demonstrated in Section 3.2, so we used HPV type-16 reference genome as bait, and were able to re-assemble the complete HPV type-33 genome sequence in a single iteration with less than 15 minutes runtime and using 1 Gb of memory. We also used genomic reads from the matched normal sample as negative control, and, as expected, Kollector did not yield any assembled HPV sequences. Because Kollector uses only sequences contributed by the reads, the assembled strain remains unbiased relative to our bait sequence.
4 Discussion
We have presented Kollector, a targeted de novo genome assembly application that takes transcript sequences and whole genome sequencing reads as input, and assembles the corresponding gene loci. Input transcripts are used to seed progressive Bloom filters, a greedy database that can efficiently expand into intronic regions using sequence overlaps. Due to our alignment free approach, we demonstrate that Kollector can scale well up to large genomes, such as those of human and spruce.
When assembling genes from transcripts, we show that Kollector successfully assembles more gene loci than iterative read recruitment methods aTRAM and GRABb in less time, and assembles more genes than both non-iterative read recruitment methods Mapsembler2 and TASR. However, we note in the latter case that this was expected since these tools were not designed for targeted gene loci assembly using RNA transcripts, and are thus unable to fill in large intronic gaps. Kollector also successfully assembles more gene loci than aTRAM when assembling the genic space of a related species.
After our evaluations, we showcased three additional use cases for Kollector. The first one showed that Kollector was able to effectively assemble gene sequences in P.glauca (white spruce), despite its large 20-Gbp genome. These gene assemblies typically included 1 kb of upstream and downstream sequence with respect to the input transcript, illustrating the utility of the approach to examine promoters and other regulatory elements around genes for downstream applications. This demonstrates that for gene-centric investigations, Kollector can be a robust substitute for de novo whole genome assembly, which remains computationally challenging at large scales.
The second use case concerned comparative genomics, where Kollector assembled M.musculus genes using orthologous H.sapiens transcripts as input. This comparative genomics approach is particularly valuable to researchers working on non-model organisms, which might not have extensive and well-annotated transcript sequences available.
Our final demonstration involved the whole genome targeted assembly of HPV in a head and neck cancer sample. Kollector solves this problem by de novo assembling reads of interest, without the risk of introducing artifacts, as typically is the case when aligning reads to a reference genome. Because Kollector can fill in missing sequences by recruiting reads with a progressive Bloom filter, it only requires a limited amount of sequence homology within the bait sequence to fully reassemble a viral sequence. We note that the extent of divergence of the seed sequence that Kollector can use was not fully explored, and thus may be interesting to investigate in future studies.
In conclusion, we expect Kollector to be a valuable addition to the current suite of targeted assembly tools. Not only does it scale to large datasets, it can be used to reconstruct orthologous sequences in non-model organisms, and will find utility in the reconstruction of large regions de novo, using only transcript sequences.
Supplementary Material
Acknowledgements
We would like to thank Golnaz Jahesh from Canada’s Michael Smith Genome Research Centre for her contributions to packaging, testing and releasing of the Kollector software. We also thank Gordon Robertson from Canada’s Michael Smith Genome Research Centre for his insights regarding HPV strain detection.
Funding
The research presented here was funded by the National Human Genome Research Institute of the National Institutes of Health (under award number R01HG007182), with additional support provided by Genome Canada, Genome British Columbia, the British Columbia Cancer Foundation and Canada Foundation for Innovation. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, or other funding organizations.
Conflict of Interest: none declared.
References
- Alkan C. et al. (2011) Genome structural variation discovery and genotyping. Nat. Rev. Genet., 12, 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J.M. et al. (2015) aTRAM – automated target restricted assembly method: a fast method for assembling loci across divergent taxa from next-generation sequencing data. BMC Bioinformatics, 16, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Birol I. et al. (2013) Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics, 29, 1492–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom B.H. (1970) Space/time trade-offs in hash coding with allowable errors. Commun. ACM, 13, 422–426. [Google Scholar]
- Brankovics B. et al. (2016) GRAbB: selective assembly of genomic regions, a new niche for genomic research. PLoS Comput. Biol., 12, e1004753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S.D. et al. (2014) Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res., 24, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas, N. (2015) Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature, 517, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J. et al. (2014) BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics, 30, 3402–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr M.G. et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol., 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn C. et al. (2013) Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res., gkt371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K.P. et al. (2014) Rates of genomic divergence in humans, chimpanzees and their lice. Proc. R. Soc. B Biol. Sci., 281, 20132174–20132174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan N., Pop M. (2013) Sequence assembly demystified. Nat. Rev. Genet., 14, 157–167. [DOI] [PubMed] [Google Scholar]
- Peng Y. et al. (2013) IDBA-tran: a more robust de novo de Bruijn graph assembler for transcriptomes with uneven expression levels. Bioinformatics, 29, i326–i334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlongo P., Chikhi R. (2012) Mapsembler, targeted and micro assembly of large NGS datasets on a desktop computer. BMC Bioinformatics, 13, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevzner P.A. et al. (2001) An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci., 98, 9748–9753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G. et al. (2010) De novo assembly and analysis of RNA-seq data. Nat. Methods, 7, 909–912. [DOI] [PubMed] [Google Scholar]
- Simpson J.T. et al. (2009) ABySS: A parallel assembler for short read sequence data. Genome Res., 19, 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranneheim H. et al. (2010) Classification of DNA sequences using Bloom filters. Bioinformatics, 26, 1595–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren R.L. et al. (2012) Derivation of HLA types from shotgun sequence datasets. Genome Med., 4, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren R.L., Holt R.A. (2011) Targeted assembly of short sequence reads. PLoS One, 6, e19816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren R.L. et al. (2015) Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. Cell Mol. Biol., 83, 189–212. [DOI] [PubMed] [Google Scholar]
- Wu T.D., Watanabe C.K. (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics, 21, 1859–1875. [DOI] [PubMed] [Google Scholar]
- Zerbino D.R., Birney E. (2008) Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res., 18, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.