

Abstract
Hydrogen sulfide (H2S) is a signaling molecule that is toxic at elevated concentrations. In eukaryotes, it is cleared via a mitochondrial sulfide oxidation pathway, which comprises sulfide quinone oxidoreductase, persulfide dioxygenase (PDO), rhodanese, and sulfite oxidase and converts H2S to thiosulfate and sulfate. Natural fusions between the non-heme iron containing PDO and rhodanese, a thiol sulfurtransferase, exist in some bacteria. However, little is known about the role of the PDO–rhodanese fusion (PRF) proteins in sulfur metabolism. Herein, we report the kinetic properties and the crystal structure of a PRF from the Gram-negative endophytic bacterium Burkholderia phytofirmans. The crystal structures of wild-type PRF and a sulfurtransferase-inactivated C314S mutant with and without glutathione were determined at 1.8, 2.4, and 2.7 Å resolution, respectively. We found that the two active sites are distant and do not show evidence of direct communication. The B. phytofirmans PRF exhibited robust PDO activity and preferentially catalyzed sulfur transfer in the direction of thiosulfate to sulfite and glutathione persulfide; sulfur transfer in the reverse direction was detectable only under limited turnover conditions. Together with the kinetic data, our bioinformatics analysis reveals that B. phytofirmans PRF is poised to metabolize thiosulfate to sulfite in a sulfur assimilation pathway rather than in sulfide stress response as seen, for example, with the Staphylococcus aureus PRF or sulfide oxidation and disposal as observed with the homologous mammalian proteins.
Keywords: enzyme kinetics, hydrogen sulfide, iron, sulfur, X-ray crystallography
Introduction
Hydrogen sulfide (H2S)2 is a gas with toxicity comparable with that of cyanide (1). It is also a signaling molecule produced by prokaryotes and eukaryotes and elicits a broad range of physiological effects (2–4). Notably, it acts as a cardioprotectant during myocardial ischemia reperfusion and is implicated in regulation of the cellular stress response, apoptosis, and inflammation (5–8). Steady-state H2S levels are governed by the rates of its production and clearance (9). In eukaryotes, H2S is produced by the transsulfuration pathway enzymes, cystathionine β-synthase and cystathionine γ-lyase, and by the cysteine catabolic pathway enzyme, mercaptopyruvate sulfurtransferase (10, 11). Sulfide clearance occurs primarily via the mitochondrial sulfide oxidation pathway (12, 13). Several enzymes are involved in the mitochondrial sulfur oxidation pathway: sulfide quinone oxidoreductase, the persulfide dioxygenase (PDO, also known as ETHE1), rhodanese, and sulfite oxidase. The main products of the sulfide oxidation pathway are thiosulfate and sulfate.
PDO is a member of the 2His–1Asp mononuclear iron-containing enzyme superfamily and catalyzes the oxidation of glutathione persulfide (GSSH) to sulfite (14, 15). Mutations in PDO result in ethylmalonic encephalopathy, an autosomal recessive disorder (16). Over 20 mutations have been described in the ethe1 gene (17). Rhodanese is a thiosulfate sulfurtransferase; in humans, it preferentially catalyzes sulfur transfer from GSSH to sulfite producing thiosulfate and GSH (Reaction 1) (18).
| (Eq. 1) |
Rhodanese domains are universal structural modules that occur in one of three variations (19). They can be found as tandem repeats as in human rhodanese, which possesses a non-catalytic N-terminal domain and a catalytically active C-terminal domain (18, 20). Alternatively, they can be found as a single domain in proteins such as human TSTD1 (21) and Escherichia coli GlpE (22) or as natural fusions with other protein domains as in the Staphylococcus aureus CstB (23).
In prokaryotes, H2S can be generated via several pathways. It is an end product of the dissimilatory sulfate-reducing pathway in which sulfate is used as a terminal electron acceptor during anaerobic respiration (24). H2S is an intermediate in the assimilatory sulfate-reducing pathway where it is produced from sulfite by sulfite reductase and is used as a substrate for cysteine synthesis (25). Additionally, H2S can be produced by orthologs of the eukaryotic H2S biosynthetic enzymes cystathionine β-synthase and cystathionine γ-lyase and mercaptopyruvate sulfurtransferase, which have been identified in several bacterial species and suggested to play a role in defense against antibiotic-induced oxidative stress (26). H2S metabolism by prokaryotes combined with its toxicity at high concentrations necessitates mechanisms for regulating its levels (27). The sulfide anion efflux transporter described in Clostridium difficile represents one mechanism for clearing sulfide via export (28). Additionally, several bacterial species possess orthologs of eukaryotic mitochondrial sulfide oxidation pathway enzymes and can presumably dispose sulfide following its oxidation (23, 29, 30).
Recently, bacterial proteins that are natural fusions between PDO and rhodanese have been identified (23). Hereafter, we refer to these fusion proteins as PRF (for PDO rhodanese fusion). Bioinformatics analysis reveals that in a subset of these PRFs, e.g. the one from Burkholderia phytofirmans PsJN, the PDO domain is fused to a single rhodanese domain, which is orthologous to human thiol sulfurtransferase, TSTD1, and distinct from the two-domain human rhodanese (Fig. 1A). In another subset, exemplified by CstB (23), the PDO domain is fused to two rhodanese domains (Fig. 1A). Of these, the first (or middle) rhodanese domain is non-catalytic, and the second (or C-terminal) domain houses the sulfurtransferase activity and oxidizes bacillithiol persulfide and coenzyme A persulfide to thiosulfate (23, 30). CstB is implicated in protecting against persulfide stress and H2S toxicity (23).
Figure 1.
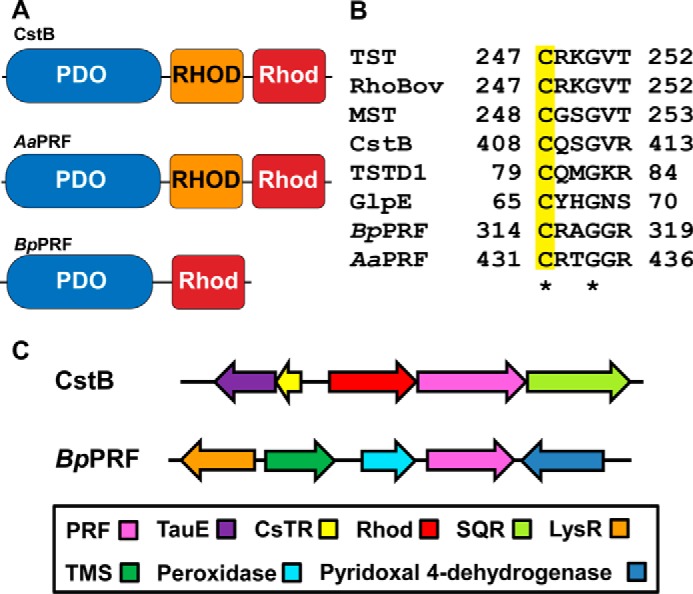
Organization of BpPRF and limited sequence comparison. A, domain organization of PRFs. PRFs form S. aureus (SaPRF) and A. acidocaldarius (AaPRF) have an N-terminal PDO domain, a non-catalytic rhodanese (RHOD) domain, and a catalytic rhodanese domain. BpPRF displays an N-terminal PDO domain and C-terminal catalytic rhodanese (Rhod) domain. B, sequence alignment of rhodanese active-site sequences from human (hTST) and bovine (RhoBov) rhodanese, human mercaptopyruvate sulfurtransferase (MST), E. coli single domain rhodanese (GlpE), human mitochondrial single-domain sulfurtransferase (TSTD1), S. aureus PRF (SaPRF or CstB), B. phytofirmans PRF (BpPRF), and A. acidocaldarius PRF (AaPRF). The conserved active-site cysteine is highlighted in yellow. C, comparison of the genomic contexts of CstB (top) and BpPRF (bottom). The operon encoding CstB (pink) includes a multidomain rhodanese (CstA, red) and an SQR homolog (light green). The adjacent locus includes the polysulfide-sensing repressor CsTR (yellow) and TauE (purple), a putative sulfite permease/transmembrane sulfur compound exporter. The loci adjacent to BpPRF (pink) include a putative peroxidase (cyan) and pyridoxal 4-dehydrogenase (blue), a putative transmembrane (TMS)-type transporter (dark green), and a putative LysR-type regulator (orange).
The existence of PRFs in nature suggests that their mitochondrial homologs, which exist as stand-alone proteins, might interact. Structures of two PRFs are available from structural genomic projects. The first is of CstB (PDB code 3R2U) in which density for the catalytic rhodanese domain is missing. The second structure is of the biochemically uncharacterized Alicyclobacillus acidocaldarius PRF (PDB code 3TP9) (referred to hereafter as AaPRF), which contains a PDO domain fused to two rhodanese domains. Interestingly, these structures reveal the presence of a cysteine-containing loop in the PDO-active site that is absent in the human PDO structure and is not predicted in the sequence of BpPRF. This additional loop is speculated to aid in substrate transfer from the PDO domain to the rhodanese domain (23). As CstB is the only PRF that has been characterized biochemically to date (23, 30), there is a paucity of information on the roles of PRFs in sulfur metabolism in bacteria.
In this study, we have characterized the PRF protein from B. phytofirmans (referred to hereafter as BpPRF), a Gram-negative endophyte originally isolated from onion roots (31). In BpPRF, the N-terminal non-heme Fe(II)-containing PDO domain is followed by a rhodanese domain containing the signature CRXGX(T/R) active-site motif (Fig. 1B). The N-terminal PDO domain of BpPRF displays high sequence identity to human PDO (56%) and displays conservation of active-site iron-binding ligands.
We demonstrate that BpPRF is a bifunctional enzyme that uses the rhodanese domain to preferentially catalyze sulfur transfer from thiosulfate to GSH to form sulfite and GSSH and uses the PDO domain to oxidize GSSH to sulfite (Reactions 2 and 3).
| (Eq. 2) |
| (Eq. 3) |
The crystal structures of BpPRF provide insights into the architecture and the relative juxtaposition of its active sites.
Results
Purification and properties of BpPRF
Recombinant wild-type and variants of BpPRF purified using a one-step protocol were obtained in >95% purity as judged by SDS-PAGE analysis (data not shown). The yield for the full-length proteins (wild type and C314S) was ∼120 mg of protein/liter of culture. The rhodanese domain was obtained with a yield of 22 mg of protein/liter of culture. BpPRF eluted from a size-exclusion column with a molecular mass corresponding to 40 kDa, consistent with it being a monomer (predicted mass = 41.5 kDa). The rhodanese domain eluted as a mixture of a monomer and a dimer (data not shown). The monomeric organization of BpPRF is distinct from that of CstB, which is tetrameric (23).
The thermal stability of the BpPRF variants was assessed in a turbidometric assay. The Tm value for the isolated rhodanese domain (55 ± 2 °C) was slightly higher than for wild type (50 ± 2 °C) and C314S (51.6 ± 0.8 °C) BpPRF. This result indicates that the stand-alone rhodanese domain is well folded and contrasts with the instability of the excised catalytic rhodanese domain of CstB (23).
Metal analysis
Plasma emission spectroscopy revealed the presence of 0.54 mol of iron/mol of monomer wild-type BpPRF. The metal content was also assessed by a colorimetric assay (32), which yielded a similar value (0.60 mol of iron/mol of monomer). The iron content of C314S BpPRF was 0.46 mol of iron/mol of monomer. Attempts to fully reconstitute the metal site with FeCl2 under anaerobic conditions were unsuccessful.
Structure of BpPRF
The crystal structure of BpPRF co-crystallized with thiosulfate was obtained at 1.79 Å resolution by molecular replacement using the AaPRF structure (PDB code 3TP9) as template (Table 1). The final model contains two chains in the asymmetric unit. Each molecule consists of an N-terminal PDO domain (residues 1–230), a 15-residue linker (residues 231–245), and a C-terminal rhodanese domain (residues 246–357) (Fig. 2A). The PDO domain displays a typical metallo-β-lactamase-type fold consisting of two central β-sheet clusters enclosed by three helices on each side (15, 29, 33). Density for eight of the 15 residues in the linker region was missing, indicating that it is disordered (Fig. 2A). The C-terminal domain displays a characteristic rhodanese fold with a five-stranded parallel β-sheet core framed by two and three α-helices on either side of the domain (34). Additionally, a β-hairpin extension is seen on the N-terminal side of the rhodanese domain (Fig. 2A). The two active sites are located at different ends of the protein and are related by a rotation angle of ∼90 ° (supplemental Fig. S1A).
Table 1.
X-ray data collection and refinement statistics
| Data collection | Wild-type PRF | C314S PRF-apo | C314S PRF-GSH |
|---|---|---|---|
| PDB code | 5VE3 | 5VE4 | 5VE5 |
| Space group | P212121 | P6522 | P6522 |
| Cell dimensions | |||
| a, b, c (Å) | 63.7, 108.3, 119.6 | 84.5, 84.5, 549.4 | 83.5, 83.5, 547.6 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 120 | 90, 90, 120 |
| X-ray source | APS 23ID-D | APS 23ID-B | APS 23ID-B |
| Wavelength (Å) | 1.033 | 1.033 | 1.033 |
| dmin (Å) | 1.79 (1.86–1.79)a | 2.65 (2.75–2.65) | 2.35 (2.43–2.35) |
| R-merge | 0.119 (1.47) | 0.126 (1.35) | 0.126 (1.56) |
| Inner-shell R-merge | 0.045 (5.3 Å)b | 0.047 (7.8 Å) | 0.050 (7.0 Å) |
| Average I/σ(I) | 14.6 (1.2) | 11.6 (1.7) | 11.0 (1.6) |
| Completeness (%) | 100 (96.0) | 100 (100) | 99.0 (99.0) |
| Multiplicity | 12.4 (9.0) | 8.6 (8.5) | 8.7 (9.2) |
| Total observations | 966,980 (67,309) | 303,925 (28,966) | 422,379 (43,869) |
| CC1/2 | 0.999 (0.537) | 0.997 (0.898) | 0.998 (0.904) |
| CC* | 1.0 (0.836) | 0.999 (0.973) | 0.999 (0.975) |
| Refinement | |||
| Data range (Å) | 41.26–1.79 | 43.94–2.65 | 41.75–2.35 |
| Reflections used in refinement | 77,982 (7448) | 35,305 (3392) | 48,680 (4773) |
| Rwork/Rfree (%) | 16.1/19.6 | 23.2/26.7 | 19.7/25.2 |
| No. of non-hydrogen atoms | 6051 | 8265 | 8471 |
| Protein | 5538 | 8153 | 8154 |
| Ligands | 2 | 24 | 74 |
| Water | 511 | 88 | 189 |
| Amino acid residues | 698 | 1047 | 1048 |
| Deviation from ideality | |||
| Bond lengths (Å) | 0.013 | 0.003 | 0.007 |
| Bond angles (°) | 1.20 | 0.70 | 0.97 |
| Average B-factor | 38.1 | 81.9 | 76.6 |
| Macromolecules | 37.6 | 82.4 | 77.0 |
| Ligands | 29.6 | 59.0 | 83.1 |
| Solvent | 44.1 | 45.6 | 58.4 |
| Ramachandran plot | |||
| Favored (%) | 98.0 | 96.0 | 97.5 |
| Allowed (%) | 2.0 | 4.0 | 2.3 |
| Outliers (%) | 0 | 0 | 0.2 |
a Values in parentheses pertain to the outermost shell of data.
b dmin inner shell is shown.
Figure 2.
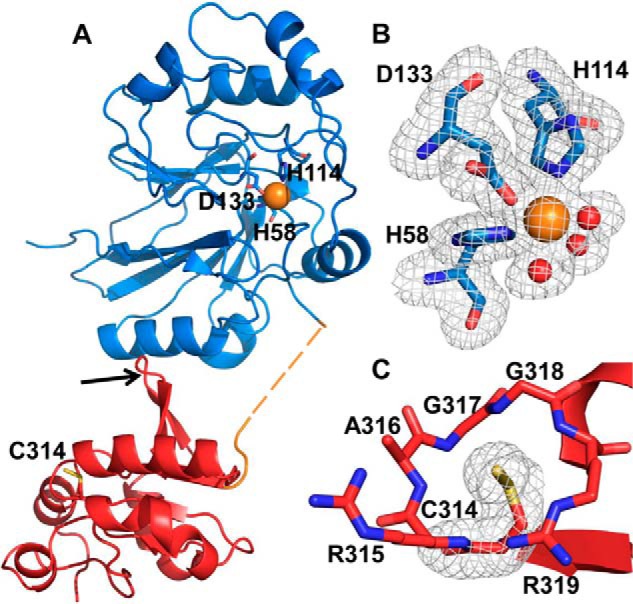
Crystal structure of BpPRF. The X-ray crystal structure of ΔCBpPRF was solved at 1.79 Å resolution by molecular replacement using the AaPRF structure (PDB code 3TP9) as a template. A, structure of the BpPRF monomer consists of an N-terminal PDO domain (blue), a 15-residue linker region (orange), and a C-terminal rhodanese domain (red). The linker region lacked density for eight residues (dashed lines) suggesting a flexible, disordered state. The iron (orange sphere) ligands in the PDO domain and the active-site cysteine, Cys-314, in the rhodanese domain are shown in stick representation. The rhodanese domain β-hairpin extension is highlighted by the red arrow. B, close-up of the PDO-active site with representative electron density (3.0σ Fo − Fc omit density; gray mesh) for side chains of His-58, His-114, Asp-133, and three water molecules (shown as red spheres) coordinated to the iron center (orange sphere). C, close-up of the rhodanese domain active site with representative electron density (3.0σ Fo − Fc omit density; gray mesh) for the Cys-314 side chain displaying the additional density for the persulfide modification.
The PDO domain active site contains a mononuclear non-heme iron coordinated by His-58, His-114, and Asp-133 comprising a 2His:1Asp facial triad. The remaining coordination sites are occupied by water molecules giving an octrahedral geometry around ferrous iron (Fig. 2B). Cys-314, located in the rhodanese-active site, was captured in its persulfidated Cys-SSH form (Fig. 2C). The PDO-active site is located at the bottom of a large pocket framed on one side by a positively charged ridge comprising residues Arg-193–Lys-216 and by Tyr-176 on the other (supplemental Fig. S1B). The rhodanese-active site is located at the bottom of a shallow pocket that is lined by the positively charged residues Arg-315 and Arg-319 on one side and hydrophobic residues Ala-316, Gly-317, and Gly-318 on the other side (supplemental Fig. S1C).
C314S BpPRF crystallized in a different space group (Table 1), but the two domains were in the same relative orientation as in wild-type BpPRF. Crystals of C314S BpPRF that had been soaked with GSH displayed additional density in two of the three PDO domains present in the asymmetric unit. GSH coordinates to iron in the PDO-active site and displaces one of the iron-coordinated waters. It is within hydrogen bonding distance of Tyr-176, Arg-193, Arg-142, and Lys-216 (Fig. 3A). Notably, in the C314S BpPRF structure, a chloride ion (from the crystallization solution) sits in place of the Cys-314 persulfide seen in the wild-type BpPRF structure. When the single-atom peak in the electron density map was modeled as a chloride ion, the Fo − Fc had no residual positive or negative density, whereas modeling as a fully occupied water resulted in strong positive Fo − Fc density.
Figure 3.
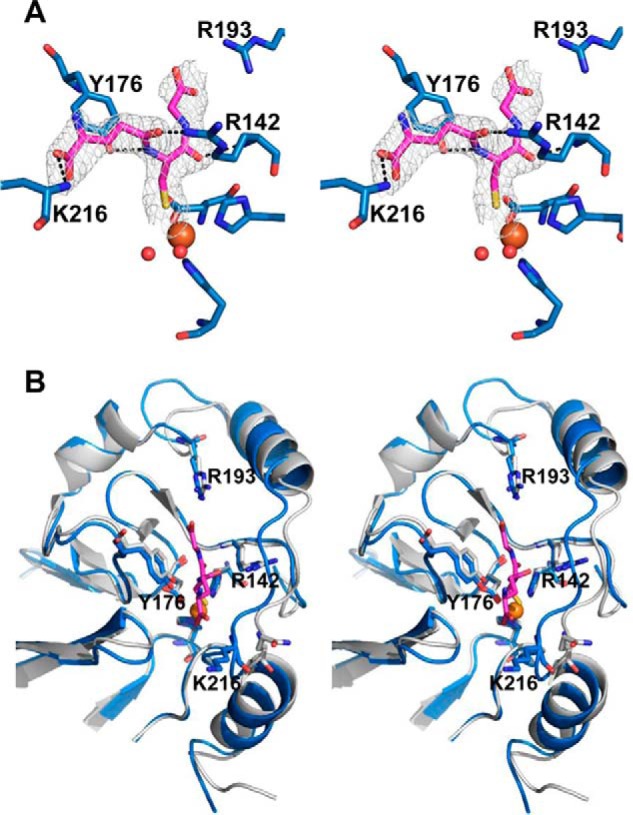
Close-up of the PDO-active site of BpPRF with GSH. A, stereo view of the C314S BpPRF PDO domain with GSH bound. The surrounding residues that may participate in substrate binding and/or stabilization are shown with electron density (3.0σ Fo − Fc omit density; gray mesh) for GSH. Tyr-176, Arg-142, and Lys-216 are hydrogen-bonded (black dashes) to GSH. Two waters (red spheres) are coordinated to the iron center (orange sphere) in addition to the sulfur atom of GSH. B, stereo view of C314S BpPRF PDO domain (blue) in complex with GSH overlaid on human PDO (15) (PDB code 4CHL) (gray). GSH (shown in green) is displayed in stick representation. Conserved residues predicted to be involved in substrate binding, stabilization, and/or positioning (15) are Tyr-176, Arg-193, Leu-212, Arg-142, and Pro-215 (BpPRF numbering). Lys-216 is not conserved in human PDO.
Structural comparisons of BpPRF and homologous human proteins
The architecture of the BpPRF PDO-active site is very similar to that of human PDO (Fig. 3B) (15). The substrate-binding pocket is framed by two α-helices on one side and by Tyr-176 on the other side. Key residues that are predicted to be involved in substrate binding in human PDO, Tyr-197, Arg-214, Arg-163, Leu-231, and Pro-234, are conserved in BpPRF (Tyr-176, Arg-193, Arg-142, Leu-212, and Pro-215) and are found in similar orientations. Lys-216, which is not conserved in human PDO, lines the entrance to the active site and might play a role in substrate positioning.
The BpPRF rhodanese domain and the catalytic domain of bovine rhodanese display very similar folds (Fig. 4A). Minor structural differences include the N-terminal β-hairpin extension in BpPRF and two extra loops in bovine rhodanese. The active sites are also very similar with the conserved residues, Cys-314, Arg-315, and Gly-318 (BpPRF numbering), being in similar orientations (Fig. 4B). However, Thr-252 and Lys-249 in bovine rhodanese occupy the same positions as Arg-319 and Ala-316, respectively, in BpPRF. These substitutions indicate differences in charge distribution that might contribute to differences in substrate specificity.
Figure 4.
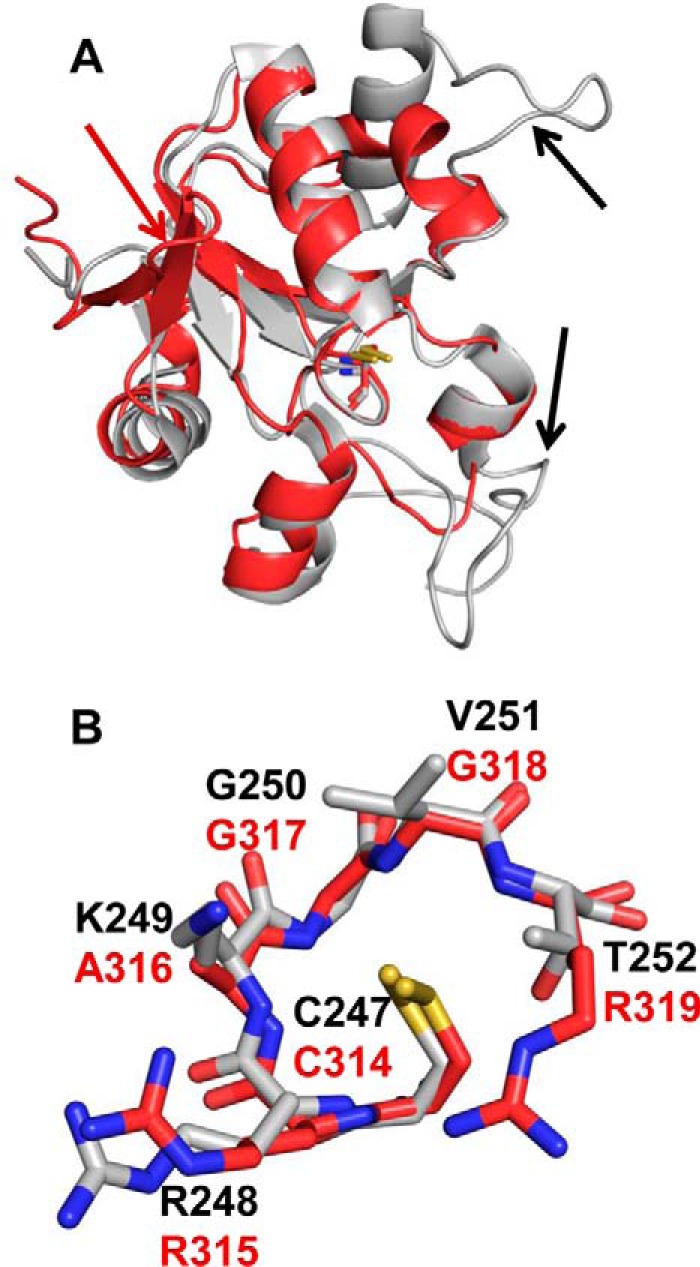
Comparison of the BpPRF rhodanese domain with rhodanese. A, structural overlay of the BpPRF rhodanese domain (red) with the catalytic domain of bovine rhodanese (PDB code 1RHD, gray (34)). The active-site cysteine residues, Cys-247 (bovine) and Cys-314 (BpPRF), are shown in stick representation Structural differences are shown with arrows: a β-hairpin extension is seen only in BpPRF (red), and two loop extensions are seen only in bovine rhodanese (black). B, structural overlay of the rhodanese active-site loops from BpPRF (red) and bovine rhodanese (gray).
PDO activity of BpPRF
The rate of O2 consumption during conversion of GSSH to sulfite (Reaction 3) was monitored using an O2 electrode (Fig. 5) and yielded the following kinetic parameters for wild-type BpPRF: Km(GSSH) = 70 ± 8 μm, and Km(O2) = 130 ± 30 μm, and kcat = 143 s−1 at 22 °C (Table 2). C314S BpPRF exhibited a 29-fold lower kcat and an ∼5-fold higher Km for GSSH than wild-type BpPRF. Addition of 2.5 mm ascorbate to the standard assay resulted in an ∼20% increase in the PDO-specific activity (data not shown). Cysteine persulfide and thiosulfate were also tested as potential substrates for the PDO domain. However, neither displayed detectable activity.
Figure 5.
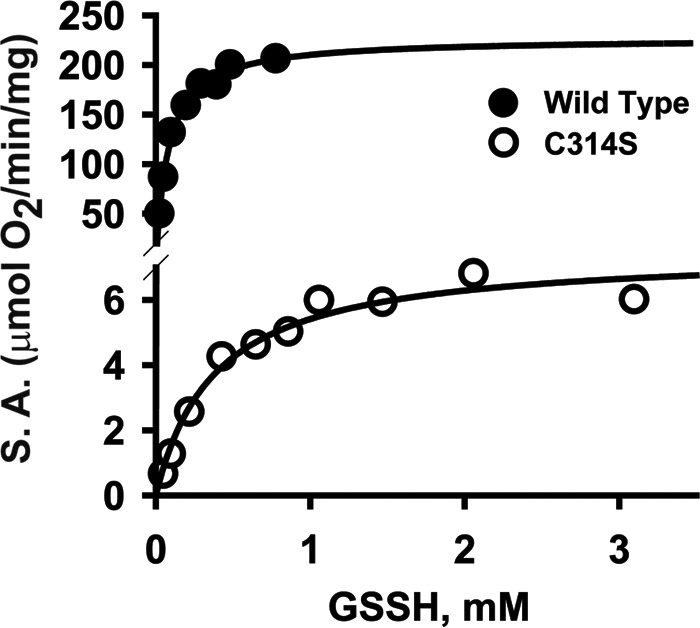
Kinetics of PDO activity of BpPRF. Dependence of PDO activity on GSSH concentration for wild-type BpPRF (solid circles) and C314S BpPRF (open circles). Oxygen consumption by BpPRF in 100 mm sodium phosphate buffer, pH 7.4, was monitored at 22 °C in the presence of varying concentration of GSSH. The data are representative of three independent experiments.
Table 2.
Comparison of the PDO activities of wild-type and C314S BpPRF
The kinetic parameters were determined by monitoring O2 consumption in the presence of GSSH at 22 °C and pH 7.4, as described under “Experimental procedures.”
| Enzyme | Iron content | Vmax | Km (GSSH) | Km (O2) | kcat | kcat/ Km (GSSH) | kcat/Km (O2) |
|---|---|---|---|---|---|---|---|
| mol of iron/mol | μmol min−1 mg−1 | mm | mm | s−1 | mm−1 s−1 | mm−1 s−1 | |
| Wild type | 0.60 ± 0.03 | 207 ± 6 | 0.070 ± 0.008 | 0.13 ± 0.03 | 143 | 2043 | 1100 |
| C314S | 0.46 ± 0.02 | 7.3 ± 0.1 | 0.37 ± 0.04 | 5 | 14 |
It is important to note that the method used to synthesize GSSH results in its contamination with an equal concentration of GSH and an ∼3-fold excess of H2S, as described previously (14). The potential effect of GSH and sulfide on PDO activity was examined in addition to the effect of sulfite (a PDO product) and thiosulfate (rhodanese substrate/product) (supplemental Table S1). Sulfite, which consumed O2, increased the background rate and was therefore used at a lower concentration (0.6 mm) than the other metabolites (5 mm each). GSH was slightly activating (1.3-fold increase in kcat/Km(GSSH)) as seen previously with human PDO (14). Thiosulfate was without effect, whereas H2S and sulfite decreased kcat/Km(GSSH) 1.6- and 2.2-fold. Hence, sulfite build up can potentially inhibit PDO activity.
Sulfurtransferase activity of BpPRF
Conversion of cyanide to thiocyanate in the presence of thiosulfate (Reaction 4) was used to monitor the sulfurtransferase activity of BpPRF (Figs. 6A and 7A).
| (Eq. 4) |
Figure 6.
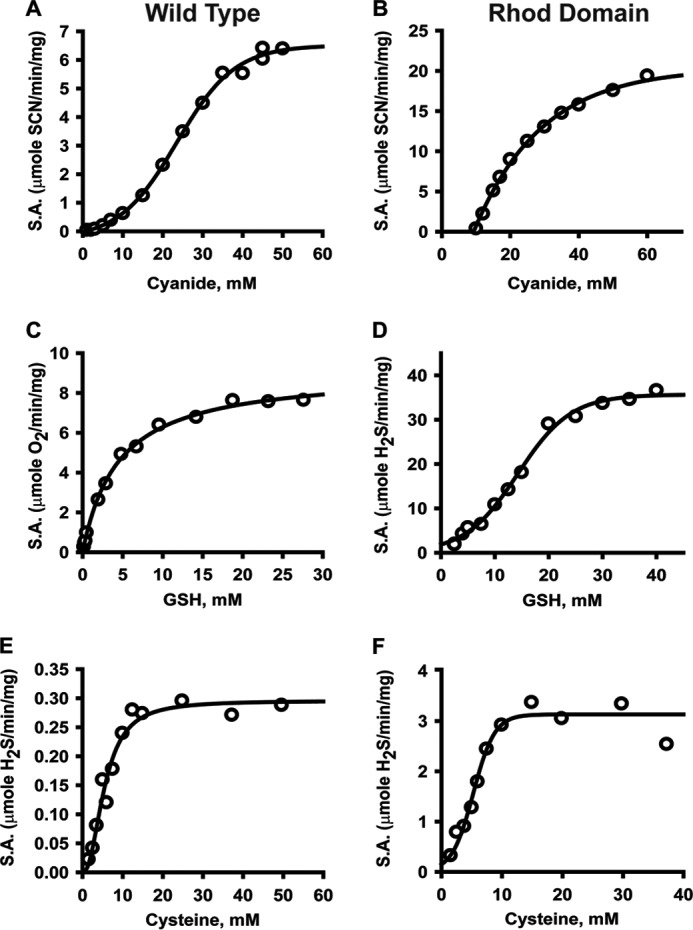
Kinetics of BpPRF-catalyzed sulfur transfer reactions. The sulfurtransferase activity associated with the rhodanese domain in wild-type BpPRF (A, C, and E) or with the isolated rhodanese domain (Rhod domain) (B, D, and F) was determined in the presence of a constant thiosulfate concentration (60 mm) and varying concentrations of cyanide (A and B), GSH (C and D), or cysteine (E and F). The data are representative of 3–4 independent experiments. The data were fitted with the Michaelis-Menten, sigmoidal, or Hill equation as described under “Experimental procedures.”
Figure 7.
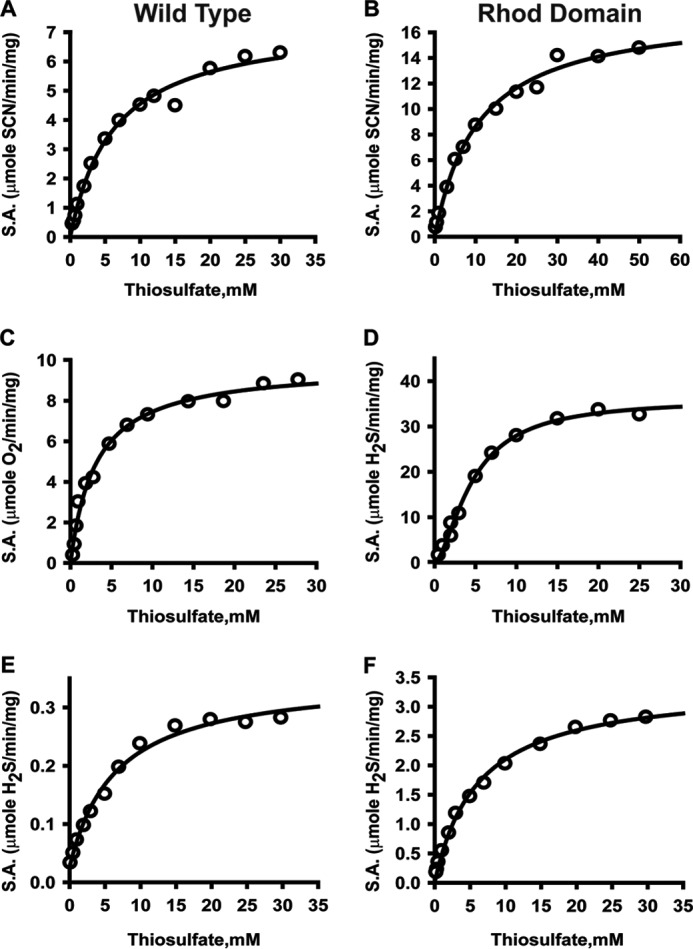
Kinetics of BpPRF-catalyzed sulfur transfer reactions. Dependence of sulfurtransferase activity on varying thiosulfate concentrations in the presence of 60 mm cyanide (A and B), 30 mm GSH (C and D), and 50 mm cysteine (E and F) for the wild-type BpPRF and the isolated rhodanese domain (Rhod Domain). The reactions were performed as described under “Experimental procedures.” The data are representative of 3–4 independent experiments. Data were fitted with either Michaelis-Menten or Hill equation.
From this analysis, Km(thiosulfate) of 5.4 ± 0.7 mm, Km(CN−) of 22.7 ± 0.7 mm, and kcat of 5.1 s−1 at 22 °C were obtained (Table 3). Under the same conditions, the sulfurtransferase activity of the C314S mutant, carrying an inactivating mutation in the rhodanese domain, was not detectable (data not shown).
Table 3.
Kinetic parameters for the sulfurtransferase activity of BpPRF sulfurtransferase
The kinetic parameters for the sulfur transfer Reactions 1–4 were determined at pH 7.4 as follows. Reaction 1 monitored thiocyanate formation (22 °C); Reaction 2 monitored thiosulfate formation (22 °C), Reaction 3 monitored sulfite formation (at 22 °C) or GSSH formation (via O2 consumption in the coupled PDO reaction at 22 °C) or H2S formation (37 °C); and Reaction 4 monitored H2S formation (37 °C), as described under “Experimental procedures.” ND is not detected.
| Donor | Acceptor | Vmax | Km donor | Km acceptor | kcat | kcat/Km donor | kcat/Km acceptor |
|---|---|---|---|---|---|---|---|
| μmol min−1 mg−1 | mm | mm | s−1 | mm−1 s−1 | mm−1 s−1 | ||
| 1) S2O32− | CN− | 7.4 ± 0.3 | 5.4 ± 0.7 | 22.7 ± 0.7 | 5.1 | 0.9 | 0.2 |
| 2) GSSH | SO32− | ND | ND | ND | ND | ND | ND |
| 3) S2O32− | GSH | 8.9 ± 0.2 | 3.1 ± 0.2 | 4.7 ± 0.4 | 6.2 | 2.0 | 1.3 |
| 4) S2O32− | Cys | 0.32 ± 0.01 | 5.8 ± 0.8 | 5.2 ± 0.4 | 0.2 | 0.04 | 0.04 |
Because GSSH, a product of the sulfurtransferase reaction (Reaction 2), is a substrate for the PDO domain (Reaction 3), this activity was monitored in a “coupled” assay, i.e. by detecting O2 consumption (Figs. 6C and 7C). This assay yielded the following parameters: Km(GSH) = 4.7 ± 0.7 mm, Km(thiosulfate) = 3.1 ± 0.2 mm, and kcat = 6.2 s−1 (Table 3). The same reaction was also monitored directly, i.e. by detecting sulfite (Reaction 2) colorimetrically, and it yielded essentially the same results (data not shown).
Cysteine was tested as an alternate sulfur acceptor in lieu of GSH, by monitoring H2S production using the lead sulfide assay (Reactions 5 and 6) (Figs. 6E and 7E).
| (Eq. 5) |
| (Eq. 6) |
This analysis yielded the following values: Km(Cys) = 5.2 ± 0.4 mm, Km(thiosulfate) = 5.8 ± 0.8 mm, and kcat = 0.2 s−1. Given the relatively low cellular concentration of cysteine versus GSH and the considerably lower kcat/Km(Cys) value (Table 3), cysteine is unlikely to be a physiologically relevant sulfur acceptor for BpPRF.
Next, we attempted to monitor sulfurtransferase activity in the opposite direction (i.e. reverse of Reaction 2) by tracking thiosulfate formation under anaerobic conditions (to inhibit GSSH consumption by PDO). However, unlike human rhodanese (18, 20), sulfur transfer activity from GSSH to sulfite was not detected with BpPRF under steady-state turnover conditions.
Sulfurtransferase activity of the rhodanese domain
The isolated rhodanese domain is active and exhibits the following kinetic parameters in the cyanide detoxification assay (Figs. 6B and 7B): Km(thiosulfate) = 9.2 ± 0.8 mm, Km(CN−) = 23 ± 1 mm, and kcat = 4.6 s−1 (Table 4), which are very similar to those for full-length BpPRF. GSSH synthesis from thiosulfate and GSH (Reaction 2) was monitored by following H2S formation (Reaction 7) (Fig. 6D and 7D).
| (Eq. 7) |
The following values were obtained from this analysis: Km(GSH) =14.9 ± 0.5 mm, Km(thiosulfate) = 3.8 ± 0.5 mm, and kcat = 9.0 s−1. As with the full-length protein, cysteine was a poorer acceptor (6-fold lower kcat/Km) than GSH (Figs. 6F and 7F), and the reverse reaction (i.e. transfer from GSSH to sulfite) was not detectable under steady-state turnover conditions (Table 4). Similar values for the kinetic parameters were obtained when the rate of sulfite production was detected (data not shown).
Table 4.
Kinetic parameters for the isolated rhodanese domain of BpPRF
The kinetic parameters of the sulfur transfer reactions catalyzed by the isolated rhodanese domain were determined as summarized in Table 3 legend and described in detail under “Experimental procedures.” ND is not detected.
| Donor | Acceptor | Vmax | Km donor | Km acceptor | kcat | kcat/Km donor | kcat/Km acceptor |
|---|---|---|---|---|---|---|---|
| μmol min−1 mg−1 | mm | mm | s−1 | mm−1 s−1 | mm−1 s−1 | ||
| 1) S2O32− | CN− | 18 ± 1 | 9.2 ± 0.8 | 23 ± 1 | 4.6 | 0.5 | 0.2 |
| 2) GSSH | S2O32− | ND | ND | ND | ND | ND | ND |
| 3) S2O32− | GSH | 35 ± 2 | 3.8 ± 0.5 | 14.9 ± 0.5 | 9.0 | 2.3 | 0.6 |
| 4) S2O32− | Cys | 3.1 ± 0.1 | 6.1 ± 0.4 | 5.4 ± 0.3 | 0.8 | 0.1 | 0.1 |
Stoichiometry of PRF reaction
Our kinetic data predict that BpPRF can catalyze a GSH-dependent net oxidation of thiosulfate to sulfite (Reactions 2 and 3). To test this prediction, the stoichiometry of thiosulfate and O2 consumed to sulfite formed was determined. As noted previously, the method used to synthesize GSSH results in an equivalent of GSH being present, which together with unreacted GSSG and an ∼3-fold excess of H2S would confound the reaction stoichiometry. To circumvent this problem, GSSH was synthesized in situ using GSH and thiosulfate as substrates (Reaction 2).
Under single-turnover conditions, the stoichiometry of sulfite formed/thiosulfate consumed/oxygen consumed was 2:1:1 (Fig. 8A) as predicted by Reactions 2 and 3. Also, as expected, the reaction stoichiometry of sulfite formed/GSH consumed/thiosulfate consumed was 1:1:1 in the absence of oxygen (Fig. 8B). However, when the BpPRF reaction was monitored under multiple turnover conditions in the presence of oxygen, the stoichiometry of sulfite formed/thiosulfate consumed/oxygen consumed switched to 1:1:1 after three turnovers (Fig. 8C). This change in stoichiometry indicates inefficient coupling between the PDO and rhodanese-active sites and is likely confounded by side reactions of the reactive products, particularly GSSH (35), as it builds up under these conditions.
Figure 8.
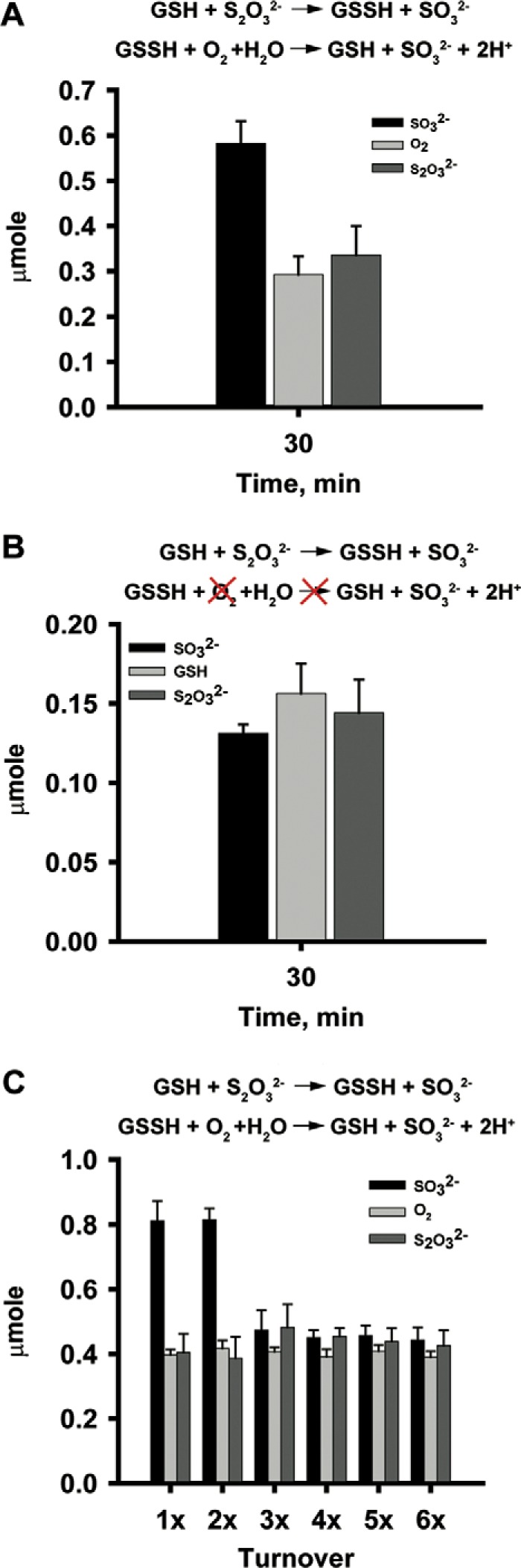
Product analysis and reaction stoichiometry of the BpPRF-The reaction stoichiometry of BpPRF under single-turnover conditions was analyzed in the presence (A) and absence (B) of oxygen. The reactions were performed in the presence of 250 μm GSH, 250 μm thiosulfate and 250 μm enzyme. C, stoichiometry of BpPRF reactions under multiple turnover conditions was analyzed in the presence of 250 μm GSH, 250 μm thiosulfate and decreasing enzyme concentrations as described under “Experimental procedures.” Data are representative of 4–5 independent experiments.
Discussion
Natural fusions of PDO and rhodanese are found in bacteria and suggest a functional interaction between them in other organisms, where they are expressed as stand-alone proteins (36). In this study, we provide the first structural and kinetic characterization of a PRF. To our knowledge, the only other PRF that has been biochemically characterized is CstB from S. aureus (23, 30, 37), but the available CstB structure is missing density for the catalytic rhodanese domain. CstB is located in the cst operon (Fig. 1C). Its operonic partners include CstA, which is a rhodanese, and a sulfide quinone oxidoreductase, which oxidizes H2S to polysulfides (30). Expression of the cst operon is regulated by the persulfide/polysulfide-sensing repressor CstR and is induced by exogenous addition of H2S or polysulfides (38). CstB catalyzes the conversion of low molecular weight persulfides to thiosulfate via the intermediate formation of sulfite (Reaction 8) (23).
| (Eq. 8) |
In contrast to CstB, BpPRF preferentially catalyzes sulfur transfer in the reverse direction, i.e. from thiosulfate to sulfite (Reaction 9), which was confirmed under single-turnover conditions (Fig. 8).
| (Eq. 9) |
GSSH serves as the intermediate sulfur carrier, transferring sulfur processed in the rhodanese-active site to the PDO-active site. Unlike CstB, which functions in the sulfide stress response, the cellular context in which BpPRF functions is not known (38). The gene encoding BpPRF does not reside in an operon, and the metabolic context in which it functions is not immediately obvious. The BpPRF-encoding gene is located downstream from a putative peroxidase and a putative LysR-type regulator, which have been shown to be involved in regulating bacteria mobility, virulence, metabolism, and quorum sensing (39) (Fig. 1C). A potential fate of sulfite, which is toxic, is its oxidation by sulfite oxidase to sulfate, which could be utilized in an assimilatory sulfate-reducing pathway. B. phytofirmans encodes a putative sulfite oxidase. Alternatively, BpPRF might be involved in sulfur assimilation from thiosulfate and sulfite could be reduced by sulfite reductase (encoded by B. phytofirmans) to H2S, which in turn can be used for cysteine synthesis. In Burkholderia cenocepacia, CysB and SsuR regulate genes encoding enzymes involved in the aliphatic sulfonate assimilation pathway (25, 40). BLAST analysis reveals that B. phytofirmans possesses two enzymes that have 23 and 26% identity to the CysB and SsuR transcriptional regulators, respectively. Bioinformatic analysis of the sequence 100 bp upstream of the BpPRF gene queried against CysB- and SsuR-binding sites was performed using the FIMO MEME suite database (25, 40, 41). Several SsuR-binding sites (supplemental Fig. S2) scored as positive motif hits in the BpPRF promoter region (p < 0.005), suggesting a role for BpPRF in sulfur assimilation.
The catalytic efficiency of BpPRF (kcat/Km(GSSH) = 2 × 106 m−1 s−1 at 22 °C) is 14-fold higher than of human PDO (1.4 × 105 m−1 s−1 at 22 ° (14)) and ∼102-fold greater than CstB with its preferred substrate, CoASSH (18 × 103 m−1 s−1 at 25 °C) (23). Mutation of the catalytic cysteine residue, Cys-314, in the rhodanese domain decreased iron content 1.3-fold and PDO activity 29-fold. This result is surprising because it indicates communication between the rhodanese- and PDO-active sites, which are, however, distant and oriented 90° away from each other in the crystal structure (supplemental Fig. S1).
The thiosulfate: cyanide sulfurtransferase activity of BpPRF (kcat = 5.1 s−1 at 22 °C) is comparable with that of CstB (8.5 s−1 at 25 °C) (23) and is 180–730-fold lower than human rhodanese (20). The thiosulfate:GSH sulfur transfer activity of BpPRF (kcat = 6.2 s−1 at 37 °C) is 9-fold higher than for human rhodanese (0.67 s−1 at 37 °C), which preferentially catalyzes the reaction in the reverse GSSH:sulfite sulfur transfer direction (kcat = 389 s−1 at 25 °C). The isolated rhodanese domain was well folded as evidenced by its high Tm (55 ± 2 °C) like that of the full-length protein (50 ± 2 °C) and its comparable kcat values in the thiosulfate:cyanide and thiosulfate:GSH sulfur transfer reactions (Table 4). The isolated rhodanese domain exhibits a sigmoidal dependence on GSH and thiosulfate concentration (Figs. 6D and 7D), in contrast to the hyperbolic dependence observed with full-length BpPRF (Figs. 6C and 7C). This difference in behavior could be due to the difference in the oligomeric state of the stand-alone rhodanese domain, which exists as a monomer/dimer mixture in solution, in contrast to the full-length protein, which is a monomer.
Unlike human rhodanese, sulfur transfer from GSSH to sulfite was not detectable with either the full-length BpPRF or the stand-alone rhodanese domain (Tables 3 and 4). However, the GSSH:sulfite sulfur transfer activity was observed for both the full-length BpPRF and the stand-alone rhodanese domain under limited turnover conditions, i.e. between 2 and 5 turnovers (data not shown). This observation indicates that under multiple turnover conditions as the concentrations of products (i.e. GSH or thiosulfate) increase, one of them inhibits the enzyme. In fact, we found that thiosulfate, but not GSH, inhibits the GSSH:sulfite sulfur transfer activity of BpPRF (data not shown) explaining the lack of activity under steady-state assay conditions. The inhibitory role of thiosulfate is consistent with studies on TSTD1 and human rhodanese, in which thiosulfate was reported to exhibit substrate inhibition, although at high concentrations, for sulfur transfer from thiosulfate to GSH (18, 21, 43).
It is possible that our structure of BpPRF represents an “open” conformation versus a “closed” conformation in which the two domains are proximal. We therefore attempted to model a putative closed conformation by superimposing the PDO and rhodanese domains on the structure of AaPRF. In the latter, the PDO and rhodanese-active sites face each other and are separated by a distance of 27 Å (Fig. 9A). AaPRF contains an additional non-catalytic rhodanese domain and a cysteine-containing loop (Cys-202, AaPRF numbering) located in the PDO domain active site. This loop, although absent in BpPRF, is also present in CstB and postulated to facilitate substrate transfer between the PDO and catalytic rhodanese domains (23). To adopt a similar domain orientation, the linker region of BpPRF, which is maximally ∼36 Å long, would have to span 47 Å requiring unfolding of a secondary structure element. Furthermore, the calculated electrostatic surface potential reveals that positively charged residues line the entrance to the PDO and rhodanese-active sites, suggesting that a direct interaction between them would be unfavorable (Fig. 9B). The structural independence of the active sites is also consistent with the high catalytic activity of the isolated rhodanese domain. This result is in contrast to CstB in which the rhodanese domain, once isolated, does not retain activity (23). Fusion of the PDO and rhodanese domains in BpPRF would serve to increase their effective local concentration and promote transfer of substrates between them.
Figure 9.
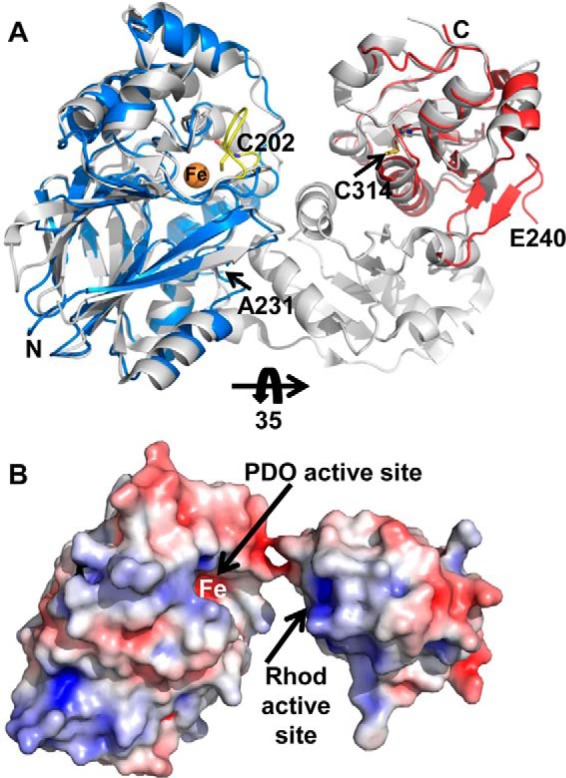
Modeling of potential BpPRF domain interaction interface. A, overlay of BpPRF PDO domain (blue) and the BpPRF rhodanese domain (red) with the PDO domain and catalytic rhodanese domain of AaPRF (PDB code 3TP9) (shown in gray). The active-site loop of AaPRF (yellow) with Cys-202 faces the iron center (orange sphere) in the PDO domain. B, electrostatic surface potential of the BpPRF domains in the same orientation as in A but rotated by 35°. The arrows highlight locations of the PDO- and rhodanese (Rhod)-active sites. Positive and negative electrical potential are shown in blue and red, respectively, and represent a range of −5 to +5 kT/e.
An overlay of five structures, i.e. two per asymmetric unit of wild-type BpPRF and three per asymmetric unit of C314S BpPRF, was used to assess significant conformational differences, if any, between them (supplemental Fig. S3). Because the electron density for eight residues in the linker region was missing in these structures, the overlay was used to assess whether the flexible linker might allow a closer approach of the rhodanese and PDO domains, permitting a closed conformation. However, only slight conformational differences were observed in the relative orientation of the two domains (supplemental Fig. S3).
In summary, the relative orientation of the two domains in BpPRF and their separation by a short linker suggest that the two active sites do not interact. There is also no support for an alternative model in which the protein dimerizes in a head-to-tail manner bringing the two active sites in proximity. In fact, the similar electrostatic potential surfaces of the two domains also suggest that the active sites are unlikely to interact directly. In light of the structural data, it is intriguing that the kinetic data provide some evidence for allosteric communication between the domains. For instance, mutation of the catalytic cysteine in the rhodanese domain adversely impacts the catalytic properties and, to a lesser extent, the iron content of the PDO domain. Similarly, isolation of the rhodanese domain improves its kcat. Our kinetic data are consistent with a role for BpPRF in sulfur assimilation rather than in a sulfide stress-response pathway.
Experimental procedures
Materials
Sodium sulfide, sodium sulfite, sodium thiosulfate, GSH, oxidized glutathione, l-cysteine, para-rosaniline hydrochloride, and 2,4,6-tripyridyl-S-triazine were purchased from Sigma. FluoroPure grade monobromobimane was purchased from Life Technologies, Inc.
Expression constructs for BpPRFs
The cDNA encoding BpPRF was amplified from genomic DNA isolated from B. phytofirmans PsjN (purchased from DSMZ, Braunschweig, Germany), using the following primers containing NdeI and HindIII restriction sites (bold font): forward 5′-TTAATTCATATGTTGATCTTCCGGCAGCTATTC-3′ and reverse 5′-TTAATTAAGCTTCTACACGCTGTTTTCAACGACGG-3′. The resulting PCR fragment was cloned into a pET28b vector. The C-terminal truncated BpPRF (ΔC-BpPRF) construct missing the last four residues (ΔC 354–357) was generated for crystallography only, using the full-length BpPRF expression construct as a template and the following primers containing NdeI and HindIII restriction sites (bold font): forward 5′-TTAATTCATATGTTGATCTTCCGGCAGCTATTC-3′ and reverse 5′-TTAATTAAGCTTCTATTCAACGACGCGGCC-3′. The resulting PCR fragment was cloned into a pET28b vector. The C314S mutant was generated using the QuickChange kit (Stratagene) and the wild-type BpPRF expression construct as a template. The isolated rhodanese domain was subcloned from the wild-type PRF expression construct using the following primers containing NdeI and HindIII restriction sites (bold font): forward 5′-TTAATTCATATGATGACCGAGCCCGATTTGGCG-3′ and reverse 5′-TTAATTAAGCTTCTACTGCGTGGGGACGCTCGC-3′. The resulting PCR fragment was cloned into a pET28b vector.
Expression and purification of BpPRF
The recombinant BpPRF protein was expressed in BL21 E. coli. A 200-ml culture in Luria Bertani medium was grown overnight at 37 °C and used to inoculate a 4× 1-liter culture in the same medium. Cultures were grown at 28 °C, and expression was induced with isopropyl β-d-thiogalactopyranoside (100 μm) when the optical density at 600 nm reached 0.5. Cultures were supplemented with ferrous ammonium sulfate at the time of induction to a final concentration of 250 μm, and growth was continued for an additional 14 h at 28 °C. Cells were harvested by centrifugation at 2683 × g for 20 min at 4 °C.
BpPRF was purified as follows. The cell pellet from a 4-liter culture was resuspended in 500 ml of 50 mm Tris buffer, pH 8, containing 0.5 m NaCl (Buffer A), 1 tablet of protease inhibitor mixture (Roche Applied Science), and 100 mg of lysozyme (Sigma). DNase (50 mg) and MgCl2 (10 mm final concentration) were added to the cell suspension and stirred at 4 °C for 60 min followed by sonication on ice with the following pulse sequence: 30-s burst, 1-min rest for a total burst time of 5 min at a power output setting of 6. The sonicate was centrifuged at 8217 × g for 15 min at 4 °C. The resulting supernatant was diluted 2-fold with Buffer A and loaded onto a 20-ml nickel-nitrilotriacetic acid column equilibrated with the same buffer. The column was washed with 500 ml of Buffer A containing 30 mm imidazole buffer. PRF was eluted from the column with a linear gradient ranging from 30 to 500 mm imidazole in Buffer A. Fractions containing the PRF protein were pooled, concentrated, and dialyzed overnight against 4 liters of Buffer A and stored at −80 °C. Protein concentration was determined using the Bradford reagent (Bio-Rad) with BSA as a standard. The ΔCBpPRF, the C314S mutant, and the isolated rhodanese domain were purified using the same protocol.
Metal analysis
Plasma emission spectroscopy was used to analyze the total metal content of wild-type BpPRF at the Chemical Analysis Laboratory (University of Georgia, Athens). Twenty metal ions were detected by this method. The iron content of the enzyme was also measured using a colorimetric assay described previously (32). This method allows for the direct measurement of total iron content, the Fe(II) content, and consequently, estimation of Fe(III) content of the enzyme. Briefly, BpPRF (10–50 μm) was denatured with 0.5 n HCl and 5% (w/v) trichloroacetic acid (final volume 1 ml), mixed for 30 s, and subsequently centrifuged for 10 min at 16,000 × g in a microcentrifuge. To determine the total iron content, the supernatant (700 μl) was mixed with 300 μl of a 1:2:1 mixture of 4 mm 2,4,6-tripyridyl-S-triazine, 50% ammonium acetate, and 10% hydroxylamine chloride, and incubated at room temperature for 5 min. Hydroxylamine is present to reduce residual Fe(III) to Fe(II). Absorbance of the resulting Fe(II)–2,4,6-tripyridyl-S-triazine complex was measured at 569 nm. The total iron concentration was calculated using an extinction coefficient of 22,600 m−1 cm−1 for the Fe(II)-2,4,6-tripyridyl–S-triazine complex (32).
Molecular mass determination
Purified BpPRF (5 ml of 3.7 mg/ml) was loaded onto a HiLoadTM 16/60 Superdex G-200 column equilibrated with Buffer A at 4 °C and calibrated with protein standards (Bio-Rad). The protein was eluted at a flow rate of 0.5 ml min−1 and was monitored by absorbance at 280 nm.
Thermal denaturation assay
The thermal stability of BpPRF and of the C314S rhodanese domain mutant was evaluated by monitoring the increase in absorbance at 600 nm with increasing temperature. For this, enzyme (100 μg) in Buffer A (final volume 200 μl) was placed in a cuvette housed in a Cary 100 Bio spectrophotometer equipped with a heating block connected to a water bath. The temperature was increased from 25 to 70 °C in 5 °C increments.
Preparation of GSSH
GSSH was prepared anaerobically as described previously (14) by reacting oxidized glutathione (GSSG) with Na2S in a Coy anaerobic chamber (atmosphere of 95:5 N2/H2) (Reaction 10).
| (Eq. 10) |
Briefly, solid Na2S was added in 4-fold excess, to an anaerobic solution of 50 mm GSSG in 350 mm sodium phosphate, pH 7.4 (final volume 5 ml). The reaction vial was immediately sealed to prevent loss of Na2S and incubated at 37 °C for 25–30 min. The concentration of GSSH was measured using the cold cyanolysis method as described previously (14). Substrate was either used immediately or stored at −20 °C until use. Substrate concentration was measured both before freezing and after thawing prior to use in the enzyme assay. Cysteine persulfide was prepared using the same procedure.
Oxygen consumption assay
The PDO activity of BpPRF was measured by monitoring O2 consumption at room temperature (22 °C). The reaction mixture consisted of 100 mm sodium phosphate, pH 7.4, and 0.5–2 μg of enzyme (final volume 1.6 ml) mixed in a Gilson-type chamber containing a Clark-type oxygen electrode and a magnetic stir bar. The reaction was initiated by addition of GSSH (0.02–3 mm). The effects of GSH, Na2S, sulfite, and thiosulfate on the PDO activity were assessed at 5 mm concentrations of each except for sulfite, which was added to a final concentration of 0.6 mm. Cysteine persulfide was tested as an alternative substrate at a final concentration of 1 mm. The dependence of the reaction velocity on O2 concentration was determined as follows. The reaction mixture containing enzyme and the GSSH substrate in 100 mm phosphate, pH 7.4, was prepared anaerobically in an inflatable glove bag (Cole-Palmer) filled with N2 (>99% purity). The reaction was started by injecting a known concentration of dissolved O2 (generated using a 100% oxygen tank). The final O2 concentration varied from 1.6 to 250 μm. The O2 concentration in oxygenated buffer was independently determined using the O2 probe.
Thiosulfate cyanide sulfurtransferase assay
The cyanide detoxification activity of BpPRF was measured in a colorimetric assay as described previously (44). The reaction mixture contained 60 mm thiosulfate, 60 mm potassium cyanide, and 5–10 μg of protein in 200 mm sodium phosphate buffer, pH 7.4 (330 μl final volume). The reaction was initiated by addition of enzyme. After incubation for 20 min at 22 °C, the reaction was stopped by addition of 100 μl of 15% (w/v) formaldehyde. Thiocyanate formation was measured by addition of 500 μl of a ferric nitrate solution containing 165 mm ferric nitrate nonahydrate and 13.3% (v/v) nitric acid. The absorbance of the resulting ferric thiocyanate complex was measured at 460 nm. The concentration of thiocyanate formed was determined using a standard curve. The concentrations of cyanide and thiosulfate were varied from 0.5 to 60 mm to determine the dependence of enzyme activity on substrate concentration.
Thiosulfate:GSH sulfurtransferase activity monitored in a coupled assay
The GSSH formed via the sulfur transfer activity of BpPRF was measured by coupling to O2 consumption during GSSH utilization by the PDO activity. For this, the reaction mixture contained 30 mm thiosulfate, 30 mm GSH, 25–30 μg enzyme, and 200 mm sodium phosphate buffer, pH 7.4 (final volume 1.6 ml), mixed in a Gilson-type chamber containing a Clark-type oxygen electrode at 22 °C. GSH and thiosulfate concentrations were varied from 0.3 to 30 mm to determine the dependence of enzyme activity on substrate concentration.
Thiosulfate:GSH sulfurtransferase activity monitored by sulfite formation
The thiosulfate:GSH sulfurtransferase activity of BpPRF and of the isolated rhodanese domain was monitored by detecting sulfite formation using a modified version of the colorimetric assay described previously (45). The reaction mixture contained 45 mm thiosulfate, 45 mm GSH, 0.7–3 μg of enzyme, and 200 mm sodium phosphate buffer, pH 7.4 (final volume of 500 μl). The reaction was initiated by addition of enzyme. After 5 min of incubation at 22 °C, the reaction was stopped by the addition of 500 μl of 0.23 m HgCl2. After centrifugation at 16,000 × g, for 1 min, the supernatant (125 μl) was mixed with 1 ml of a p-rosaniline solution containing 2:1 0.04% p-rosaniline prepared in 0.72 m HCl (w/v), 0.2% formaldehyde. The absorbance of the resulting p-rosaniline sulfonic acid complex was measured at 570 nm. The concentration of sulfite formed was determined using a standard curve. The concentrations of thiosulfate and GSH were varied from 0.3 to 50 mm to determine the dependence of enzyme activity on substrate concentration.
Thiosulfate-thiol sulfurtransferase activity monitored by H2S formation
The sulfur transfer activities of BpPRF and of the isolated rhodanese domain was measured by detecting H2S formation using the lead acetate assay described previously (18). The reaction mixture contained thiosulfate (0.5–25 mm) and either GSH (2–40 mm) or cysteine (1–50 mm) in 200 mm sodium phosphate buffer, pH 7.4 (final volume of 1 ml), and was preincubated at 37 °C for 10 min. The reaction was initiated by addition of 0.7–15 μg of enzyme. Formation of lead sulfide was measured at 390 nm, and the concentration of H2S formed was calculated using an extinction coefficient of 5500 m−1 cm−1 for lead sulfide (18).
GSSH:sulfite sulfurtransferase activity
The reactions were performed in a Coy anaerobic chamber to prevent oxidation of GSSH by the PDO domain. The standard reaction mixture contained 1 mm GSSH and 1 mm sulfite, 0.5–50 μg of enzyme, and 20 mm sodium phosphate buffer, pH 7.4 (200 μl final volume). The derivatization reaction was initiated by the addition of enzyme. After 5 min of incubation, monobromobimane was added to a final concentration of 3 mm and incubated at room temperature for 15 min. The derivatization reaction was terminated by addition of 100 μl of 0.2 m sodium citrate, pH 2.0. Production of thiosulfate from GSSH and sulfite was monitored by HPLC. For this, a 60 mm stock solution of monobromobimane was prepared in DMSO and protected from light during preparation and handling. Bimane adducts of GSSH, Na2S, GSH, sulfite, and thiosulfate were prepared in 20 mm sodium phosphate, pH 7.4. Each standard (final concentration 250 μm) was incubated with a final concentration of 3 mm monobromobimane (200 μl final volume) for 10 min at 22 °C.
The derivatized samples were centrifuged at 10,000 × g for 10 min at 4 °C, and the supernatants were separated on a 4.6 × 150-cm C18 reverse-phase HPLC column (3 μm packing, SunFire) using an Agilent 1100 series HPLC system equipped with a multisignal fluorescence detector. The column was equilibrated with the following solution: 80% solvent A (897.5 ml of water, 100 ml of methanol, and 2.5 ml of acetic acid) and 20% solvent B (97.5 ml of water, 900 ml of methanol, and 2.5 ml of acetic acid). Samples (50 μl) were injected onto the column and resolved using the following gradient of solvent B: 20% from 0 to 5 min; 20–45% from 5 to 10 min; 45% isocratic from 10 to 15 min; 45–50% from 15 to 25 min; 50% isocratic from 25 to 28 min; 50–100% from 28 to 30 min; 100% isocratic from 30 to 39 min; 100–20% from 39 to 42 min; and 20% isocratic from 42 to 48 min. The flow rate was 0.75 min/ml. Fluorescence of the bimane adducts was detected using 340 nm excitation and 450 nm emission. The concentration of thiosulfate was determined using the peak areas and a standard curve.
Stoichiometry of O2 consumption and sulfite formation
The stoichiometry of coupling between the PDO and rhodanese-active sites was monitored using wild-type and C314S BpPRF as follows. The reaction mixture contained 1 mm GSSH, 75 nm enzyme, and 200 mm sodium phosphate buffer, pH 7.4 (1.6 ml final volume), in a Gilson-type chamber containing a Clark-type oxygen electrode. The reaction was initiated by addition of GSSH, and O2 consumption was recorded. After 5 min at 22 °C, an aliquot of the reaction mix (50 μl) was added to 60 mm monobromobimane (20 μl) to a final concentration of 17 mm. After incubation for 15 min at room temperature, the reaction was quenched with 100 μl of 0.2 m sodium citrate, pH 2.0. The derivatized samples were processed and separated by HPLC as described above to determine the concentrations of sulfite formed and thiosulfate consumed.
Reaction stoichiometry under single and multiple turnover conditions
The reaction stoichiometry of wild-type and C314S BpPRF under single-turnover conditions was monitored as described above with the following variation. The reaction mixture contained 250 μm thiosulfate, 250 μm GSH, and varying concentrations of enzyme (250, 125, 83, 62.5, 50, and 42 μm). The reaction was initiated by addition of GSH. After incubation for 30 min at 22 °C, an aliquot of the reaction mix (50 μl) was mixed with 20 μl of 60 mm monobromobimane and processed as described above. For analysis of the reaction stoichiometry under single-turnover conditions in the absence of O2, the enzymatic assay and derivatization reactions were performed in a Coy anaerobic chamber (atmosphere of 95:5 N2/H2).
Protein crystallization
The C-terminal truncated BpPRF (ΔCBpPRF) and C314S BpPRF were crystallized by sitting drop vapor diffusion method from a 0.75-μl/0.75-μl mixture of protein stock and well solution. ΔCBpPRF (5 mg/ml in 50 mm Tris, pH 8, 0.25 m NaCl, 10% glycerol, 5 mm thiosulfate) crystallized in 3 days at 20 °C using a well solution of 0.2 m zinc acetate, 0.1 m MES, pH 6, 10% PEG-8000. A single rhombohedral rod-shaped crystal was harvested directly from the drop and flash-cooled in liquid nitrogen. C314S BpPRF (5 mg/ml in 50 mm Tris, pH 8.0, 0.25 m NaCl, 10% glycerol) crystallized in 2 days using a well solution of 0.12 m NaCl, 0.1 m imidazole, pH 8.0, 25% PEG-8000. To obtain structural data for C314S BpPRF in complex with GSH, 5 mm GSH was added to the drop overnight. The hexagonal rod-shaped crystals were cryo-protected by passing harvested crystals through 2 μl of reservoir solution with an additional 15% glycerol followed by flash cooling in liquid nitrogen.
Data collection and crystal structure determination
Diffraction data were collected at the Advanced Photon Source (APS, Argonne National Laboratory) on the GM/CA beamline 23ID-D or 23ID-B (Table 1). For ΔCBpPRF, data were collected on a single crystal to 1.79 Å resolution. For the apo- and GSH-soaked C314S BpPRF crystals, data were collected to 2.65 and 2.35 Å, respectively. For the C314S BpPRF crystals, data quality was limited by high mosaicity along the c axis. All data were processed using XDS (46). The truncated ΔCBpPRF structure was solved by molecular replacement using BALBES (47) with the AaPRF (PDB code 3TP9), which shares 28% sequence identity with BpPRF, as a search model. Phenix AUTOBUILD was used to build ∼70% of the model. To complete the model, consecutive rounds of model building and refinement were performed using Coot (48) and Phenix Refine (49). The structures were validated using MolProbity (50). Electron density is complete for all BpPRF residues except for eight residues in the linker region between the PDO and rhodanese domains and the N-terminal histidine tag. The C314S structure was solved via molecular replacement using Phaser within the Phenix software suite with the complete ΔCBpPRF model as a search model. Molecular replacement successfully placed three PDO domains and two rhodanese domains. The third rhodanese domain is poorly ordered and was built manually. Density is poor for this domain compared with the rest of the structure. The GSH-bound structure was solved via rigid body refinement in Phenix Refine. Electrostatic surface potentials were calculated using the APBS program in PyMOL (42, 51, 52). Figures were prepared using PyMOL.
Author contributions
N. M. designed, performed, and analyzed the experiments and wrote the manuscript. N. M. and M. A. S. solved the crystal structures. M. A. S. and J. L. S. assisted with the crystallography experiments, data collection, and analysis. O. K. helped design and perform the O2 consumption experiments. R. B. helped conceive the experiments, analyzed the data, and co-wrote the manuscript. All authors approved the final version of the manuscript.
Supplementary Material
This work was supported in part by National Institutes of Health Grant GM112455 (to R. B.) and NIGMS Training Grant T32GM008353 (to N. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs S1–S3 and Table S1.
The atomic coordinates and structure factors (codes 5VE3, 5VE4, and 5VE5) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- H2S
- hydrogen sulfide
- PDO
- persulfide dioxygenase (also known as ETHE1)
- GSSH
- glutathione persulfide
- PRF
- PDO–rhodanese fusion
- PDB
- Protein Data Bank.
References
- 1. Beauchamp R. O. Jr., Bus J. S., Popp J. A., Boreiko C. J., and Andjelkovich D. A. (1984) A critical review of the literature on hydrogen sulfide toxicity. Crit. Rev. Toxicol. 13, 25–97 [DOI] [PubMed] [Google Scholar]
- 2. Kimura H. (2010) Hydrogen sulfide: from brain to gut. Antioxid. Redox Signal. 12, 1111–1123 [DOI] [PubMed] [Google Scholar]
- 3. Kabil O., and Banerjee R. (2010) The redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kabil O., Motl N., and Banerjee R. (2014) H2S and its role in redox signaling. Biochim. Biophys. Acta 1844, 1355–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhao W., Zhang J., Lu Y., and Wang R. (2001) The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 20, 6008–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Elrod J. W., Calvert J. W., Morrison J., Doeller J. E., Kraus D. W., Tao L., Jiao X., Scalia R., Kiss L., Szabo C., Kimura H., Chow C. W., and Lefer D. J. (2007) Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. U.S.A. 104, 15560–15565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wallace J. L., Vong L., McKnight W., Dicay M., and Martin G. R. (2009) Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology 137, 569–578 [DOI] [PubMed] [Google Scholar]
- 8. Gao X. H., Krokowski D., Guan B. J., Bederman I., Majumder M., Parisien M., Diatchenko L., Kabil O., Willard B., Banerjee R., Wang B., Bebek G., Evans C. R., Fox P. L., Gerson S. L., et al. (2015) Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. eLife 4, e10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vitvitsky V., Kabil O., and Banerjee R. (2012) High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid. Redox Signal. 17, 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Banerjee R. (2017) Catalytic promiscuity and heme-dependent redox regulation of H2S synthesis. Curr. Opin. Chem. Biol. 37, 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh S., and Banerjee R. (2011) PLP-dependent H2S biogenesis. Biochim. Biophys. Acta 1814, 1518–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hildebrandt T. M., and Grieshaber M. K. (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 275, 3352–3361 [DOI] [PubMed] [Google Scholar]
- 13. Mishanina T. V., Libiad M., and Banerjee R. (2015) Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 11, 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kabil O., and Banerjee R. (2012) Characterization of patient mutations in human persulfide dioxygenase (ETHE1) involved in H2S catabolism. J. Biol. Chem. 287, 44561–44567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pettinati I., Brem J., McDonough M. A., and Schofield C. J. (2015) Crystal structure of human persulfide dioxygenase: structural basis of ethylmalonic encephalopathy. Hum. Mol. Genet. 24, 2458–2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tiranti V., Briem E., Lamantea E., Mineri R., Papaleo E., De Gioia L., Forlani F., Rinaldo P., Dickson P., Abu-Libdeh B., Cindro-Heberle L., Owaidha M., Jack R. M., Christensen E., Burlina A., and Zeviani M. (2006) ETHE1 mutations are specific to ethylmalonic encephalopathy. J. Med. Genet. 43, 340–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tiranti V., D'Adamo P., Briem E., Ferrari G., Mineri R., Lamantea E., Mandel H., Balestri P., Garcia-Silva M. T., Vollmer B., Rinaldo P., Hahn S. H., Leonard J., Rahman S., Dionisi-Vici C., et al. (2004) Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am. J. Hum. Genet. 74, 239–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Libiad M., Yadav P. K., Vitvitsky V., Martinov M., and Banerjee R. (2014) Organization of the human mitochondrial H2S oxidation pathway. J. Biol. Chem. 289, 30901–30910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bordo D., and Bork P. (2002) The rhodanese/Cdc25 phosphatase superfamily. Sequence-structure-function relations. EMBO Rep. 3, 741–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Libiad M., Sriraman A., and Banerjee R. (2015) Polymorphic variants of human rhodanese exhibit differences in thermal stability and sulfur transfer kinetics. J. Biol. Chem. 290, 23579–23588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Melideo S. L., Jackson M. R., and Jorns M. S. (2014) Biosynthesis of a central intermediate in hydrogen sulfide metabolism by a novel human sulfurtransferase and its yeast ortholog. Biochemistry 53, 4739–4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Spallarossa A., Forlani F., Carpen A., Armirotti A., Pagani S., Bolognesi M., and Bordo D. (2004) The “rhodanese” fold and catalytic mechanism of 3-mercaptopyruvate sulfurtransferases: crystal structure of SseA from Escherichia coli. J. Mol. Biol. 335, 583–593 [DOI] [PubMed] [Google Scholar]
- 23. Shen J., Keithly M. E., Armstrong R. N., Higgins K. A., Edmonds K. A., and Giedroc D. P. (2015) Staphylococcus aureus CstB is a novel multidomain persulfide dioxygenase-sulfurtransferase involved in hydrogen sulfide detoxification. Biochemistry 54, 4542–4554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barton L. L., Fardeau M.-L., and Fauque G. D. (2014) in The Metal-driven Biogeochemistry of Gaseous Compounds in the Environment (Kroneck P. M., and Torres M. E., eds) pp. 237–277, Springer, Dordrecht, Netherlands [Google Scholar]
- 25. Iwanicka-Nowicka R., Zielak A., Cook A. M., Thomas M. S., and Hryniewicz M. M. (2007) Regulation of sulfur assimilation pathways in Burkholderia cenocepacia: identification of transcription factors CysB and SsuR and their role in control of target genes. J. Bacteriol. 189, 1675–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shatalin K., Shatalina E., Mironov A., and Nudler E. (2011) H2S: a universal defense against antibiotics in bacteria. Science 334, 986–990 [DOI] [PubMed] [Google Scholar]
- 27. Motl N., Yadav P. K., and Banerjee R. (2013) in Hydrogen Sulfide and Its Therapeutic Applications (Kimura H., ed), pp. 1–36, Springer, New York [Google Scholar]
- 28. Czyzewski B. K., and Wang D. N. (2012) Identification and characterization of a bacterial hydrosulphide ion channel. Nature 483, 494–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sattler S. A., Wang X., Lewis K. M., DeHan P. J., Park C. M., Xin Y., Liu H., Xian M., Xun L., and Kang C. (2015) Characterizations of two bacterial persulfide dioxygenases of the metallo-β-lactamase superfamily. J. Biol. Chem. 290, 18914–18923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen J., Peng H., Zhang Y., Trinidad J. C., and Giedroc D. P. (2016) Staphylococcus aureus sqr encodes a type II sulfide:quinone oxidoreductase and impacts reactive sulfur speciation in cells. Biochemistry 55, 6524–6534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sessitsch A., Coenye T., Sturz A. V., Vandamme P., Barka E. A., Salles J. F., Van Elsas J. D., Faure D., Reiter B., Glick B. R., Wang-Pruski G., and Nowak J. (2005) Burkholderia phytofirmans sp. nov., a novel plant-associated bacterium with plant-beneficial properties. Int. J. Syst. Evol. Microbiol. 55, 1187–1192 [DOI] [PubMed] [Google Scholar]
- 32. Fischer D. S., and Price D. C. (1964) A simple serum iron method using the new sensitive chromogen tripyridyl-s-triazine. Clin. Chem. 10, 21–31 [PubMed] [Google Scholar]
- 33. McCoy J. G., Bingman C. A., Bitto E., Holdorf M. M., Makaroff C. A., and Phillips G. N. Jr. (2006) Structure of an ETHE1-like protein from Arabidopsis thaliana. Acta Crystallogr. D Biol. Crystallogr. 62, 964–970 [DOI] [PubMed] [Google Scholar]
- 34. Ploegman J. H., Drent G., Kalk K. H., Hol W. G., Heinrikson R. L., Keim P., Weng L., and Russell J. (1978) The covalent and tertiary structure of bovine liver rhodanese. Nature 273, 124–129 [DOI] [PubMed] [Google Scholar]
- 35. Yadav P. K., Martinov M., Vitvitsky V., Seravalli J., Wedmann R., Filipovic M. R., and Banerjee R. (2016) Biosynthesis and reactivity of cysteine persulfides in signaling. J. Am. Chem. Soc. 138, 289–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tiranti V., Viscomi C., Hildebrandt T., Di Meo I., Mineri R., Tiveron C., Levitt M. D., Prelle A., Fagiolari G., Rimoldi M., and Zeviani M. (2009) Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 15, 200–205 [DOI] [PubMed] [Google Scholar]
- 37. Higgins K. A., Peng H., Luebke J. L., Chang F. M., and Giedroc D. P. (2015) Conformational analysis and chemical reactivity of the multidomain sulfurtransferase, Staphylococcus aureus CstA. Biochemistry 54, 2385–2398 [DOI] [PubMed] [Google Scholar]
- 38. Luebke J. L., Shen J., Bruce K. E., Kehl-Fie T. E., Peng H., Skaar E. P., and Giedroc D. P. (2014) The CsoR-like sulfurtransferase repressor (CstR) is a persulfide sensor in Staphylococcus aureus. Mol. Microbiol. 94, 1343–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maddocks S. E., and Oyston P. C. (2008) Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154, 3609–3623 [DOI] [PubMed] [Google Scholar]
- 40. L̸ochowska A., Iwanicka-Nowicka R., Zielak A., Modelewska A., Thomas M. S., and Hryniewicz M. M. (2011) Regulation of sulfur assimilation pathways in Burkholderia cenocepacia through control of genes by the SsuR transcription factor. J. Bacteriol. 193, 1843–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grant C. E., Bailey T. L., and Noble W. S. (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jarabak R., and Westley J. (1974) Human liver rhodanese. Nonlinear kinetic behavior in a double displacement mechanism. Biochemistry 13, 3233–3236 [DOI] [PubMed] [Google Scholar]
- 44. Sörbo B. H. (1953) Rhodanese. Acta Chem. Scand. 7, 1137–1145 [Google Scholar]
- 45. Chauncey T. R., Uhteg L. C., and Westley J. (1987) Thiosulfate reductase. Methods Enzymol. 143, 350–354 [DOI] [PubMed] [Google Scholar]
- 46. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Long F., Vagin A. A., Young P., and Murshudov G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 49. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen V. B., Arendall W. B. 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dolinsky T. J., Czodrowski P., Li H., Nielsen J. E., Jensen J. H., Klebe G., and Baker N. A. (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35, W522–W525 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.