

Abstract
Kindlin-2 (K2), a 4.1R-ezrin-radixin-moesin (FERM) domain adaptor protein, mediates numerous cellular responses, including integrin activation. The C-terminal 15-amino acid sequence of K2 is remarkably conserved across species but is absent in canonical FERM proteins, including talin. In CHO cells expressing integrin αIIbβ3, co-expression of K2 with talin head domain resulted in robust integrin activation, but this co-activation was lost after deletion of as few as seven amino acids from the K2 C terminus. This dependence on the C terminus was also observed in activation of endogenous αIIbβ3 in human erythroleukemia (HEL) cells and β1 integrin activation in macrophage-like RAW264.1 cells. Kindlin-1 (K1) exhibited a similar dependence on its C terminus for integrin activation. Expression of the K2 C terminus as an extension of membrane-anchored P-selectin glycoprotein ligand-1 (PSGL-1) inhibited integrin-dependent cell spreading. Deletion of the K2 C terminus did not affect its binding to the integrin β3 cytoplasmic tail, but combined biochemical and NMR analyses indicated that it can insert into the F2 subdomain. We suggest that this insertion determines the topology of the K2 FERM domain, and its deletion may affect the positioning of the membrane-binding functions of the F2 subdomain and the integrin-binding properties of its F3 subdomain. Free C-terminal peptide can still bind to K2 and displace the endogenous K2 C terminus but may not restore the conformation needed for integrin co-activation. Our findings indicate that the extreme C terminus of K2 is essential for integrin co-activation and highlight the importance of an atypical architecture of the K2 FERM domain in regulating integrin activation.
Keywords: cell adhesion, integrin, membrane, protein chemistry, talin, kindlin
Introduction
The three human kindlins, kindlin-1 (K1),2 kindlin-2 (K2), and kindlin-3 (K3), are ∼50% identical in primary amino acid sequence (1, 2). They are FERM domain proteins composed of a unique N-terminal F0 subdomain that precedes the F1, F2, and F3 subdomains that typify FERM domain proteins (3–5). A distinctive feature of the FERM domains of kindlins is the presence of a PH domain that transects the F2 subdomain (1, 5–8).
The most extensively characterized function of kindlins relates to their ability to regulate the ligand binding and cellular responses mediated by integrins (1, 9–13). Integrins can transit between lower and higher affinity conformational states for their cognate ligands. Such transitions are particularly important on circulating blood cells, where such activation of integrins is necessary for rapid responses, such as thrombus formation and inflammatory cell recruitment. Integrin activation depends on the binding of talin, which also contains a FERM domain in its N-terminal region, talin-H, with extensive homology to the FERM domains of kindlins (2, 14). Kindlin and talin bind to adjacent NXX(Y/F) motifs in the cytoplasmic tails of integrins (13). Central to the integrin binding site in K2 are residues 614QW615 within its F3 subdomain. Mutation of these two residues to alanine greatly diminishes integrin binding activity of K2 and its capacity to cooperate with talin to induce integrin activation (9–11, 16). Nevertheless, optimal cooperation with talin-H to support co-activation of the platelet integrin αIIbβ3 requires each subdomain of kindlin-2 (17).
In addition to the integrin binding site in the F3 subdomain, other known binding partners of kindlins include an actin binding site in the F0 subdomain (18), an ILK binding site in the F2 subdomain (19–21), and a clathrin and an integrin binding site in the F3 subdomain (9, 11, 22–24). The PH subdomain contains a charged surface with lipid binding properties that contribute to the membrane localization of the kindlins (25, 26). Lipid binding properties are also associated with the F0 and F1 subdomains of kindlins (27, 28). The aforementioned binding sites have been localized primarily in studies of K2, but many are likely to be conserved in the other two kindlin family members.
In the present study, we identify a small segment of K2 that was previously unknown to be involved in its co-activator function. This segment resides at the extreme C terminus of K2 and is absent in canonical FERM domain proteins, including talin. Deletion of as few as the last seven amino acids from K2, truncation at residue 673 of the 680-amino acid K2, resulted in a complete loss of co-activator function. Indeed, deletion of the last two amino acids was sufficient to cause a 50% decline in its capacity to cooperate with talin. The deletion/mutation of the C-terminal segment did not affect the ability of K2 to bind to the integrin β3 subunit but rather interacted intramolecularly with the F2 subdomain within K2 itself to control its co-activator activity.
Results
The K2 C-terminal region as a regulator of integrin αIIbβ3 co-activation
The kindlins are evolutionarily conserved adapter proteins with at least one kindlin predicted to exist in all sequenced metazoan genomes (5, 29–31). A BLAST search (www.ncbi.nlm.nih.gov/blast/) was performed to consider the conservation of the 660–680 sequence of K2 across species. The C-terminal sequence for the top 100 matches was 100%, even in species distant from humans, such as Gekko japonicus or Atlantic salmon. In a lower organism, Drosophila melanogaster fermitin 1, transcript variant A (Fit1), the K2 homolog was still 69% identical and 89% conserved compared with the human K2 C-terminal sequence (Fig. 1A). The sequence is absent in canonical FERM domains, including talin-H. Speculating that such selective conservation might be indicative of a unique function of this segment, we constructed a K2 truncation mutant terminating at residue 660, K2Δ660. This mutant as well as several other C terminus truncation and point mutations and full-length (FL) K2 were expressed as enhanced green fluorescent protein (EGFP) constructs together with DS-Red talin-H in αIIbβ3-CHO cells. The CHO cell system has been widely used in integrin studies (32, 32–34), including the demonstration of the co-activator activity of K2 + talin-H (10, 35–37). It has the advantage of providing a facile means to quantify integrin αIIbβ3 activation based upon binding of the activation-specific mAb PAC-1 in a cell that can readily express several vectors without changing its surface expression of integrin αIIbβ3. The dot plots and resulting histograms from the flow cytometric analyses for selected K2 mutants are shown in supplemental Fig. S1, and the mean fluorescence intensities (MFI) from the histograms were used to calculate activation indices of each K2 form tested (Fig. 1B). As reported previously (9, 10), talin-H alone induces significant activation of αIIbβ3. FL K2 alone induces only minimal activation, but when talin-H and K2 are co-transfected, a significant increase in integrin activation well above that induced by talin-H alone was observed (Fig. 1B). This synergism, the co-activator activity of talin-H + K2, was not observed with K2 truncated at residue 660; the activation of cells transfected with the K2Δ660 vector was similar to that observed with talin-H alone (Fig. 1B). As shown in Fig. 1, K2Δ666, K2Δ671, and K2Δ673 exhibited no co-activator activity; expression of these constructs did not affect the activity of talin-H alone. Truncation of the last two amino acids, K2Δ679, led to a ∼50% loss in the co-activator activity calculated as integrin activation index of ((talin-H + K2Δ679) − (talin-H alone)/(talin-H + FL K2) − talin-H alone).
Figure 1.

Conserved residues in the C-terminal region of kindlin-2 support αIIbβ3 activation in αIIbβ3-CHO cells. A, alignment of C-terminal sequences of human K2 and Fit1, the K2 homolog of D. melanogaster. B and C, αIIbβ3-CHO cells were transiently transfected with plasmids encoding EGFP-fused FL, wild-type K2 or its mutants and DsRed talin-H, and activation of αIIbβ3 integrin was determined at 24 h by flow cytometry by staining the cells with the activation-specific antibody PAC-1 (see “Experimental procedures”). Error bars, S.D. Intermediate flow cytometry data (dot plots and histograms) are shown in supplemental Fig. S1.
We next focused on Tyr-673 for a potential involvement in co-activator activity. When this residue was mutated to Phe to preclude most post-translational modifications, such as phosphorylation, the K2Y673F mutant retained co-activator activity (not shown). However, K2Y673A reduced co-activator activity by ∼50% (Fig. 1C and supplemental Fig. S1). We then combined the two sets of mutations that each partially inhibited co-activator activity, K2Y673A and K2Δ679 (referred to subsequently as the double mutant); this combination failed to support co-activator activity of K2 (Fig. 1C). Thus, as little as three residue changes, a two-amino acid truncation and a point mutation, within the C-terminal region of K2 was sufficient to ablate its ability to co-activate integrin αIIbβ3 in CHO cells. With all of the K2 mutants shown in Fig. 1 (B and C) and supplemental Fig. S1, αIIbβ3 expression differed by <4%.
Influence of the K2 C-terminal region on αIIbβ3 function in other cells and on other integrins
To assess the importance of the K2 C-terminal segment in αIIbβ3 integrin regulation in cells other than CHO cells, we tested its function in HEL erythroleukemic cells. Like megakaryocytes and platelets, these cells express αIIbβ3 endogenously (38). They do not adhere spontaneously to fibrinogen, an αIIbβ3 ligand, but, when stimulated with phorbol 12-myristate 13-acetate (PMA), they bind PAC-1 and adhere and spread on fibrinogen via their αIIbβ3 (blocked by EDTA, RGD peptides, and mAbs) (39). We have also recently shown that when these cells are transfected with K3, they enhance their adhesion to fibrinogen and binding of soluble fibrinogen (40). As shown in Fig. 2A, when HEL cells were transfected with talin-H alone, their binding of PAC-1 increased significantly. (The dot plots and histograms for the data of key mutants in Fig. 2 are shown in supplemental Fig. S2A.) Co-transfection with K2 and talin-H enhanced their capacity to bind PAC-1 significantly compared with talin-H alone, although the co-activation was not as extensive as in αIIbβ3-CHO cells. With truncations of the C terminus of K2Δ660 or K2Δ666, co-activation was lost, whereas the K2Δ673 partially lost co-activator activity. The K2Y673A also partially lost co-activator activity, and the double mutant was similar to talin-H alone. In short, although the co-activator activity of K2 + talin-H was not as great in HEL cells as in αIIbβ3-CHO cells, the behavior of the deletion mutants in HEL cells was very similar to that exerted in αIIbβ3-CHO cells. When HEL cells are stimulated with PMA, they also increase their adhesion and spreading on fibrinogen (40). The stimulus-dependent spreading of these cells is supported by endogenous K3, as demonstrated by siRNA knockdown experiments (40), and was not affected by expressing K2 in these cells (Fig. 2B). Expression of K2Δ666 in HEL cells did, however, reduce spreading, and the inhibition was almost as extensive as observed with the integrin binding–defective mutant, K2Q614A/W615A (9).
Figure 2.

The kindlin-2 C-terminal segment supports integrin function in HEL megakaryotic and RAW 264.7 macrophage-like cells. A, HEL cells were transiently transfected with plasmids encoding EGFP-fused K2 or its mutants and DsRed talin-H, and the extent of αIIbβ3 activation in DsRed and EGFP double-positive cells was quantified by flow cytometry with the activation-specific antibody PAC-1 (see “Experimental procedures”). The experiments were performed three times. Error bars, S.D. The total αIIbβ3 expression, measured with an mAb unaffected by the activation status of the receptor, in the presence and absence of the various K2s with or without talin-H was unaffected. The expression levels of the K2 mutants were similar. B, HEL cells were transiently transfected with plasmids encoding EGFP alone and EGFP-K2 constructs. After 24 h, EGFP expression levels in HEL cells were determined by flow cytometry; transfection efficiency was 70–80%. The transfected HEL cells were treated with PMA and allowed to adhere to fibrinogen-coated coverslips, and cell spreading was measured after 30 min. The adherent cells were fixed and stained with Alexa 568 phalloidin, and the areas of cells were measured using ImageJ software; 300 cells were quantified per construct. Error bars, S.D. (p < 0.001). C, mouse RAW 264.7 cells were transiently transfected with plasmids encoding EGFP alone or EGFP-K2 constructs. The transfected cells were stained with 9EG7 monoclonal antibody to assess β1 integrin activation. Flow cytometry was used to measure 9EG7 binding. Error bars, S.E. of three independent experiments (p < 0.001). Intermediate flow cytometry data (dot plots and histograms) are shown in supplemental Fig. S2.
The importance of the C-terminal region was also noted with another cell line and with a different target integrin. K2 overexpression in RAW 264.7 macrophage-like cells leads to activation of β1 integrins as monitored with 9EG7, an mAb specific for the activated conformation of these integrins (41). As originally reported by Moser et al. (22) for kindlin-3/β1 integrin, we found that β1 integrin activation by K2 did not require expression of exogenous talin in these cells. The intermediate histograms are shown in supplemental Fig. S2B and quantified in Fig. 2C. Expression of K2 enhanced 9EG7 binding to these cells, but K2Δ666 had a much lower effect (Fig. 2C). The increase in 9EG7 binding by the K2Δ666 mutant was only 50% greater than the EGFP control compared with the 400% increase induced by full-length K2.
Role of the C terminus of other kindlins in integrin activation
The K1 C-terminal segment displays extensive similarity to the K2 C-terminal sequence (Fig. 3A), 60% identity and 80% conservation over the last 20 residues. To address the role of the K1 C-terminal aspect, the αIIbβ3-CHO cells were again used. In these cells, co-transfection of EGFP–K1 and Ds-Red talin-H led to enhanced activation of αIIbβ3 compared with talin-H alone (Fig. 3B) with the intermediate dot plots and histograms shown in supplemental Fig. S3. This co-activator activity of K1 was greatly diminished (80%) with a construct encoding of K1 lacking the last 8 amino acids, K1Δ670, although it remained greater than talin-H alone. Deletion of the last two amino acids of K1 (676QD677) and mutation of H670A, corresponding to Y673A in K2, each led to partial reduction in co-activator activity compared with full-length K1. In short, C-terminal mutations in K1 had effects similar to those observed in K2.
Figure 3.

The C-terminal segment of kindlin-1 is necessary to maintain the co-activator function of K1 in talin-H mediated integrin activation in αIIbβ3-CHO cells. A, alignment of C-terminal sequences of human K1 and human K2. B, αIIbβ3-CHO cells were transiently transfected with plasmids encoding EGFP-fused K1, wild type or its mutants, and DsRed talin-H. After 24 h, activation of the integrin was quantified by flow cytometry as in Fig. 1. Error bars, S.D. Intermediate flow cytometry data (dot plots and histograms) are shown in supplemental Fig. S3.
The K3 C-terminal region shows some conservation of sequence compared with K2, 40% identity and 50% conservative substitutions (Fig. 4A). However, K3 does not exhibit co-activator activity in αIIbβ3-CHO (Fig. 4B), confirming the absence of co-activator activity of FL K3 in the αIIbβ3-CHO as reported previously by us (40) and other laboratories (37, 42). To compare the role of the C-terminal end of K3 in integrin activation, we used two approaches. First, we created a chimeric K3 expression vector in which the C-terminal 20 residues of K3 were replaced with the C-terminal 20 residues of K2. When co-transfected with talin-H, no co-activation by the chimeric kindlin was observed (Fig. 4B). The dot plots and histograms for Fig. 4B are shown in supplemental Fig. S4. The EGFP fluorescence of cells transfected with the chimera, FL K3, and FL K2 were similar, indicating that all constructs were expressed at similar levels. Second, HEL cells were transfected with FL K3 and with a K3 truncated of its last 20 amino acids. When HEL cells were transfected with truncated K3 and stimulated with PMA, their spreading on fibrinogen was not affected; it was comparable with that of EGFP-transfected cells (data not shown).
Figure 4.

The C-terminal segment of kindlin-3 is dispensable for K3 functions. A, alignment of C-terminal sequences of human K2 and K3; B, αIIbβ3 integrin co-activation analysis using αIIbβ3-CHO cells transfected with EGFP-fused wild-type K3 or K3 chimera and talin-H; the activation of the integrin was determined after 24 h by flow cytometry with the activation-specific antibody PAC-1 (see “Experimental procedures”). Error bars, S.D. Intermediate flow cytometry data (dot plots and histograms) are shown in supplemental Fig. S4.
Mechanistic studies
If the K2 C terminus were to regulate binding of the β3 CT, it would control integrin activation. To address possible influences of the K2 C terminus in integrin binding, pull-down experiments were performed. FL K2 and K2Δ666 were transfected as EGFP constructs in αIIbβ3-CHO cells, and interaction of FL K2 and K2Δ666 with β3 was assessed by co-immunoprecipitation. The eluate from GFP-nAb beads was analyzed by Western blotting with anti-β3 and anti-EGFP. The results shown in Fig. 5A (top left) indicate that the K2 Δ666C-terminal deletion did not reduce the capacity of K2 to interact with the β3 CT. Densitometric scanning of the gels from three independent experiments showed that the K2Δ666 pulled down 0.97 ± 0.07 as much β3 CT as FL K2 (assigned a value of 1.0). The bottom panel in Fig. 5A shows that similar amounts of FL K2 and K2Δ666 were loaded on the gels as detected with an anti-EGFP. Thus, the C-terminal deletion did not prevent co-activation by precluding interaction of K2 with the integrin cytoplasmic tail. In a recent publication (20), we localized an ILK binding site to K2(339–358). K2Δ666 and the K2 double mutant retained their capacities to immunoprecipitate ILK (Fig. 5B). The bottom panel demonstrates that similar amounts of GST-tagged proteins were subjected to immunoprecipitation.
Figure 5.

Effects of the C-terminal segment of kindlin-2 on its association with known binding partners. A, GFP-fused FL K2 and K2Δ666 were transiently expressed in αIIbβ3-CHO cells. After 24 h, lysates of these cells were immunoprecipitated with GFP-nAb. The association of β3 subunit with the K2 forms was assessed by gel electrophoresis followed by Western blots with an anti-β3. The bottom panel shows an equal amount of EGFP-tagged K2 in immunoprecipitates. β3 levels in total lysates (TL) are also shown, with actin used as a loading control. B, GST-fused K2, K2Δ666, K2-double mutant, and GST alone were used in pulldown assays to precipitate ILK from αIIbβ3-CHO cell lysates. The binding of ILK to K2 variants was detected by Western blotting using anti-ILK antibodies. The bottom panel shows Coomassie Blue–stained GST and GST–K2 constructs used for pulldown assays.
Although less K2 double mutant was present, it still immunoprecipitated a similar amount of ILK. K2Δ666 also retained the capacity to pull down actin, which binds to K2 via a site in F0 (not shown) (18). Thus, the C-terminal deletion did not lead to global loss of K2 binding functions.
A vector for PSGL-1 was modified to replace its natural intracellular region with several K2 C-terminal extensions. We had previously used this approach to express various β3 CT mutants (10, 36); these β3 CT mutants that bound to talin-H or K2 competed with αIIbβ3 in αIIbβ3-CHO cells and inhibited spreading of the cells on fibrinogen, whereas mutants that did not bind either of the co-activators failed to prevent cell spreading. In the present set of experiments, the PSGL tag serves to anchor the selected K2 C-terminal peptides to the membrane, and expression of the constructs could be detected with KPL-1, an mAb to the extracellular domain of PSGL-1 (43). In the current experiment, the PSGL-1 constructs with sequences corresponding to K2(666–680), K2(666–678), K2(666–680) with an Y673A substitution, and K2(666–678) with the Y673A substitution (the double mutant) were expressed in αIIbβ3-CHO cells. PSGL-1 with no K2 sequence and PSGL-1 with the β3 CT at its C terminus served as negative and positive controls, respectively. Twenty-four hours after transfection, some cells were lysed and subjected to Western blot analysis. With respect to expression of the constructs, Western blots with anti-PSGL of cell lysates indicated that all four constructs were expressed, although the expression of PSGL-K2Y673A was somewhat lower (Fig. 6A). Some of the cells were spread on fibrinogen, and spreading was quantified after 2 h. Representative immunofluorescent microscopic images of the cells are shown in Fig. 6B. Clear differences in the size of the cells are evident. Cells expressing the PSGL-1–K2CT were smaller than the PSGL-1 (monomer) construct and comparable in size with the PSGL-1–β3CT construct. By only quantifying spreading of cells strongly expressing PSGL-1, effects of differences in transfection efficiencies with the constructs were minimized. The results in Fig. 6C show that K2 C-terminal peptide inhibited cell spreading significantly. The inhibition was less extensive than with the β3 CT but still quite marked, ∼50% inhibition. The ΔWV680 K2 mutant and the K2Y673A were less inhibitory than the K2(666–680) peptide but still produced significant (p < 0.001) inhibition compared with the PSGL-1 control, and the incremental increase in the series of K2CT > K2CTY673A > K2CTΔ679 > K2CT double mutant was significantly different (p = 0.001). However, the double mutant was not significantly different from the PSGL-1 control (p = 0.2). The results shown in Fig. 6C are from one experiment with each construct, measuring the area of >80 cells, and are representative of results from three independent experiments.
Figure 6.

The kindlin-2 C-terminal peptide is sufficient to influence cell spreading. αIIbβ3-CHO cells were transiently transfected with plasmids encoding PSGL-1 alone, PSGL-1–β3 (β3 CT-containing chimera as a positive control), and K2CT chimeras: PSGL-1–K2CT, PSGL-1–K2CTY673A, PSGL-1–K2CTΔ679, and PSGL-1–K2CT double mutant. A, the expression of PSGL-1 constructs was assessed by gel electrophoresis followed by Western blotting with anti-PSGL-1 antibodies. The bottom panel shows actin as an internal loading control. B, the cells were allowed to adhere to fibrinogen-coated coverslips 12 h after transfection, and cell spreading was measured by confocal microscopy after an additional 2 h. The adherent cells were fixed and stained with the anti-PSGL-1 mAb, KPL-1 followed by Alexa Fluor 488 (green, left) for visualization of the expressing cells by confocal microscopy, and Alexa Fluor 568-phalloidin for actin visualization (red, middle), The merged images are shown on the right. Bar, 20 μm. C, the areas of PSGL-1–positive cells were measured using ImageJ software, and 100 cells were quantified. The graph is representative of three independent experiments. Error bars, S.E.
The cells expressing the PSLG-1–K2CT constructs (Fig. 6) were lysed, immunoprecipitated with anti-PSGL-1 monoclonal antibody KPL-1, and then Western blotted with either anti-K2 or anti-PSGL-1 polyclonal antibodies. Remarkably, the PSGL-1 construct fused to the full-length K2CT contained FL K2 (Fig. 7, top); the shorter C-terminal constructs, PSGL-1-K2CTΔ679 and PSGL-1-K2CT double mutant contained lesser or negligible amounts of FL K2 (Fig. 7, top). This impression was verified by densitometric scanning of the gels. The intensity of the FL K2 band immunoprecipitating with PSGL-1-K2Δ679 was 6.5% of that associated with the PSGL-1-K2CT band, and no FL K2 bands were detected with the PSGL-1 double mutant or PSGL-1 alone. Anti-PSGL-1 antibodies immunoprecipitated comparable amounts of PSGL-1 constructs from cell lysate (Fig. 7) Furthermore, PSGL-1 antibody blotted all constructs as expected, and PSGL-1, K2, and actin (loading control) were present in similar amounts in total cell lysates (Fig. 7, TL panels). These results suggest that the C-terminal K2 peptides might interact with K2 itself. Analysis of a previously proposed computational model of full-length K2 (44) indeed suggested that the C terminus interacts with the F2 subdomain, which encompasses the PH domain of K2. Such an intramolecular interaction would explain the capacity of the PSGL-K2CT peptide to immunoprecipitate intact K2, where the peptide competes with intramolecular interaction in intact K2. To gain direct experimental evidence of this intramolecular interaction, we performed an NMR-based HSQC experiment with 15N-labeled (labeled Leu675) K2 C-terminal peptide (residues 666–680) in the absence or presence of unlabeled K2 F2. Fig. 8A shows that the K2 peptide does indeed specifically interact with F2, causing a shift in the signal obtained from the labeled Leu675 residue. As a control, a peptide from RIAM (45), which had two 15N-labeled residues, Phe12 and Leu22, did not undergo a chemical shift change when K2 F2 was added (Fig. 8B), indicating that the K2 C terminus peptide interaction with K2 F2 is highly specific.
Figure 7.

Kindlin-2 interacts with K2 C-terminal peptide. Lysates of αIIbβ3-CHO cells transfected with PSGL-1 alone, PSGL-1–K2CT, PSGL-1–K2CTΔ679, or PSGL-1–K2CT double mutant were used for co-IP assays. After incubating with A/G-agarose and PSGL-1 antibody, full-length K2 bound to PSGL-1 constructs was evaluated by SDS-PAGE and Western blotting using anti-K2 (Cell Signaling), which does not detect the C-terminal end of K2. PSGL-1 and K2 levels in total lysates (TL) are also shown, with actin used as a loading control.
Figure 8.
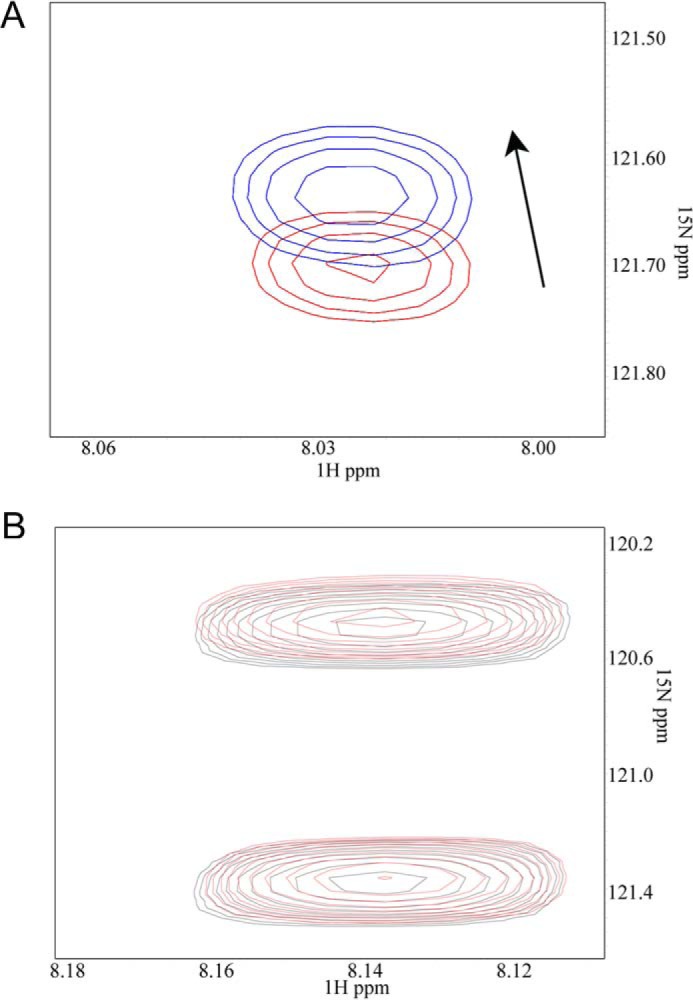
The kindlin-2 F3 C-terminal region has intramolecular interaction with the F2 subdomain of K2. A, 2D 1H-15N HSQC spectra of 0.05 mm 15N-labeled Leu675 of the peptide Ser666–Val680 in the absence (red) and presence (blue) of 0.25 mm kindlin-2 F2, providing experimental evidence that Ser666–Val680 interacts with the F2 subdomain. Note that the binding between isolated peptide and F2 is relatively weak yet detectable by this sensitive NMR technique (15), but the binding is expected to be stronger in intact K2. B, control HSQC spectra of 0.05 mm 15N-labeled Phe12 and Leu22 of the RIAM peptide (7DIDQMFSTLLGEMDLLTQSLGVDT30) in the absence (black) and presence (red) of 0.25 mm kindlin-2 F2, showing that K2 F2 does not induce chemical shifts.
Discussion
In this paper, we have identified a previously unrecognized site in K2 that is essential for its role in integrin activation. This site is contained within the C-terminal aspects of K2. Deletion of as few as seven residues from the K2 C terminus ablated the capacity of the entire N-terminal region to support integrin co-activation. Previous studies have implicated every subdomain of K2 in its co-activator function (17), but our study provides critical new insight to show that an atypical FERM domain topology is necessary for K2 to activate integrin (for further discussion, see below). The reduction in K2 co-activator function associated with deletion of the C terminus is as dramatic as deletion of any of its individual subdomains, including mutation of the integrin binding site in the F3 subdomain (17). The requirement for an intact C terminus was not only observed with αIIbβ3-CHO cells but also in HEL cells, which express their αIIbβ3 endogenously.
The importance of the C terminus in integrin-activating function is not restricted to K2 but also extends to K1. Truncation of the corresponding positions in K2 blunted the integrin-activating functions of K1 in a system, αIIbβ3-CHO cells, where K1 and K2 can be directly compared. Although the residue corresponding to K2 Tyr673 is K1 His676, the single point mutation of these corresponding residues led to a significant reduction in co-activator activity. Deletion of the last two C-terminal residues of K1 also led to a significant reduction of its co-activator activity. The dependence on the C terminus did not extend to K3. It had been previously shown that differential localization of K2 and K3 in focal adhesion was not dependent on the K3 C terminus (31). In the present study, when we replaced the K3 C terminus with the K2 C terminus, the chimera did not acquire co-activator activity. This observation does not mean that the C terminus of K3 is not important for integrin activation, but this region might react selectively with the F2 subdomain of K3. Nevertheless, the observation adds to several examples that indicate that, despite their similarity in sequence and their role in integrin activation, there are basic functional distinctions between K2 and K3 (5, 13, 46).
In our experiments using the PSGL-1–K2 peptide chimera expressed in CHO cells, the suppression of cell spreading by the various K2 chimera tested paralleled their loss of co-activator activity in αIIbβ3-CHO cells, with the K2 CTΔ679 and the K2Y673A being less suppressive than the K2 CT peptide but still retaining some suppressive activity. None of the PSGL-1–K2 chimera were as suppressive as the PSGL-1–β3CT fusion, which inhibits K2 as well as talin binding to the integrin (10). This difference may be due to the more modest affinity of binding of PSGL-1–CT peptides for the F2 subdomain of intact K2 (Fig. 8) as compared with the more potent binding of β3 CT binding to K2 or talin.
In considering the mechanism by which the K2 C terminus regulates integrin activation, one obvious possibility was that this segment regulated binding of K2 to the integrin β cytoplasmic tail. However, the binding strengths of K2Δ666 and FL K2 to β3CT were very similar. A second possible role for of the K2 C terminus was its interaction with a binding partner, although no binding partner for this segment was known. Two approaches were used to search for such a partner. First, selective binding of a protein in CHO cell extracts to FL K2 versus K2Δ666, was explored. However, when eluates from the affinity beads were analyzed on SDS-PAGE, no selective and consistent enrichment of a Coomassie Blue staining band from the FL K2 eluate was noted. Second, αIIbβ3-CHO cells expressing PSGL-1–K2 constructs were immunoprecipitated with anti-PSGL-1 antibodies. Western blots identified K2 in the immunoprecipitate of PSGL-1–K2CT chimera, but not in the immunoprecipitate of PSGL-1 alone. This was surprising and led us to consider an intermolecular interaction between the C terminus and K2.
To consider how the K2 C terminus might interact with K2, we examined a structural model of K2 (44). This model was generated by submitting full-length K2 (residues 1–680) to the structure prediction server ROBETTA (47). The lowest-energy model of K2 predicted that the K2 C terminus contains a short helix (Ser666–Val680) that inserts into a groove between the folded C-terminal half of F2 (F2-C) and the PH domain and thus becomes an integral part of the K2 F2–F3 configuration. Deletion of the C terminus may not unfold the individual subdomains in K2 or dramatically alter the overall fold of K2, but it could alter/destabilize the F2–F3 configuration, especially the juxtapositioning of F2 and F3, causing a defect in the receptor activation. Such destabilization does not lead to a gross collapse of the tertiary structure of K2; the integrin binding site in the F3 subdomain, the ILK binding site in in the N-terminal segment of F2 (Fig. 5), and the actin binding site in F0 (not shown) remained functional. One may argue that the C terminus of K2 might insert into a second K2 molecule to form a K2 dimer and that such multimerization is necessary for co-activation. Militating against this possibility is the lack of evidence for a tendency of K2 to dimerize or multimerize during its isolation by gel filtration (18). Also, ultracentrifugation experiments provided no evidence of dimerization (48).
The K2 C terminus, when expressed as a PSGL-1 chimera, suppressed integrin-mediated cell spreading; PSGL-1–tagged K2 C terminus may compete with the internal C terminus to disrupt the F2–F3 configuration within intact K2. The competition would not only displace the C terminus but also perturb the positioning of F2–F3 (e.g. the relative orientation of F3 containing the C terminus versus F2 may be affected upon binding of an external C-terminal peptide (Fig. 8) that is necessary for the co-activator function of K2. The suppression of cell spreading by the various K2 chimeras tested closely paralleled their loss of co-activator activity in αIIbβ3-CHO cells when deleted or mutated in K2, with the K2Δ679 and the K2Y673A being less suppressive than the FL K2 but still retaining some suppressive activity. None of the PSGL-1–K2 chimeras were as suppressive as the PSGL-1–β3CT fusion, which inhibits K2 as well as talin binding to the integrin (10). This difference may be due to the more modest affinity of binding of the PSGL-1–tagged C terminus for the F2 subdomain of intact K2 (Fig. 8) as compared with the potent binding of β3 CT binding to K2 or talin. The free K2 C-terminal peptide may be capable of displacing the intramolecular association of the region into F2, but this displacement may inhibit the function of K2, and replacement with free K2 C-terminal peptide may not support or restore the conformation of K2 necessary for its co-activator activity. Although we favor this insertional model, we cannot exclude the possibility that an as yet unidentified binding partner might interact with the C-terminal region. Implicit in our findings is the conclusion that binding of K2 to the integrin β subunit is not in itself sufficient for integrin activation; an additional downstream event is required. Defining this downstream event could provide new insights into the integrin activation process. The conclusion that the appropriate alignment of lipid binding and integrin binding is necessary for optimal integrin activation also applies to talin (49, 50) and is now extended to kindlins, although talin and other FERM domain proteins lack this C-terminal extension.
Experimental procedures
Reagents
The following primary antibodies were used: PAC1, which reacts with the activated but not the resting conformation of integrin αIIbβ3 (51) (BD Biosciences); 9EG7, which reacts selectively with the activated conformer of β1 integrins (41); mouse anti-EGFP (Takara Clontech, Mountain View, CA); mouse anti-GFP (clone B2) (Santa Cruz Biotechnology, Inc., Dallas, TX); mouse anti-β1 integrin (BD Transduction Laboratories); mouse anti-human CD41b (BD Biosciences); mouse anti-PSGL-1; clone KPL-1 (EMD Millipore, Temecula, CA); rabbit anti-integrin β3 integrin (Cell Signaling); mouse anti-GST (EMD Millipore); rabbit anti-PSGL-1(Santa Cruz Biotechnology); rabbit anti-kindlin 2 (Cell Signaling); rabbit anti-actin (Cell Signaling); and rabbit anti-ILK (Cell Signaling). Secondary antibodies for Western blots were donkey anti-mouse HRP and goat anti-rabbit HRP (both from Santa Cruz Biotechnology). For confocal microscopy, detecting reagents were Alexa Fluor 568 goat anti-rabbit Ig, Alexa Fluor 488 goat anti-mouse Ig, Alexa Fluor 488 phalloidin, Alexa Fluor 633 phalloidin, and Alexa Fluor 568 phalloidin, all from Thermo Fisher Scientific (Waltham, MA). For flow cytometry, Alexa Fluor 647 goat anti-mouse IgM (Jackson Immunoresearch, West Grove, PA) and PE-goat anti-rat IgG (Thermo Fisher Scientific) were used to detect PAC1 and 9E7 binding, respectively. All other chemical reagents were analytical grade.
Protein expression and purification
GST-fused FL K2 and its mutants were expressed, purified, and quantified as described previously (35). Briefly, GST-fused K2 and mutants were expressed in E. coli BL21 cells (Stratagene, La Jolla, CA) and purified using glutathione-Sepharose 4B resin. The eluates from the resin were then subjected to gel filtration on a HiLoad Superdex200 10/300 column (GE Healthcare). The K2 eluted from the column as a major peak with an estimated molecular mass of ∼103,000 Da for GST–K2 and ∼72,000 for K2 (18). For some experiments, the GST tag was removed from K2 by factor Xa cleavage as described (36). The purified proteins were dialyzed against 50 mm Tris, pH 8.0, containing 150 mm NaCl, quantified using Bio-Rad protein assay kits, and assessed for homogeneity by SDS-PAGE and their elution from the Superdex columns (18). The hexahistidine-tagged human kindlin-2 F2 subdomain (residues 281–569) was bacterially expressed and purified as described previously (20).
Plasmid construction
For mammalian expression, the cDNAs encoding for FL K2, FL K1, or talin-H were inserted into pEGFP or pDsRed-monomer vectors, respectively. Mutations, either truncations or substitutions, were introduced into the kindlin expression vectors using QuikChange mutagenesis kits (Agilent Technologies, Santa Clara, CA). The full nucleotide sequences of all mutant kindlins were confirmed in the Genomics Core of the Cleveland Clinic. The PSGL-1–K2CT chimera was constructed in pcDNA3.1 vector, in which nucleotide sequences encoding various segments of the C terminus of K2 were fused onto N-terminal and transmembrane regions of human PSGL-1 as described previously (10, 36). All indicated mutations of PSGL-1 chimera were introduced using QuikChange site-directed mutagenesis kits (Agilent Technologies), and their authenticity was confirmed by DNA sequencing.
Cell culture and transfections
CHO-K1 cells and a CHO cell line stably expressing αIIbβ3 (αIIbβ3-CHO) were cultured in DMEM/F-12 medium with 10% fetal bovine serum at 37 °C in 5% CO2. The cells were passaged using 0.05% trypsin, 0.53 mm EDTA for dissociation at ∼80% confluence. For vector transfections of these CHO lines, Lipofectamine 2000 (Thermo Fisher Scientific) was used. HEL and RAW 264.7 were obtained from American Type Culture Collection (Manassas, VA). For transfections of these cell lines, nucleofection with kit V from Lonza was used with program X-005 (HEL) or D-032 (RAW 264.7). Specific functional assays were performed with these cells 24 h after transfection.
Pull-down assays, immunoprecipitation, and Western blotting
Pull-down assays were performed using GST fusion proteins. Equal amounts of GST-fused K2 and its mutants were added together with glutathione–Sepharose 4B (GE Healthcare) to aliquots of the lysates of αIIbβ3-CHO cells. After overnight incubation at 4 °C, the precipitates were washed and boiled in Laemmli sample buffer. The eluates were analyzed on gradient acrylamide gels under reducing conditions, and interactions of ILK with GST–K2 or its mutants were determined by Western blotting with anti-ILK antibody. The gels were also stained with Coomassie Blue to verify that sample loadings were similar. GFP co-IP assays were performed using GFP-nAb (Allele Biotechnology) according to the manufacturer's instructions. After 2 h at 4 °C, the precipitates from co-IP were collected by centrifugation, washed, and boiled in Laemmli sample buffer. The eluates were then analyzed on 4–20% gradient acrylamide gels under reducing conditions, and interactions of the β3 with EGFP, EGFP-K2, and EGFP-K2Δ666 were determined by Western blotting with rabbit anti-β3 antibody (Cell Signaling) and anti-EGFP. For antibody-mediated co-precipitation of K2 and PSGL-1 constructs, αIIbβ3-CHO cells were transfected with the indicated PSGL-1 constructs, and after 24 h, cells were collected, and samples were lysed in 50 mm Tris-HCl, pH 7.4, 150 mm sodium chloride, 1% Nonidet P-40, 1 mm calcium chloride, containing protease and phosphatase inhibitors. Lysates were held on ice for 30 min prior to centrifugation at 12,000 × g for 15 min. Aliquots of the detergent-soluble material were precleared on A/G protein–agarose for 1 h at 4 °C. Precleared lysates were incubated with 2 μl of anti-PSGL-1 antibody (KPL-1 clone) antibodies and protein A/G–agarose for 16 h at 4 °C. Immunoprecipitated proteins were solubilized in Laemmli buffer and analyzed on Western blots with PSGL-1 (Santa Cruz Biotechnology) and K2 antibodies.
Densitometry was performed using ImageJ software, and band intensities were quantified relative to the intensity of the PSGL-1–K2CT band.
Integrin activation assays
Integrin αIIbβ3 activation was evaluated using PAC-1 as described previously (10, 35). Talin-H domain tagged with DsRed monomer or EGFP-tagged K2 and its mutants were expressed in CHO cells stably expressing αIIbβ3 (αIIbβ3-CHO) by transient transfection using Lipofectamine 2000 (Life Technologies, Inc.). PAC-1 binding to the different transfectants (EGFP and DsRed double-positive cells) was analyzed by flow cytometry after incubating the transfected cells with 10 μg/ml anti-PAC1 mAb in HBSS buffer containing 0.1% BSA, 0.5 mm CaCl2, 0.5 mm MgCl2 for 30 min at room temperature followed by fixation with 1% PFA for 10 min at room temperature, washing, and incubation with 5 μg/ml Alexa Fluor 647–conjugated F(ab′)2 anti-mouse IgM secondary antibody for 30 min on ice. To consider whether expression of various constructs in the αIIbβ3-CHO cells affected surface expression levels of the integrin, we evaluated reactivity with an mAb (2G12) that reacts with αIIbβ3 independent of its activation state (52), and surface expression of the integrin varied by <4% among all transfectants. To assess activation of endogenous αIIbβ3, HEL cells were co-transfected with the EGFP K2 and DsRed-talin-H constructs using Nucleofector kit V (Lonza). Doubly transfected (EGFP-positive and DsRed-positive) cells were quantified after 24 h by adding PAC-1 (10 μg/ml) in HBSS buffer containing 0.1% BSA, 0.5 mm CaCl2, and 0.5 mm MgCl2 for 30 min at room temperature followed by fixation with 1% PFA for 10 min, washing, and incubation with 5 μg/ml Alexa Fluor 647–conjugated F(ab′)2 anti-mouse IgM secondary antibody for 30 min on ice. Integrin αIIbβ3 expression was assessed in parallel by staining with anti-CD41 antibody (BD Pharmingen). Surface expression of αIIbβ3 varied by <5% among all transfectants.
PAC-1 binding to αIIbβ3-CHO cells or HEL cells was analyzed using LSRFortessa flow cytometer and FlowJo software (BD Biosciences). MFI of PAC-1 binding were normalized based on the basal level of PAC-1 binding to cells transfected with the EGFP/DsRed vectors alone to obtain relative MFI (RFI) values (10, 40).
As a second assay to assess activation of αIIbβ3 on HEL cells, nontransfected HEL cells or HEL cells transfected with various K2 constructs were stimulated with 800 nm PMA for 5 min and then plated onto immobilized fibrinogen (20 μg/ml) in triplicate for 30 min at 37 °C in serum-free medium. After extensive washing with PBS, the adherent cells were fixed with 4% PFA and stained with Alexa Fluor 568 phalloidin to visualize actin within the adherent cells. The cells were photographed at ×40 on a Leica K microscope, and the adherent cells were counted in 20 randomly selected fields. Cell area was determined from measurements of 300 cells using ImageJ software (40).
Raw 264.7 cells, a murine macrophage-like cell line (53), were transfected with the indicated K2 constructs. 9EG7 binding to the different transfectants was analyzed by gating only on the EGFP-positive cells. MFI of 9EG7 binding were normalized based on the basal level of 9EG7 binding to cells transfected with the EGFP vector alone to obtain relative RFI. Cells (2 × 105) were incubated with 10 μg/ml 9EG7 in HBSS buffer containing 0.1% BSA, 0.5 mm CaCl2, 0.5 mm MgCl2. After incubation, cells were fixed with 1% PFA for 10 min in room temperature. After washing, cells were incubated with 10 μg/ml R-PE-labeled secondary antibody for 30 min at room temperature, washed, fixed with 1% PFA, and washed with PBS. Cells were resuspended in 400 μl of PBS for flow cytometric analysis.
NMR spectroscopy
A peptide corresponding to K2 Ser666–Val680 was synthesized at the Lerner Research Institute Biotechnology Core with [15N]leucine at position 675. To detect its binding to the K2 F2 subdomain, HSQC experiments were performed on 0.05 mm 15N-labeled peptide mixed with and without 0.25 mm unlabeled F2. The samples were prepared in 20 mm Tris, 150 mm NaCl, 2 mm DTT, pH 7.0, 10% D2O. All NMR experiments were performed on a Bruker Advance 600-MHz instrument at 25 °C, and the data were processed as described (36, 54).
Cell spreading assays
To test the function of the K2 C-terminal peptides in cells, a series of chimeric PSGL-1–K2 constructs were expressed in αIIbβ3-CHO cells, and their effects on cell spreading were assessed. The chimera used the extracellular and transmembrane domains of PSGL-1 (mPSGL-1) and anchors and fused these to selected segments of the C-terminal aspects of K2. This approach, described previously (10, 36), allows expression of functional segments of K2 under investigation while tethered to PSGL-1, which allows for monitoring of expression levels with an mAb to the extracellular domain of PSGL-1. The transiently transfected cells were allowed to adhere and spread on fibrinogen-coated (20 μg/ml) glass coverslips. After incubation at 37 °C for 2 h, the wells were washed three times with PBS, and the adherent cells were fixed with 4% PFA and stained with Alexa Fluor 647 phalloidin (Invitrogen). To identify PSGL-1–expressing cells, the fixed cells were stained by anti-PSGL-1 mAb, KPL-1 (BD Biosciences), followed by goat anti-mouse IgG conjugated with Alexa Fluor 488 (Invitrogen), which stains actin filaments and enhances the microscopic visualization of the cells. As controls, nontransfected cells were included in each experiment and always showed no PSGL-1 staining. The positively stained (green) cells were visualized with a ×40 oil immersion objective using a Leica SP1 laser-scanning confocal microscope (Imaging Core, Cleveland Clinic). Areas of the PSGL-positive cells were analyzed with ImageJ software. For quantitation, the areas of >80 cells from >10 random fields were measured.
Statistical analysis
Means and S.E. are reported. Statistical analyses were performed using two-tailed Student's t tests in SigmaPlot (version 11). Differences were considered to be significant with p < 0.05.
Author contributions
K. M. B, K. B., and J. H. performed experiments and edited the manuscript. J. L. performed NMR experiments and K. F. purified K2 F2 and edited the manuscript. J. Q. designed NMR experiments and wrote the paper and E. F. P. planned experiments and wrote the paper.
Supplementary Material
Acknowledgments
We thank the Imaging Core and the Biotechnology Core of the Lerner Research Institute for assistance. The Leica SP8 confocal microscope utilized in this work was purchased with funding from National Institutes of Health SIG Grant 1S10OD019972-01.
This work was supported in part by National Institutes of Health Grants R01 HL096062, PO1 HL073311, and P01 HL076491. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S4.
- K1
- kindlin-1
- K2
- kindlin-2
- K3
- kindlin-3
- EGFP
- enhanced green fluorescent protein
- FERM
- 4.1R-ezrin-radixin-moesin
- HEL
- human erythroleukemia
- ILK
- integrin-linked kinase
- PMA
- phorbol 12-myristate 13-acetate
- PSGL-1
- P-selectin glycoprotein ligand-1
- talin-H
- head domain of talin
- PE
- phycoerythrin
- nAb
- nano antibody
- PH
- pleckstrin homology
- MFI
- mean fluorescence intensities
- RFI
- relative MFI
- FL
- full-length
- IP
- immunoprecipitation
- HBSS
- Hanks' balanced salt solution
- PFA
- paraformaldehyde
- HSQC
- heteronuclear single quantum coherence.
References
- 1. Larjava H., Plow E. F., and Wu C. (2008) Kindlins: essential regulators of integrin signalling and cell-matrix adhesion. EMBO Rep. 9, 1203–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Plow E. F., Qin J., and Byzova T. (2009) Kindling the flame of integrin activation and function with kindlins. Curr. Opin. Hematol. 16, 323–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ma Y. Q., Qin J., and Plow E. F. (2007) Platelet integrin α(IIb)β(3): activation mechanisms. J. Thromb. Haemost. 5, 1345–1352 [DOI] [PubMed] [Google Scholar]
- 4. Ye F., and Petrich B. G. (2011) Kindlin: helper, co-activator, or booster of talin in integrin activation? Curr. Opin. Hematol. 18, 356–360 [DOI] [PubMed] [Google Scholar]
- 5. Rognoni E., Ruppert R., and Fässler R. (2016) The kindlin family: functions, signaling properties and implications for human disease. J. Cell Sci. 129, 17–27 [DOI] [PubMed] [Google Scholar]
- 6. Moser M., Legate K. R., Zent R., and Fässler R. (2009) The tail of integrins, talin, and kindlins. Science 324, 895–899 [DOI] [PubMed] [Google Scholar]
- 7. Morse E. M., Brahme N. N., and Calderwood D. A. (2014) Integrin cytoplasmic tail interactions. Biochemistry 53, 810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Plow E. F., Das M., Bialkowska K., and Sossey-Alaoui K. (2016) Of kindlins and cancer. Discoveries 10.15190/d.2016.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shi X., Ma Y. Q., Tu Y., Chen K., Wu S., Fukuda K., Qin J., Plow E. F., and Wu C. (2007) The MIG-2/integrin interaction strengthens cell-matrix adhesion and modulates cell motility. J. Biol. Chem. 282, 20455–20466 [DOI] [PubMed] [Google Scholar]
- 10. Ma Y. Q., Qin J., Wu C., and Plow E. F. (2008) Kindlin-2 (Mig-2): a co-activator of β3 integrins. J. Cell Biol. 181, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Montanez E., Ussar S., Schifferer M., Bösl M., Zent R., Moser M., and Fässler R. (2008) Kindlin-2 controls bidirectional signaling of integrins. Genes Dev. 22, 1325–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meves A., Stremmle C., Gottschalk K., and Fässler R. (2009) The Kindlin protein family: new members to the club of focal adhesion proteins. Trends Cell Biol. 19, 504–513 [DOI] [PubMed] [Google Scholar]
- 13. Plow E. F., Meller J., and Byzova T. V. (2014) Integrin function in vascular biology: a view from 2013. Curr. Opin. Hematol. 21, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kloeker S., Major M. B., Calderwood D. A., Ginsberg M. H., Jones D. A., and Beckerle M. C. (2004) The Kindler syndrome protein is regulated by transforming growth factor-β and involved in integrin-mediated adhesion. J. Biol. Chem. 279, 6824–6833 [DOI] [PubMed] [Google Scholar]
- 15. Vaynberg J., Fukuda T., Chen K., Vinogradova O., Velyvis A., Tu Y., Ng L., Wu C., and Qin J. (2005) Structure of an ultraweak protein-protein complex and its crucial role in regulation of cell morphology and motility. Mol. Cell 17, 513–523 [DOI] [PubMed] [Google Scholar]
- 16. Xu Z., Chen X., Zhi H., Gao J., Bialkowska K., Byzova T. V., Pluskota E., White G. C. 2nd, Liu J., Plow E. F., and Ma Y. Q. (2014) Direct interaction of Kindlin-3 with integrin αIIbβ3 in platelets is required for supporting arterial thrombosis in mice. Arterioscler. Thromb. Vasc. Biol. 34, 1961–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu Z., Gao J., Hong J., and Ma Y. Q. (2013) Integrity of kindlin-2 FERM subdomains is required for supporting integrin activation. Biochem. Biophys. Res. Commun. 434, 382–387 [DOI] [PubMed] [Google Scholar]
- 18. Bledzka K., Bialkowska K., Sossey-Alaoui K., Vaynberg J., Pluskota E., Qin J., and Plow E. F. (2016) Kindlin-2 directly binds actin and regulates integrin outside-in signaling. J. Cell Biol. 213, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qadota H., Moerman D. G., and Benian G. M. (2012) A molecular mechanism for the requirement of PAT-4 (integrin-linked kinase (ILK)) for the localization of UNC-112 (Kindlin) to integrin adhesion sites. J. Biol. Chem. 287, 28537–28551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fukuda K., Bledzka K., Yang J., Perera H. D., Plow E. F., and Qin J. (2014) Molecular basis of kindlin-2 binding to integrin-linked kinase pseudokinase for regulating cell adhesion. J. Biol. Chem. 289, 28363–28375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huet-Calderwood C., Brahme N. N., Kumar N., Stiegler A. L., Raghavan S., Boggon T. J., and Calderwood D. A. (2014) Differences in binding to the ILK complex determines kindlin isoform adhesion localization and integrin activation. J. Cell Sci. 127, 4308–4321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moser M., Nieswandt B., Ussar S., Pozgajova M., and Fässler R. (2008) Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14, 325–330 [DOI] [PubMed] [Google Scholar]
- 23. Moser M., Bauer M., Schmid S., Ruppert R., Schmidt S., Sixt M., Wang H. V., Sperandio M., and Fässler R. (2009) Kindlin-3 is required for β2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 15, 300–305 [DOI] [PubMed] [Google Scholar]
- 24. Pluskota E., Ma Y., Bledzka K. M., Bialkowska K., Soloviev D. A., Szpak D., Podrez E. A., Fox P. L., Hazen S. L., Dowling J. J., Ma Y. Q., and Plow E. F. (2013) Kindlin-2 regulates hemostasis by controlling endothelial cell-surface expression of ADP/AMP catabolic enzymes via a clathrin-dependent mechanism. Blood 122, 2491–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu J., Fukuda K., Xu Z., Ma Y. Q., Hirbawi J., Mao X., Wu C., Plow E. F., and Qin J. (2011) Structural basis of phosphoinositide binding to Kindlin-2 pleckstrin homology domain in regulating integrin activation. J. Biol. Chem. 286, 43334–43342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qu H., Tu Y., Shi X., Larjava H., Saleem M. A., Shattil S. J., Fukuda K., Qin J., Kretzler M., and Wu C. (2011) Kindlin-2 regulates podocyte adhesion and fibronectin matrix deposition through interactions with phosphoinositides and integrins. J. Cell Sci. 124, 879–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bouaouina M., Goult B. T., Huet-Calderwood C., Bate N., Brahme N. N., Barsukov I. L., Critchley D. R., and Calderwood D. A. (2012) A conserved lipid-binding loop in the kindlin FERM F1 domain is required for kindlin-mediated αIIbβ3 integrin coactivation. J. Biol. Chem. 287, 6979–6990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perera H. D., Ma Y. Q., Yang J., Hirbawi J., Plow E. F., and Qin J. (2011) Membrane binding of the N-terminal ubiquitin-like domain of Kindlin-2 is crucial for its regulation of integrin activation. Structure 19, 1664–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Malinin N. L., Plow E. F., and Byzova T. V. (2010) Kindlins in FERM adhesion. Blood 115, 4011–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ali R. H., and Khan A. A. (2014) Tracing the evolution of FERM domain of Kindlins. Mol. Phylogenet. Evol. 80, 193–204 [DOI] [PubMed] [Google Scholar]
- 31. Meller J., Rogozin I. B., Poliakov E., Meller N., Bedanov-Pack M., Plow E. F., Qin J., Podrez E. A., and Byzova T. V. (2015) Emergence and subsequent functional specialization of kindlins during evolution of cell adhesiveness. Mol. Biol. Cell 26, 786–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ylänne J., Chen Y., O'Toole T. E., Loftus J. C., Takada Y., and Ginsberg M. H. (1993) Distinct functions of integrin α and β subunit cytoplasmic domains in cell spreading and formation of focal adhesions. J. Cell Biol. 122, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma Y. Q., Yang J., Pesho M. M., Vinogradova O., Qin J., and Plow E. F. (2006) Regulation of integrin α(IIb)β(3) activation by distinct regions of its cytoplasmic tails. Biochemistry 45, 6656–6662 [DOI] [PubMed] [Google Scholar]
- 34. Harburger D. S., Bouaouina M., and Calderwood D. A. (2009) Kindlin-1 and -2 directly bind the C-terminal region of β integrin cytoplasmic tails and exert integrin-specific activation effects. J. Biol. Chem. 284, 11485–11497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bledzka K., Bialkowska K., Nie H., Qin J., Byzova T., Wu C., Plow E. F., and Ma Y. Q. (2010) Tyrosine phosphorylation of integrin β3 regulates kindlin-2 binding and integrin activation. J. Biol. Chem. 285, 30370–30374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bledzka K., Liu J., Xu Z., Perera H. D., Yadav S. P., Bialkowska K., Qin J., Ma Y. Q., and Plow E. F. (2012) Spatial coordination of kindlin-2 with talin head domain in interaction with integrin β cytoplasmic tails. J. Biol. Chem. 287, 24585–24594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kasirer-Friede A., Kang J., Kahner B., Ye F., Ginsberg M. H., and Shattil S. J. (2014) ADAP interactions with talin and kindlin promote platelet integrin αIIbβ3 activation and stable fibrinogen binding. Blood 123, 3156–3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bray P. F., Rosa J.-P., Lingappa V. R., Kan Y. W., McEver R.-P., and Shuman M. A. (1986) Biogenesis of the platelet receptor for fibrinogen: evidence for separate precursors for glycoproteins IIb and IIIa. Proc. Natl. Acad. Sci. U.S.A. 83, 1480–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boudignon-Proudhon C., Patel P. M., and Parise L. V. (1996) Phorbol ester enhances integrin αIIbβ3-dependent adhesion of human erythroleukemic cells to activation-dependent monoclonal antibodies. Blood 87, 968–976 [PubMed] [Google Scholar]
- 40. Bialkowska K., Byzova T. V., and Plow E. F. (2015) Site-specific phosphorylation of kindlin-3 protein regulates its capacity to control cellular responses mediated by integrin αIIbβ3. J. Biol. Chem. 290, 6226–6242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lenter M., Uhlig H., Hamann A., Jenö P., Imhof B., and Vestweber D. (1993) A monoclonal antibody against an activation epitope on mouse integrin chain β1 blocks adhesion of lymphocytes to the endothelial integrin α6β1. Proc. Natl. Acad. Sci. U.S.A. 90, 9051–9055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kahner B. N., Kato H., Banno A., Ginsberg M. H., Shattil S. J., and Ye F. (2012) Kindlins, integrin activation and the regulation of talin recruitment to αIIbβ3. PLoS One 7, e34056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Snapp K. R., Ding H., Atkins K., Warnke R., Luscinskas F. W., and Kansas G. S. (1998) A novel P-selectin glycoprotein ligand-1 monoclonal antibody recognizes an epitope within the tyrosine sulfate motif of human PSGL-1 and blocks recognition of both P- and L-selectin. Blood 91, 154–164 [PubMed] [Google Scholar]
- 44. Perera H. D. D. (2010) Molecular Basis of the Role of Kindlin 2 in Cell Adhesion. Ph.D. dissertation, Cleveland State University [Google Scholar]
- 45. Yang J., Zhu L., Zhang H., Hirbawi J., Fukuda K., Dwivedi P., Liu J., Byzova T., Plow E. F., Wu J., and Qin J. (2014) Conformational activation of talin by RIAM triggers integrin-mediated cell adhesion. Nat. Commun. 5, 5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao Y., Malinin N. L., Meller J., Ma Y., West X. Z., Bledzka K., Qin J., Podrez E. A., and Byzova T. V. (2012) Regulation of cell adhesion and migration by Kindlin-3 cleavage by calpain. J. Biol. Chem. 287, 40012–40020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim D. E., Chivian D., and Baker D. (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yates L. A., Füzéry A. K., Bonet R., Campbell I. D., and Gilbert R. J. (2012) Biophysical analysis of Kindlin-3 reveals an elongated conformation and maps integrin binding to the membrane-distal β-subunit NPXY motif. J. Biol. Chem. 287, 37715–37731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Song X., Yang J., Hirbawi J., Ye S., Perera H. D., Goksoy E., Dwivedi P., Plow E. F., Zhang R., and Qin J. (2012) A novel membrane-dependent on/off switch mechanism of talin FERM domain at sites of cell adhesion. Cell Res. 22, 1533–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moore D. T., Nygren P., Jo H., Boesze-Battaglia K., Bennett J. S., and DeGrado W. F. (2012) Affinity of talin-1 for the β3-integrin cytosolic domain is modulated by its phospholipid bilayer environment. Proc. Natl. Acad. Sci. U.S.A. 109, 793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shattil S. J., Cunningham M., and Hoxie J. A. (1987) Detection of activated platelets in whole blood using activation-dependent monoclonal antibodies and flow cytometry. Blood 70, 307–315 [PubMed] [Google Scholar]
- 52. Woods V. L. Jr., Wolff L. E., and Keller D. M. (1986) Resting platelets contain a substantial centrally located pool of glycoprotein IIb-IIIa complex which may be accessible to some but not other extracellular proteins. J. Biol. Chem. 261, 15242–15251 [PubMed] [Google Scholar]
- 53. Raschke W. C., Baird S., Ralph P., and Nakoinz I. (1978) Functional macrophage cell lines transformed by Abelson leukemia virus. Cell 15, 261–267 [DOI] [PubMed] [Google Scholar]
- 54. Vinogradova O., Velyvis A., Velyviene A., Hu B., Haas T., Plow E., and Qin J. (2002) A structural mechanism of integrin αIIbβ3 “inside-out” activation as regulated by its cytoplasmic face. Cell 110, 587–597 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.